

1. Marine Natural Products as a Source of Chemical Diversity and Drug Leads
The marine environment is among the most diverse and prolific source of natural products. The unmatched chemical diversity of secondary metabolites from invertebrates (sponges, tunicates, nudibranchs, etc.) and marine microorganisms have led to the discovery of pharmacologically promising bioprobes and exciting drug candidates.i Presently, three marine derived compounds are approved for use as therapeutic agents; ziconotide (Prialt®, 1), a ‘cysteine knot’ peptide isolated by Olivera and coworkersii from the cone snail Conus magus with potent analgesic properties for neuropathic pain in patients who no longer tolerate morphine. Yondelis® (Trabectidin, ET-743, 2), a complex tetrahydroisoquinoline alkaloid reported by the Rinehart group in 1990 from the Caribbean tunicate Ecteinascidia turbinata,iii was approved in Europe in 2007 for soft tissue sarcoma. The polyether toxin, halichondrin B (3) reported by Uemura from a marine sponge Halichondria okadai,iv provided a scaffold for synthetic refinements by truncation to the newly licensed drug, eribulin mesylate (Halaven®, 4) a molecule half its size but retaining most of its potency. In late 2010, Halaven® was approved in the USA for treatment of solid tumors.
These three examples illustrate an underappreciated fact: the structure of therapeutic natural products with stereochemical complexity have been solved mostly with integrated chemical approaches requiring detailed, painstakingly acquired data, sometimes over the course of several years, often with limited sample size. Modern natural product discovery efforts are now focused on rare or scarce materials from niche sources. The available amounts of many of these ‘nanomole-scale’ natural products make their structures impossible to solve using historical approaches. The issues of procurement for biological evaluation and scale-up production are another matter (e.g. the commercially available Yondelis® is produced by ~14 steps from cyanosafracin B (5), a compound readily available in kg-scale by fermentation of Pseudomonas fluorescens.v
In natural product drug discovery, the structure is primal. The information content of a natural product– whether it be used for structure-activity relationships, or informing retrosynthetic design for procurement by total synthesis – is embodied in its 3D structure and revealed by structure elucidation. Finally, the holy grail of intellectual property is the New Chemical Entity (NCE) or New Active Substance (NAS)ib – a bioactive molecule representing an unprecedented structural type.
Although modern developments in spectroscopic techniques enable the identification of the structures of microgram-samples of complex small molecules, there is no general solution to the absolute configuration (AC) of a compound, those even containing only one or a few stereocenters; absolute stereostructures are solved on a case-by-case basis. The scope of this review will cover briefly the current state of instrumentation for the determination of RC and AC. Instrumental developments have been covered extensively in previous reviewsvi and mentioned here only in passing. Second, case studies will be presented where chemical approaches to structure elucidation have been integrated with spectroscopy to solve stereochemical problems. The strategy for configurational assignment is outlined in the first figure for each example. Several acronyms or abbreviations appear in these figures and throughout the remaining text: relative configuration, RC; absolute configuration, AC; modified Mosher method, MMM; J-based configurational analysis, JBCA; universal NMR database, UDB. The dual purposes of economy of pages and contemporary interest are served by restriction of the scope of the review to examples since about 2000; the time during which NMR technologies (mainly sensitivity improvements through cryoprobes) had undergone significant evoluation. The majority of the RC and AC assignments were completed only after the planar structure – a 2D structure lacking stereochemical assignments – had been proposed. In most cases, the natural product is a rare entity, generally procured from uncommon marine organisms instead of sustainable fermentation methods. It may be of interest to readers to be informed of the breadth of the technical challenges associated with the scale of the chemical operations and, so, the corresponding quantities are given in some of the Figures (most chemical transformations were carried out on the microscale, if not nanomole-scale). We have tried to select a variety of molecules of different structural types that often dictate a unique approach to a particular problem, but they do not necessarily constitute the ‘most difficult’ cases. The examples for this review subscribe to one of the following criteria: those that (a) push the limit to spectroscopic and chemical methods, (b) demonstrate applicability and limitations of new techniques, (c) resolve configurational assignment of particularly difficult or marine natural products with potent biological activity. Finally, we hope the selected case studies provide some inspiration to a readership with broad interests: natural products chemists and synthetic organic chemists with passionate interests in the art of structure determination and synthesis of marine natural products.
2. NMR Based Methods for Structure Determination and Stereochemical assignment
2.1. Current State of NMR
Nuclear magnetic resonance spectroscopy is still the most important tool for modern structure determination. Over the past 20 years, NMR-sensitivity has steadily increased to provide a refined tool to ‘view’ unexplored regimes of natural products. Around 1992, 500 MHz 3 mm inverse-detect probes were introduced reducing the sample volume from ~600 μL (5 mm tube) to ~140 μL and effectively doubling the S/N.vii The applicability of the 500 MHz 3 mm inverse detect probe was demonstrated by measurements of a sample of brevetoxin-C (800 μg, 0.95 μmole) and complete 1H and 13C assignments based on homo– and heteronuclear 2D experiments.viii A substantial advancement came with the advent of commercial 500 MHz 3 mm cryogenic probes that appeared around 2000 and showed an increase of S/N of ~3.5 times compared to a room temperature 500 MHz 3 mm probe.ix Current generation 1 mmx and 1.7 mm microcryoprobe NMR spectrometers allow the acquisition of conventional 2D NMR data with as little sample as a few nanomole. The least sensitive 2D H-C correlation experiment commonly used (gHMBC) can be acquired on 15 nmol (5.4 μg) of strychnine over a weekend (43 h).xi These and other contemporary developments in NMR are having profound effects on the conduct of natural products structure elucidation.
The complete structure elucidation of complex natural products on micrograms of material have been typically carried out with assistance of a parent compound. Pteriatoxins A–C (6–8)xii reported by Uemura and coworkers from the Okinawan bivalve Pteria penguin are derivatives of pinnatoxin A (9)xiii and the full structures (except the configuration of the sidechains) of 6–8 were established by homonuclear based (1H, COSY, and HOHAHA) experiments. Phorbaside F (10)xiv and hemi-phorboxazole (11)xv from the marine sponge Phorbas sp. are derivatives of phorbaside A (12)xvi and phorboxazole A (13),xvii respectively.
The structure determination of these two compounds were assisted by comparative analysis of NMR data of the corresponding parent compounds, but the structure of 10 also drew upon new data obtained using a 600 MHz 1.7 mm microcryoprobe NMR (decoupled and 1H-coupled HSQC and HMBC). These case studies illustrate the power of modern NMR with microgram samples, even acquisition of heteronuclear (1H, 13C) 2D NMR data, particularly those obtained from the least sensitive – but indispensible – pulse sequences (e.g. HMBC). Connectivity and planar structures are revealed from these data;xi the RC is solved from NOESY and ROESY data combined with 2,3JCH data obtained from inherently less sensitive experiments (e.g. HETLOC, HSQC-HECADE, HSQMBC).
Often, NMR alone is insufficient to secure RC or AC – or both – and degradation strategies are employed. In cases of stereochemical assignments, empirical chemical manipulations become essential. These efforts are nontrivial; no one general method is applicable and solutions emerge on a case-by-case basis that test the expertise of the natural product chemist who is often confined to working with μg-sample amounts for each chemical transformation. NMR structure elucidation, complemented by degradation-synthesis at the microgram level, atests to the skill and tenacity of the practicing natural products chemist from who is demanded, not just great instrumental proficiency, but exceptional powers of observation, deduction and extreme perserverance.
Determination of molecular structures by analysis of 2D NMR data and bond connectivity networks is subject to a fundamental limitation; a sufficient number of assignable 1H signals that relay to 13C or 15N nuclei of the underlying molecular skeleton. From the so-called ‘Crews rule’xviii – a guideline for successful 2D NMR analysis that requires a minimum ratio of H/C ~2 – it is easily appreciated that compounds depauperate in H (e.g. polycyclic alkaloids with high heteroatom content and many sp2 carbons; often crystallinexix) are the most challenging. A bias in this review is selection of polyketide structures; compounds biosynthesized by head-to-tail additions of acetate or other ketides with high H/C ratios, and ornate, stereochemically complex carbon skeletons (usually non-crystalline) that lend themselves nicely to NMR structure elucidation.
2.2. Nuclear Overhauser Effect (NOESY) and J coupling (1H–1H)
Interpretations of dipolar coupling (1D and 2D NOESY and ROESY) and 1H-1H scalar coupling are widely exploited for solving the RC of small molecules. NOESY experiments are also useful for acyclic spin systems where constraints placed by 1H-1H and 1H-13C scalar couplings lead to unique solutions.xx Complications arise with slowly interconverting rotamers that contribute to conformational heterogeneity, however judicious application of molecular modeling or ab initio calculations often resolves equivocal solutions.
2.3. The 13C NMR Acetonide Method (Rychnovsky)xxi
First described in 1990, the 13C acetonide method developed by Rychnovsky, provided a valuable empirical method to assign the RC of syn- and anti-1,3-diols. Diols are converted to the corresponding acetonides, and 13C chemical shifts of the geminal methyl groups can be used to differentiate the two diastereomers. The method was based on symmetry principles and conformational preferences. The meso-like syn-1,3-diol acetonide (SDA), adopts a chair 1,3-dioxolane conformation with axial and equatorial methyl groups. The anti-1,3-diol acetonide (ADA) with C2-like symmetry, prefers the twist-boat conformation to minimize 1,3-diaxial interactions; and the methyl groups are almost equivalent. In the SDA, the axial and equatorial methyl groups resonate at ~δ 30 ppm and ~δ 20 ppm, respectively. In the ADA case, the methyl group chemical shifts are almost identical to each other (~δ 25 ppm). As the size difference of substituents at C4 and C6 increase, the 1,3- dioxane ring tends to adopt a chair conformation.xxii It should be noted that the symmetry elements in 1,2-diol acetonides are also useful for assignment purposes.xxiii
The Evans group extended the 13C acetonide method to polypropionate chains containing branched methyl groups at C5.xxiv The chemical shift of the acetonide quaternary carbon is also diagnostic for configuration (SDA, ~98.1 ppm; ADA, ~100.6 ppm).
Three standard 2D NMR experiments: the NOESY/ROESY, HSQC/HMQC, and HMBC experiments allow extension of the method to more complex polyacetonide systems. In the SDA, the axial methyl group shows nOe correlations to H4 and H6 axial protons. In the ADA, one acetonide methyl shows an nOe correlation to H4, and the other methyl shows an nOe to H6. Sensitivity is greatly improved by the simple expedient of employing 1,3-{13C2}–labeled acetone for acetonide preparation.xxv
2.4. Chiral Derivatizing Agents (Mosher’s Method)
Originally proposed by Dale and Mosher in 1973xxvi, and refined by Ohtani and coworkers in 1991xxvii, the “Mosher’s method” and “modified Mosher’s method” (MMM) represent the most widely used tool for determining the AC of secondary alcohols. Optically pure 2-methoxy-2-phenyl-2-trifluoromethyl acetic acid (MTPA), or the corresponding acid chloride (MTPA-Cl), are the most commonly used chiral derivatizing agents (CDAs). Differential chemical shifts are aligned for each group L1 and L2, and fitted to configurational models (Figure 4). The generally accepted model, conformer a (Figure 4), suggests that the carbinol proton, ester carbonyl, and trifluoromethyl group lie in the same plane. However, conformational studies carried out by Riguera and coworkers demonstrate MTPA esters are populated by three major conformers (a–c)..xxviii In conformer b, the phenyl ring is twisted and deshields L2. Conformer c has the trifluoromethyl group antiperiplanar to the carbinol proton and aryl group twisted which deshields L1. These opposing shielding and deshielding effects contribute to the relatively small net magnitudes of Δδ values and may even give anomalous alternation in sign.xxix In any case, the MMM has a built in ‘self-examination mechanism’ where anomalous values of Δδs, inconsistent with the model, diminish its reliance and indicate a need for an alternate assignment method.
Figure 4.
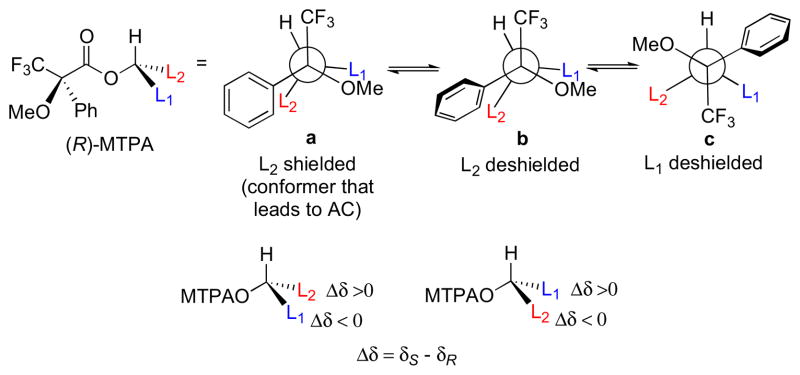
Conformers for (R)-MTPA esters (a) major conformer leading to reliable configurational assignment, other conformers (b and c) also present.
The methoxyphenylacetic acid (MPA) may be a more reliable CDA because only two major ester conformers (a and b, Figure 5.) are present leading to Δδ values of greater magnitude. The model for MPA places the methoxy group, ester carbonyl, and carbinol proton in the same plane (a, Figure 5). L1 is shielded in the (R)-MPA ester and L2 is unaffected, and the opposite effects are observed with the (S)-MPA esters. Note that the phenyl group lies on the opposite side of the C=O plane with respect to the MTPA esters. For this reason, Δδ values are defined by a different formula (Δδ = δR − δS).
Figure 5.
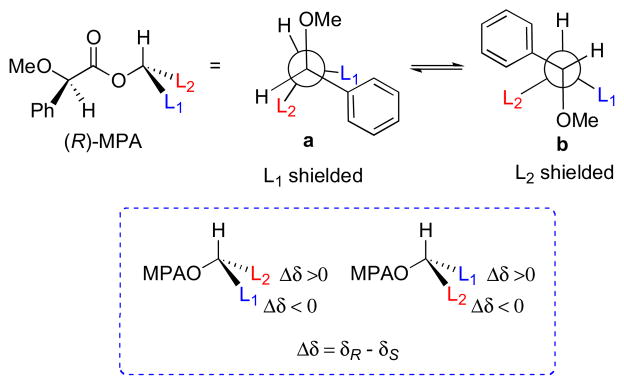
Conformers for (R)-MPA esters. (a) Major conformer used for configurational assignment. (b) Minor conformer.
The anisotropic differences between MTPA and MPA esters in addition to derivatives of other CDAs have been recently quantified by Hoye’s group. (−)-Menthol was converted to both (R) and (S) diastereomeric esters and Δδ values = δR−δS were acquired for each derivative. The absolute average for all the values, reflects the discriminating power of the CDA, were calculated for each pair. These comparative absolute values of Δδ may be useful when choosing a CDA to assign the configuration of a secondary hydroxyl group flanked by one or more CH2 chains. 2-NMA gives rise to particularly large anisotropy. Successful configurational assignments have been achieved for challenging compounds such as ginnolxxx and shishidemniols (section 7.15) using 2-NMA (2-naphthylmethoxyacetic acid).
MTPA esters are also used to ‘fingerprint’ diastereomers from degradation or chemical conversion for comparison with optically pure standards synthesized for comparison by NMR. The properties of CDAs and applications have been the subject of extensive reviews by Riguera,xxxi Kusumi and Ohtanixxxii, and Wenzel.xxxiii
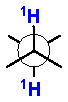
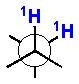
2.4 J-Based Configurational Analysis (Murata’s Method)
In 1990 Murata and coworkers published a seminal paper on ‘J-based configurational analysis’ (JBCA).xxxiv This method exploits both 1H-1H and 1H-13C coupling constants in order to assign anti or gauche relationships of vicinally substituted chains. This integrated JBCA method, combines information from homonuclear and heteronuclear coupling with NOE, and is used frequently for the determination of the RC of ‘contiguous’ or 1,3-skipped stereogenic centers in acyclic molecules.
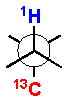
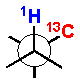
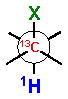
The 3JH-H and 2,3JH-C coupling constants are measured indirectly through a combination of NMR experiments. 1H-1H coupling constants can be extracted from 1D-1H NMR spectra, 1D-TOCSY, or absolute value cross–peaks in E-COSY type experiments. 1H-13C coupling constants are typically measured from HETLOC, HSQC-HECADE, PS HMBC, J-resolved HMBC, or HSQMBC experiments. The HETLOC and HSQC-HECADE experiments are the most sensitive and easily interpretable, but limited to spin systems with contiguous TOCSY coherence transfer. For subunits that contain quaternary centers or small 1H-1H couplings, PS-HMBC, J-HMBC or HSQMBC experiments are used. The advantages and disadvantages of each of these experiments, in addition to methods of interpretation of data, are outlined in a detailed review by Williamson.xxxv The extracted coupling constants are ordered into either ‘small’ or ‘large’ ranges and fit to an empirical model that reports the RC of the attached substituents (Table 1). Nevertheless, limitations to the method are underscored by examples where application of JBCA, alone, has lead to anomalous assignments.
Table 1.
1H-1H and 1H-13C coupling constants for the assignment of RC for vicinally-disubstituted chains using JBCA. Values obtained from refs. 34, 36, and 38.
|
|
||||||
|---|---|---|---|---|---|---|
| 3J 1H-1H coupling constants | 3J 1H-13C coupling constants | 2J 1H-13C coupling constants | ||||
|
| ||||||
| rotamer (magnitude of coupling) | ||||||
| anti (large) | gauche (small) | anti (large) | gauche (small) | anti (small) | gauche (large) | |
|
|
|
|
|
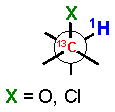
|
|
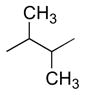
|
9 – 12 Hz | 2 – 4 Hz | 6 – 8 Hz | 1 – 3 Hz | – | – |
|
| ||||||
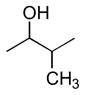
|
8 – 11 Hz | 1 – 4 Hz | 6 – 8 Hz | 1 – 3 Hz | 0 – −2 Hz | −5 – −7 Hz |
|
| ||||||
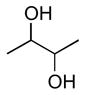
|
7 – 10 Hz | 0 – 4 Hz | 5 – 7 Hz | 1 – 3 Hz | 0 – 2 Hz | −4 – −6 Hz |
|
| ||||||
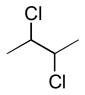
|
7.5 – 10.5 Hz | 1 – 3.5 Hz | 4 – 5 Hz | 0 – 3 Hz | −0.5 – 4 Hz | −3.5 – −6.5 Hz |
Recently, Carreira and coworkersxxxvi disclosed a detailed report on the coupling constant values for synthetic polychlorinated contiguous stereogenic systems commonly encountered in chlorinated sulfolipidsxxxvii. The study verified the JBCA method is applicable to polychlorinated natural products, however subtle differences in coupling constant values should be taken into account.
Prior to the JBCA method, the only reliable way to elucidate the RC of contiguous stereogenic centers was by a combination of the 13C acetonide method (see above), and multi-step partial or total synthesis followed by spectroscopic comparison with the natural product. The use of NMR in the assignment of RC have been reviewed by Riccio and coworkers.xxxviii
The task of assignment of molecules with isolated ‘stereosegments’ where lack of NMR correlations prevent the relay of configurational information remains a challenge, even with integrated techniques. The magnitude of the problem can be stated simply: for a molecule with n isolated defined stereo-segments, the maximum possible number of stereoisomers is 2n. Methods for connnecting isolated ‘islands of stereochemistry’ within complex molecules is one of the outstanding problems in natural product structure elucidation, but one that has inspired innovative and imaginative solutions.
2.5 Universal NMR Database (Kishi)
Kishi observed, through observation of numerous examples of configurational assignments in complex polyketides prepared by synthesis, that small systematic patterns of 1H NMR and 13C NMR chemical shift differences are associated with different diastereomers. Expanding on this observation, Kishi’s group compiled NMR data that came to be known as as the ‘universal database’ (UDB) to assign the RC of contiguous stereogenic units.xxxix The UDB is also useful for the relative and absolute configurational assignment of complex polyketides.
The UDB works under the assumption that: “(1) the structural properties of these clusters are inherent to the specific stereochemical arrangement of the (small) substituents on the carbon backbone and (2) the structural properties of these clusters are independent from the rest of molecule, when they are sufficiently separated from each other.”xl In practice, stereogenic subunits need only be separated by two or more methylene groups so that they may be treated independently. The 1H or 13C NMR chemical shifts of the carbon framework in the molecule are averaged, and these values are subtracted from the respective chemical shifts of the molecule under examination. For a given diastereomer, these aggregated deviations (±Δδ) are characteristic of the RC of each diastereomer and can be compared to the deviations of other diastereomers.
Not surprisingly, the most straightforward analysis is comparison from a database where the side chains closely resemble that of the compound in question. However, it may be possible to adjust the chemical shifts appropriately of a given database to apply it to a substrate not identical to the database. The UDB has also been extened to AC assignment with the use of chiral anisotropic NMR reagents.xli
An extension of UDBxlii matches overlapping contiguous triads of 1H-1H coupling constants with those of synthetic diastereomers of defined configuration for the purpose of assignment in polyol and polyacetoxy compounds. This is advantageous over chemical shift comparison because it is less influenced by solvent and substituents on the side chain and mainly influenced by the local conformation. Application of the UDB approach is illustrated in the configurational assignment of sagittamides (Section 7.12).xliii
3. Chiroptical methods
3.1. Polarimetry
Optical rotation [α]D is widely used for chiroptical characterization and determination of optical purity of organic compounds. Modern commercial digital polarimeters employing bright light sources, short-path microcells (as low as 0.1 dm, 100 μL) and sensitive polarizers capable of measuring optical rotations of ~0.1 millidegree. Compounds with a relatively large [α]D value (>20) can be measured with samples of less than a milligram. Assignments are often made on degradation products of natural compounds by chiroptical comparisons with synthetic standards, but at the cost of sacrificing a large amount of the parent compound; a luxury not always granted in cases where the sample is rare and mass-limited. It is now possible, as shown by the Wipf group, that accurate ab initio calculation of molar rotations by time-dependent DFT methods by van Hoff’s superposition principle can discriminate between diastereomers. This approach was used in the absolute stereoassignment of the bistramides (section 7.5).cxv Calculated molar rotations complement the UDB approach, but have another limitation. In absence of independent measurements of the [α]D of the natural product, the method largely rests on trust and reliance that the reported literature values were measured accurately on very pure samples; a condition that, regrettably, has not always prevailed. A review of current literature shows that the ab initio molar rotation approach to stereochemistry has not been largely adopted yet.
3.2 UV-Vis and Electronic Circular Dichroism
Electronic circular dichroism (ECD) arises from differential absorption of left and right circularly polarized light; it represents one of the most useful techniques available for stereochemical and conformational analysis of chiral molecules.xliv ECD, like UV-vis, is a molar quantity that obeys the Beer-Lambert law, and its measurement can be inherently more sensitive than [α]D, IR, or NMR. and amenable to very small samples (i.e. μg) when the compound contains one or more chromophores (λmax >200 nm) or where suitable chromophores can be introduced. Rather than exhaustively reviewing sector rules, two examples pertinent to natural products – the octant rule for cyclohexane and the Mo(OAc)4 method for vicinal diols – will be illustrated.
3.3. The Octant Rule
Pioneering work by Moskiewitzxlv, Crabbé,xlvi Djerassixlvii, Lightnerxlviii and others lead to development of a series of empirical ‘sector rules’ for assignment of configuration, the best known of which is the ‘octant rule’ for cyclic ketones. For example, the qualitative contributions to the Cotton effect due to the forbidden n-π* transition of the C=O group (λmax 284 nm) in 3R or 3S-methylcyclohexanonexlix can be predicted by considering contributions of atoms within each of the eight sectors (Figure 9) formed by three intersecting planes: the nodal plane at the C=O goup and two orthogonal planes bisecting the C=O bond. The octant rule was supported by measurements of ORD and CD of numerous cyclohexanones and largely conforms to the simple predictive factors.
Figure 9.
Applications of the exciton chirality method for the assignment of configuration. Signs of helicity angles, θ, correlate with the sign of the split Cotton effect. See Ref. li.
3.4 Snatzke’s Method
Snatke’s method employs ECD of molybdate esters for the assignment of the corresponding 1,2-diols. Dimolybdenum tetraacetate (Mo2(OAc)4) is added to the diol and the resultant conformationally restricted complex gives rise to an induced CD (ICD). The resulting Cotton effect (~λ305 nm) correlates with the sign of the O–C–C–O torsional angle which is related to the configuration of the diol. The torsional angle is influenced by the size of the substituents (RL and RS); bulkier substituents (RL and R’L) will orient in the pseudo-equatorial postion to avoid non-bonded interactions. Th simplicity of this empirical method is evident in the procedure; the reagent is simply mixed with the diol (10 μmol) in DMSO, and CD spectra are acquired at room temperature after a few minutes.l
3.5 The Exciton Chirality Method (Nakanishi)
The exciton chirality CD method (ECCD), is a non-empirical configurational assignment by CD, largely developed and popularized by Nakanishi and Harada.li A pair of degenerate or near-degenerate chromophores undergo exciton coupling, with resultant Davydov splitting of the transition, that is dependent upon oscillator strength, the inter-chromophoric distance and the angle, θ, subtended by their respective electronic transition dipole moments. In ECCD, chiral molecules exhibit strong biphasic Cotton effects whose signs are directly correlated with the sign of θ. For example, the ‘dibenzoate method’ can be applied to the corresponding dibenzoate of a vicinal diol by observation of the sign of the split Cotton effect and correlation with the absolute sign of the O-C-CO angle. Many variants of this example of bi-chromophoric ECCD have been described (see Figure 9) li
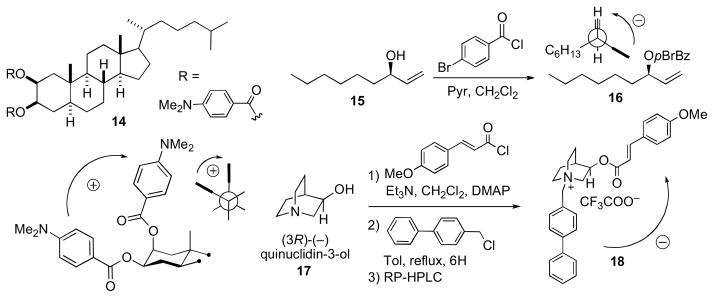
The bis-dimethylamino benzoate of 5α-cholestane-2β,3β (14) shows a positive split Cotton effect between the C2 axial and C3 equatorial benzoate chromophores. The configuration of allylic alcohols (cyclic and acyclic) are readily assigned by the allylic benzoate method.lii (R)-Non-1-en-3-ol (15) was converted to the corresponding p-bromobenzoate (16), which is assigned the R configuration based on observation of a positive split Cotton effect resulting from exciton coupling between the benzoate (λ~230 nm) and ene chromophores. The addition of two chromophores to two different functional groups was demonstrated in the configurational assignment of (3R)-quinuclidinol.liii After esterification (p-methoxycinnamoyl-chloride) of the secondary hydroxyl, the teriary amine was alkylated with phenylbenzylchloride to give the bis-chromophoric derivative (18).
The exciton chirality method has been exploited in more complex multichromophoric systems of triols, higher polyols, and aminopolyols.liv The sensitivity of ECD lends a great advantage to assignments of chromophoric derivatives of complex molecules. A limitation of ECD to simple interpretations may be interference from other chromphores that reside in the molecule. More recently, prediction of the ECD has been made possible by ab initio calculations using time-dependent DFT methods, although these have been largely applied to molecules with rigidly oriented chromophores, or more flexible molecules where the orientations of the transition dipole moments in each conformer, along with their Boltzmann distributions, can be also reliably calculated.lv
3.6 Infrared and Vibrational Circular Dichroism
Infrared spectroscopy (IR) has been the traditional tool for functional group identification in unknown organic compounds. Sadly, the use of IR has declined in recent years or is relegated to fulfil pro forma reporting requirements with little interpretation, even though modern Fourier transform IR (FTIR) instrumentation, with attenuated total reflectance (ATR), simplifies measurement of samples of less than 50 μg with full sample recovery.
Vibrational circular dichroism (VCD) arises from differential absorption of left and right circularly polarized IR and can be used for assignment of AC. The advantages of VCD include richly detailed bands, even from the ‘fingerprint region’, and ease of ab initio calculation of VCD spectra. Several assignments of natural products by VCD have been reported.lvi Wider usage of VCD may be impeded by relative insensitivity compared to other spectroscopic methods (samples of several milligrams and hours of acquisition time are required), and the need access to expensive, uncommon instrumentation.
4. Inferences from Biosynthesis or Bioinformatics
Stereochemically defined natural products are useful for the configurational assignment of derivatives and structurally related metabolites. The construction of similar compounds are often carried out by analogous enzymes in a conserved stereospecific manner. Biosynthetic inferences were successfully used in combination with spectroscopic methods for the stereochemical assignments of psymberin (100, Section 7.8) and dictyostatin (98, Section 7.7). Genetic and bioinformatics–based approaches are increasingly used for structure and configurational assignment.lvii
5. X-Ray Crystallography
X-ray crystallography is the ultimate method for structural determination,lviii however it is singularly dependent upon good quality diffracting crystals that exceed critical minimum dimensions (10–100 μm). While this is routinely achieved for overexpressed biomacromolecules using high-throughput crystal optimization techniques, it is quite a different story for a natural product that is only available in microgram amounts – one has limited options for making saturated solutions with only μL amounts of solvent. The power of X-ray crystallography becomes most apparent with highly functionalized alkaloids where NMR fails; when the formula is so depaurperate in hydrogen (H/C ratio <2, the so-called Crews rule), that 2D NMR experiments provide few useful cross-peaks. This review will only cover some representative examples of X-ray structures of optically active compounds where NMR-based structure determination failed. See determination of spirastrellolide B by microscale two-step chemical degradation leading to an X-ray quality crystal – all from a 100 μg sample!
6. Chemical Synthesis
The ideal solution to a stereochemical problem would be to synthesize all possible stereoisomers for comparison of spectroscopic data. From a practical standpoint, this task is neither tractable nor necessary. The most favorable option is to carry out chemical degradation to simpler compounds that are more amenable to synthesis and or spectroscopic analysis. Often, synthesis of ‘key’ segments for comparison suffice. All cases require expert and refined skills that are the traits of well–trained – and fearless! – natural product chemists.
7. Selected Examples
7.1. Amphidinols
Marine dinoflagellates are the source of some of the most biologically potent and structurally complex metabolites isolated to date.lix The amphidinols, first disclosed in 1991 by the Yasumoto group are antifungal polyhydroxy polyene metabolites from the marine dinoflagellate Amphidinium klebsii.lx The stereochemical determination of amphidinol 3 (18) was one of the initial reports that demonstrated the effectiveness of the JBCA method applied to a complex natural product.lxi
The configurational assignment was preceded by a division of the molecule into three sterochemical subunits: (a) C2–C14, (b) C20–C27, and (c) C32–C52. The RC of subunits (b) and (c) were assigned using JBCA, and relevant coupling constants (2,3JHC and 2JHH) were extracted from HETLOC, PS-HMBC, and E–COSY experiments. The RC between C39 and C44 was assigned from coupling constant and NOE data. In addition, stereoassignments of the diastereotopic methylene protons (CH2-22 and CH2-26) allowed complete assignment of the RC of the C20–C27 segment.
The ACs at C6, C10, C14, C23 and C39 were assigned from degradation products. Amphidinol 3 (19) was oxidized (HIO4), reduced (NaBH4), esterified (S- or R-MTPA-Cl), and purified (RP-HPLC) which gave esters (20–22, Scheme 2). The MMM was used for both sets of MTPA esters 20ab and 22ab to assign the 6R, 10R, 14R, and 39R configurations. The 1H chemical shift differences for C11 (+0.01, 0.0) were small and approach the limits of mutual interactions between the two MTPA groups compromising interpretation. The (R)-MTPA ester 21a was compared to both (R)- and (S)- MTPA esters of authentic (R)-methyl-1,4-butanediol which revealed the 23S configuration.
Scheme 2.
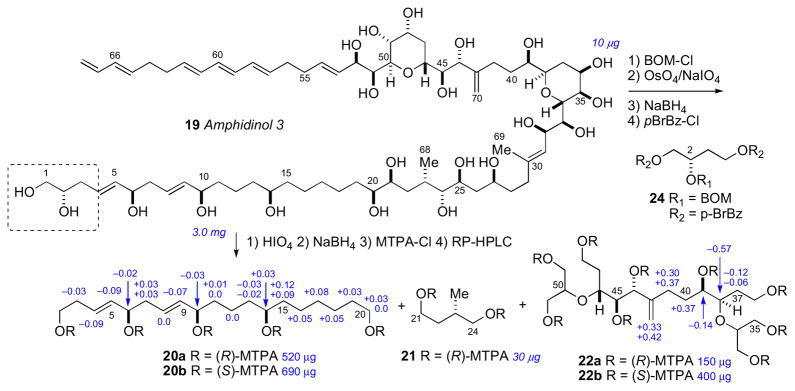
Degradation of amphidinol 3 (19). Note the 2S configuration depicted here was revised to 2R. See Ref. lxvi.
To assign the C2 configuration, a degradation approach reminiscent of the configurational assignment of C2 in ciguatoxin was adopted.lxii Amphidinol 3 (19) was subjected to protection (BOM-Cl), dihydroxylation (OsO4), oxidation (NaIO4), reduction (NaBH4), and acylation (p-BrBz-Cl) to give the protected ester 24. Comparison of the latter product with optically pure standards by chiral HPLC (Chiralpak AD) supportive of the 2S configuration. However, a re-examination of the C2 configuration using an alternative approach based on synthesis of diastereomers of a longer-chain terminal segment of 19 and comparison of 1H NMR data lead to reassignment to C2 to R (see below).
The reported configurational assignment of amphidinol 3 (19) was one of the first reports using JBCA on a complex natural product. The full stereochemical assignment was made with only 3 mg of material for degradation experiments and 8 mg of 13C enriched material for NMR experiments. It should be recalled that previous studies to gain this type of stereochemical information would have required much more material (> 100 mg) for chemical conversion or degradations studies; for example configurational assignments of complex natural products included mycoticins by Schreiber’s group,lxiii nystatin by Beau’s group,lxiv and roflamycoin by Rychnovsky’s group.lxv
In 2008, Oishi and coworkers synthesized all diastereomeric models of amphidinol 3 (19) encompassing C2, C6, and C10 (25–28) and compared 13C chemical shifts with 19, and showed protected pentaol 26 was the closest match and that the C2 stereocenter had been assigned incorrectly.lxvi To verify the C2 configuration, vinyl alcohol 29 was cleaved from amphidinol 3 (19, 50 μg) by cross metathesis using Grubbs’ second generation catalyst under an atmosphere of ethylene (Scheme 3). The cross metathesis product was compared with authentic standards by chiral GLC, which lead to the conclusion that the C2 configuration should be revised from S to R. It was uncertain how the misassignment was made, but an HPLC peak corresponding to the protected ester 24 may have contaminanted the degradation product. Amphidinol 3 (19) has been the subject of intense synthetic efforts by research groups led by Cossy,lxvii Rychnovsky,lxviii Roush,lxix Paquette,lxx Oishi,lxxi Crimmins,lxxii and Marko.lxxiii
Scheme 3.

