

Abstract
Two new series of aryl SMAMPs (synthetic mimics of antimicrobial peptides) with facially amphiphilic (FA) and disrupted amphiphilic (DA) topologies were designed and synthesized to directly assess the role of amphiphilicity on their antimicrobial activity against Gram-positive and Gram-negative bacteria in closely related structures. The FA SMAMPs displayed broad spectrum antimicrobial activity against both Gram-positive S. aureus and Gram-negative E. coli, whereas the DA SMAMPs, which contained a polar amide bond in between the hydrophobic moieties, only exhibited activity toward S. aureus with increasing hydrophobicity. The integy moment (IW) was used to quantify the amphiphilicity of the SMAMPs and confirmed that it is critical for the design of SMAMPs with Gram-negative activity.
Keywords: antimicrobial peptides (AMPs), synthetic mimics of antimicrobial peptides (SMAMPs), amphiphilicity, hydrophobicity, facially amphiphilic, disrupted amphiphilic, Suzuki coupling, integy moment (IW), Volsurf software, Gram-negative, Gram-positive
The design of novel antibacterial agents with reduced bacterial resistance has become imperative due to the emergence of antibiotic-resistant bacteria like methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus (VRE).1 In the past two decades, significant research on naturally occurring antimicrobial peptides (AMPs) has demonstrated their potential as therapeutic candidates.2 AMPs constitute an essential component of the innate immune system of all multicellular organisms. AMPs have multiple biological functions including direct broad spectrum activity against a variety of pathogens and modulation of the immune response.3 One plausible mechanism of action involves interaction with and permeabilization of the bacterial cell membrane, leading to cell death.4 This unique cell membrane interaction of AMPs makes bacterial resistance evolution difficult, giving them a strong advantage over conventional antibiotics.5 However, their clinical development has been hampered due to their toxicity, poor bioavailability, and low proteolytic stability.4,6 This has driven the development of synthetic mimics of antimicrobial peptides (SMAMPs), with the goals of overcoming the problems associated with AMPs while retaining the essential characteristics that are vital for antimicrobial activity.6,7
One of the main design parameters in the development of SMAMPs is facial amphiphilicity, since most AMPs fold into amphiphilic structures upon interaction with the bacterial cell membrane.3 Natural AMPs typically have both cationic charges and hydrophobic groups that segregate into the amphiphilic structure. The positive charge, ranging from +2 to +9, is the driving force for the electrostatic interaction between AMPs and the negatively charged bacterial membrane, while the hydrophobic residues help insertion into the hydrophobic core.3 However, the influence