Synthetic model compounds 25–28, and cross metathesis of amphidinol 3 (19).
Since the initial reports of the amphidinols, additional families of related metabolites have been isolated: the luteophanolslxxiv, lingshuiols,lxxv and karatungiols,lxxvi from Amphidinium sp. and most recently the karlotoxins from Karlodinium veneficum.lxxvii Blooms of K. veneficum have been implicated in massive fish kill events. In 2010, the Hamann group reported the full structure for karlotoxin 2 (30) by a similar approach applied to amphidinol 3 (Scheme 4).lxxviii The JBCA method secured C14–C18, C21–C24, and C28–C49. The ACs of C6, C10, C14, C21, and C28 were derived from NMR analysis of degradation (HIO4/NaBH4/(R)- or (S)-MPA) products 31–33. The Δδ values (δR–δS) were used for 31ab, and 32–33 in comparison with authentic standards. It is worth noting that although the Δδ values for 31ab are larger in magnitude than those observed for similar segments derived from amphidinol 3 (19ab), overlapping anisotropies of the MPA esters are observed and the assignmetnt at C6 becomes equivocal. Interestingly, amphidinol 3 (19) and karlotoxin 2 (30) share almost identical structural features in the C30–C52 region (amphidinol numbering), but the reported AC was antipodal. The stereochemical fidelity of these assignments should be revealed by total synthesis.
Scheme 4.
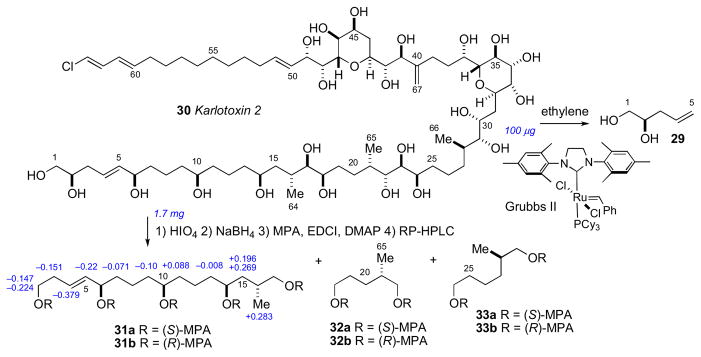
Structure and configurational assignment of karlotoxin 2 (30) by Hamann.
Trisoxazole macrolides belong to a structurally unique group of sponge and nudibranch derived natural products that have received attention from the chemists and biologists. The ulapualides A and Blxxix and kabiramide Clxxx, published simultaneously in 1986 by the Scheuer and Fusetani groups, respectively were the first reported members of the trisoxazole class. Additional related trisoxazole macrolides including the halichondramides,lxxxi mycalolides,lxxxii and the jaspisamideslxxxiii appeared in the following decade.
The trisoxazole macrolides exhibit potent antifungal and cytotoxic activities due to their property of binding tightly to G-actin, and inducing depolymerization of F-actin, and disruption of actin filament formation and organization.lxxxiv Because of their structural complexity, the trisoxazole macrolides eluded complete stereochemical assignment until the report of an elegant chemical correlation study of various mycalolide congeners. For the first time, the stereochemistry of a trisoxazole was defined by the Fusetani and Panek groups in 1999.lxxxv
Mycalolide B (35), like other trisoxazoles exhibit conformational isomerism in the NMR spectra due to slow interconversion of the N–formyl amide. Consequently, NMR experiments were hampered and stereochemical assignments were secured through chemical conversion to compounds more suitable for spectroscopic analysis. In an effective reaction sequence, mycalolide B (35) was subjected to oxidation (RuO4),lxxxvi methanolysis and lactonization to provide bislactone 40 (Scheme 4) in sufficient yield for 2D NMR experiments. Extensive NMR analysis of 40 suggested both lactone rings were in the boat conformation.lxxxvii NOESY and coupling constant data were used to assign the RC of both lactone rings, and the anti orientation between H24 and H26, however the RC between C26/C27, and C27/C30 remained ambiguous.
The secondary hydroxyl groups located throughout the mycalolide structures were useful for assignment by the MMM. The configuration at these positions as well as other segments were likely conserved among all mycalolide congeners based on chemical interconversion.lxxxiic Therefore, the MMM was independently applied to C3, C30 and C32 in mycalolide B (35), 30-hydroxymycalolide A (38), and 32-hydroxymycalolide A (39), respectively (Scheme 5). Oxidative degradation of mycalolide C (36) and protection of the product gave alcohol (45), and the C24 center assigned by MMM (Scheme 6).
Scheme 5.

Degradation and MMM on mycalolide B (35), 30-hydroxymycalolide A (38), and 32-hydroxymycalolide A (39).
Scheme 6.

Degradation of mycalolide C (36).
The side chain RC was confirmed after perruthenate–catalyzed oxidative degradation of 38-hydroxymycalolide B (37) to acetate 47. The NMR data for the degradation product (47) was indistinguishable from synthetic 47, but different from that of epimeric 48 (Scheme 7). This comparison unambiguously allowed the assignment of the relative and absolute configuration of the C22–C33 of the mycalolides.
Scheme 7.

Oxidative degradation of 38-hydroxymycalolide B (37).
The 8R and 9S ACs of 37 were assigned by perruthenate degradation and conversion to bis-p-bromophenacyl derivative 49 (Scheme 7) and comparison to authentic standards by chiral HPLC (Chiralcel OD). Finally, the configuration at C37 of the natural products 35–37 were assigned by saponification, derivatization (p-bromophenacylbromide) to 50–52, and comparison with authentic standards by chiral HPLC (Scheme 8).
Scheme 8.

Hydrolysis of the side chain in mycalolides B (35), C (36), and 38-hydroxymycalolide B (37).
The mycalolide assignments were the first reported stereochemical assignments of any trisoxazole macrolide. Shortly after, total synthesis of (−)-mycalolide A (34) by the Panek group, verified the stereochemical assignment.lxxxviii In 2004, Rayment and coworkers acquired an X-ray crystal structure of structurally related ulapualide A bound to actin, which showed the stereochemistry is conserved between the mycalolides and the ulapualides.lxxxix The side chain of the trisoxazole macrolides bears structural resemblance to other actin-binding marine macrolides including reidispongiolide A (53),xc sphinxiolide (54),xci aplyronine (55),xcii and the terrestrially-derived scytophycin (56)xciii (Figure 13). Rayment and coworkers have reported X–ray crystal structures of a number of these macrolides demonstrating a powerful benefit of stereocomplex small molecules bound to proteins: total stereochemical assignment.xciv This is significant for trisoxazoles as none produce X–ray quality crystals.
Figure 13.

Actin-binding marine macrolides.
7.3 Oceanapiside
α-ω-Functionalized sphingolipids (Figure 14) from marine sponges are C28–C30 long chain lipids that are terminated as a 2-amino-3-alkanol or 2-amino-1,3-alkanediol. These lipids represented an interesting challenge for configurational assignment because the stereosegments are separated by a long hydrocabon chain and effectively insulated making NMR correlations of the chain termini impossible. The first configurational assignment of this family was carried out on oceanapiside (57a)xcv from Oceanapia sp. collected from Port Phillip Bay, Australia. Oceanapiside showed good antifungal activity against fluconazole resistant Candida glabrata (MIC = 10 μg/mL); the aglycone (oceanin; 57b) is more active (MIC = 3 μg/mL) presumably due to improved cell permeability.
Figure 14.
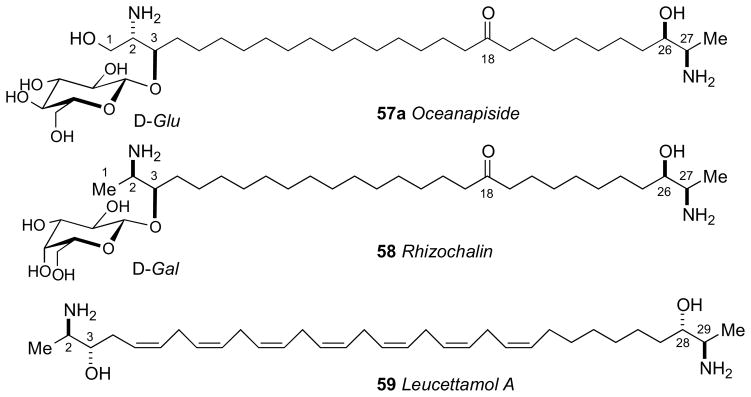
Structures of dimeric C28 and C30 α,ω–functionalized sphingolipids from sponges.
The initial report by Molinski established a planar structure for oceanapiside (57a)xcv and the configuration of the D–glucose residue (note the position of the keto-group was later revised to C18xcvc). The position of the carbonyl group relied on MALDI-MS-MS measurements of the C10, C12-d4 isotopomer obtained upon standing in MeOH-d4 (23°C, 2 months) (however this was later revised). Acidic methanolysis (HCl/MeOH, 80°C, 2h) of 57a gave an anomeric mixture of α,β-1-O-methyl-D-glucopyranosides, identical with authentic material by high-performance TLC, and the aglycone, oceanin (57b).
A follow–up report described the solution of the C2, C3, C26, C27 configurations of oceanin by fitting ‘hybrid’ ECCD spectra from perbenzoylated synthetic aminoalcohols to that of perbenzoyl–oceanin. This general approach to α,ω–functionalized sphingolipids was reliant upon the assumption that the CD spectra can be treated as a superposition of exciton couplets. Local vicinal benzoyl groups give rise to ECCD but not between separate terminal benzoyl groups.
The advantage of this approach is only four model compounds (60–61) were necessary and sufficient to create all 16 stereoisomeric permutations by simple linear combinations of model CD spectra. Perbenzoyl–oceanin (62) showed an ECCD spectrum uniquely superimposable upon the combination erythro-60b + threo-61a leading to the 2S,3R,26R,27R configuration (Scheme 9).
Scheme 9.

Synthetic models (60–61) used to generate hybrid CD spectra, and conversion of oceanapiside to perbenzoylated aglycon 62. CD spectra of 62 (dotted line), and 60b+61a (solid line) (Adapted with permission from Nicholas, G. M.; Molinski, T. F. Enantiodivergent Biosynthesis of the Dimeric Sphingolipid Oceanapiside from the Marine Sponge Oceanapia phillipensis. Determination of Remote Stereochemistry. J. Am. Chem. Soc. 2000, 122(17), 4011–4019. © 2000. American Chemical Society (ref. 95b)). Note, the position of the C=O group was later revised from C11 to C18. Ref. xcvc.
The successful configurational assignment of oceanapiside represents a relatively simple and concise solution to a difficult stereochemical problem, one that would not be resolved by total synthesis since the optical rotary strengths of chiral amino alcohols are generally weak and discrimination of the diastereomers by NMR would be equivocal. The hybrid ECCD method has also been successfully applied to rhizochalin (58)xcvi from Rhizochalina incrustata, leucettamol A (59)xcvii from Leucetta microrhaphis, and additional α,ω–functionalized sphingolipids.xcviii It is notable that leucettamol A (59) was assumed first to be racemic because the [α]D was ~0, however, ECCD studies by the Molinski group showed leucettamol A (59) is optically active and has a pseudo–C2 configuration.
7.4 Amphidinolide E
The Kobayashi group have revealed dinoflagellates of the genus Amphidinium separated from flatworm Amphiscolops spp. collected in Okinawa, to be a highly productive source of cytotoxic polyketide marine macrolides.xcix Collectively known as the amphidinolides, over 34 macrolides have been described to date. Structurally, the macrolide varies in ring size and frequently embody tetrahydrofuran or pyran rings, with variable levels of unsaturation, hydroxylation, and methylation as typically observed in small molecules constructed by Type I PKS biosynthetic machinery. As most of these macrolides are often functionally and stereochemically rich, they have provided challenges for structural assignment by integrated methods. At the same time, the potent cytotoxicities of these compounds have instigated several synthetic efforts.c A full account of all meticulous assignments, and reassignments by total synthesis is beyond the scope of this review, but we will consider amphidinolide E as a technically challenging, representative amphidinolide target.
The isolation (0.9 mg) and planar structure of amphidinolide E was first reported in 1990. Amphidinolide E (63) showed mild cytotoxicity against murine leukemia cell lines: L1210 (IC50 2.0 μg/mL) and L5178Y (IC50 4.8 μg/mL).ci By 2002, a larger amount of 63 (2 mg) was secured by repetitive cultivation from hundreds of liters, which allowed the full stereochemical assignment to be made. The strategy for configurational assignment integrated the use of NMR, CD, chemical conversion, and synthesis of optically active model compounds and standards.
The RC of 63 was assigned by a combination of JBCA, NOESY and chemical conversion. The cis orientation of the tetrahydrofuran ring system was identified by NOESY correlations between H13/H16. Formation of the 7,8-isopropylidene analog 64 (Scheme 10) allowed the threo assignment between H7 and H8. The C16 to C19 RC was assigned by JBCA and NOESY data in addition to chemical conversion. The orientation between H17 and H18 was confirmed from interpretation of NMR of bis–acetonide derivative. Amphidinolide E was hydrogenated (H2, Rh/Al2O3), further reduced (LiAlH4), and the resultant alcohol (65) converted to bis-isopropylidene 66 (Scheme 10).
Scheme 10.

Chemical conversion of amphidinolide E (63).
The AC of amphidinolide E (63) was assigned by a combination of the MMM, exciton chirality method, and degradation. The threo orientation of the H7/H8 diol allowed for application of the di-benzoate method.51 Compound 63 was converted to the p-methoxycinnamate derivative (70) to avoid overlap with lower wavelength chromophores (Scheme 11). The resulting ECCD spectra showed a negative split–Cotton effect, consistent with the 7R,8R configuration. Application of the MMM to the tri-MTPA esters (69ab) gave the 17R configuration (Scheme 11). The remote C2 methyl group was assigned by periodate cleavage–borohydride reduction (Scheme 10) to attain diols 67ab. 1H NMR signals of diastereotopic C1 methylene group are diagnostic for the configuration of the methyl group, and was assigned the R configuration.cii Finally, tetrahydrofuran degradation products (68ab) were compared to authentic standards prepared by synthesis.
Scheme 11.
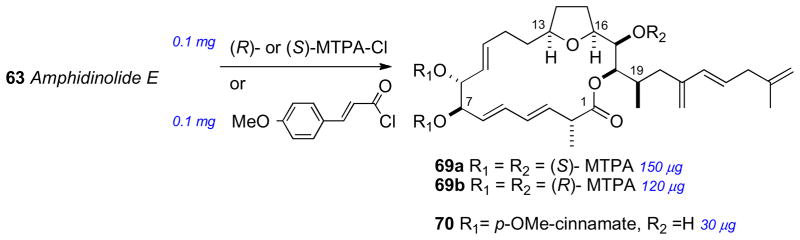
Preparation of MTPA esters (69) and p-methoxycinnamate derivatives (70).
This work represents a tour de force modern structure determination where the full configurational assignment was completed on a sample of less than 2 mg. Total syntheses of amphidinolide E (63) have been completed by the Roushciii and Leeciv groups, and confirmed the configurational assignment proposed by Kobayashi.
7.5 Bistramides
In 1988, bistramide A (71) the first member of a family of cytotoxic spiroketals, was reported from the tunicate Lissoclinum bistratum by the Verbist group in New Caledonia,cv The complete planar structure was disclosed by Ireland and coworkers by 1992.cvi Later, a number of analogs were reported.cvii The bistramides have attracted interest due to their broad, potent antiproliferative effects arising from inhibition of actin polymerization through tight covalent binding to G-actin, and disruption of the microfilament cytoskeleton.cviii A high resolution X-ray crystal structure of an actin-bistramide A complex revealed that the binding site of bistramide A (71) has little overlap with other small molecule actin inhibitors (e.g. swinholide A,cix kabiramide A,cx and other structurally related macrolidescxi,cxii). Rationally designed analogs and a fluorescent probe showed the enone functionality participates in covalent modification of the protein target.cxiii
Although the bistramides were reported in 1988, the first partial RC did not appear for more than a decade until the proposal by the Sollidie group in 2000. NOESY data acquired on the acetate derivative (72) of bistramide A established the RCs of both the tertrahydropyran (C6–C11) and spiroketal segments (C22–C31) (Figure 19).cxiv
Figure 19.
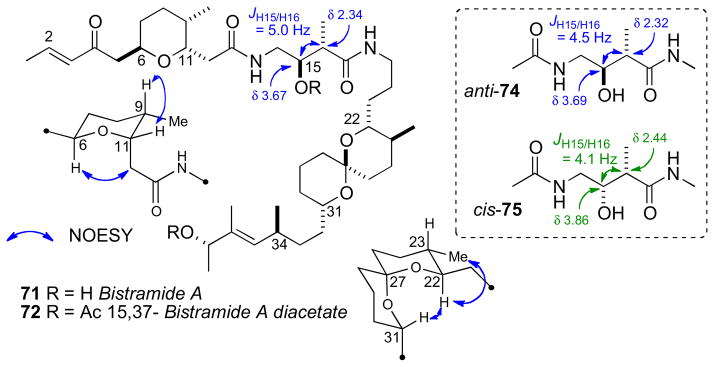
RC of bistramide A (71).
With a proposed RC of these two fragments, the Wipf group initiated efforts toward defining the relative and AC of bistramide C (73).cxv Wipf’s strategy synthesized the three stereosegments of bistramide C and analysis of their contributions to the molar rotation of the natural product.
The stereochemically less–complex bistramide C (73) with one less stereocenter (C37) relative to bistramide A (71), was chosen as the target. Two models, anti–74 and syn–75 were prepared to model the C15/C16 stereocenters of the amide. Comparison of 1H NMR to bistramide A, showed a closer match to anti–74. The synthetic strategy narrowed the number of possible diastereomers to 16 based on Sollide’s RC for the tetrahydropyran (C6–C11), C15,C16 stereocenters, and spiroketal subsections (C22–C31). Despite the arbitrary choice of the target diastereomer 76, with only a 1 in 16 probablility being correct, the convergent strategy provided a representative stereo-fragment along with chiroptical data (molar rotations) that, collectively, informed their proposal for the complete AC of 73.
Bistramide diastereomer 73 was synthesized by a two-segment (77 and 78) coupling strategy. The NMR data for 77 matched that reported for bistramide C (73), except for the 13C chemical shift of C34. Since the NMR data for the C1–C15 portion of 77 closely matched those of bistramide C (73), the RC between the tetrahydropyran and the amide linkage was correctly assigned in the synthetic diastereomer. Therefore, the discrepancy in 13C NMR shifts was a result of mismatch between configurations of the amide and spiroketal, the spiroketal and C34, or a combination of the two. With several synthetic intermediates comprising all stereochemical elements of bistramide C, a synthetic diastereomer, and molar rotations for bistramide C, the chiroptical analysis could be completed.
The molar rotations of each subunit were summed according to Van’t Hoff’s principle of optical superposition.cxvi Molar rotations of the synthetic model compounds were calculated from measured optical rotations and summed to give molar rotation data for the completely assembled bistramide C diastereomer (Table 1.2 and Figure 20). The tetrahydropyran/amide (77) containing the 6S,9R,11R,15S,16R configuration constituted a diastereomer with [M]D +119. The C34–contatining fragment was represented by (+)-normanicone (79) previously reported by Bestmann.cxvii The molar rotation contribution for the spiroketal (C22–C31) portion was calculated by subtracting the molar rotation of (−)-normanicone ([M]D –51) from synthetic 78 ([M]D +105) to give a value of [M]D +156. These values were summed to return the calculated value of [M]D +224 for bistramide diastereomer 76 that closely matched the experimental measurement of [M]D +211. The same analysis was applied to bistramide C (Figure 21). Combinations of all permutations of chiral subunits showed that natural bistramide C (73) with an experimental molar rotation of [M]D +70 should have the 6R,9S,11S,15R,16R,22R,23S, 27S,31S,34S configuration (calculated [M]D +88).
Figure 20.

Assignment of AC of bistramide diastereomer (76) by Van’t Hoff’s principle of optical superposition.
Figure 21.

Van’t Hoff principles to assign AC of natural bistramide C (73).
In a subsequent report, the Wipf group calculated [M]D values for each subunit from Boltzmann weighted energy minimized conformations, and summed as before to obtain reasonable values for each diastereomer and natural product.cxviii The benefits of the superpositions of molar rotations are obvious: none of the original material was required for chemical conversions for the assignment of configuration. The disadvantage is the necessity of synthesis of fairly advanced intermediates and, of course, accurately reported [α]D for natural product free of strongly rotating contaminants. Fortunately, all subunit contribution to the [M]D were relatively large (> 50) giving combinations with significantly different [M]D values. A cautionary note is approrpiate: the superposition method is probably less suitable for molecules whose synthetic fragments show only weak rotatory power (low [MD]) or similar [MD] where the combinations may not be sufficiently discriminated.
Subsequently, the Kozmin group successfully synthesized bistramide A (71), including both 37R and 37S diastereomers, the latter showed 13C NMR data identical to the natural product and confirmed the stereochemical predictions by the Wipf group were correct.cxix In 2005, the Wipf group successfully synthesized natural bistramide C, identical to the proposed configuration.cxx Since then, several total syntheses of the bistramide class have been reported.cxxi
7.6 Schulzeines
The Fusetani group reported schulzeines A–C (80–82), α-glucosidase inhibitors from the marine sponge Penares schulzei from Hachijo-island, Japan.cxxii The schulzeines are characterized by a 9,11-dihydroxytetrahydroisoquinoline unit connected to a sulfated fatty acid amide. The enzyme inhibitory activity of schulzeines A–C (80–82) range from 48–170 nM.
Methanolysis of 80 provided four cleavage products (83–86, Scheme 11). Analysis of FAB-MS/MS fragmentation data of the ring–C opened product (83) located the sulfate groups and, consequently, the full planar structure of schulzeine A (80).
Methanolysis product 84 was used to assign the configuration of C3 and C11 in the dihydroisoquinoline ring system. NOE correlations verified stereochemical fidelity of 84 a configuration unchanged from the parent compound. The free amine was reacted with (R)- or (S)-MTPA-Cl (pyr, 1h) followed by saponification to amides 90ab (Scheme 12). The Δδ chemical shift differences were fairly small, however the 3S,11R configuration could be assigned based on Mosher type analysis.cxxiii
Scheme 12.

Acid hydrolysis of schulzeine A (80).
Conversion of the triol 85 (Scheme 13) to the isopropylidene protected alcohol and esterification with (S)- and (R)-MTPA-Cl gave MTPA esters 89ab. leading to the 14’S configuration. The trans-relationship between H17′ and H18′ was evident from ROESY crosspeaks observed between H17′ and one methyl group (δ 1.34 ppm) and H18′ and the methyl group on the opposite face (δ 1.32 ppm).
Scheme 13.
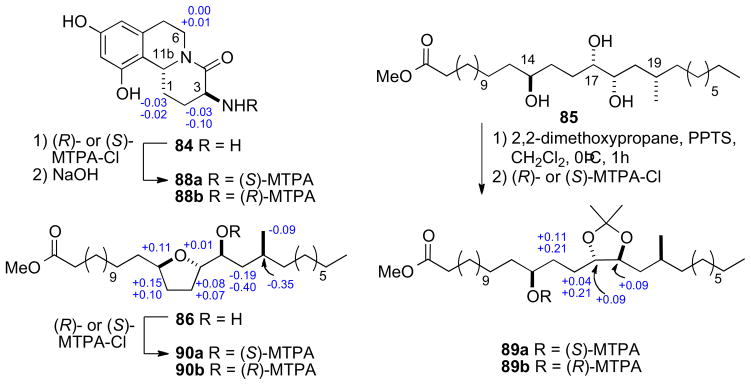
MTPA esters from hydrolysis products of schulzeine A (80).
The RC of the tetrahydrofuran 86 from the methanolysis was assigned by comparison of 13C chemical shifts to synthetic derivative 87 (Scheme 12)cxxiv. Conversion to both (R)- and (S)-MTPA esters 90ab, and analysis of 1H NMR gave the C18’S configuration. The stereochemical outcome upon formation of the tetrahydrofuran 86 is explained by initial hydrolysis of the C17′ sulfate followed by SN2 displacement of the C14′ sulfate.
The final stereocenter (C20′) was be assigned by degradation and comparison to optically pure standards (Scheme 14). Schulzeine A (80) was subjected to desulfation under mild acidic conditions (TsOH),cxxv oxidation–reduction (NaIO4, NaBH4) to give the primary alcohol, which was converted to the corresponding (R)-MTPA ester (91). The 1H NMR data of 91 was compared with both (S)- and (R)-MTPA esters (93ab) of (S)-3-methyl-1-pentanol which was consistent with the 20’S configuration. Thus, completion of the full stereochemical assignment of schulzeine A.
Scheme 14.
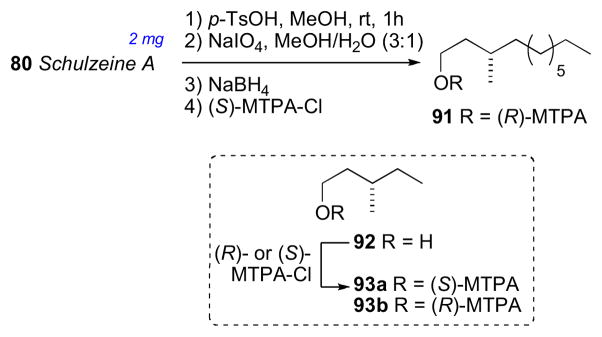
Degradation of schulzeine A (80) for configurational assignment of C19.
Schulzeine B (81) was subjected to a similar analysis as 80 (Scheme 15). NMR and FABMS data of the desulfation product showed absence of the methyl branch. Correlations from a ROESY experiment placed H3 and H11b were on the same face of the fused ring system. The AC of 81 was assigned from the methanolysis products (94–96), and showed schulzeine B to be epimeric to sschulzeine A at C11b.
Scheme 15.

Acid hydrolysis of schulzeine B (81).
It is also worth mentioning that this work is reminiscent of previous studies carried out on α-glucodisase inhibitors penarolide sulfates A1 and A2 (proline containing sulfated macrolides)cxxvi and penasulfate A (sulfated lipids).cxxvii Total synthesis of schulzeines A and B has been reported by the Gurjarcxxviii and Romocxxix groups. The Wardrop group synthesized schulzeines A-Ccxxx, and showed the configurations at C20′ in 80 should be inverted. The reason for the anomaly in assignments is uncertain.
7.7 Dictyostatin
In 1994, Pettit and coworkers disclosed the structure of a highly potent antimitotic marine macrolide, dictyostatin from Spongia sp. collected in Maldives.cxxxi The yield of dictyostatin was extremely low (1.35 mg from 400 kg wet. wt of sponge), and only a partial assignment (98a) of the RC was disclosed.
A decade later, following reisolation of (−)-dictyostatin from a North Jamaican lithistid sponge, a collaborative effort between the Paterson and Wright groups lead to an assignment (98b) for the AC (which differed considerably from 98a proposed by Pettit) based on high-field (700 and 800 MHz) NMR experiments, molecular modeling, and biosynthetic considerations.cxxxii Detailed analysis of NMR data (J coupling and NOESY), showed the C1–C16 to be relatively rigid, adopting only one preferred conformation (Figure 25).
Figure 25.
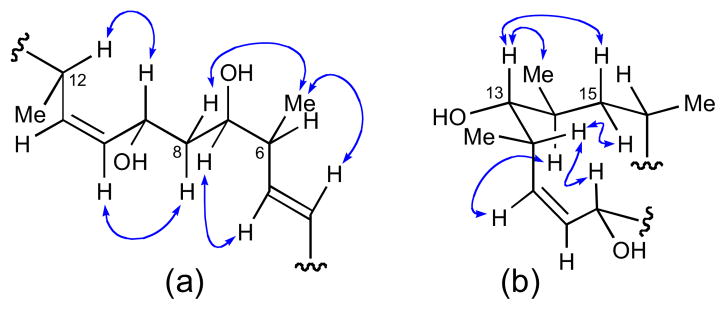
Configurational assignments made for (a) C6 to C12 and (b) C9 to C16 by NOESY and JBCA.
On the otherhand, NMR data (NOESY and J coupling) for the C16– C26 segment were consistent with two rapidly interconverting conformations (Figure 26). Evidence for this phenomenon was provided by intermediate coupling constants observed between: H-19 to H- 18a, H-19 to H-20, H-19 to Me-20 and H-20 to H-21. The two conformers differed in the orientation of C1/C2 (s-cis or s-trans), but were formulated as (a) and (b) to satisfy NOESY data. Molecular modeling (Macromodel, MM2, MonteCarlo search) gave a low energy conformation consistent with the s-trans configuration and fully consistent with NMR data.
Figure 26.
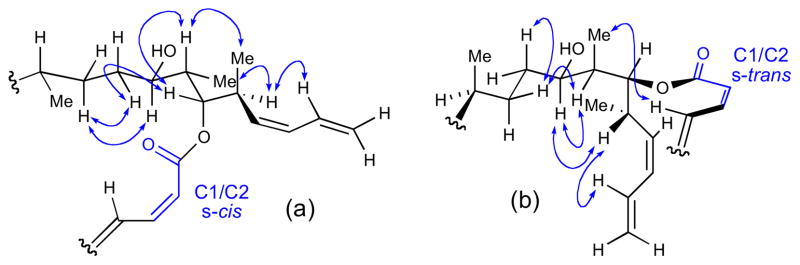
Two conformations consistent with NMR data: (a) C1/C2 s-cis and (b) C1/C2 s-trans.
The structural features of dictyostatin bear strong resemblance to discodermolide (99, Figure 27)cxxxiii another highly potent anti–cancer polyketidecxxxiv reported by Gunasekera and coworkers from the deep water marine sponge Discodermia dissolute. Although the two natural products derive from different sponges, the RC of 98 mapped to the corresponding segments in discodermolide (99). Therefore, it was assumed that the AC of dictyostatin was likely the same based on a similar biogenesis to discodermolide (99) the AC of which was defined by total synthesis of ent–discodermolide by Schreiber.cxxxv
Figure 27.
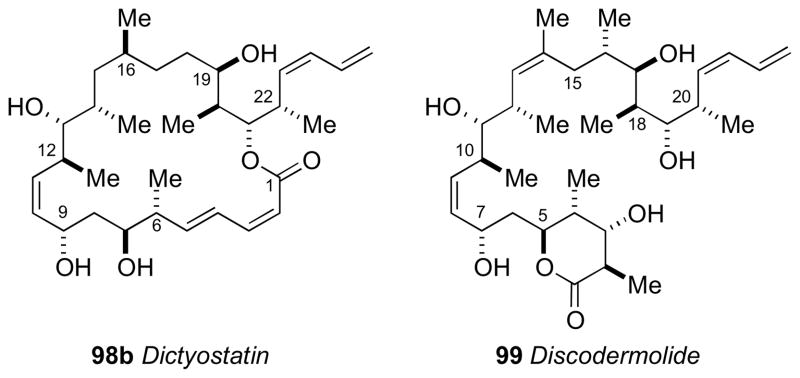
Structures for dictyostation (98) and discodermolide (99).
This represents a prime example of the power of high field NMR and biosynthetic inferences to arrive at the complete stereostructure for a complex natural product. Shortly after the proposed configurational assignment for dictyostatin (98), total syntheses by the Patersoncxxxvi and Currancxxxvii groups reported that the proposed stereostructure was correct. The material provided by total synthesis is the only viable source of this rare natural product for biological testing. Currently, efforts are driven towards SAR studies of synthetic derivatives, further biological testing, and optimization of a total synthesis to provide material for pre–clinical trials.
7.8 Psymberin
Psymberincxxxviii (also known as irciniastatin A,cxxxix 100) is a highly cytotoxic polyketide from the marine sponges Psammocinia reported independently by the Crews and Pettit groups, respectively. In the NCI 60-cell line screen, psymberin showed selectivity for several melanoma, breast, and colon cancer cell lines (LC50 < 2.5 nM) over leukemia cell lines (LC50 > 25 mM). The structure composition of psymberin was intriguing because it resembled the compound pederin (101)cxl isolated from the beetle Paederus sp. and other sponge metabolites (e.g. onnamide A (102)cxli mycalamide,cxlii and theopederincxliii from Theonella sp). Metagenomic analysis of whole-sponge DNA by Piel and coworkers have identified the putative genes responsible for the production of psymberin.cxliv
NMR spectroscopic analysis of 100 by Crews was more detailed and is discussed below. The planar structure was assigned by a combination of MS, IR, NMR and chemical conversions (methylation and acetylation), and stereochemical analysis proceeded by independent consideration of three subunits a–c. JBCA for the C4 and C5 stereocenters (subunit a) was unsuccessful due to complications from intermediate 2JHC couplings (3 Hz) between H4/C5 and H5/C4.cxlv
The tetrahydropyran ring system (subunit b) was deduced by coupling constant and NOESY correlations (Figure 29). The C8/C9 configurational assignment was made based on large 1H-1H coupling between H8/H9 (J = 8.0 Hz) that orients the two protons anti to each other. NOESY correlation between OMe-8/H10eq and H8/H13ax established the rotamer depicted in Figure 29. NOESY correlations of the C15–C17 stereotriad (c) established two rotamers in both C15/C16 and C16/C17 (Figure 27c).
Figure 29.

Configurational analysis of psymberin (100): (a) C5 assigned by analogy to pederin (101) and onnamide (102). (b) Preferred conformation of the tetrahydopyran and relevant nOes, and (c) two conformational rotamers about C15/C16 and C16/C17.
The AC of the C17 stereocenter was assigned by comparative CD analysis. The CD spectrum of psymberin showed a strong positive Cotton effect at λ 280 nm, and that of dihydrocoumarin 103 (R-configuration)cxlvi shows a negative Cotton effect at λ 275 nm assigned to the “n→π*” transition. Therefore psymberin (100) was assigned the opposite configuration (C17R) to that of 103 (Figure 29). The remainder of the molecule was assigned from assumptions of a similar biogenesis to pederin and related sponge metabolites (i.e. onnamide).
Following the initial report of psymberin, two synthetic approaches by the Williamscxlvii and Floreancigcxlviii groups to the diastereomeric models of psymberin established the relative and ACs of the amide side chain. The Floreancig group stereospecifically synthesized the four diastereomers (105–108, Scheme 16). PMB ether 104 was deprotected with ceric ammonium nitrate and, under acidic conditions (MeOH, H2SO4, 60°C), gave tetrahydrofuran 105 without racemization. The other three diastereomers (106–108) were prepared by similar procedures to give the four tetrahydrofuran derivatives needed for analysis. Separation of the four diastereomers was achieved by chiral GC (Chiraldex G–TA). Methanolic hydrolysis of psymberin (100) under identical conditions provided tetrahydrofuran 105 that was identical to the compound derived from 104 (Scheme 15). Therefore, the side chain of psymberin has the 4S,5S configuration. Psymberin has been the subject of many total and fragment syntheses, which have verified the stereo–assignment.cxlix
Scheme 16.
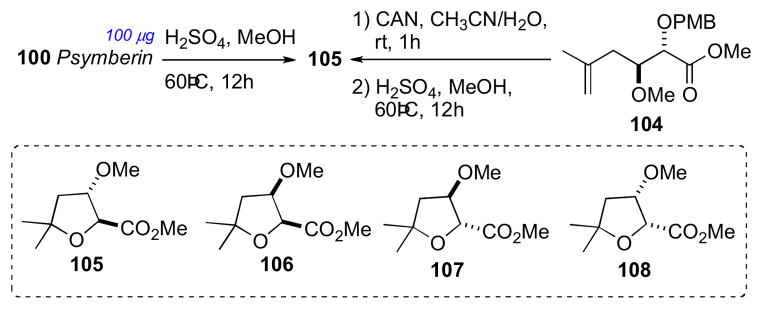
Stereochemical determination of the amide side chain in psymberin (100).
7.9 Caminoside A
Caminoside A (109),cl a gycolipid from the marine sponge Caminus spareoconia from Dominica was the first active natural product identified in a screening effort to identify small molecule inhibitors of a type III bacterial secretory pathway. Compound 109 exhibits an IC50 = 20μM in the assay.
Structurally, caminoside A (109) contains an oligosaccharide glycone comprised of four sugars (two equivalents of D–glucose, D–6-deoxytalose, and L–quinovose) appended to an oxygenated lipid aglycone that were identified by NMR and MS data. Compound 109 was subjected to methanolic acid hydrolysis (Scheme 16), to yield the aglycone (110), as well as a mixture of the mono–methyl glycosides, which were separated. The glycosidic fraction was acetylated and purified by HPLC to give D-α-1-methoxy-2,3,4,6-O-tetraacetylglucose (111) and D-β-1-methoxy-2,3,4-O-triacetyl-6-deoxyglucose (112), both of which matched authentic samples by specific rotation. The third product, 113 (1-methoxy-2,3,4-O-triacetyl-6-deoxytalose) was assigned by NMR, and then subjected to acid hydrolysis before comparison with authentic standards.
The configuration of the aglycone portion was assigned by Molinski and coworkers after conversion of 110 to its corresponding bis-TPP ester (118), measurement of liposomal exciton coupled circular dichroism (LECCD), and comparison to optically active models (Scheme 18).cli Previous CD studies on meso 1,5-, 1,7-, and 1,9-glycol bis-TPP esters showed that the AC of these systems are readily assigned by interpretation of the resulting positive or negative bisignate ECCD spectra obtained in liposomally–ordered media.clii In isotropic media, the latter compounds show only baseline spectra. Caminoside aglycone (110) was converted to the naphthoate derivative, and the ketone was reduced to the corresponding alcohol (115) as a C2 epimeric mixture (1:1). The mixture of diastereomers was subjected to kinetic resolution (Novozym 435, vinyl acetate), to give pure 2R–acetate diastereomer 116a. Removal of acyl groups by Ammoniolysis of 116a gave the diol 117, which was converted to the bis–TPP ester 118 (TPP-piv, Et3N).
Scheme 18.

Molinski’s configurational assignment of caminoside A aglycon. Conversion of aglycon 110 to bis-TPP ester 118.
The CD spectrum for 118 (MeOH) showed no significant Cotton effects. However, when CD spectra of 118 was acquired in DSPC liposomes, a bisignate positive ECCD spectra was observed. Therefore caminsoside A (109) has the 10R configuration. LECCD methodology should be useful for assignment of other natural products containing 1,n– diols (n= odd; 3, 5, 7, 9, etc.).
7.10 Polytheonamides
The highly cytotoxic polytheonamides A (119) and B (120) show a remarkable array of alternating L- and D-tert-alkyl glycine residues. They were first reported in 1992cliii by Fusetani and coworkers, and revised in 2005cliv The amino acid composition was established by amino acid analysis, and NMR analysis of the whole acid–hydrolysate. The N-terminus of polytheonamide B was originally assigned as a carbamoyl group, but later revised to 5,5-dimethyl-2-oxo-hexanoyl group based on reduction of 120 with sodium borohydride (Scheme 19) to give secondary alcohol epimers 121. Subsequently, the 44th residue, first formulated as a γ-hydroxy-t-leucine was revised to a β,β-dimethylmethionine sulfoxide based careful interpretation of MS and NMR data.
Scheme 19.

Chemical conversions of polytheonamide B.
The sequence assignment of amino acids was carried out by NOESY (n to n+1 crosspeaks of NH to Hα). In DMSO-d6, polytheonamide B (120) is in a random-coil conformation which facilitated sequential assignment.
The AC of the constituent amino acids were secured by chiral GCMS and Marfey’s analysisclv of the total acid hydrosylate. Initial analysis showed the L-amino acids were: Thr, Ile, Glu, Val, and βMeIle, and D-amino acids consisted of: HO–Asp, Ser, and αThr. The remaining amino acids: Ala, t-Leu, Asp, HO–Val were mixed D and L configurations and a sequence specific stereochemical method was used to address this problem.
Partial hydrolysis of polytheonamide B (120) (HCl/EtOH, 70°C, 30 min) afforded a complex mixture of peptide fragments. The composition of several fragments were identified by FABMS, and the N-terminal amino acids of each fragment were identified by dansylation, partial hydrolysis, and chiral GC or Marfey’s analysis, followed by Edman degradation. Exhaustive analysis of the peptide fragments gave the complete configuration of all amino acid residues except for the sulfoxide containing amino acid. The β,β-dimethylmethionine sulfoxide did not survive acidic hydrolysis, therefore 120 was reduced to the corresponding β,β-dimethyl methionine analog (122) and cleaved with cyanogen bromide (Scheme 18) to the corresponding L-β,β-dimethylhomoserine lactone (123) which was compared with authentic samples by GC analysis.
Finally, the structure of polytheonamide B (120) showed the same gross structure as polytheonamide A (119). This led to the proposal that the difference between the two compounds was either a change in configuration of one or more amino acids or the sulfoxide stereocenter. Adventitious autooxidation of 122, obtained from either polytheonamides A or B gave a 1:1 mixture of polytheonamides A (119) and B (120). In addition, separate oxidation of polytheonamides A (119) or B (120) (oxone, Scheme 18), provided the same sulfone (124) confirming that the two peptides were epimeric at S.
The heroic effort of structure elucidation of polytheonamides A and B, has now been followed by the first total synthesis of these peptides by the Inoue group.clvi In addition, a solution conformation based on NMR and molecular modeling has also been reported which shows that the potent cytotoxicity associated with these peptides is attributed to their pore forming abilities through a unique β-helix motif.clvii A single molecule of polytheonamide B spans 45Å, or about three times longer than gramicidin A, another pore-forming peptide. It is interesting to note that the polytheonamides are more effective pore-forming peptides than synthetic peptides with non-natural alternating configurations (D- and L-form).
7.11 Citrinadin A
Citrinadin A (125) obtained by fermentation of Penicillin citrinadin, separated from a red alga is a pentacyclic spirooxindole alkaloid containing 7 stereogenic centers.clviii It exhibits modest activity against murine leukemia L1210 and human epidermoid carcinoma KB cells (IC50 6.2 and 10 mg/mL, respectively). The planar structure was assembled by 2D NMR data.
The RC of the pentacyclic core of citrinadin A was established by ROESY data and 1H-1H coupling constants (Figure 32a). The relative orientation of the spirooxindole system was secured from ROESY correlations from H4 to both NMe26 and Me29.
Figure 32.

NOESY correlations for (a) citrinadin A (125) and (b) citrinadin A chlorohydrin (126).
The AC L-N,N-dimethyl valine residue was assigned by acid hydrolysis (1N HCl) and chiral HPLC (Sumichiral OA-5000, 1 mM CuSO4 aq).clix In a subsequent report, the Kobayashi group assigned the C14 AC of the pentacyclic core by ROESY correlations relayed from the 2S-N,N-dimethylvaline residue (Figure 32b).clx The chlorohydrin derivative (126) obtained by treatment of citrinadin A with HCl (50 mM in MeOH).
No standard methods to assign the isolated C21 stereocenter in the epoxide ring were available. Consequently, vibrational circular dichroism (VCD) spectra of the natural product was measured and compared to those of enantiomeric aryl keto-epoxides, 2R-(+)-127 and 2S-(−)-127 that were synthesized in five steps from benzaldehyde (Figure 33). Although the VCD spectra of the model compounds did not show exact mirror images as expected, the Cotton effect at 1230 cm−1, was attributed to symmetrical stretching of the epoxide ring, and the Cotton effects of the model spectra were opposite. This band was used to assign the configuration in the natural product. Therefore the complete assignment of citrinadin A was assigned as 3S,8S,2R,14R,16S,18R,21S. The use of VCD by comparative methods should gain popularity as instrumentation becomes more available and sensitivity improves.
Figure 33.