of amphiphilicity on the overall activity of AMPs is not fully understood. Gellman and co-workers studied the antimicrobial activity of several β-peptides and found that the non-amphiphilic scrambled peptide was inactive, confirming that a facially amphiphilic (FA) helix is necessary for their antibacterial activity.8 Similarly, in a different study, increasing the amphiphilicity and helicity of cecropin A-melittin hybrids improved the activity and lowered the toxicity as compared to the natural peptide, melittin.9 On the other hand, an increase in the amphiphilicity of some peptides (measured in terms of the hydrophobic moment) has been shown to increase hemolytic activity.10−12
Focus has more recently turned to the interfacial partitioning of both AMPs and SMAMPs into the membrane and their ability to disrupt lipid organization within the bilayer.13,14 Wimley has further proposed that the peptide must have disrupted amphiphilicity; i.e., the peptide must have imperfect segregation of polar and hydrophobic groups with the hydrophobic region broken up by the presence of at least one polar residue (lysine or arginine).13 Several studies have been reported that support this theory.11,15,16 For example, the incorporation of a lysine residue into the hydrophobic region of a peptide, D1 (V13), led to a significant reduction in toxicity (32-fold) and a 3-fold enhancement in the antimicrobial activity against Gram-negative bacteria.11 In another study, the pore-forming ability of melittin was reduced when a charged residue in the hydrophobic region was replaced by leucine (hydrophobic).15
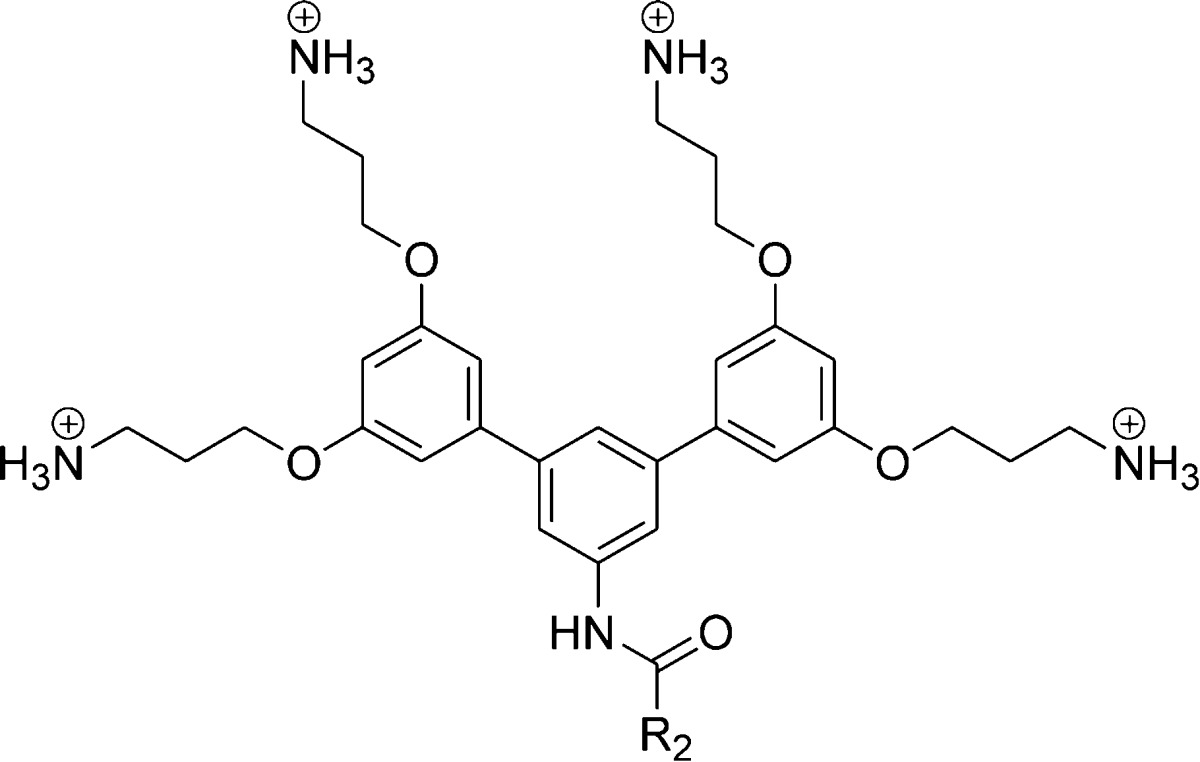
Previously, our research group designed several aromatic oligomers based on arylamide,17,18 urea,19 and phenylene-ethynylene (PE)20 backbones. These aromatic oligomers were FA with broad spectrum antimicrobial activity and selectivity. In the case of arylamides and ureas, rigidifying the conformation via hydrogen bonding led to a more FA topology, which improved the antimicrobial activity. The PE oligomers adopted a FA topology at the oil–water interface via proper placement of the amine groups and rotation around the single bonds.21 However, to the best of our knowledge, there are no reports evaluating the role of amphiphilicity and/or imperfect hydrophobicity on the antimicrobial activity of oligomeric SMAMPs. Herein, we report two new series of aryl SMAMPs based on Suzuki coupling, designed to directly evaluate the role of amphiphilicity on their activity. The first series of SMAMPs is FA, and the second series has a disrupted amphiphilic (DA) topology, with a polar amide linker incorporated in the hydrophobic region to disrupt the amphiphilicity (Figure 1). The structure–activity relationship (SAR) studies of these series of SMAMPs reveal that an amphiphilic topology is critical for broad spectrum activity, especially against Gram-negative bacteria.
Figure 1.