Synthetic epoxides (−)-127 and (+)-127 used for comparison of VCD to citrinadin A (125).
7.12 Sagittamide A
The sagittamides,clxi reported by Lievens and Molinski from an unidentified tunicate collected in Micronesia are polyacetoxy long–chain α,ω-dicarboxcylic acids terminated as amides of ornithine and valine.
L-ornithine and L-valine were identified by Marfey’s analysisclv of the acid hydrolysate of 128. Two different configurational assignments by the Molinskiclxii and Kishiclxiii groups were proposed for the contiguous hexa–acetoxy segment, however the Kishi configuration was verified by total synthesis as described.clxiv
The UDB approach was advanced beyond 13C chemical shift profiles to 3JHH coupling constant profiles derived from synthetic compounds of known configuration reported in the literature.clxv Profiles were generated for all the diastereomers of a contiguous tetracetate for a total of eight subgroup profiles (SSS, AAA, ASA, SAS, SSA, ASS, SAA, and AAS) (Figure 35). Polyacetates containing greater than four contiguous stereocenters can be assigned by overlap of adjacent coupling constant profiles.
Figure 35.
3JHH profiles for tetraol peracetates. (A = anti, S = syn). Adapted with permission from Seike, H.; Ghosh, I.; Kishi, Y. Attempts to assemble a universal NMR database without synthesis of NMR database compounds Org. Lett. 2006, 8, 3861–3864. © 2006 American Chemical Society (ref. clxv).
The hexa–acetoxy profile (A) of 128 was divided into three subgroups: (D) C5–C8, (C) C6–C9, and (B) C7–C10. The profile for each subgroup was compared to the subgroup profiles generated for the tetraol peracetates. The closest match for subgroups D, C, and B were SAA, ASA, and SAS, respectively. The RC of the hexa–acetoxy segment to be defined as 5S*,6S*,7S*,8S*,9S*,10R*.
The Kishi group proceeded to synthesize two diastereomers 128 and the antipode of 129 (Figure 37). 1H NMR mixing experiments showed antipode I was identical to natural sagittamide, while the antipode of 129 showed slight differences in the acetoxy 1H signals.
Figure 37.

Diastereomers synthesized by Kishi.
Griesinger and coworkers reported the use of residual dipolar couplings (RDCs) for the assignment of the RC of the hexa–acetoxy segment of sagittamide A.clxvi A combination of JBCA and NOE data narrowed the RC to four diastereomers. Measurement of RDCs and comparison to Boltzman–weighted calculated values gave only one diastereomer (with the same RC as 128) with data consistent with the measured values. RDCs, up until now an undervalued NMR parameter in natural products structure determinations, should find expanded use in the future, particularly for resolving ambigous solutions from JBCA.
7.13 Gymnocins
Gymnocin A (130), a highly cytotoxic polyether toxin was isolated from the red tide dinoflagellate Karenia (formerly Gymnodinium) mikimotoi by Satake and coworkers in 2002.clxvii The structure of 130 was assembled through analysis of 2D NMR and detailed analysis by FAB collision–induced dissociation (CID) MS. The RC of the polyether system (all rings oriented in the trans cisoid fashion) was assigned from NOE and coupling constant data. The C50S AC of gymnocin A was addressed by application of the MMM to esters 131ab. Soon after the isolation, asymmetric synthesis of (+)-gymnocin A (130) was completed by Sasaki, verifying the absolute stereostructure.clxviii
In 2005 Satake and coworkers reported another polyether derivative related to 130 they named gymnocin B (132).clxix Gymnocin B is the largest contiguous polyether compound reported to date. The structure determination (planar and relative conformation) of 132 was carried out in a similar fashion as gymnocin A. Unfortunately, the secondary hydroxyl groups were unreactive towards MTPA-Cl, and the MMM was not applicable. That same year, Berova and coworkers reported a tour de force effort to assign the AC of gymnocin B by chemical chiroptical methods.clxx
Long range exciton coupled CD between chromophores separated by 50Å was described by Nakanishi using p-(meso-triphenylporphyrin)-carboxylic acid (O-TPP) esters of brevetoxin B as chromophores.clxxi The brevetoxin polyether system adopts a preferred conformation in polar solvents (MeOH/H2O) that leads to a strong bisignate exciton coupled CD spectrum. These principles were applied to the assignment of gymnocin B (132).
TPP chromophores were attached to the less reactive –OH groups in relatively high yield by a combination esterification/cross metathesis protocol (Scheme 20). This method was perhaps inspired by previous successes in alkene metathesis with styrenes to install benzenoid chromophoresclxxii to allylic alcohols and allylic amines.clxxiii Gymnocin B (132, 300 μg) was converted to the acetonide (2,2 dimethoxypropane, PPTS), and acylated (acryloyl chloride) to give an acrylate ester 133 that was subjected to cross metathesis with vinyl–TPP 135 in the presence of Grubbs’ second generation catalyst (134) which gave bis-TPP-cinnamate ester 136 (~10 μg). A positive split Cotton effect (λ (MeOH) 419 nm (ΔΣ +11), 414 nm (ΔΣ −15)) was observed in the CD spectra of 136 due to long-range exciton coupling, which confirmed attachement of both TPP groups.
Scheme 20.

Conversion of gymnocin A (130) to MTPA esters (131ab), and conversion of gymnocin B (132) to bis-TPP ester (136).
The relative orientation of the hydroxyl groups (pseudoaxial or pseudoequatorial) in 136 were critical for defining the direction of electronic transition dipoles. Evaluation of coupling constants and NOE data for gymnocin B (132) established that both hydroxyl groups (C10 and C37) are oriented in the pseudoaxial position (Figure 39). Conformational analysis (MMFF94) on a series of truncated monocinnamate (rings systems including: A–C, I–K, H–K, G–K, and F–K) and biscinnamate models (A–O) verified that the cinnamate esters orient in the axial position and with an s-cis conformation of the double bond. Conformational analysis of the complete gymnocin B TPP cinnamate system showed that while the C10 ester remains in the axial position, the C37 ester adopted an equatorial position. The Boltzman distribution of the lowest energy conformations (< 7 kcal/mol), showed the projection angle is positive in sign and should give rise to a positive split Cotton effect if gymnocin B (132) contained the 10S and 37S configuration. Finally, quantitative calculated CD spectra of the three lowest energy conformers showed that the magnitude of the Cotton effects were of substantial magnitude.
Figure 39.
NOESY correlations establishing the RC of the BC and JK ring systems of gymnocin B (132).
7.14 Spirastrellolides
The think–red encrusting sponge, Spirastrella coccinea from Dominica has been the source of a group of highly antimitotic spiroketal macrolides named the spirastrellolides, isolated by the Andersen group.clxxiv To facilitate purification, the spirastrellolides were converted to their corresponding methyl esters by treatment of either the crude extract or partially purified fractions with TMS-diazomethane. Spirastrellolide A methyl ester is the most abundant, and showed high anti-mitotic activity against MCF7 human cancer cells (80 nM)clxxv. The planar structure and partial RC for spirastrellolide A was depicted as 137 (Figure 40) in the initial report based on HRCIMS and 2D NMR analysis. The RC of the C13–C21 spiroketal and C27–C29 portion of the second spiroketal were assigned by coupling constant and ROESY correlations.
Figure 40.
Originally proposed structure for spirastrellolide A (137), and corrected structure for spirastrellolide A (138).
The following year, larger amounts of spirastrellolide A methyl ester (45.1 mg) were purified which allowed chemical conversions to be carried out. ESIFTMS of peracetylated spirastrellolide A suggested that the originally assigned structure was incorrect due to an error in the original molecular formula; the correct molecular formula should be C53H83O17Cl.clxxvi Further NMR analysis showed spirastrellolide A is the structure depicted 138. Following revision of the planar structure, a thorough investigation to assign the RC was carried out by extensive analysis of J coupling and ROESY data of the diacetonide derivative 140 (Figure 40). This analysis led to stereochemical assignments of the three isolated stereogenic units (C3–C7, C9–C21, and C27–C38) that reduced the number of possible diastereomers for 133 to 16.
The RC and AC of spirastrellolide B (141), except C46, were solved by X-ray diffraction by chemical conversion of a dihydro, dechloro derivative of 138.clxxvii Detailed NMR analysis of spirastrellolide B methyl ester (142) and comparison to spirastrellolide A methyl ester (139) revealed that the RC was conserved between both macrolides. The bisacetonide derivative (143) of spirastrellolide B was subjected to perruthenate cleavage (RuCl3/NaClO4) and alkylation with p-bromo-α-bromoacetophenone to provide phenacyl ester 144 (Scheme 21). Under these conditions, the C22/C23 acetonide had been cleaved and the C23 hydroxyl oxidized to the C23 ketone.
Scheme 21.
Structure for spirastrellolide B (142) and conversion to p-bromophenacyl derivative (144).
Slow evaporation of 144 from CH3CN gave crystals suitable for X-ray diffraction studies. X-ray data showed the RCs for the three subunits in spirastrellolide A were identical to those in spirostrellolide B, and the presence of a heavy atom (bromine) provided the anomalous dispersion required to solve the absolute stereostructure. The unique structure of spirastrellolide and its potent biological activity has attracted significant attention in the synthetic community. Several fragment syntheses of 141 have been reported by De Brabander,clxxviii Forsyth,clxxix Paterson,clxxx Hsung,clxxxi Furstner,clxxxii, Smith,clxxxiii and Phillips.clxxxiv Total syntheses of spirastrellolide A methyl ester and spirastrellolide F methyl ester have been accomplished by the Patersonclxxxv and Furstnerclxxxvi groups, respectively.
7.15 Shishididemniols
In 2007, Matsunaga and coworkers disclosed the complete structures of long chain lipids, shishididemniols A (145) and B (146) from a tunicate of the family Didemnidae. Compounds 145 and 146 showed antibacterial activity against the fish pathogenic bacterium Vibrio anguillarum (20 μg/6.5 mm φ disk zone of inhibition; 8 and 7 mm for 145 and 146, respectively). The shishididemniols resemble serinolipids previously reported from Didemnid tunicates (e.g. didemniserinolipid B (160)clxxxvii and cyclodidemniserinolipid A (159)clxxxviii).
The planar structures of the shishididemniols were established by 2D NMR and MS. The location of the C16 hydroxyl was assigned by analysis of fragmentation ions observed by FAB-MS/MS data. The structures of the shishididemniols contain several isolated stereochemical elements, and presented several challenges for configurational assignment. Each isolated stereo segment was independently assigned by a combination of chemical conversions, and several different uses of CDAs.
The cis RC of the epoxide was assigned from observation of large vicinal coupling (J = 4.1 Hz). The C6/C7 RC was assigned by conversion of 145 to the C6/C7 acetonide 149 (Scheme 22). CBz-protected shishididemniol A (147), was subjected to epoxide ring opening (MgBr2• Et2O) which gave bromohydrin (148), that was converted to the acetonide 149.
Scheme 22.

Conversion of shishididemniol A (145) to acetonide (149), Δδ’s (ppm) from application of the MMM to C2, C6 and C30.
NOESY crosspeaks for acetonide (149) revealed the trans orientation. The 2S, 6S, and 30S stereochemical assignments were assigned by the MMM from MTPA esters 151ab. Acetonide (149) was also converted to its MTPA esters (150ab), which showed the configurational assignment at C2 was consistent with the MTPA esters (151ab) derived from 145.
The AC of the tyramine portion of the molecule was addressed by cleavage of the amide bond, conversion to the per-MTPA esters and comparison to model compounds (Scheme 23). Acidic hydrolysis of shishididemniol A (145) gave a mixture of epimers at the C2′ position, however hydrazinolysis gave only one isomer which was treated with both (R)- and (S)- MTPA-Cl and gave esters 153ab. For 1H-NMR comparisons, two sets of diatereomeric esters were synthesized: 154ab for comparison of H9′ to H11′ and 155ab for comparison to H1′ and H2′. Comparative NMR analysis showed the tyramine portion to contain the 2’R and 10’S ACs.
Scheme 23.

Conversion of shishisididemniol A (145) to tyramine MTPA esters (153ab), and selected 1H NMR chemical shifts for 153ab, and synthetic models 154ab and 155ab.
Chiral derivatizing agent, 2-naphthylmethoxyacetic acid (2-NMA) was used to assign the remaining isolated C16 stereocenter (Scheme 24). Anisotropic studies have shown that 2-NMA shows favorable anisotropic effects over long distances, and 2-NMA was been used previously for the assignment of remote stereocenter in ginnol.xxx Bromohydrin 148 was subjected to periodate cleavage followed by reduction (NaBH4) to the primary diol 156, which was protected as the TBDPS ether 157. (R)- and (S)-2-NMA were separately coupled to the secondary alcohol 157. Differential 1H NMR analysis of the derived esters 158a,b showed C16 was consistent with the R configuration.
Scheme 24.

Conversion of bromohydrin 148 to (R)- and (S)- 2-NMA esters (158ab) and Δδ values.
The complete stereochemical assignment of shishididemniol A included a number of chemical conversions, and various incorporations of MTPA esters to assign the AC of the six isolated stereocenters. The C16 stereocenter was assigned by 1H NMR of the (S)– and (R)– 2-NMA esters, which showed differential 1H chemical shifts ten carbons removed from the stereocenter. The methodology used in this investigation should be useful for other serinol lipids of this class.
7.16 Phorbasides
The phorbasides A (12), B–E, and F (10)xvi from the marine sponge Phorbas sp. are recent additions to a group of cytotoxic glycosidic 14-membered ring macrolides from various marine organisms; others include aurisides A (161)clxxxix and B and dolastatin 19 (162)cxc from the sea slug Dolabella auricularia, callipeltoside A (163) from the sponge Callipelta sp.,cxci and lyngbyaboulloside (164) from the cyanobacteria Lyngbya bouillonii.cxcii Configurational assignment of auriside A (161) by combined J coupling and NOESY analysis which established the complete RC. The AC was assigned by degradation, and comparison with authentic standards of 165 and ent-165. The configurational assignment of the aurisides was verified by total synthesis by Paterson.cxciii
The callipeltosides and phorbasides contain 3′-O-methyl evalose, a C–methyl sugar, and an additional chromophoric stereochemical element, a diene-yne chlorocyclopropane and ene-yne chlorocyclopropane, respectively. Although the RC of the cyclopropane has been assigned by coupling constant and NOE data, no general method has been presented to establish the relative and AC between the macrolide ring and chlorocyclopropane. Prior to the report of the phorbasides, configurational assignment of the chlorocyclopropane ring of callipeltoside A had been achieved only after total synthesis reported by Trostcxciv and Evans.cxcv
The Molinski group developed a quantitative approach to simultaneously solve the AC of both the configuration of the side chain and C13 of the macrolide ring using circular dichroism (Figure 45). Both UV and CD spectra of phobasides A and B were dominated by the ene-yne chlorocyclopropane chromophore, and displayed a moderately intense positive CE [λmax 232 nm (Δε +9.1), 241 (+8.1)]. The contribution of the ene-yne chlorocyclopropane lacking the C13 stereocenter was observed by synthesis of two stereospecific model compounds 166 and ent-166. The sign of the CEs for model compounds 166 and ent-166 was informative of the configuration of the cyclopropane, but the magnitude was less than expected. The additional contributions to the Cotton effects were attributed to the chiral C13 stereocenter and confirmed by comparison with two additional synthetic diastereomers 167 and 168. The CD spectrum of 167 was almost identical to that phorbaside A (12) showing, unexpectedly, that the AC of the yne-chlorocyclopropane of 11 is opposite to that of callipeltoside A (163).
Figure 45.
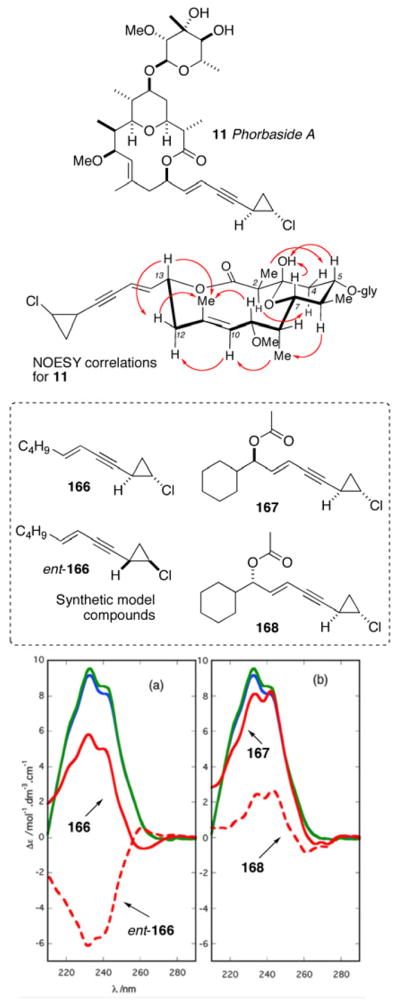
NOESY correlations used to establish the relative conformation in phorbaside A, and synthetic models (166–168) used for quantative CD comparison. (a) CD spectra of phorbaside A (12), 166, and ent–166. (b) CD spectra for 12, 167, and 168. Reproduced with permission from Skepper, C. K.; MacMillan, J. B.; Zhou, G-X.; Masuno, M. N.; Molinski, T. F. Chlorocyclopropane Macrolides from the Marine Sponge Phorbas sp. Assignment of the Configurations of Phorbasides A and B by Quantitative CD. J. Am. Chem. Soc. 2007, 129(14), 4150–4151. © 2007 American Chemical Society. (Ref. xvia.
The assignment of the AC of the 2-O-methylevalose glycone of phorbaside A was developed.xvi Assignment of sugar configuration by exciton coupled CD of dibenzoates was described by Nakanishi,cxcvi however this approach less practical for the tertiary OH in the C–methyl sugar, O–methyl evalose. Consequently, a more reactive aryl-isocyanate was chosen for derivatization. Phorbaside A (12) was subjected to a three-step degradation sequence: methanolysis, naphthoylation, and addition of 2-isocyanato-6-methoxynaphthalene to give carbamate 170 (Scheme 25). The CD spectrum of 170, derived from L-rhamnose gave a weak positive bisignate Cotton effect [λmax 241 nm (Δε +2.6), 226, (−1.5); A = 4.1] identical in sign and magnitude to that of authentic 170 prepared from L-rhamnose.
Scheme 25.
Conversion of phorbaside A (12) to dichromophoric (170). CD spectra for (a) synthetic–170 and (b) 170 derived from phorbaside A (taken from Ref. xvib).
The configurational assignment was verified by the total synthesis of phorbaside A (12) by Paterson and Paquetcxcvii and measurement of the CD spectrum of the product.
7.17 Goniodomin
Goniodomin was purified first by Burkholder and coworkers in 1968 from the marine dinoflagellate Goniodoma sp. collected in La Parguera, Puerto Rico.cxcviii At the time, limited structural information was provided. Murakami and coworkers, working a decade later on a different sample of goniodomin A obtained from Goniodoma pseudogoniaula, provided additional configurational detail through analysis of extensive NMR experiments (COSY, COLOC, PSNOESY).cxcix
Almost two decades passed before the Fujiwara group disclosed the synthesis of model compounds to establish the RC of goniodomin A (171, Figure 47).cc Synthesis of A-ring models 172 and 173, and comparison of NMR data revealed consistency with goniodomin A. In particular, the C32/C33 and C33/C34 RCs are assigned to the cis and trans configurations, respectively. Macrocyclic model compounds 176 and 177 were synthesized to represent the D/E rings of goniodomin A (171). Compound 176, with a trans C21/C22 relative confiuguration, was more consistent with the coupling constant observed for goniodomin A.
Figure 47.

Model compounds synthesized by Fujiwara.
In 2008, just prior to Fujiwara’s disclosure of the RC of the DE ring system, the Sasaki group proposed the relative and AC of goniodomin A (171) based on high-field NMR (500, 600, and 900 MHz) experiments in different solvents, chemical conversion of the natural product, and comparison with synthetic model compounds.cci High-field NMR experiments, J-coupling and NOESY correlations were used to secure the RC of the relatively rigid macrolide ring containing A through E (Figure 48(a and b)). The threo orientation of the C26/C27 diol was assigned by conversion to the corresponding acetonide (178, Scheme 26) and NOESY correlations from one methyl group to H26 and the other methyl group to H27. The RC of the macrolide portion was relayed to the C31 sterocenter by NOESY correlations (Figure 48(c)).
Figure 48.

Selected NOESY correlations for the assignment of RC of the macrolide ring and sidechain (ring F) for goniodomin A (171).
Scheme 26.

Converision of goniodomin A to acetonide 178 and di-aldehyde 179, and synthetic model aldehydes 181 and 182.
The RC between C31 and C32 as well as the AC were assigned by chemical methods (Scheme 26). After considerable experimentation, it was found that cleavage of the C26, C27 diol of 171 with Pb(OAc)4 gave labile di-aldehyde 179 of sufficient stablility for immediate NMR analysis before spontaneous elimination to aldehyde 180. Synthesis of model compounds 181 and 182 provided chemical shift data that was indicative of the RC between H31 and H32. The NMR data for di-aldehyde 179 showed a closer match to 181.
The AC of goniodomin (171) was secured from conjugated ketone 183a, prepared by treatment of 180 with (S)-MTPA-Cl (Scheme 27). Authentic diastereomeric MTPA esters 183ab were formed from 181, which showed goniodomin (171) contained 33R and 34S configurations.
Scheme 27.

Conversion of model ketone (S)- or (R)- MTPA-Cl.
7.18 Plakinic Acids I – L
Relatively few methods exist for solving the absolute stereochemistry of remote or isolated methyl branched stereocenters. The Molinski group applied liposomal CD (L-CD) of naphthamides to assign remote methyl branched stereocenters in plakinic acids I (184) and J (185)ccii, endoperoxides from the marine sponge Plakortis halichondroides, collected in the Bahamas. ω-Phenyl polyketide peroxides have received interest due to their sub-micromolar activity against parasites, including the malaria vector Plasmodium falciparum.cciii
The relative and and AC of the endoperoxide (1,2-dioxane) was solved by conventional methods (NOESY measurements and the MMM). Plakinic acid I (184) was methylated (CH2N2, ether), and subjected to hydrogenolysis to the ring opened diol (188), which was transformed into both MTPA esters (189ab). Conventional analysis (nOe, Mosher’s ester analysis) led the 3S, 4S, 6R configurations (Scheme 28).
Scheme 28.

Configurational assignment at C3 of plakinic acid I.
The stereochemistry at C8 in the acyclic chain of plakinic acids I (184) or J (185) was not readily assignable by standard NMR methods that typically relay stereo–information only over a limited number of bonds removed from a chiral center of known configuration (C6). Plakinic acid I (184) was reduced with aqueous Fe(II)Cl2 under oxygen-free conditions to give three products, 190, 191, and 192 (Scheme 29) by intermolecular radical fission. Primary alkyl chloride (190), which arises from radical ‘rebound’ to the Fe(III)-Cl coordination sphere, was converted to the azide (193) and the product reduced to the free amine (194), which was derivatized with 6-methoxy–2–naphthoyl chloride to furnish naphthamide 195.
Scheme 29.

Conversion of plakinic acid I (184) to naphthamide 195.
The CD spectrum of 195 in MeOH showed no significant Cotton effects due to conformational averaging, however CD spectra acquired in ordered arrays (DSPC liposomes) revealed strong Cotton effects of the opposite sign and magnitude as synthetic (S)-195 (Figure 51). Therefore the configuration at C8 in plakinic acid I was assigned as R. The same approach was used for plakinic acid J (185) also revealed the 8R configuration. Plakinic acids K (186) and L (187), containing two remotely-spaced methyl branches, were assigned by liposomal CD after a similar degradation protocol to dimethyl naphthamide 196 and comparison to optically active synthetic models (197–199).cciv The liposomal CD spectrum of 196 was dominated by Cotton effects of similar sign, but subtle differences in magnitude and fine-structure were attributed to diastereomeric differences of extended chromophores in the liposomal bilayer.
Figure 51.

CD spectra of napthamides derived from plakinic acids: L–CD of (d) (S)-195, (e) (±)-195, (f) (R)-195; (b) L-CD of (2R, 6S)-196 from 186 (solid line), (2S,6R)-197 (dotted line); (c) L–CD of (2R,6R)-199 (solid line), (2S,6S)-198 (dotted line). Adapted with permission from Dalisay DS, Quach T, Nicholas GN, Molinski TF. Amplification of the Cotton effect of a single chromphore through liposomal ordering-stereochemical assignment of plakinic acids I and J. Angew Chem Int Ed Engl 2009; 48(24):4367–71 and Dalisay S. D., Quach T., Molinski T. F. Liposomal Circular Dichroism. Assignment of Remote Stereocenters in Plakinic Acids K and L from a Plakortis -Xestospongia Sponge Association. Org. Lett. 2010, 12, 1524–1527. © 2010 American Chemical Society (references ccii and cciv).
7.19 Biselyngbyaside
Biselyngbyaside (200), a glycosidic polyketide macrolide from the marine cyanobacteria Lyngbya sp. from Okinawa that was reported by Suenaga and coworkers in 2009. Biselyngbyaside showed a mean GI50 of 0.60 μM against 39 human cancer cell lines. The 2D structure was established in a straight forward manner by MS and 2D NMR, and the RC of the 3-O-methyl glucoside was assigned by NOESY and 1H-1H couplings, all protons showed axial orientation.
The remote stereocenters insulated by sp2 carbons precluded assignment by J-based methods. The configurations at C3 and C7 were addressed by chemical degradation, and the MMM. Biselyngbyaside was subjected to hydrolysis under basic conditions to give methyl ester 201, and was converted to both (S)- and (R)- MTPA esters (202ab, Scheme 30). The Δδ values were consistent with the 18R configuration.
Scheme 30.
Hydrolysis of biselyngbyaside, and configurational assignment of C16 by the MMM.
Since the methanolysis induced elimination of the C3 oxygen, an approach was chosen to reduce this problem. Hydrogenolysis under acidic conditions provided methyl ester 203 and an anomeric mixture of O-methylglucosides 204 (Scheme 31). The MMM was applied to 205ab derived from 203 to give the R configuration for C3. The AC of the glucoside 204 was assigned by the CD spectra after conversion to both tri-p-bromo-benzoate derivatives 206 and 207 which were separated by HPLC. The tribromobenzoate derivative 207 showed identical CD spectra (λmax 238 nm (Δε +7.2), 254 nm (Δε −17.2) to the tribromobenzoate derived from 1-O-methyl-D-glucose.
Scheme 31.
Hydrogenolysis of biselyngbyside.
The final two stereocenters in biselyngbyaside (200) were identified by a three-step degradation, followed by NMR and chromatographic comparison to optically pure standards (Scheme 32). Ozonolysis of 200 followed by reduction gave the corresponding diol, which was immediately derivatized with p-bromophenylisocyanate gave 208 in good yield. Optically enriched standards 208 and 209 were prepared for comparison of NMR data to differentiate diastereomers. Synthetic and naturally-derived 208 were identical by NMR. To identify the AC of natural 208, its corresponding enantiomer was synthesized for comparison by HPLC analysis. Chiral HPLC (Chiralpak IA) showed natural and synthetic 208 to have identical retention times (tR = 6.5 min) when compared to ent-126 (8.1 min), which corroborated the 7S and 10S configuration for biselyngbyaside (200).
Scheme 32.
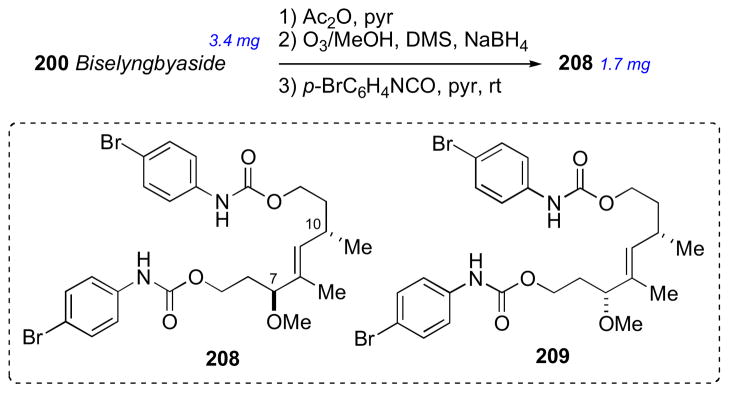
Oxidative degradation of biselyngbyside (200), and synthetic models (208–209).
7.20 Muironolide
Muironolide A (210) is the third class of macrolides along with phorboxazoles and phorbasides co-occurring in the marine sponge Phorbas sp from Western Australia. Three structural features set muironolide A (210) apart from other marine macrolides: a hexahydro-1H-isoindolinone-triketide ring, a trans-2-chlorocyclopropyl ketide (CCK), and trichloromethylcarbinol ester. The entire structure determination and preliminary biological testing was carried out on the total available sample – 90 μg of isolated compound.
The planar structure was established by MS and NMR data. The trichlorocarbinol ester was confirmed by comparison of 1H and 13C spectra to model compound 211.ccv The RC of the isoindolone ring and macrocycle were addressed by coupling constant and NOESY data (Figure 54).
Figure 54.
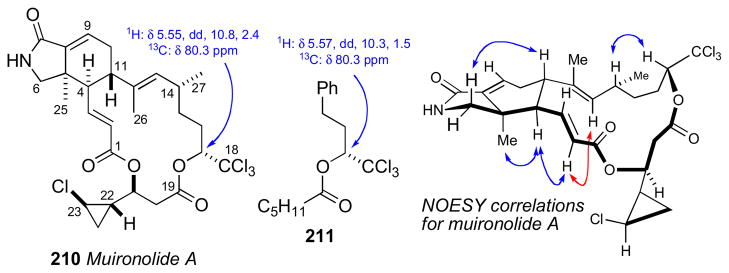
Muironolide A and NOESY correlations.
Attempted assignment of the RC by NOESY or J-based methods were unsuccessful in relaying the stereochemistry of the CCK element to the other macrolide ring stereocenters. Consequently, a microscale degradative approach was used for 211, and correlation of the products with standards of known configuration – both enantiomers of diastereomeric 212 and 213 (Scheme 33). Muironolide A (112, 30 μg) was subjected to alkaline hydrolysis followed derivatization to the naphthacyl ester 212 that was shown to be identical retention time (Chiralpak AD) to authentic standard.
Scheme 33.

Degradation of Muironolide A (210) and configurational assignment of the CCK unit.
The CD spectra for 210 [λmax 186 (Δε +58.5), 225 (−37.2)] revealed a negative exciton split Cotton effect due to coupled π→π* transitions of two unsaturated carbonyl chromophores: the enamide C7–C9 and the enoate C1–C3. This was interpreted as arising from the negative torsional angle of −116° (obtained from NOESY correlations and molecular modeling) for the π→π* transition dipole and the 4R, 5R, 11S, 14R, and 17R configuration of muironolide.
7.21 Enigmazole
Enigmazoles A (214) and B (215) were isolated from a extracts of a the marine sponge Cinachyrella enigmata from Papua New Guinea, that showed differential activity against a c-Kit mutant of murine mast cells.ccvi They were identified using standard bioassay guided fractionation of the cytotoxic n-butanol extract. Several stereochemical assignment problems of these structurally unique macrolides were successfully solved through an elegant combination of microscale chemical conversions and capillary NMR measurements.
The planar structure for enigmazole A (214) was assigned by 2D NMR. Unique features included a phosphate ester and a 2,4-disubstituted oxazole side chain. Enigmazole A (214) was divided into four sections for stereochemical analysis: (a) C1–C5, (b) THP: C7–C11, (c) C15–C17, and (d) C23. Each subunit was adderessed independently, and then relayed together. A trans-disustituted tetrahydropyran (THP) ring was assigned from syn-diaxial protons with mutual NOESY crosspeaks and coupling constant data. The AC of 15S was determined by the MMM after methylation of the phosphate group (TMS-CH2N2) and conversion to both (R)- or (S)- MTPA-esters 216ab (Scheme 34).
Scheme 34.

Configurational assignment at C15 of enigmazole A (214).
Compound 214 was converted to 217 by phosphoramidation (CH3NH2, HOBt), base hydrolysis (KOH/MeOH), and methylation (Scheme 34). The RC of the syn-1,3-diol (C15/C17) was established by the 13C acetonide method on 218.xxi
Phenylglycine methyl ester (PGME) was chosen as a suitable CDA for the assignment of C2 (Scheme 35). Compound 217 was subjected to methanolysis and coupled to each of (R)- and (S)- 2-phenylglycine methyl ester (PGME, HOBt) to give 219ab.ccvii Analysis of the 1H NMR anisotropic shifts revealed the 2S configuration.
Scheme 35.

Ring opening to methyl ester 217, conversion to acetonide 218, and conversion to (R)- and (S)-PGME esters 219ab.
The phosphate group was resistant to enzymatic hydrolysis,ccviii but succumbed to mild basic conditions (wet DMSO, potassium acetate) to give lactone 220 (Scheme 36).ccix The C4S,C5S configurations established by NOESY correlations.ccx Only the 7R,11R diastereomer was consistent with J coupling, NOESY data, and molecular modeling.
Scheme 36.

Formation of δ-lactone 220, and conversion of side chain to dinitrophenylhydrazone derivative 221.
The final stereocenter in enigmazole A was assigned by chemical conversion and comparison with optically pure (R)-acetoin (Scheme 36). Perruthenate cleavage of 214 (RuCl3-NaIO4) gave 3-methoxy-2-butanone, which was converted to the 2,4-dinitrophenylhydrazone (221),ccxi and compared to that of an authentic sample obtained from (R)-acetoin.ccxii The Cotton effects in the CD spectra of naturally-derived and synthetic 221 were identical, therefore enigmazole A (214) has the 23R configuration, and the complete stereostructure is as depicted 214.
The structure elucidation of enigmazole A represents a tour de force; in total, 13 chemical manipulations were successfully executed on ~4.5 mg of enigmazole A or derivatives. The stereochemical assignment relied solely on chemical conversions, in contrast to JBCA analysis typically used for polyketide type compounds. The assigned stereostructure of 214 was identical to that of enigmazole A prepared by Skepper and Molinski through total synthesis.ccxiii
7.22 Xestoproxamines A–C
In 2011, Molinski and coworkers disclosed the structures of three bis-piperidine alkaloids xestoproxamines A–C (222–224) from the marine sponge Neopetrosia proxima.ccxiv The structures of 222–224 are related to halicyclamine A (225)ccxv and haliclonacyclamines A (226) and B (227).ccxvi
The 2D structures were established from MS and NMR data. NOESY and coupling constant data for the bispiperidine ring systems in 222 and 224 revealed the relative configuration of the four stereocenters. No known methods were reported for the AC assignment of polycyclic diamine alkaloids except for MMM when a secondary hydroxyl is present or X-ray crystallography. Double quaternization of xestoproxamine C (224) with two p-bromophenacyl groups and application of the exciton chirality method, and a Hoffman degradation/cross-metathesis protocol to detemine the stereochemistry of the remote branched methyl at C23.
Hydrogenation of xestoproxamine C (224) and conversion of the product 228 to the bis-methiodide salt 229 (Scheme 37), followed by ion-exchange to the ammonium hydroxid double salt followed by to microwave-promoted Hofmann elimination, afforded one major alkene product, 230. Alkene 230 was subjected to cross metathesis with excess 2-methoxy-6-vinylnaphthaleneccxvii (231) in the presence of Grubbs’ second generation catalyst (134),ccxviii and selectively engaged only the less hindered vinyl group derived from ring B to give the conjugated naphthalene 232. While the CD spectrum of 233 derived from xestoproxamine A showed only baseline, the Cotton effects observed in 232 were identical to those of optically active synthetic 234.
Scheme 37.
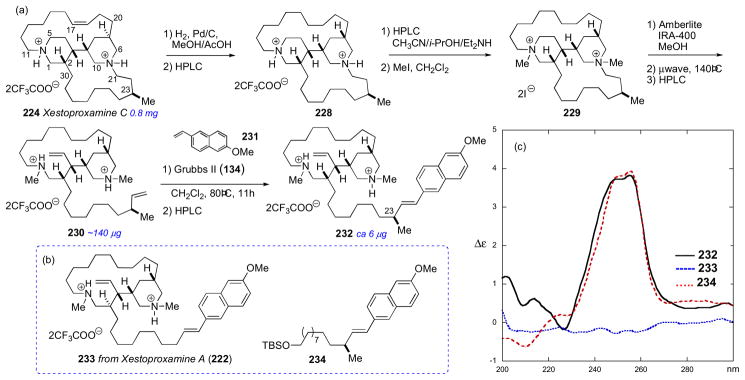
(a) Hoffman degradation/cross-metathesis of xestoproxamine C. (b) Hoffman degradation/cross-metathesis product from xestoproxamine A and optically enriched model compound 234. (c) CD spectra of 232, 233, and 234 in MeOH at 23°C. Reproduced with permission from Morinaka B..I, Molinski T.F. Xestoproxamines A-C from Neopetrosia proxima. Assignment of absolute stereostructure of bis-piperidine alkaloids by integrated degradation-CD analysis. J Nat Prod. 2011 Mar 25;74(3):430–40. Epub 2011 Feb 22. (Ref. ccxiv).
The absolute configuration of the bis-piperidine ring system was addressed by double quaternization with p-bromophenacyl bromide to provide bis-chromophoric derivative 235 (Scheme 38). Conformational analysis by NMR allowed assignment of the negative exciton couplet (λ 273 nm, Δε −5.8; 253 (Δε +2.4)) between the chromophores when N-CH2-(C=O)-C bond is in the preferred S-trans conformation. Similar transformations of (+)-haliclonacyclamine B and the enantiomorphic (−)-perhydrohaliclonacyclamine gave 236 and 237 that showed equal and opposite split Cotton effects – the former identical with 235 – consistent with their ACs.
Scheme 38.

(a) Double quaternization of xestoproxamine C and conformational analysis. (b) Bis-p-bromophenacyl derivatives of haliclonacyclamines of known configuration. (c) CD spectra of 235, 236, and 237 (MeOH, 23 °C). Taken from Ref. ccxiv.
8 Current Challenges to Configurational Assignment
The above examples demonstrate the execution of complete solutions to stereocomplex marine natural products using modern NMR/CD instrumentation, often augmented by synthesis, requiring only a few milligrams or even micrograms of a natural product,. Nevertheless, outstanding problems remain; the structures of several reported complex marine natural products are only partially solved or require re-evaluation.
Nigricanosides A (222) and B (Figure 59), unusual ether-linked glycoglycerolipids from the green alga Avrainvillea nigrans collected in Dominica, were purified by the Andersen group from.ccxix after extraction of over 20 kg of alga. The sub-milligram amounts of nigricanosides (A, 800 μg; B, 400 μg) available for structure elucidation were sufficient only for determination of the 2D structure and initial biological evaluation. Nigricanosides A and B methyl esters showed potent activity against MCF-7 and HCT-116 cells (~3 nM), and the free carboxylic acids acids were speculated to possess greater activity. The potent activity and unique structure prioritizes the nigricanosides as a key leads however, limited amounts of material precludes further stereocemical investigations, and completion of the nigricanoside stereostrutures awaits procurement of additional sample, or synthesis of appropriate diastereomeric models for spectroscopic comparisons. Nigricanosides stand as prime targets for total synthesis.
Figure 59.

Current challenging molecules for configurational assignment.
Caylobolide (224),ccxx a macrolide from the cyanobacterium Lyngbya majuscula, contains a series of 1,5-diols that are not readily assignable by standard methods. The Molinski group has developed a method of assignment of 1, n-diols (n = 5, 7, 9) using liposomal CD and TPP esters for,ccxxi however, the application to polyols requires better understanding of the fundamental photophysics of long-range exciton coupled CD of TPP and other chromophores withinin ordered liposomal bilayers.
Symbiodinolide (225),ccxxii a 62-membered polyol macrolide from the dinoflagellate Symbiodimnium sp. is a representative of the family of so-called ‘super carbon chain’ molecules that include palytoxin and maitotoxin.ccxxiii At nanomolar concentrations, symbiodinolide has been shown to induce significant increase in [Ca2+] in human neuroblastoma cells. The complete configurational assignment of symbiodinolide is ongoing, and the subject of intense stereochemical efforts by the Uemura group.ccxxiv
The oxazole-containing polyketide macrolide theonezolide A was isolated by Kobayashi in 1995 from the marine sponge Theonella swinhoei.ccxxv Although ozonolysis of 223 successfully yielded a number of degradation products, containing multiple isolated stereogenic centers. These degradation products have not yet yielded to stereochemical analysis; the problem is outstanding and the subject of ongoing investigations.
Marine sponges have been a significant source of polycyclic amine alkaloids. The RCs of these compounds are often easily addressed by NMR (NOESY experiments), however the AC of these molecules remain a challenge. The configuration of the secondary hydroxyl in polycyclic amine alkaloid upenamide (226),ccxxvi reported by Scheuer, is readily assigned by MMM, but the RC between the two rings is a significant challenge. Madangamine (227) is another polycyclic alkaloid that does not contain functional groups for chemical conversion, and awaits complete configurational assignment. Finally, relatively simple rigid macrolides still pose difficulties for structural assignment. For example, the structure of iriomoteolide-1a (228),ccxxvii a macrolide obtained from cultured Amphidium sp. and related molecules, was assigned based on integrated NMR techniques, particularly J-based analysis and nOe. The first synthesisccxxviii of the proposed structure 228 gave a product with the NMR spectral properties different from those reported for the natural product. A subsequent synthesis of the same structure by Horne and coworkers lead to the same result, “calling into question the original structural assignment”.ccxxix Spectroscopic data of several other synthetic diastereomers of 228 also failed to match those of natural iriomoteolide 1a; consequently, at this time, the true structure of iriomoteolide-1a is in need of reappraisal.
9 Conclusions
Natural products (NPs) are a significant and important source for new small-molecule drug leads. Stereocomplexity often distinguishes the structures of natural products from synthetic medicinal drugs, however, many complex natural products are listed in the US and other pharmacopeia, and new structures are likely to find to their way into the pages of these compendia. The deficit in new drugs for treatments for many disease conditions, particularly neurodegenerative disease such as Alzheimers, diabetes and complications that arise from metabolic syndrome, and antiinfectives against resistant pathogens (e.g MRSA) make it likely that natural products will continue to provide leads to supply pipelines of drug discovery. The foregoing examples illustrate the power of integrated structure analysis by spectroscopic techniques and chemical methods augmented by rational asymmetric synthesis. Reliable solutions to their structures are likely to be refined as current needs remain and new rare chemical entities are discovered.
Figure 1.
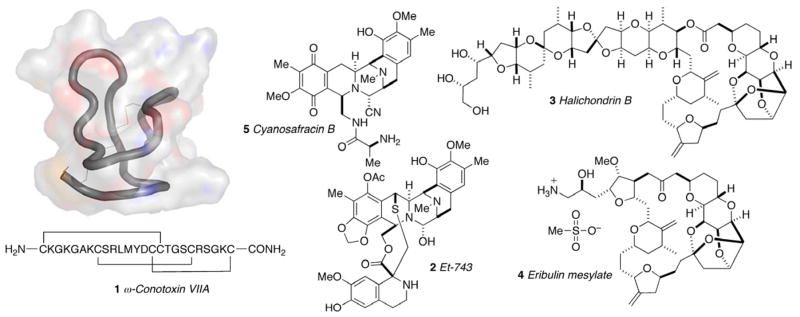
Currently approved marine-derived pharmaceuticals: ω-conotoxin MVIIA (Prialt®, 1), Et-743 (Yondelis®, 2), halichondrin B (3), and eribulin mesylate (Halaven®, 4).
Figure 2.
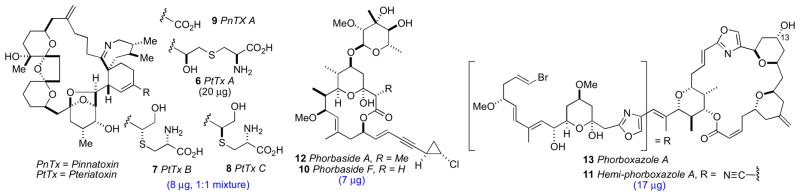
Examples of marine natural products characterized at the microgram scale.
Figure 3.

Conformations, chemical shifts, and nOes for (a) syn-1,3-diol acetonide (SDA) and (b) anti-1,3-diol acetonide (ADA).
Figure 6.
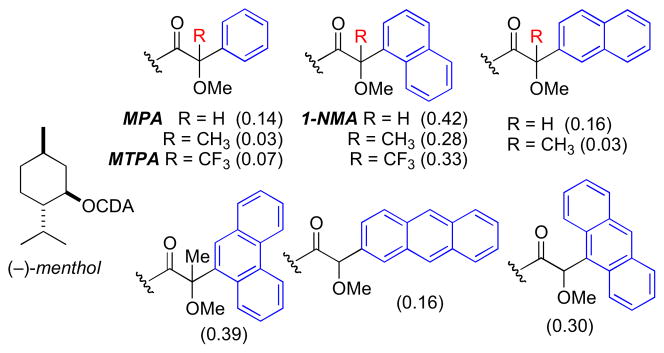
Discriminating power of various CDAs. Mean ⊗™ values of menthyl esters. (Δδ = δR – δS).
Figure 7.
Synthetic databases included in the Universal NMR Database (UDB) by Kishi.
Figure 8.
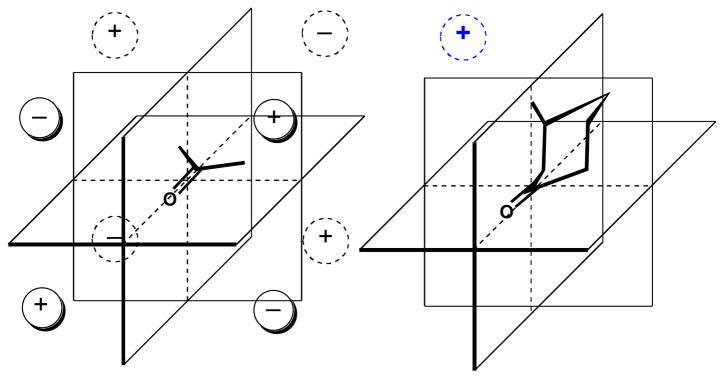
Octant rule for saturated ketones and the origin of the net positive Cotton effect observed of (3R) methylcyclohexanone.
Figure 10.
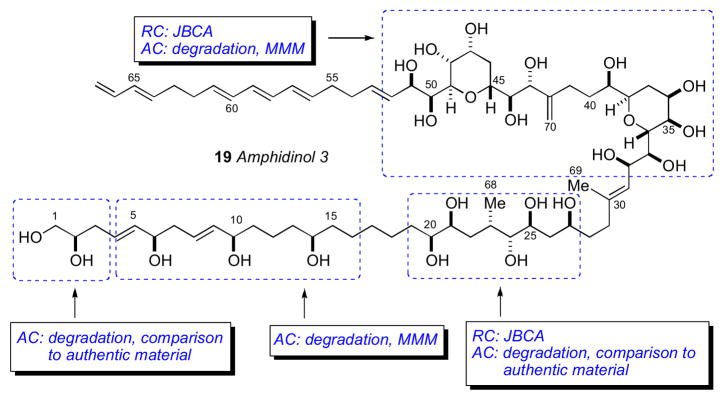
Murata’s strategy for configurational assignment of amphidinol 3 (19).
Figure 11.

Structures of mycalolides (34–39).
Figure 12.

Fusetani/Panek strategy for configurational assignment of the mycalolides.
Figure 15.
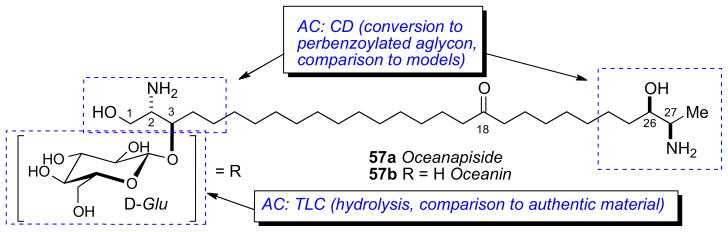
Molinski’s strategy for configurational assignment of oceanapiside. The position of the keto-group was revised from C11 to C18. See Ref. xcvc.
Figure 16.
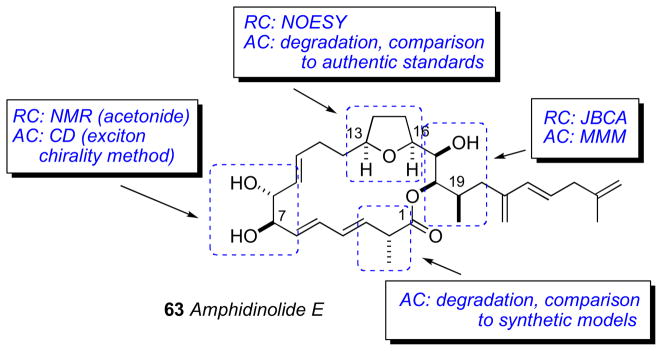
Kobayashi’s strategy for configurational assignment of amphidinolide E.
Figure 17.

Structures for Bistramides A (71) and C (72).
Figure 18.
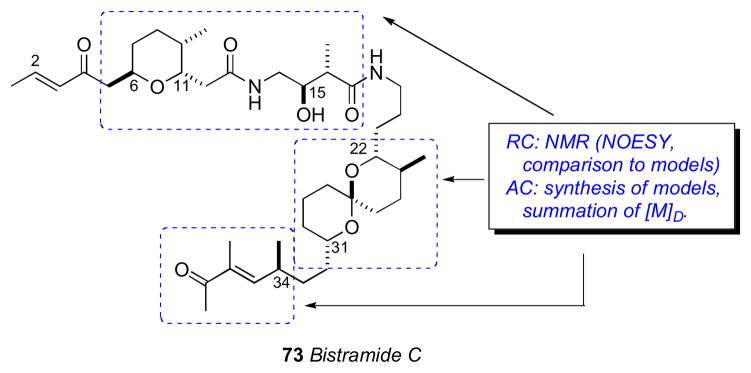
Wipf’s strategy for the configurational assignment of bistramide C (73).
Figure 22.

Schulzeines A-C (80–82).
Figure 23.
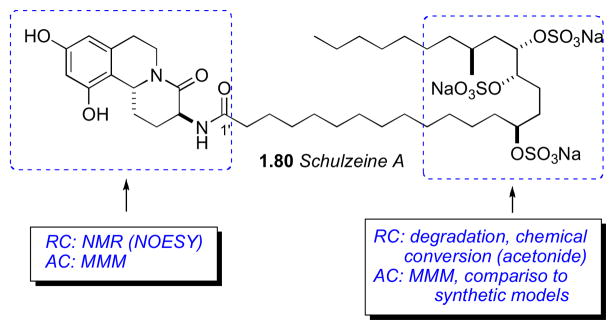
Fusetani’s strategy for configurational assignment of schulzeine A (80).
Figure 24.
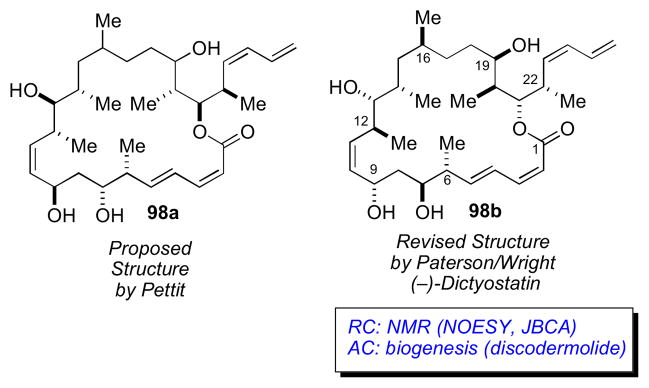
Pettit’s proposed structure and Paterson/Wright revised structure for dictyostatin.
Figure 28.

Structures for psymberin (100), pederin (101), and onnamide (102).
Figure 30.
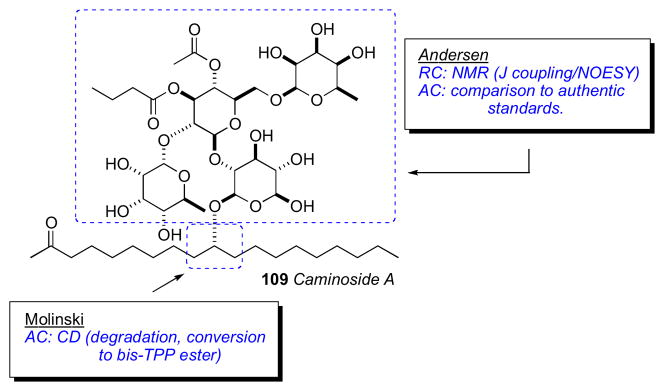
Strategy for configurational assignment of caminoside A (109).
Figure 31.
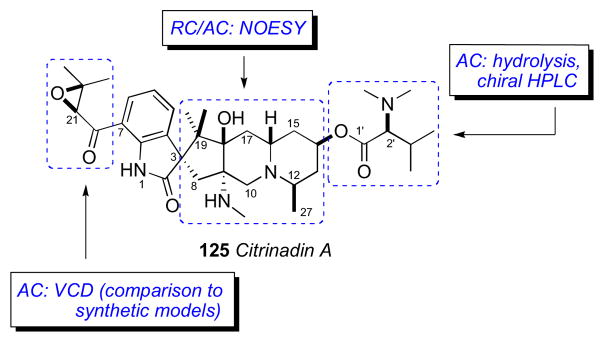
Kobayashi’s strategy for stereochemical assignment of citrinadin A (125).
Figure 34.

Strategies by Molinski and Kishi for the configurational assignment of sagittamide A (128).
Figure 36.

UDB approach for assigning the hexahydroxy acetate portion of sagittamide A. (A = anti, S = syn). Adapted with permission from Seike H, Ghosh I, Kishi Y. Stereochemistry of sagittamide A: prediction and confirmation. Org Lett. 2006 Aug 17;8(17):3865–8. © 2006 American Chemical Society (ref. 163).
Figure 38.
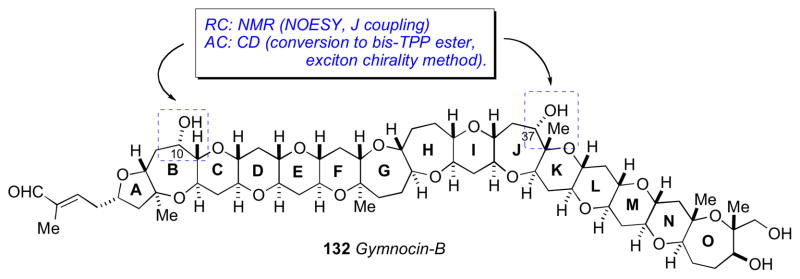
Berova’s strategy for configurational assignment of gymnocin B (132).
Figure 41.

Structures of shishididemniols A (145) and B (146), didemniserinolipid B, and cyclodidemniserinol sulfate.
Figure 42.
Matsunaga’s strategy for configurational assignment of shishididemniol A (145).
Figure 43.

Structures and activities for auriside A (161), callipeltoside A (163), lyngbouilloside (164), dolastatin 19 (162), and phorbaside A (12).
Figure 44.
Molinski’s strategy for configurational assignment of phorbaside A (12).
Figure 46.
Strategies by Fujiwara and Sasaki for the configurational assignment of goniodomin A (171).
Figure 49.
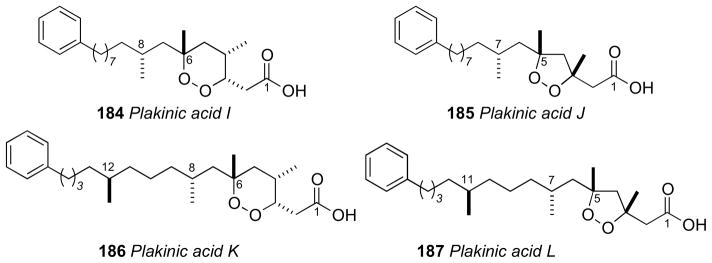
Structures for plakinic acids I – J (184–187).
Figure 50.
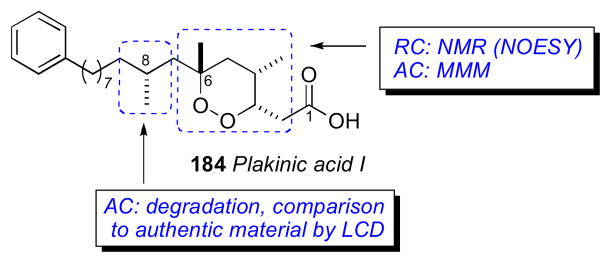
Molinski’s strategy for the configurational assignment of the plakinic acids.
Figure 52.

Suenaga’s strategy for configurational assignment of biselyngbyaside (200).
Figure 53.
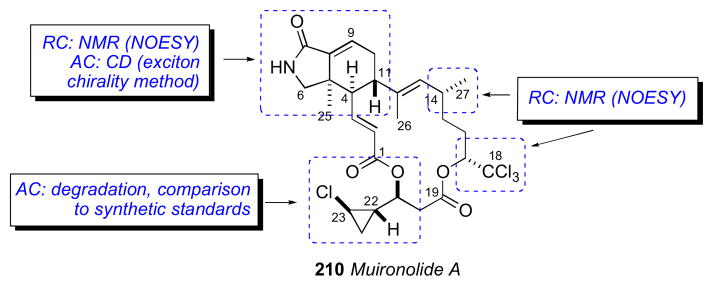
Molinski’s stategy for configurational assignment of muironolide A (210).
Figure 55.

Structure of enigmazoles A (214) and B (215).
Figure 56.
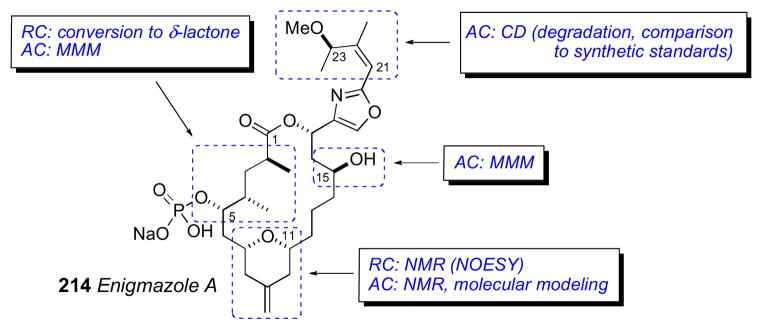
Gustafson’s strategy for the configurational assignment of enigmazoles A (214).
Figure 57.

Structures of xestoproxamines A–C (222–224), halicyclamine A (225), and haliclonacyclamines A (226) and B (227).
Figure 58.

Molinski’s strategy for the configurational assignment of xestoproxamine C (224).
Scheme 1.
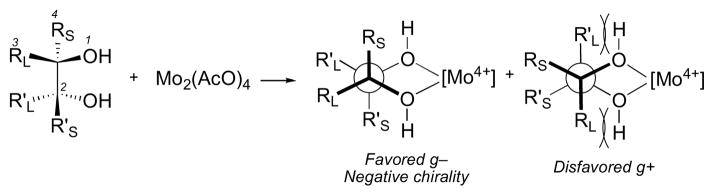
Preferred conformations of Mo(OAc)4–diol complexes. RL, R’L = larger substituent. RS, R’S = smaller substituent.
Scheme 17.
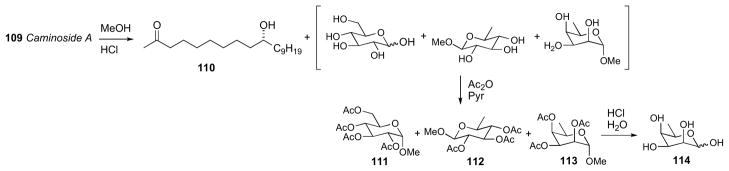
Table 2.
Molar rotations of bistramide diastereomers.
| Stereogenic segments composing bistramide | [M]D | ||
|---|---|---|---|
| C6–C11 THP/amide | C22–C31 Spiroketal | C34 ketone | |
| (6S, 9R, 11R, 15S, 16R) [M]D = +119a | (22S, 23R, 27R, 31R) [M]D = −156 | (34R) [M]D = −51 | −88 |
|
| |||
| (34S) [M]D = +51d | +14 | ||
|
| |||
| (22R, 23S, 27S, 31S) [M]D = +156b | (34R) [M]D = −51 | +224 | |
|
| |||
| (34S) [M]D = +51d | +326 | ||
|
| |||
| (6R, 9S, 11S, 15R, 16S) [M]D = −119 | (22S, 23R, 27R, 31R) [M]D = −156 | (34R) [M]D= −51 | −326 |
|
| |||
| (34S) [M]D = +51d | −224 | ||
|
| |||
| (22R, 23S, 27S, 31S) [M]D = +156b | (34R) [M]D = −51 | −14 | |
|
| |||
| (34S) [M]D= +51d | +88 | ||
synthetic
derived from synthetic 78.
from normanicone (+)-79.
Acknowledgments
The authors gratefully acknowledge research support the NIH (AI039987, CA122256 to TFM) and a Ruth L. Kirschstein National Research Service Award (NIH/NCI T32 CA009523 to BIM).
Biographies

Tadeusz F. Molinski (b. 1957, Victoria Australia) earned his B.Sc. (Hons) (1978) from Monash University, Melbourne, and Ph.D. (1985) in organic chemistry from the Australian National University, Research School of Chemistry, Canberra. He carried out postdoctoral research at Scripps Institution of Oceanography, University of California, San Diego with the late D. John Faulkner, and Chris Ireland in the Department of Medicinal Chemistry, University of Utah. Professor Molinski joined the faculty of the Department of Chemistry, University of California, Davis, in 1989 as Assistant Professor and rose to his current rank of Full Professor in 1997. In 2005, he returned to UC San Diego where he currently holds joint professorial appointments in the Department of Chemistry and Biochemistry and the Skaggs School of Pharmacy and Pharmaceutical Sciences. His research interests include structure, synthesis and biological activity of marine natural products, and stereochemical analysis. Professor Molinski is the recipient of the 2006 Albert Hofmann Award (University of Zürich). (Photo credit: J. A. Molinski)

Brandon I. Morinaka (b. 1981, Los Angeles, CA) graduated with a B.S. in chemistry from University of California, Santa Cruz (2003). After a brief interlude as a research technician in the laboratory of Professor Phillip Crews in the Department of Chemistry and Biochemistry, UC Santa Cruz he entered the Ph.D. program at UC Davis, then moved with Professor Tadeusz F. Molinski to UC San Diego where he completed his Ph.D. (2011). He then moved to Germany and is currently a postdoctoral fellow studying the molecular genetics of natural products biosynthesis under the guidance of Professor Joern Piel at the University of Bonn. His research interests include isolation and configurational assignment of marine natural products, and genome-based approaches for identifying new biosynthetic pathways. He was the recipient of a Ruth L. Kirschstein National Research Service Award (2009), and currently holds an Alexander von Humboldt Research Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- i.(a) Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]; (b) Cragg GM, Grothaus PG, Newman DJ. Chem Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]; (c) Molinski TF, Dalisay DS, Lievens SC, Saludes JP. Nat Rev Drug Discov. 2009;8:59–85. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- ii.(a) Olivera BM, Gray WR, Zeikus R, McIntosh JM, Varga J, Rivier J, de Santos V, Cruz LJ. Science. 1985;230:1338–1343. doi: 10.1126/science.4071055. [DOI] [PubMed] [Google Scholar]; (b) Terlau H, Olivera BM. Physiol Rev. 2004;84:41–68. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- iii.Rinehart KL, Holt TG, Fregeau NL, Stroh JG, Keifer PA, Sun F, Li LH, Martin DG. J Org Chem. 1990;55:4512–4515. [Google Scholar]
- iv.Hirata Y, Uemura D. Pure Appl Chem. 1986;58:701–710. [Google Scholar]
- v.(a) Cuevas C, Perez M, Martin MJ, Chicharro JL, Fernandez-Rivas C, Flores M, Francesch A, Gallego P, Zarzuelo M, de la Calle F, Garcia J, Polanco C, Rodriguez I, Manzanares I. Org Lett. 2000;2:2545–2548. doi: 10.1021/ol0062502. [DOI] [PubMed] [Google Scholar]; (a) Cuevas C, Perez M, Francesch A, Fernandez C, Chicharro JL, Gallego P, Zarzuelo M, de la Calle F, Manzanares I. WO 69862. PCT Int Appl. 2000 doi: 10.1021/ol0062502. [DOI] [PubMed]; (c) Cuevas C, Perez M, Francesch A, Fernandez C, Chicharro JL, Gallego P, Zarzuelo, Manzanares I, Martin MJ, Munt S. WO 87895 PCT Int Appl. 2001
- vi.(a) Molinski TF. Curr Opin Biotech. 2010;21:819–826. doi: 10.1016/j.copbio.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Molinski TF. Curr Opin Drug Discov Develop. 2009;12:197–206. [PubMed] [Google Scholar]
- vii.Crouch RC, Martin GE. Magn Reson Chem. 1992;30:S66–S70. [Google Scholar]
- viii.Crouch RC, Martin GE, Dickey RW, Baden DG, Gawley RE, Rein KS, Mazzola EP. Tetrahedron. 1995;51:8409–8422. [Google Scholar]
- ix.Russell DJ, Hadden CE, Martin GE, Gibson AA, Zens AP, Carolan JL. J Nat Prod. 2000;63:1047– 1049. doi: 10.1021/np0003140. [DOI] [PubMed] [Google Scholar]
- x.Brey WW, Edison AS, Nast RE, Rocca JR, Saha S, Withers RS. J Magn Reson. 2006;179:290–293. doi: 10.1016/j.jmr.2005.12.008. [DOI] [PubMed] [Google Scholar]
- xi.Hilton BD, Martin GE. J Nat Prod. 2010;73:1465–1469. doi: 10.1021/np100481m. [DOI] [PubMed] [Google Scholar]
- xii.Takada N, Umemura N, Suenaga K, Uemura D. Tetrahedron Lett. 2001;42:3495–3497. [Google Scholar]; (a) Matsuura F, Peters R, Ananda M, Harried SS, Hao J, Kishi Y. J Am Chem Soc. 2006;128:7463–7465. doi: 10.1021/ja0618954. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hao J, Matsuura F, Kishi Y, Kita M, Uemura D, Asai N, Iwashita T. J Am Chem Soc. 2006;128:7742–7743. doi: 10.1021/ja061893j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xiii.(a) Uemura D, Chuo T, Haino T, Nagatsu A, Fukuzawa S, Zheng S, Chen H. J Am Chem Soc. 1995;117:1155–1156. [Google Scholar]; (b) Chuo T, Kamo O, Uemura D. Tetrahedron Lett. 1996;37:4023–4026. [Google Scholar]; (c) McCauley JA, Nagasawa K, Lander PA, Mischke SG, Semones MA, Kishi Y. J Am Chem Soc. 1998;120:7647–7648. [Google Scholar]
- xiv.Dalisay DS, Molinski TF. J Nat Prod. 2009;72:739–744. doi: 10.1021/np900009b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xv.(a) Dalisay DS, Molinski TF. Org Lett. 2009;11:1967–1970. doi: 10.1021/ol9004189. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Liu Z, Hogan AML, Dalisay DS, Molinski TF. Org Lett. 2009;11:3766–3769. doi: 10.1021/ol9014317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xvi.(a) Skepper CK, MacMillan JB, Zhou GX, Masuno MN, Molinski TF. J Am Chem Soc. 2007;129:4150–4151. doi: 10.1021/ja0703978. [DOI] [PubMed] [Google Scholar]; (b) MacMillan JB, Xiong-Zhou G, Skepper CK, Molinski TF. J Org Chem. 2008;73:3699– 3706. doi: 10.1021/jo702307t. [DOI] [PubMed] [Google Scholar]
- xvii.(a) Searle PA, Molinski TF. J Am Chem Soc. 1995;117:8126–8131. [Google Scholar]; (b) Searle PA, Molinski TF, Brzezinski LJ, Leahy JW. J Am Chem Soc. 1996;118:9422–9423. [Google Scholar]; (c) Molinski TF. Tetrahedron Lett. 1996;37:7879–7880. [Google Scholar]
- xviii.After Phil Crews (University of California, Santa Cruz) who first proposed the limiting H/C guideline. White KN, Amagata T, Oliver AG, Tenney K, Wenzel PJ, Crews P. J Org Chem. 2008;73:8719–8722. doi: 10.1021/jo800960w.Molinski TF. Curr Opin Biotechnol. 2010;21:819–826. doi: 10.1016/j.copbio.2010.09.003.
- xix.For example, petrosamine, C21H17BrClN3O2, H/C = 0.81. The structure was solved by X-ray crystallography. Molinski TF, Fahy E, Faulkner DJ, Van Duyne GD, Clardy J. J Org Chem. 1988;53:1340–1341.
- xx.(a) Hoye TR, Hanson PR, Vyvyan JR. J Org Chem. 1994;59:4096–4103. [Google Scholar]; (b) Hoye TR, Zhao H. J Org Chem. 2002;67:4014–4016. doi: 10.1021/jo001139v. [DOI] [PubMed] [Google Scholar]
- xxi.(a) Rychnovsky SD, Skalitzky DJ. Tetrahedron Lett. 1990;31:945–948. [Google Scholar]; (b) Rychnovsky SD, Rogers BR, Richardson TI. Acc Chem Res. 1998;31:9–17. [Google Scholar]
- xxii.(a) Rychnovsky SD, Powers JP, Lepage TJ. J Am Chem Soc. 1992;114:8375–8384. [Google Scholar]; (b) Rychnovsky SD, Rogers B, Yang G. J Org Chem. 1993;58:3511–3515. [Google Scholar]; (c) Rychnovsky SD, Yang G, Powers JP. J Org Chem. 1993;58:5251–5255. [Google Scholar]
- xxiii.Dana G, Danechpajouh H. Bull Soc Chim Fr. 1980;II:395–399. [Google Scholar]
- xxiv.Evans DA, Rieger DL, Gage JR. Tetrahedron Lett. 1990;31:7099–7100. [Google Scholar]
- xxv.Rychnovsky SD, Skalitzky DJ, Pathirana C, Jensen PR, Fenical W. J Am Chem Soc. 1992;114:671–677. [Google Scholar]
- xxvi.Dale JA, Mosher HS. J Am Chem Soc. 1973;95:512–519. [Google Scholar]
- xxvii.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- xxviii.Latypov ShK, Seco JM, Quinoa E, Riguera R. J Org Chem. 1996;61:8569–8577. [Google Scholar]
- xxix.(a) Joshi BS, Newton MG, Lee DW, Barber AD, Pelletier SW. Tetrahedron: Asymmetry. 1996;7:25–28. [Google Scholar]; (b) Kusumi T, Fujita Y, Ohtani I, Kakisawa H. Tetrahedron Lett. 1991;32:2923–2926. [Google Scholar]; (c) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Org Chem. 1991;56:1296–1298. [Google Scholar]; (d) Molinski TF, Brzezinski LJ, Leahy JW. Tetrahedron: Asymmetry. 2002;13:1013–1016. [Google Scholar]; (e) Yang L, Andersen RJ. J Nat Prod. 2002;65:1924–1926. doi: 10.1021/np020297+. [DOI] [PubMed] [Google Scholar]
- xxx.Kusumi T, Takahashi H, Hashimoto T, Kan Y, Asakawa Y. Chem Lett. 1994;6:1093–1094. [Google Scholar]
- xxxi.(a) Seco JM, Quinoa E, Riguera R. Tetrahedron: Asymmetry. 2001;12:2915–2925. [Google Scholar]; (b) Seco JM, Quinoa E, Riguera R. Chem Rev. 2004;104:17–117. doi: 10.1021/cr2003344. [DOI] [PubMed] [Google Scholar]
- xxxii.(a) Kusumi T, Ohtani II. In: The Biology – Chemistry Interface A Tribute to Koji Nakanishi. Cooper R, Snyder JK, editors. CRC Press; New York, New York: 1999. p. 103. [Google Scholar]; (b) Kusumi T, Ooi T, Ohkubo Y, Yabuuchi T. Bull Chem Soc Jpn. 2006;79:965–980. [Google Scholar]
- xxxiii.(a) Wenzel TJ. Discrimination of Chiral Compounds Using NMR spectroscopy. Hoboken, NJ: Wiley; 2007. [Google Scholar]; (b) Wenzel TJ, Chisholm CD. Chirality. 2011;23:190–214. doi: 10.1002/chir.20889. [DOI] [PubMed] [Google Scholar]
- xxxiv.Matsumori N, Kaneno D, Murata M, Nakamura H, Tachibana K. J Org Chem. 1999;64:866–876. doi: 10.1021/jo981810k. [DOI] [PubMed] [Google Scholar]
- xxxv.Marquez BL, Gerwick WH, Williamson RT. Magn Reson Chem. 2001;39:499–530. [Google Scholar]
- xxxvi.Nilewski C, Geisser RW, Ebert MO, Carreira EM. J Am Chem Soc. 2009;131:15866–15876. doi: 10.1021/ja906461h. [DOI] [PubMed] [Google Scholar]
- xxxvii.(a) Ciminiello P, Fattorusso E, Forino M, Magno S, Di Rosa M, Ianaro A, Poletti R. J Org Chem. 2001;66:578–582. doi: 10.1021/jo001437s. [DOI] [PubMed] [Google Scholar]; (b) Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, Di Rosa M, Ianaro A, Poletti R. J Am Chem Soc. 2002;124:13114–13120. doi: 10.1021/ja0207347. [DOI] [PubMed] [Google Scholar]; (c) Chen JL, Proteau PJ, Roberts MA, Gerwick WH, Slate DL, Lee RH. J Nat Prod. 1994;57:524–527. doi: 10.1021/np50106a015. [DOI] [PubMed] [Google Scholar]; (b) Pereira AR, Byrum T, Shibuya GM, Vanderwal CD, Gerwick WH. J Nat Prod. 2010;73:279–283. doi: 10.1021/np900672h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxviii.Bifulco G, Dambruoso P, Gomez-Paloma L, Riccio R. Chem Rev. 2007;107:3744–3779. doi: 10.1021/cr030733c. [DOI] [PubMed] [Google Scholar]
- xxxix.(a) Kobayashi Y, Lee J, Tezuka K, Kishi Y. Org Lett. 1999;1:2177–2180. doi: 10.1021/ol9903786. [DOI] [PubMed] [Google Scholar]; (b) Lee J, Kobayashi Y, Tezuka K, Kishi Y. Org Lett. 1999;1:2181–2184. doi: 10.1021/ol990379y. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi Y, Tan CH, Kishi Y. Helv Chim Acta. 2000;83:2562–2571. [Google Scholar]; (d) Kobayashi Y, Tan CH, Kishi Y. J Am Chem Soc. 2001;123:2076–2078. doi: 10.1021/ja004154q. [DOI] [PubMed] [Google Scholar]; (e) Fidanze S, Song F, Szlosek-Pinaud M, Small PLC, Kishi Y. J Am Chem Soc. 2001;123:10117–10118. doi: 10.1021/ja011824z. [DOI] [PubMed] [Google Scholar]; (f) Higashibayashi S, Kishi Y. Tetrahedron. 2004;60:11977–11982. [Google Scholar]
- xl.Kishi Y. Tetrahedron. 2002;58:6239–6258. [Google Scholar]
- xli.(a) Kobayashi Y, Hayashi N, Tan HH, Kishi Y. Org Lett. 2001;3:2245–2248. doi: 10.1021/ol010108z. [DOI] [PubMed] [Google Scholar]; (b) Hayashi N, Kobayashi Y, Kishi Y. Org Lett. 2001;3:2249–2252. doi: 10.1021/ol010109r. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi Y, Hayashi N, Kishi Y. Org Lett. 2002;4:411–414. doi: 10.1021/ol0171160. [DOI] [PubMed] [Google Scholar]; (d) Kobayashi Y, Czechtizky W, Kishi Y. Org Lett. 2003;5:93–96. doi: 10.1021/ol0272895. [DOI] [PubMed] [Google Scholar]
- xlii.(a) Higashibayashi S, Czechtizsky W, Kobayashi Y, Kishi Y. J Am Chem Soc. 2003;125:14379–14393. doi: 10.1021/ja0375481. [DOI] [PubMed] [Google Scholar]; (b) Seike H, Ghosh I, Kishi Y. Org Lett. 2006;8:3861–3864. doi: 10.1021/ol061580t. [DOI] [PubMed] [Google Scholar]
- xliii.Seike H, Ghosh I, Kishi Y. Org Lett. 2006;8:3865–3868. doi: 10.1021/ol061582d. [DOI] [PubMed] [Google Scholar]
- xliv.Nakanishi K, Berova N, Woody RW, editors. Circular Dichroism: Principles and Applications. 2. Wiley-VCH; New York: 2000. [Google Scholar]
- xlv.Moscowitz A. Tetrahedron. 1961;13:48–56. [Google Scholar]
- xlvi.Crabbé P. Holden-Day. 1965. Optical rotatory dispersion and circular dichroism in organic chemistry. [Google Scholar]
- xlvii.Djerassi C. Optical rotatory dispersion: applications to organic chemistry. McGraw-Hill; 1960. [Google Scholar]
- xlviii.Lightner DA, Gurst JE. Organic Conformational Analysis and Stereochemistry from Circular Dichroism Spectroscopy. Wiley VCH; New York: 2000. [Google Scholar]
- xlix.Wellman KM, Bunnenberg E, Djerassi C. J Am Chem Soc. 1963;85:1871–1873. [Google Scholar]
- l.Review: Frelek J, Geiger M, Voelter W. Curr Org Chem. 1999;3:117–146.and references therein. In particular: Snatzke G, Wagner U, Wolff HP. Tetrahedron. 1981;37:349–361.Frelek J, Snatzke G. Fresenius’ J Anal Chem. 1983;316:261–264.Frelek J, Perkowska A, Snatzke G, Tima M, Wagner U, Wolff HP. Spectrosc Int J. 1983:274–295.Diener W, Frelek J, Gerards M, Majer Z, Perkowska A, Snatzke G, Wagner U. Proceedings of the FECS International Conference of Circular Dichroism; Sofia. Sept. 21–25, 1985; pp. 10–33.Frelek J, Pakulski Z, Zamojski A. Tetrahedron: Asymmetry. 1996;7:1363–1372.Frelek J, Ikekawa N, Takatsuto S, Snatzke G. Chirality. 1997;9:578–582.Bari LD, Pescitelli G, Pratelli C, Pino D, Salvadori P. J Org Chem. 2001;66:4819–4825. doi: 10.1021/jo010136v.Gorecki M, Jablonska E, Kruszewska A, Suszczynska A, Urbanczyk-Lipkowaka Z, Gerards M, Morzycki JW, Szczepek WJ, Frelek J. J Org Chem. 2007;72:2906– 2916. doi: 10.1021/jo062445x.
- li.Harada N, Nakanishi K. Circular Dichroic Scectroscopy – Exciton Coupling in Organic Stereochemistry. University Science Books; Mill Valley: 1983. [Google Scholar]
- lii.(a) Harada N, Iwabuchi J, Yokota Y, Uda H, Nakanishi K. J Am Chem Soc. 1981;103:5590–5591. [Google Scholar]; (b) Gonnella NC, Nakanishi K, Martin VS, Sharpless KB. J Am Chem Soc. 1982;104:3775–3776. [Google Scholar]; (c) Tanaka K, Nakanishi K, Berova N. J Am Chem Soc. 2003;125:10802–10803. doi: 10.1021/ja036847n. [DOI] [PubMed] [Google Scholar]
- liii.Zhao N, Kumar N, Neuenschwander K, Nakanishi K, Berova N. J Am Chem Soc. 1995;117:1844– 1845. [Google Scholar]
- liv.(a) Wiesler WT, Nakanishi K. J Am Chem Soc. 1989;111:9205–9213. [Google Scholar]; (b) Zhao P, Berova N, Nakanishi K, Rohmer M. J Chem Soc, Chem Commun. 1991:256–258. [Google Scholar]; (c) Zhao P, Berova N, Nakanishi K, Knani M, Rohmer M. J Am Chem Soc. 1991;113:4040–4042. [Google Scholar]; (d) Zhao P, Zhou N, Rele DN, Berova N, Nakanishi K. J Am Chem Soc. 1993;115:9313–9314. [Google Scholar]; (e) Zhao P, Berova N, Wiesler WT, Nakanishi K. Tetrahedron. 1993;49:9343–9352. [Google Scholar]; (f) Zhao N, Zhou P, Berova N, Nakanishi K. Chirality. 1995;7:636–651. doi: 10.1002/chir.530070815. [DOI] [PubMed] [Google Scholar]; (g) Zhao N, Berova N, Nakanishi K, Rohmer M, Mougenot P, Jurgens UJ. Tetrahedron. 1996;52:2777–2788. [Google Scholar]; (h) Rele DN, Zhao N, Nakanishi K, Berova N. Tetrahedron. 1996;52:2759–2776. [Google Scholar]
- lv.Stephens PJ, Harada N. Chirality. 2010;22:229–233. doi: 10.1002/chir.20733. [DOI] [PubMed] [Google Scholar]
- lvi.Freedman TB, Cao XL, Dukor RK, Nafie LA. Chirality. 2003;15:743–758. doi: 10.1002/chir.10287. [DOI] [PubMed] [Google Scholar]; (b) Stephens PJ, Devlin FJ, Pan JJ. Chirality. 2008;20:643–663. doi: 10.1002/chir.20477. [DOI] [PubMed] [Google Scholar]
- lvii.Moore BS. Nat Prod Rep. 2005;22:580–593. doi: 10.1039/b404737k. [DOI] [PubMed] [Google Scholar]; (b) Moore BS. Nat Prod Rep. 2006;23:615–629. doi: 10.1039/b508781n. [DOI] [PubMed] [Google Scholar]; (c) Lane AL, Moore BS. Nat prod Rep. 2011;28:411–428. doi: 10.1039/c0np90032j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lviii.(a) Ladd MFC, Palmer RA. Structure Determination by X-ray Crystallography. 2. Plenum Press; New York: 1985. [Google Scholar]; (b) Harada N. Chirality. 2008;20:691–723. doi: 10.1002/chir.20478. [DOI] [PubMed] [Google Scholar]
- lix.Shimizu Y. Chem Rev. 1993;93:1685–1698. [Google Scholar]; (b) Yasumoto T, Murata M. Chem Rev. 1993;93:1897–1909. [Google Scholar]; (c) Kobayashi J, Kubota T. J Nat Prod. 2007;70:451–460. doi: 10.1021/np0605844. [DOI] [PubMed] [Google Scholar]
- lx.(a) Satake M, Murata M, Yasumoto T, Fujita T, Naoki H. J Am Chem Soc. 1991;113:9859–9861. [Google Scholar]; (b) Paul GK, Matsumori N, Murata M, Tachibana K. Tetrahedron Lett. 1995;36:6279–6282. [Google Scholar]; (c) Morsy N, Matsuoka S, Houdai T, Matsumori N, Adachi S, Murata M, Iwashita T, Fujita T. Tetrahedron. 2005;61:8606–8610. [Google Scholar]; (d) Echigoya R, Rhodes L, Oshima Y, Satake M. Harmful Algae. 2005;4:383–389. [Google Scholar]; (e) Morsy N, Houdai T, Matsuoka S, Matsumori N, Adachi S, Oishi T, Murata M, Iwashita T, Fujita T. Bioorg Med Chem. 2006;14:6548–6554. doi: 10.1016/j.bmc.2006.06.012. [DOI] [PubMed] [Google Scholar]
- lxi.Murata M, Matsuoka S, Matsumori N, Paul GK, Tachibana K. J Am Chem Soc. 1999;121:870–871. [Google Scholar]
- lxii.Satake M, Morohashi A, Oguri H, Oishi T, Hirama H, Harada N, Yasumoto T. J Am Chem Soc. 1997;119:11325–11326. [Google Scholar]
- lxiii.Schreiber SL, Goulet MT. Tetrahedron Lett. 1987;28:6001–6004. [Google Scholar]
- lxiv.Lancelin JM, Beau JM. Tetrahedron Lett. 1989;30:4521–4524. [Google Scholar]
- lxv.Rychnovsky SD, Griesgraber G, Schlegel R. J Am Chem Soc. 1995;117:197–210. [Google Scholar]
- lxvi.Oishi T, Kanemoto M, Swasono R, Matsumori N, Murata M. Org Lett. 2008;10:5203–5206. doi: 10.1021/ol802168r. [DOI] [PubMed] [Google Scholar]
- lxvii.(a) BouzBouz S, Cossy J. Org Lett. 2001;3:1451–1454. doi: 10.1021/ol0157095. [DOI] [PubMed] [Google Scholar]; (b) Cossy J, Tsuchiya T, Ferrie L, Reymond S, Kreuzer T, Colobert F, Jourdain P, Marko IE. Synlett. 2007:2286–2288. [Google Scholar]; (c) Colobert F, Kreuzer T, Cossy J, Reymond S, Tsuchiya T, Ferrie L, Marko IE, Jourdain P. Synlett. 2007:2351–2354. [Google Scholar]; (d) Cossy J, Tsuchiya T, Reymond S, Kreuzer T, Colobert F, Marko IE. Synlett. 2009;16:2706–2710. [Google Scholar]
- lxviii.(a) de Vicente J, Betzemeier B, Rychnovsky SD. Org Lett. 2005;7:1853–1856. doi: 10.1021/ol050477l. [DOI] [PubMed] [Google Scholar]; (b) de Vicente J, Huckins JR, Rychnovsky SD. Angew Chem, Int Ed. 2006;45:7258–7262. doi: 10.1002/anie.200602742. [DOI] [PubMed] [Google Scholar]; (c) Huckins JR, de Vicente J, Rychnovsky SD. Org Lett. 2007;9:4757–4760. doi: 10.1021/ol7020934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxix.(a) Flamme EM, Roush WR. Org Lett. 2005;7:1411–1414. doi: 10.1021/ol050250q. [DOI] [PubMed] [Google Scholar]; (b) Hicks JD, Flamme EM, Roush WR. Org Lett. 2005;7:5509–5512. doi: 10.1021/ol052322j. [DOI] [PubMed] [Google Scholar]; (c) Hicks JD, Roush WR. Org Lett. 2008;10:681–684. doi: 10.1021/ol703042q. [DOI] [PubMed] [Google Scholar]
- lxx.(a) Paquette LA, Chang SK. Org Lett. 2005;7:3111–3114. doi: 10.1021/ol0511833. [DOI] [PubMed] [Google Scholar]; (b) Chang SK, Paquette LA. Synlett. 2005:2915–2918. [Google Scholar]; (c) Bedore MW, Chang S-K, Paquette LA. Org Lett. 2007;9:513–516. doi: 10.1021/ol062875+. [DOI] [PubMed] [Google Scholar]
- lxxi.Kanemoto M, Murata M, Oishi T. J Org Chem. 2009;74:8810–8813. doi: 10.1021/jo901793f. [DOI] [PubMed] [Google Scholar]
- lxxii.Crimmins MT, Martin TJ, Martinot TA. Org Lett. 2010;12:3890–3893. doi: 10.1021/ol1015898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxxiii.Dubost C, Markó IE, Bryans J. Tetrahedron Lett. 2005;46:4005–4009. [Google Scholar]
- lxxiv.Kobayashi J, Kubota T. J Nat Prod. 2007;70:451–460. doi: 10.1021/np0605844. [DOI] [PubMed] [Google Scholar]
- lxxv.(a) Huang XC, Zhao D, Guo YW, Wu HM, Lin LP, Wang ZH, Ding J, Lin YS. Bioorg Med Chem Lett. 2004;14:3117–3120. doi: 10.1016/j.bmcl.2004.04.029. [DOI] [PubMed] [Google Scholar]; (b) Huang XC, Zhao D, Guo YW, Wu HM, Trivellone E, Cimino G. Tetrahedron Lett. 2004;45:5501–5504. [Google Scholar]
- lxxvi.Washida K, Koyama T, Yamada K, Kita M, Uemura D. Tetrahedron Lett. 2006;47:2521–2525. [Google Scholar]
- lxxvii.Van Wagoner RM, Deeds JR, Tatters AO, Place AR, Tomas CR, Wright JLC. J Nat Prod. 2010;73:1360–1365. doi: 10.1021/np100158r. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxxviii.Peng J, Place AR, Yoshida W, Anklin C, Hamann MT. J Am Chem Soc. 2010;132:3277–3279. doi: 10.1021/ja9091853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxxix.Roesener JA, Scheuer PJ. J Am Chem Soc. 1986;108:846–847. [Google Scholar]
- lxxx.Matsunaga S, Fusetani N, Hashimoto K, Koseki K, Noma M, Noguchi H, Sankawa U. J Org Chem. 1989;54:1360–1363. [Google Scholar]
- lxxxi.(a) Kernan MR, Faulkner DJ. Tetrahedron Lett. 1987;28:2809–2812. [Google Scholar]; (b) Kernan MR, Molinski TF, Faulkner JD. J Org Chem. 1988;53:5014–5020. [Google Scholar]
- lxxxii.(a) Fusetani N, Yasumuro K, Matsunaga S, Hashimoto K. Tetrahedron Lett. 1989;30:2809–2812. [Google Scholar]; (b) Rashid MA, Gustafson KR, Cardellina JH, II, Boyd MR. J Nat Prod. 1995;58:1120–1125. doi: 10.1021/np50121a025. [DOI] [PubMed] [Google Scholar]; (c) Matsunaga S, Sugawara T, Fusetani N. J Nat Prod. 1998;61:1164–1167. doi: 10.1021/np980102r. [DOI] [PubMed] [Google Scholar]
- lxxxiii.Kobayashi J, Murata O, Shigemori H. J Nat Prod. 1993;56:787–791. doi: 10.1021/np50095a021. [DOI] [PubMed] [Google Scholar]
- lxxxiv.Klenchin VA, Allingham JS, King R, Tanaka J, Marriott G, Rayment I. Nat Struct Biol. 2003;10:1058–1063. doi: 10.1038/nsb1006. [DOI] [PubMed] [Google Scholar]
- lxxxv.Matsunaga S, Liu P, Celatka CA, Panek JS, Fusetani N. J Am Chem Soc. 1999;121:5605–5606. [Google Scholar]
- lxxxvi.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J Org Chem. 1981;46:3936–3938. [Google Scholar]
- lxxxvii.Walaszek Z, Horton D, Ekiel I. Carbohydr Res. 1982;106:193–201. [Google Scholar]
- lxxxviii.(a) Liu P, Panek JS. J Am Chem Soc. 2000;122:1235–1236. [Google Scholar]; (b) Panek JS, Liu P. J Am Chem Soc. 2000;122:11090–11097. [Google Scholar]
- lxxxix.Allingham JS, Tanaka J, Marriott G, Rayment I. Org Lett. 2004;6:597–599. doi: 10.1021/ol036458y. [DOI] [PubMed] [Google Scholar]
- xc.(a) D’Auria MV, Gomez-Paloma L, Minale L, Zampella A, Verbist JF, Roussakis C, Debitus C, Patissou J. Tetrahedron. 1994;50:4829–4834. [Google Scholar]; (b) Bassarello C, Bifulco G, Zampella A, D’Auria MV, Riccio R, Gomez-Paloma L. Eur J Org Chem. 2001:39–44. [Google Scholar]; (c) Zampella A, Bassarello C, Bifulco G, Gomez-Paloma L, D’Auria MV. Eur J Org Chem. 2002:785–790. [Google Scholar]; (d) Paterson I, Britton R, Ashton K, Knust H, Stafford J. Proc Natl Acad Sci USA. 2004;101:11986–11991. doi: 10.1073/pnas.0401548101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xci.Guella G, Mancini I, Chiasera G, Pietra F. Helv Chim Acta. 1989;72:237–246. [Google Scholar]
- xcii.(a) Yamada K, Ojika M, Ishigaki T, Yoshida Y, Ekimoto H, Arakawa M. J Am Chem Soc. 1993;115:11020–11021. [Google Scholar]; (b) Ojika M, Kigoshi H, Ishigaki T, Yamada K. Tetrahedron Lett. 1993;34:8501–8504. [Google Scholar]; (c) Ojika M, Kigoshi H, Ishigaki T, Nisiwaki M, Tsukada I, Mizuta K, Yamada K. Tetrahedron Lett. 1993;34:8505–8508. [Google Scholar]; (d) Ojika M, Kigoshi H, Ishigaki T, Tsukada I, Tsuboi T, Ogawa T, Yamada K. J Am Chem Soc. 1994;116:7441–7442. [Google Scholar]
- xciii.(a) Ishibashi M, Moore RE, Patterson GML, Xu C, Clardy J. J Org Chem. 1986;51:5300–5306. [Google Scholar]; (b) Moore RE, Patterson GML, Mynderse JS, Barchi J, Jr, Norton TR, Furusawa E, Furusawa S. Pure Appl Chem. 1986;58:263–271. [Google Scholar]
- xciv.Allingham JS, Zampella A, D’Auria MV, Rayment I. Proc Natl Acad Sci USA. 2005;102:14527–14532. doi: 10.1073/pnas.0502089102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xcv.(a) Nicholas GM, Hong TW, Molinski TF, Lerch ML, Cancilla MT, Lebrilla CB. J Nat Prod. 1999;62:1678–1681. doi: 10.1021/np990190v. [DOI] [PubMed] [Google Scholar]; (b) Nicholas GM, Molinski TF. J Am Chem Soc. 2000;122:4011–4019. [Google Scholar]; (c) Makarieva TN, Dmitrenok PS, Zakarenko AM, Denisenko VA, Guzzi AG, Li R, Skepper CK, Molinski TF, Stonik VA. J Nat Prod. 2007;70:1991–1998. doi: 10.1021/np0704811. [DOI] [PubMed] [Google Scholar]
- xcvi.(a) Makarieva TN, Denisenko VA, Stonik VA, Milgrom YuN, Rashkes YaW. Tetrahedron Lett. 1989;30:6581–6584. [Google Scholar]; (b) Makarieva TN, Guzii AG, Denisenko VA, Dmitrenok PS, Santalova EA, Pokanevich EV, Molinski TF, Stonik VA. J Nat Prod. 2005;68:255–257. doi: 10.1021/np049710z. [DOI] [PubMed] [Google Scholar]; (c) Molinski TF, Makarieva TN, Stonik VA. Angew Chem, Int Ed. 2000;39:4076–4079. [PubMed] [Google Scholar]
- xcvii.(a) Kong FH, Faulkner DJ. J Org Chem. 1993;58:970–971. [Google Scholar]; (b) Dalisay DS, Tsukamoto S, Molinski TF. J Nat Prod. 2009;72:353–359. doi: 10.1021/np800549n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xcviii.Makarieva TN, Dmitrenok PS, Zakharenko AM, Denisenko VA, Guzii AG, Li R, Skepper CK, Molinski TF, Stonik VAJ. Nat Prod. 2007;70:1991–1998. doi: 10.1021/np0704811. [DOI] [PubMed] [Google Scholar]
- xcix.(a) Kobayashi J. J Antibiot. 2008;61:271–284. doi: 10.1038/ja.2008.39. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi J, Kubota T. J Nat Prod. 2007;70:451–460. doi: 10.1021/np0605844. [DOI] [PubMed] [Google Scholar]
- c.Kobayashi J, Tsuda M. Nat Prod Rep. 2004;21:77–93. doi: 10.1039/b310427n. [DOI] [PubMed] [Google Scholar]; (d) Ishibashi M, Kobayashi J. Heterocycles. 1997;44:543–572. [Google Scholar]; (c) Fürstner A. Isr J Chem. 2011;51:329–345. [Google Scholar]
- ci.Kobayashi J, Ishibashi M, Murayama T, Takamatsu M, Iwamura M, Ohizumi Y, Sasaki T. J Org Chem. 1990;55:3421–3423. [Google Scholar]
- cii.(a) Finamore E, Minale L, Riccio R, Rinaldo G, Zollo F. J Org Chem. 1991;56:1146–1153. [Google Scholar]; (b) De Riccardis F, Minale L, Riccio R, Giovannitti B, Iorizzi M, Debitus C. Gazz Chim Ital. 1993;123:79–86. [Google Scholar]; (c) Tsuda M, Toriyabe Y, Endo T, Kobayashi J. Chem Pharm Bull. 2003;51:448–451. doi: 10.1248/cpb.51.448. [DOI] [PubMed] [Google Scholar]
- ciii.(a) Va P, Roush WR. J Am Chem Soc. 2006;128:15960–15961. doi: 10.1021/ja066663j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Va P, Roush WR. Tetrahedron. 2007;63:5768–5796. doi: 10.1016/j.tet.2007.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- civ.(a) Kim CH, An HJ, Shin WK, Yu W, Woo SK, Jung SK, Lee E. Angew Chem. 2006;118:8187–8189. doi: 10.1002/anie.200603363. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:8019–8021. doi: 10.1002/anie.200603363. [DOI] [PubMed] [Google Scholar]; (b) Kim CH, An HJ, Shin WK, Yu W, Woo SK, Jung SK, Lee E. Chem Asian J. 2008;3:1523–1534. doi: 10.1002/asia.200800062. [DOI] [PubMed] [Google Scholar]
- cv.(a) Gouiffes D, Moreau S, Helbecque N, Bernier JL, Henichart JP, Barbin Y, Laurent D, Verbist JF. Tetrahedron. 1988;44:451–459. [Google Scholar]; (b) Degnan BM, Hawkins CJ, Lavin MF, McCaffrey EJ, Parry DL, Watters DJ. J Med Chem. 1989;32:1354–1359. doi: 10.1021/jm00126a035. [DOI] [PubMed] [Google Scholar]
- cvi.Complete planar structure: Foster MP, Mayne CL, Dunkel R, Pugmire RJ, Grant DM, Kornprobst J-M, Verbist J-F, Biard J-F, Ireland CM. J Am Chem Soc. 1992;114:1110–1111.
- cvii.(a) Biard JF, Roussakis C, Kornprobst JM, Gouffes-Barbin D, Verbist JF. J Nat Prod. 1994;57:1336–1345. doi: 10.1021/np50112a002. [DOI] [PubMed] [Google Scholar]; (b) Murphy BT, Cao S, Brodie P, Maharavo J, Andriamanantoanina H, Ravelonandro P, Kingston DGI. J Nat Prod. 2009;72:1338–1340. doi: 10.1021/np900072k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- cviii.Statsuk AV, Bai R, Baryza JL, Verma VA, Hamel E, Wender PA, Kozmin SA. Nat Chem Biol. 2005;1:383–388. doi: 10.1038/nchembio748. [DOI] [PubMed] [Google Scholar]
- cix.Klenchin VA, King R, Tanaka J, Marriott G, Rayment I. Chem Biol. 2005;12:287–291. doi: 10.1016/j.chembiol.2005.02.011. [DOI] [PubMed] [Google Scholar]
- cx.Klenchin VA, Allingham JS, King R, Tanaka J, Marriott G, Rayment I. Nat Struct Biol. 2003;10:1058–1063. doi: 10.1038/nsb1006. [DOI] [PubMed] [Google Scholar]
- cxi.(a) Allingham JS, Tanaka J, Marriott G, Rayment I. Org Lett. 2004;6:597–599. doi: 10.1021/ol036458y. [DOI] [PubMed] [Google Scholar]; (b) Allingham JS, Zampella A, D’Auria MV, Rayment I. Proc Natl Acad Sci USA. 2005;102:14527–14532. doi: 10.1073/pnas.0502089102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- cxii.Rizvi SA, Tereshko V, Kossiakoff AA, Kozmin SA. J Am Chem Soc. 2006;128:3882. doi: 10.1021/ja058319c. [DOI] [PubMed] [Google Scholar]
- cxiii.Rizvi SA, Courson DS, Keller VA, Rock RS, Kozmin SA. Proc Natl Acad Sci USA. 2008;105:4088–4092. doi: 10.1073/pnas.0710727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- cxiv.Solladie G, Baudet C, Biard JF. Tetrahedron Lett. 2000;41:7747–7750. [Google Scholar]
- cxv.Wipf P, Uto Y, Yoshimura S. Chem—Eur J. 2002;8:1670–1681. doi: 10.1002/1521-3765(20020402)8:7<1670::aid-chem1670>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- cxvi.(a) Ribe S, Kondru RK, Beratan BN, Wipf P. J Am Chem Soc. 2000;122:4608–4617. [Google Scholar]; (b) Wipf P, Methot JL. Org Lett. 2000;2:4213–4216. doi: 10.1021/ol006759x. [DOI] [PubMed] [Google Scholar]; (c) Kondru RK, Lim S, Wipf P, Beratan DN. Chirality. 1997;9:469–477. doi: 10.1002/(SICI)1520-636X(1997)9:5/6<469::AID-CHIR13>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]; (d) Kondru RK, Wipf P, Beratan DN. J Phys Chem. 1999;103:6603–6611. [Google Scholar]; (e) Kondru RK, Wipf P, Beratan DN. J Am Chem Soc. 1998;120:2204–2205. [Google Scholar]; (f) Kondru RK, Wipf P, Beratan DN. Science. 1998;282:2247–2250. doi: 10.1126/science.282.5397.2247. [DOI] [PubMed] [Google Scholar]; (g) Specht KM, Nam J, Ho DM, Berova N, Kondru RK, Beratan DN, Wipf P, Pascal RA, Kahne D. J Am Chem Soc. 2001;123:8961–8966. doi: 10.1021/ja0104406. [DOI] [PubMed] [Google Scholar]
- cxvii.(a) Martischonok V, Melikyan GG, Mineif A, Vostrowsky O, Bestmann HJ. Synthesis. 1991:560–564. [Google Scholar]; (b) Melikyan GG, Mineif A, Vostrowsky O, Bestmann HJ. Synthesis. 1991:633–636. [Google Scholar]
- cxviii.Zuber G, Goldsmith MR, Hopkins TD, Beratan DN, Wipf P. Org Lett. 2005;7:5269–5272. doi: 10.1021/ol052154v. [DOI] [PubMed] [Google Scholar]
- cxix.Statsuk AV, Liu D, Kozmin SA. J Am Chem Soc. 2004;126:9546–9547. doi: 10.1021/ja046588h. [DOI] [PubMed] [Google Scholar]
- cxx.Wipf P, Hopkins TD. Chem Commun. 2005:3421–3423. doi: 10.1039/b505100b. [DOI] [PubMed] [Google Scholar]
- cxxi.For other synthetic approaches of bistramides, see: Crimmins MT, DeBaillie AC. J Am Chem Soc. 2006;128:4936–4937. doi: 10.1021/ja057686l.Lowe JT, Wrona IE, Panek JS. Org Lett. 2007;9:327–330. doi: 10.1021/ol062957y.Yadav JS, Chetia L. Org Lett. 2007;9:4587–4589. doi: 10.1021/ol702095n.Wrona IE, Lowe JT, Turbyville TJ, Johnson TR, Beignet J, Beutler JA, Panek JS. J Org Chem. 2009;74:1897–1916. doi: 10.1021/jo802269q.
- cxxii.Takada K, Uehara T, Nakao Y, Matsunaga S, van Soest RW, Fusetani N. J Am Chem Soc. 2004;126:187–193. doi: 10.1021/ja037368r. [DOI] [PubMed] [Google Scholar]
- cxxiii.Seco JM, Latypov ShK, Quinoa E, Riguera R. J Org Chem. 1997;62:7569–7574. [Google Scholar]
- cxxiv.Fujimoto Y, Murasaki C, Shimada H, Nishioka S, Kakinuma K, Singh S, Singh M, Gupta YK, Sahai M. Chem Pharm Bull. 1994;42:1175–1184. [Google Scholar]
- cxxv.Singh SB. Tetrahedron Lett. 2000;41:6973–6976. [Google Scholar]
- cxxvi.Nakao Y, Maki T, Matsunaga S, van Soest RWM, Fusetani N. Tetrahedron. 2000;56:8977–8987. [Google Scholar]
- cxxvii.Nakao Y, Maki T, Matsunaga S, van Soest RWM, Fusetani N. J Nat Prod. 2004;67:1346–1350. doi: 10.1021/np049939e. [DOI] [PubMed] [Google Scholar]
- cxxviii.Gurjar MK, Pramanik C, Bhattasali D, Ramana CV, Mohapatra DK. J Org Chem. 2007;72:6591–6594. doi: 10.1021/jo070560h. [DOI] [PubMed] [Google Scholar]
- cxxix.Liu G, Romo D. Org Lett. 2009;11:1143–1146. doi: 10.1021/ol802992m. [DOI] [PubMed] [Google Scholar]
- cxxx.Bowen EG, Wardrop DJ. J Am Chem Soc. 2009;131:6062–6063. doi: 10.1021/ja9005755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- cxxxi.(a) Pettit GR, Cichacz ZA, Gao F, Boyd MR, Schmidt JM. J Chem Soc, Chem Commun. 1994;9:1111. [Google Scholar]; (b) Pettit GR, Cichacz ZA. 5,430,053. US Patent. 1995
- cxxxii.Paterson I, Britton R, Delgado O, Wright AE. Chem Commun. 2004:632–633. doi: 10.1039/b316390c. [DOI] [PubMed] [Google Scholar]
- cxxxiii.Gunasekera SP, Gunasekera M, Longley RE, Schulte GK. J Org Chem. 1990;55:4912–4915. (Corrigendum: J. Org. Chem. 1991, 56, 1346) [Google Scholar]
- cxxxiv.(a) Kalesse M. ChemBiochem. 2000;1:171–175. doi: 10.1002/1439-7633(20001002)1:3<171::AID-CBIC171>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]; (b) De Souza MV. Scientific World Journal. 2004;4:415–436. [Google Scholar]
- cxxxv.Nerenberg JB, Hung DT, Somers PK, Schreiber SL. J Am Chem Soc. 1993;115:12621–12622. [Google Scholar]
- cxxxvi.Paterson I, Britton R, Delgado O, Meyer A, Poullennec KG. Angew Chem, Int Ed. 2004;43:4629–4633. doi: 10.1002/anie.200460589. [DOI] [PubMed] [Google Scholar]
- cxxxvii.Shin Y, Fournier JH, Fukui Y, Brückner AM, Curran DP. Angew Chem, Int Ed. 2004;43:4634–4637. doi: 10.1002/anie.200460593. [DOI] [PubMed] [Google Scholar]
- cxxxviii.Cichewicz RH, Valeriote FA, Crews P. Org Lett. 2004;6:1951–1954. doi: 10.1021/ol049503q. [DOI] [PubMed] [Google Scholar]
- cxxxix.Pettit GR, Xu J-P, Chapuis J-C, Pettit RK, Tackett LP, Doubek DL, Hooper JNA, Schmidt JM. J Med Chem. 2004;47:1149–1152. doi: 10.1021/jm030207d. [DOI] [PubMed] [Google Scholar]
- cxl.(a) Cardani C, Ghiringhelli D, Mondelli R, Quilico A. Tetrahedron Lett. 1965;6:2537–3545. [Google Scholar]; (b) Bonamartini Corradi A, Mangia A, Nardelli M, Pelizzi G. Gazz Chim Ital. 1971;101:591. [Google Scholar]; (c) Matsumoto T, Yanagiya M, Maeno S, Yasuda S. Tetrahedron Lett. 1968;9:6297–6300. doi: 10.1016/s0040-4039(01)98905-1. [DOI] [PubMed] [Google Scholar]; (d) Furusaki A, Watanabe T, Matsumoto T, Yanagiya M. Tetrahedron Lett. 1968;9:6301–6304. [Google Scholar]
- cxli.Isolation: Sakemi S, Ichiba T, Kohmoto S, Saucy G, Higa T. J Am Chem Soc. 1988;110:4851–4853.Absolute configuration by total synthesis: Hong CY, Kishi Y. J Am Chem Soc. 1991;113:9693–9694.
- cxlii.Isolation: Perry NB, Blunt JW, Munro MHG, Pannell LK. J Am Chem Soc. 1988;110:4850–4851.Perry NB, Blunt JW, Munro MHG, Thompson AM. J Org Chem. 1990;55:223–227.Absolute configuration by total synthesis: Hong CY, Kishi Y. J Org Chem. 1990;55:4242–4245.
- cxliii.(a) Fusetani N, Sugawara T, Matsunaga S. J Org Chem. 1992;57:3828–3832. [Google Scholar]; (b) Tsukamoto S, Matsunaga S, Fusetani N, Toh-e A. Tetrahedron. 1999;55:13697–13702. [Google Scholar]; (c) Paul GK, Gunasekera SP, Longley RE, Pomponi SA. J Nat Prod. 2002;65:59–61. doi: 10.1021/np0103766. [DOI] [PubMed] [Google Scholar]
- cxliv.Fisch KM, Gurgui C, Heycke N, van der Sar SS, Anderson SA, Webb VL, Taudien S, Platzer M, Rubio BK, Robinson SJ, Crews P, Piel J. Nat Chem Biol. 2009;5:494–501. doi: 10.1038/nchembio.176. [DOI] [PubMed] [Google Scholar]
- cxlv.Although the structure of the amide side chain for mycalamide A is quite different. Two rotamers of the C6/C7 bond have been proposed. Perry NB, Blunt JW, Munro MHG, Thompson AW. J Org Chem. 1990;55:223–227.
- cxlvi.Krohn K, Bahramsari R, Florke U, Ludewig K, Kliche-Spory C, Michel A, Aust H-J, Draeger S, Schulz B, Antus S. Phytochemistry. 1997;45:313–320. doi: 10.1016/s0031-9422(96)00854-0. [DOI] [PubMed] [Google Scholar]
- cxlvii.Kiren S, Williams LJ. Org Lett. 2005;7:2905–2908. doi: 10.1021/ol0508375. [DOI] [PubMed] [Google Scholar]
- cxlviii.Green ME, Rech JC, Floreancig PE. Org Lett. 2005;7:4117–4120. doi: 10.1021/ol051396s. [DOI] [PubMed] [Google Scholar]
- cxlix.(a) Jiang X, Garcia-Fortanet J, De Brabander JK. J Am Chem Soc. 2005;127:11254–11255. doi: 10.1021/ja0537068. [DOI] [PubMed] [Google Scholar]; (b) Huang X, Shao N, Palani A, Aslanian R, Buevich A. Org Lett. 2007;9:2597–2600. doi: 10.1021/ol071068n. [DOI] [PubMed] [Google Scholar]; (c) Smith AB, III, Jurica JA, Walsh SP. Org Lett. 2008;10:5625–5628. doi: 10.1021/ol802466t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Crimmins MT, Stevens JM, Schaaf GM. Org Lett. 2009;11:3990–3993. doi: 10.1021/ol901655e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shangguan N, Kiren S, Williams LJ. Org Lett. 2007;9:1093–1096. doi: 10.1021/ol063143k. [DOI] [PubMed] [Google Scholar]; (f) Watanabe T, Imaizumi T, Chinen T, Nagumo Y, Shibuya M, Usui T, Kanoh N, Iwabuchi Y. Org Lett. 2010;12:1040–1043. doi: 10.1021/ol1000389. [DOI] [PubMed] [Google Scholar]
- cl.Linington RG, Robertson M, Gauthier A, Finlay BB, van Soest RWM, Andersen RJ. Org Lett. 2003;5:4089–4092. doi: 10.1021/ol0268337. [DOI] [PubMed] [Google Scholar]
- cli.MacMillan JB, Linington RG, Andersen RJ, Molinski TF. Angew Chem Int Ed. 2004;43:5946–5951. doi: 10.1002/anie.200461158. [DOI] [PubMed] [Google Scholar]
- clii.MacMillan JB, Molinski TF. J Am Chem Soc. 2004;126:9944–9945. doi: 10.1021/ja047741a. [DOI] [PubMed] [Google Scholar]
- cliii.(a) Hamada T, Sugawara T, Matsunaga S, Fusetani N. Tetrahedron Lett. 1994;35:719–720. [Google Scholar]; (b) Hamada T, Sugawara T, Matsunaga S, Fusetani N. Tetrahedron Lett. 1994;35:609–612. [Google Scholar]
- cliv.Hamada T, Matsunaga S, Yano G, Fusetani N. J Am Chem Soc. 2005;127:110–118. doi: 10.1021/ja045749e. [DOI] [PubMed] [Google Scholar]
- clv.Marfey P. Carlsberg Res Commun. 1984;49:591–596. [Google Scholar]
- clvi.(a) Inoue M, Shinohara N, Tanabe S, Takahashi T, Okura K, Itoh H, Mizoguchi Y, Iida M, Lee N, Matsuoka S. Nat Chem. 2010;2:280–285. doi: 10.1038/nchem.554. [DOI] [PubMed] [Google Scholar]; (b) Inoue M, Matsuoka S. Isr J Chem. 2011;51:346–358. [Google Scholar]
- clvii.Hamada T, Matsunaga S, Fujiwara M, Fujita K, Hirota H, Schmucki R, Güntert P, Fusetani N. J Am Chem Soc. 2005;127:110–118. doi: 10.1021/ja104616z. [DOI] [PubMed] [Google Scholar]
- clviii.Tsuda M, Kasai Y, Komatsu K, Sone T, Tanaka M, Mikami Y, Kobayashi J. Org Lett. 2004;6:3087–3089. doi: 10.1021/ol048900y. [DOI] [PubMed] [Google Scholar]
- clix.Bowman RE, Stroud HH. J Chem Soc. 1950:1342–1345. [Google Scholar]
- clx.Mugishima T, Tsuda M, Kasai Y, Ishiyama H, Fukushi E, Kawabata J, Watanabe M, Akao K, Kobayashi J. J Org Chem. 2005;70:9430–9435. doi: 10.1021/jo051499o. [DOI] [PubMed] [Google Scholar]
- clxi.(a) Lievens SC, Molinski TF. Org Lett. 2005;7:2281–2284. doi: 10.1021/ol050717x. [DOI] [PubMed] [Google Scholar]; (b) Lievens SC, Morinaka BI, Molinski TF. Aus J Chem. 2010;63:935–941. [Google Scholar]
- clxii.Lievens SC, Molinski TF. J Am Chem Soc. 2006;128:11764–11765. doi: 10.1021/ja063735y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- clxiii.Seike H, Ghosh I, Kishi Y. Org Lett. 2006;8:3865. doi: 10.1021/ol061582d.[corrigendum: Org Lett. 2006;8:5177.]. Seike H, Ghosh I, Kishi Y. Org Lett. 2006;8:3865–3868. doi: 10.1021/ol061582d.
- clxiv.An error in the Molinski approach (Ref. clxii) can be traced to some mis-measured 1JCH values. See reference clxvi for a corrected configuration based on measurement and interpretation of residual dipolar couplings (RDCs).
- clxv.Seike H, Ghosh I, Kishi Y. Org Lett. 2006;8:3861–3864. doi: 10.1021/ol061580t. [DOI] [PubMed] [Google Scholar]
- clxvi.Schuetz A, Junker J, Leonov A, Lange OF, Molinski TF, Griesinger C. J Am Chem Soc. 2007;129:15114–15115. doi: 10.1021/ja075876l. [DOI] [PubMed] [Google Scholar]
- clxvii.Satake M, Shoji M, Oshima Y, Naoki H, Yasumoto T. Tetrahedron Lett. 2002;43:5829–5832. [Google Scholar]
- clxviii.Tsukano C, Sasaki M. J Am Chem Soc. 2003;125:14294–14295. doi: 10.1021/ja038547b. [DOI] [PubMed] [Google Scholar]
- clxix.Satake M, Tanaka Y, Ishikura Y, Oshima Y, Naoki H, Yasumoto T. Tetrahedron Lett. 2005;46:3537–3540. [Google Scholar]
- clxx.Tanaka K, Itagaki Y, Satake M, Naoki H, Yasumoto T, Nakanishi K, Berova N. J Am Chem Soc. 2005;127:9561–9570. doi: 10.1021/ja0512677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- clxxi.(a) Chen SML, Harada N, Nakanishi K. J Am Chem Soc. 1974;96:7352–7354. [Google Scholar]; (b) Canceill J, Collet A, Jacques J. J Chem Soc, Perkin II. 1982:83–89. [Google Scholar]; (c) Matile S, Berova N, Nakanishi K, Fleischhauer J, Woody RW. J Am Chem Soc. 1996;118:5198–5206. [Google Scholar]
- clxxii.(a) Connon SJ, Blechert S. Angew Chem, Int Ed. 2003;42:1900–1923. doi: 10.1002/anie.200200556. [DOI] [PubMed] [Google Scholar]; (b) Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- clxxiii.(a) Tanaka K, Nakanishi K, Berova N. J Am Chem Soc. 2003;125:10802–10803. doi: 10.1021/ja036847n. [DOI] [PubMed] [Google Scholar]; (b) Tanaka K, Pescitelli G, Di Bari L, Xiao TL, Nakanishi K, Armstrong DW, Berova N. Org Biomol Chem. 2004;2:48–58. doi: 10.1039/b312542d. [DOI] [PubMed] [Google Scholar]
- clxxiv.Williams DE, Keyzers RA, Warabi K, Desjardine K, Riffell JL, Roberge M, Andersen RJ. J Org Chem. 2007;72:9842–9845. doi: 10.1021/jo7018174. [DOI] [PubMed] [Google Scholar]
- clxxv.Williams DE, Roberge M, Van Soest R, Andersen RJ. J Am Chem Soc. 2003;125:5296–5297. doi: 10.1021/ja0348602. [DOI] [PubMed] [Google Scholar]
- clxxvi.Williams DE, Lapawa M, Feng X, Tarling T, Roberge M, Andersen RJ. Org Lett. 2004;6:2607–2610. doi: 10.1021/ol0490983. [DOI] [PubMed] [Google Scholar]
- clxxvii.Warabi K, Williams DE, Patrick BO, Roberge M, Andersen RJ. J Am Chem Soc. 2007;129:508–509. doi: 10.1021/ja068271i. [DOI] [PubMed] [Google Scholar]
- clxxviii.Pan Y, De Brabander JK. Synlett. 2006:853–856. [Google Scholar]
- clxxix.(a) Wang C, Forsyth CJ. Org Lett. 2006;8:2997–3000. doi: 10.1021/ol0609457. [DOI] [PubMed] [Google Scholar]; (b) Wang C, Forsyth C. Heterocycles. 2007;72:621–632. [Google Scholar]
- clxxx.(a) Paterson I, Anderson EA, Dalby SM, Loiseleur O. Org Lett. 2005;7:4125–4128. doi: 10.1021/ol051405x. [DOI] [PubMed] [Google Scholar]; (b) Paterson I, Anderson EA, Dalby SM, Loiseleur O. Org Lett. 2005;7:4121–4124. doi: 10.1021/ol051403c. [DOI] [PubMed] [Google Scholar]; (c) Paterson I, Anderson EA, Dalby SM. Synthesis. 2005:3225–3228. [Google Scholar]; (d) Paterson I, Anderson EA, Dalby SM, Lim JH, Maltas P, Moessner C. Chem Commun. 2006:4186–4188. doi: 10.1039/b612697a. [DOI] [PubMed] [Google Scholar]; (e) Paterson I, Anderson EA, Dalby SM, Genovino J, Lim JH, Moessner C. Chem Commun. 2007:1852–1854. doi: 10.1039/b700827a. [DOI] [PubMed] [Google Scholar]
- clxxxi.(a) Liu J, Hsung RP. Org Lett. 2005;7:2273–2276. doi: 10.1021/ol050653q. [DOI] [PubMed] [Google Scholar]; (b) Ghosh SK, Ko C, Liu J, Wang J, Hsung RP. Tetrahedron. 2006;62:10485–10496. [Google Scholar]; (c) Liu J, Yang JH, Ko C, Hsung RP. Tetrahedron Lett. 2006;47:6121–6123. [Google Scholar]; (d) Yang JH, Liu J, Hsung RP. Org Lett. 2008;10:2525–2528. doi: 10.1021/ol8008057. [DOI] [PubMed] [Google Scholar]
- clxxxii.(a) Furstner A, Fenster MDB, Fasching B, Godbout C, Radkowski K. Angew Chem, Int Ed. 2006;45:5506–5510. doi: 10.1002/anie.200601654. [DOI] [PubMed] [Google Scholar]; (b) Furstner A, Fenster MDB, Fasching B, Godbout C, Radkowski K. Angew Chem, Int Ed. 2006;45:5510–5515. doi: 10.1002/anie.200601655. [DOI] [PubMed] [Google Scholar]; (c) Furstner A, Fasching B, O’Neil GW, Fenster MDB, Godbout C, Ceccon J. Chem Commun. 2007:3045–3047. doi: 10.1039/b707835h. [DOI] [PubMed] [Google Scholar]
- clxxxiii.Smith AB, III, Kim DS. Org Lett. 2007;9:3311–3314. doi: 10.1021/ol071282b. [DOI] [PubMed] [Google Scholar]; Smits H, Kim D-S. Tetrahedron. 2010;66:6597–6605. doi: 10.1016/j.tet.2010.01.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- clxxxiv.Keaton KA, Phillips AJ. Org Lett. 2008;10:1083–1086. doi: 10.1021/ol702955m. [DOI] [PubMed] [Google Scholar]
- clxxxv.(a) Paterson I, Anderson EA, Dalby SM, Lim JH, Genovino J, Maltas P, Moessner C. Angew Chem Int Ed. 2008;47:3016–3020. doi: 10.1002/anie.200705565. [DOI] [PubMed] [Google Scholar]; (b) Paterson I, Anderson EA, Dalby SM, Lim JH, Genovino J, Maltas P, Moessner C. Angew Chem Int Ed. 2008;47:3021–3025. doi: 10.1002/anie.200705566. [DOI] [PubMed] [Google Scholar]
- clxxxvi.O’Neil GW, Ceccon J, Benson S, Collin MP, Fasching B, Furstner A. Angew Chem Int Ed. 2009;48:9940–9945. doi: 10.1002/anie.200906121. [DOI] [PubMed] [Google Scholar]; (b) Benson S, Collin MP, O’Neil GW, Ceccon J, Fasching B, Fenster MDB, Godbout C, Radkowski K, Goddard R, Furstner A. Angew Chem Int Ed. 2009;48:9946–9950. doi: 10.1002/anie.200906122. [DOI] [PubMed] [Google Scholar]
- clxxxvii.Isolation: Gonzalez N, Rodriguez J, Jimenez C. J Org Chem. 1999;64:5705–5707. doi: 10.1021/jo9903914.Total synthesis and structure revision: Kiyota H, Dixon DJ, Luscombe CK, Hettstedt S, Ley SV. Org Lett. 2002;4:3223–3226. doi: 10.1021/ol026421y.
- clxxxviii.Mitchell SS, Rhodes D, Bushman FD, Faulkner DJ. Org Lett. 2000;2:1605–1607. doi: 10.1021/ol005866o. [DOI] [PubMed] [Google Scholar]
- clxxxix.Sone H, Kigoshi H, Yamada K. J Org Chem. 1996;61:8956–8960. doi: 10.1021/jo961302f. [DOI] [PubMed] [Google Scholar]
- cxc.Pettit GR, Xu J-P, Doubek DL, Chapuis J-C, Schmidt JM. J Nat Prod. 2004;67:1252–1255. doi: 10.1021/np030198b. [DOI] [PubMed] [Google Scholar]
- cxci.(a) Zampella A, D’Auria MV, Minale L, Debitus C, Roussakis C. J Am Chem Soc. 1996;118:11085–11088. [Google Scholar]; (b) Zampella A, D’Auria MV, Minale L, Debitus C. Tetrahedron. 1997;53:3243–3248. [Google Scholar]
- cxcii.Tan LT, Marquez BL, Gerwick WH. J Nat Prod. 2002;65:925–928. doi: 10.1021/np010526c. [DOI] [PubMed] [Google Scholar]
- cxciii.Paterson I, Florence GJ, Heimann AC, Mackay AC. Angew Chem, Int Ed. 2005;44:1130–1133. doi: 10.1002/anie.200462267. [DOI] [PubMed] [Google Scholar]
- cxciv.(a) Trost BM, Dirat O, Gunzner JL. Angew Chem, Int Ed. 2002;41:841–843. doi: 10.1002/1521-3773(20020301)41:5<841::aid-anie841>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Gunzner JL, Dirat O, Rhee YH. J Am Chem Soc. 2002;124:10396–10415. doi: 10.1021/ja0205232. [DOI] [PubMed] [Google Scholar]
- cxcv.Evans DA, Hu E, Burch JD, Jaeschke GJ. Am Chem Soc. 2002;124:5654–5655. doi: 10.1021/ja026235n. [DOI] [PubMed] [Google Scholar]
- cxcvi.(a) Harada N, Sato H, Nakanishi K. J Chem Soc D, Chem Commun. 1970:1691–1693. [Google Scholar]; (b) Chang M, Meyers HV, Nakanishi K, Ojika M, Park JH, Park MH, Takeda R, Vázquez JT, Wiesler WT. Pure Appl Chem. 1989;61:1193–1200. [Google Scholar]
- cxcvii.Paterson I, Paquet T. Org Lett. 2010;12:2158–2161. doi: 10.1021/ol100693c. [DOI] [PubMed] [Google Scholar]
- cxcviii.Sharma GM, Michaels L, Burkholder PR. J antibiot. 1968;21:659–664. doi: 10.7164/antibiotics.21.659. [DOI] [PubMed] [Google Scholar]
- cxcix.Murakami M, Makabe K, Yamaguchi K, Konosu S, Walchli MR. Tetrahedron Lett. 1988;29:1149–1152. [Google Scholar]
- cc.(a) Fujiwara K, Naka J, Katagiri T, Sato D, Kawai H, Suzuki T. Bull Chem Soc Jpn. 2007;80:1173–1186. [Google Scholar]; (b) Katagiri T, Fujiwara K, Kawai H, Suzuki T. Tetrahedron Lett. 2008;48:233–237. [Google Scholar]
- cci.Takeda Y, Shi J, Oikawa M, Sasaki M. Org Lett. 2008;10:1013–1016. doi: 10.1021/ol8000377. [DOI] [PubMed] [Google Scholar]
- ccii.Dalisay DS, Quach T, Nicholas GM, Molinski TF. Angew Chem, Int Ed. 2009;48:4367–4371. doi: 10.1002/anie.200900888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- cciii.(a) Higgs MD, Faulkner DJ. J Org Chem. 1978;43:3454–3457. [Google Scholar]; (b) Fattorusso C, Campiani G, Catalanotti B, Persico M, Basilico N, Parapini S, Taramelli D, Campagnuolo DC, Fattorusso E, Romano A, Taglialatela-Scafati O. J Med Chem. 2006;49:7088–7094. doi: 10.1021/jm060899g. [DOI] [PubMed] [Google Scholar]; (c) Davidson BS. J Org Chem. 1991;56:6722–6724. [Google Scholar]
- cciv.Dalisay DS, Quach T, Molinski TF. Org Lett. 2010;12:1524–1527. doi: 10.1021/ol100249v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ccv.Corey EJ, Link JO, Shao Y. Tetrahedron Lett. 1992;33:3435. [Google Scholar]
- ccvi.Oku N, Takada K, Fuller RW, Wilson JA, Peach ML, Pannell LK, Mcmahon JB, Gustafson KR. J Am Chem Soc. 2010;132:10278–10285. doi: 10.1021/ja1016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ccvii.Nagai Y, Kusumi T. Tetrahedron Lett. 1995;36:1853–1856. [Google Scholar]
- ccviii.Boger DL, Hikota M, Lewis BM. J Org Chem. 1997;62:1748–1753. [Google Scholar]
- ccix.(a) Grzyska PK, Czyryca PG, Purcell J, Hengge AC. J Am Chem Soc. 2003;125:13106–13111. doi: 10.1021/ja036571j. [DOI] [PubMed] [Google Scholar]; (b) Grzyska PK, Czyryca PG, Golightly J, Small K, Larsen P, Hoff RH, Hengge AC. J Org Chem. 2002;67:1214–1220. doi: 10.1021/jo016104p. [DOI] [PubMed] [Google Scholar]
- ccx.Brandänge S, Färnbäck M, Leijonmarck H, Sundin A. J Am Chem Soc. 2003;125:11942–11955. doi: 10.1021/ja036002b. [DOI] [PubMed] [Google Scholar]
- ccxi.Behforouz M, Bolan JL, Flynt MS. J Org Chem. 1985;50:1186–1189. [Google Scholar]
- ccxii.Crout DHG, Morrey SM. J Chem Soc, Perkin Trans 1. 1983:2435–2440. [Google Scholar]
- ccxiii.Skepper CK, Quach T, Molinski TF. J Am Chem Soc. 2010;132:10286–10292. doi: 10.1021/ja1016975. [DOI] [PubMed] [Google Scholar]
- ccxiv.Morinaka BI, Molinski TF. J Nat Prod. 2011;74:430–440. doi: 10.1021/np1008637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ccxv.Jaspars M, Pasupathy V, Crews P. J Org Chem. 1994;59:3253–3255. [Google Scholar]
- ccxvi.a) Charan RD, Garson MJ, Brereton IM, Willis AC, Hooper JNA. Tetrahedron. 1996;52:9111–9120. [Google Scholar]; b) Clark RJ, Field KL, Charan RD, Garson MJ, Brereton IM, Willis AC. Tetrahedron. 1998;54:8811–8826. [Google Scholar]; c) Mudianta IW, Garson MJ, Bernhardt PV. Aust J Chem. 2009;62:667–670. [Google Scholar]
- ccxvii.2-Methoxy-6-vinylnaphthalene was synthesized from 2-bromo-6-methoxynaphthalene. Lindh J, Savmarker J, Nilsson P, Sjoberg PJR, Larhed M. Chem Eur J. 2009;15:4630–4636. doi: 10.1002/chem.200802744.
- ccxviii.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- ccxix.Williams DE, Sturgeon CM, Roberge M, Andersen RJ. J Am Chem Soc. 2007;129:5822–5823. doi: 10.1021/ja0715187. [DOI] [PubMed] [Google Scholar]
- ccxx.MacMillan JB, Molinski TF. Org Lett. 2002;4:1535–1538. doi: 10.1021/ol025759p. [DOI] [PubMed] [Google Scholar]
- ccxxi.MacMillan JB, Molinski TF. J Am Chem Soc. 2004;126:9944–9945. doi: 10.1021/ja047741a. [DOI] [PubMed] [Google Scholar]
- ccxxii.Kita M, Ohishi N, Konishi K, Kondo M, Koyama T, Kitamura M, Yamada K, Uemura D. Tetrahedron. 2007;63:6241–6251. [Google Scholar]
- ccxxiii.Kita M, Uemura D. Chem Rec. 2010;10:48–52. doi: 10.1002/tcr.200900030. [DOI] [PubMed] [Google Scholar]
- ccxxiv.(a) Takamura H, Ando J, Abe T, Murata T, Kadota I, Uemura D. Tetrahedron Lett. 2008;49:4626–4629. [Google Scholar]; (b) Takamura H, Kadonaga Y, Yamano Y, Han C, Aoyama Y, Kadota I, Uemura D. Tetrahedron Lett. 2009;50:863–866. [Google Scholar]; (c) Murata T, Sano M, Takamura H, Kadota I, Uemura D. J Org Chem. 2009;74:4797–4803. doi: 10.1021/jo900546k. [DOI] [PubMed] [Google Scholar]; (d) Han C, Yamano Y, Kita M, Takamura H, Uemura D. Tetrahedron Lett. 2009;50:5280–5282. [Google Scholar]; (e) Takamura H, Kadonaga Y, Yamano Y, Han C, Kadota I, Uemura D. Tetrahedron. 2009 [Google Scholar]; (f) Takamura H, Kadonaga Y, Kadota I, Uemura D. Tetrahedron. 2010;66:7569–7576. [Google Scholar]
- ccxxv.Kobayashi J, Kondo K, Ishibashi M, Wiilchli MR, Nakamura TJ. Am Chem Soc. 1993;115:6661–6665. [Google Scholar]; (b) Kondo K, Ishibashi M, Kobayashi J. Tetrahedron. 1994;50:8355–8362. [Google Scholar]
- ccxxvi.Jimenez JI, Goetz G, Mau CMS, Yoshida WY, Scheuer PJ, Williamson RT, Kelly M. J Org Chem. 2000;65:8465–8469. doi: 10.1021/jo000789w. [DOI] [PubMed] [Google Scholar]
- ccxxvii.Tsuda M, Oguchi K, Iwamoto R, Okamoto Y, Kobayashi Ji, Fukushi E, Kawabata J, Ozawa T, Masuda A, Kitaya Y, Omasa K. Iriomoteolide-1a, a Potent Cytotoxic 20-Membered Macrolide from a Benthic Dinoflagellate Amphidinium Species. J Org Chem. 2007;72:4469–4474. doi: 10.1021/jo070414b. [DOI] [PubMed] [Google Scholar]
- ccxxviii.Fang L, Yang J, Yang F. Org Lett. 2010;12:3124–3127. doi: 10.1021/ol1011423. [DOI] [PubMed] [Google Scholar]
- ccxxix.Xie J, Ma Y, Horne DA. J C S Chem Comm. 2010;46:4770–4772. doi: 10.1039/c0cc00628a. [DOI] [PubMed] [Google Scholar]