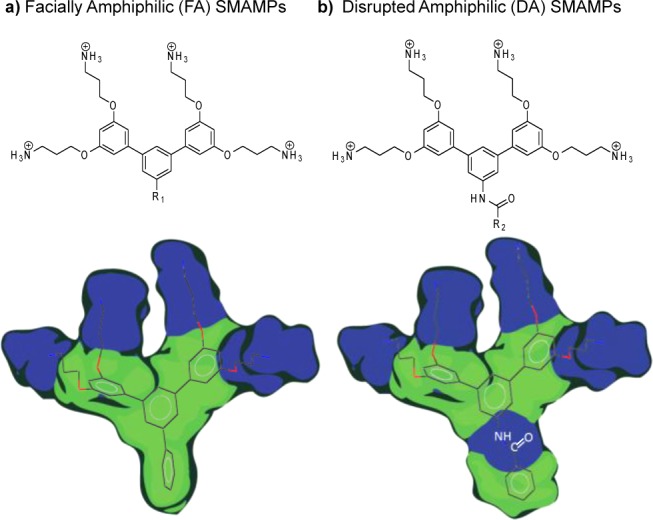
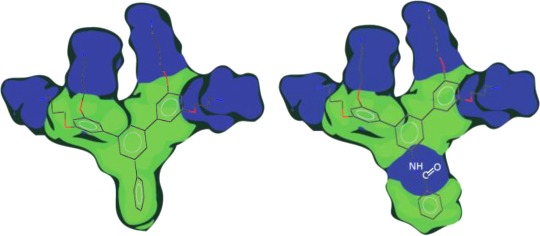
Design of aryl SMAMPs with pendant aromatic groups. (a) SMAMPs with a FA topology and (b) SMAMPs with a DA topology. R1 and R2 represent the pendant aromatic groups (see Tables 1 and 2 for detailed structures). Blue and green colors represent the hydrophilic and hydrophobic regions, respectively.
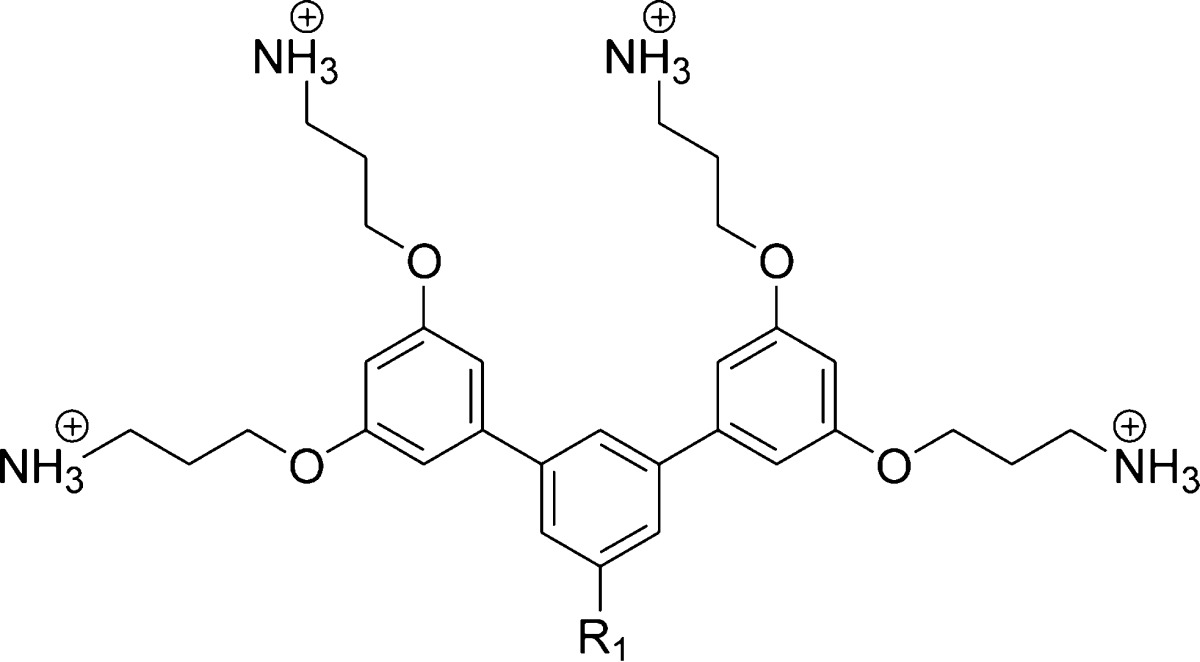
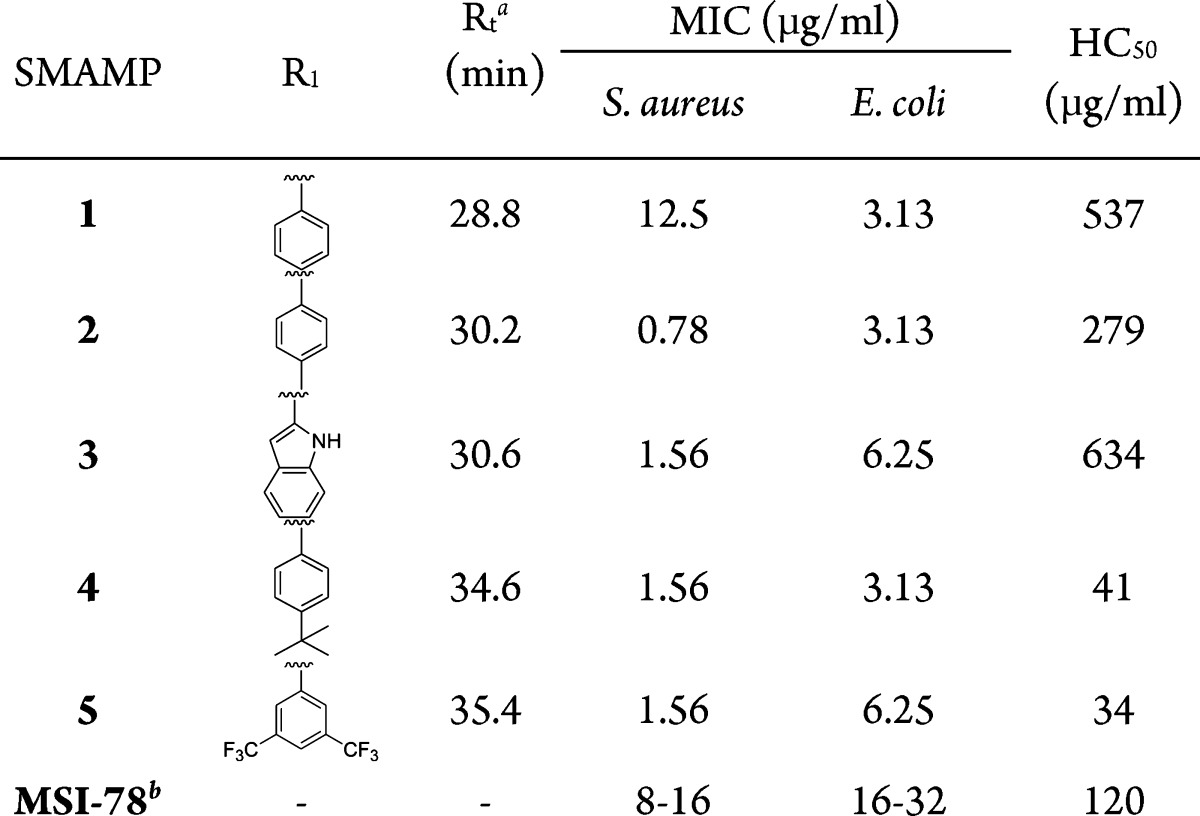
Previous SAR studies of aryl SMAMPs synthesized via Suzuki coupling showed that the addition of a pendant aromatic ring (SMAMP 1) significantly improved the activity of an otherwise inactive SMAMP (R1 = H) toward Gram-negative E. coli.22 Moving forward with this strategy, a new series of SMAMPs were designed with different pendant aromatic groups of increasing hydrophobicity directly attached to the central ring (Table 1, SMAMPs 2–5), thus retaining their FA structure. To gain insight on the role of amphiphilicity on the antimicrobial activity, a second series were developed, where an amide moiety was used to append the pendant hydrophobic aromatic groups (Table 2, SMAMPs 6–9). The polar amide group functioned as a disruptive linker (hydrophilic patch) that broke up the hydrophobic region, resulting in a DA topology. The synthesis and characterization of all the SMAMPs are described in the Supporting Information. The antimicrobial activity of these SMAMPs was determined by their minimum inhibitory concentration (MIC) against four pathogens (Tables S1, S2 in the Supporting Information), including both Gram-negative and Gram-positive bacteria, and the toxicity toward human red blood cells (RBCs) was evaluated in terms of the HC50 (the concentration that causes 50% hemolysis of the RBCs). The relative hydrophobicity of the SMAMPs was measured by reverse-phase HPLC retention time (Rt).
Table 1. Antimicrobial and Hemolytic Activities of the SMAMPs with FA Topologies.
Measured by HPLC using a C8 column with a gradient of 1% acetonitrile/min starting with 100% water.
Literature values (reference (22) and references therein).
Table 2. Antimicrobial and Hemolytic Activities of the SMAMPs with DA Topologies.
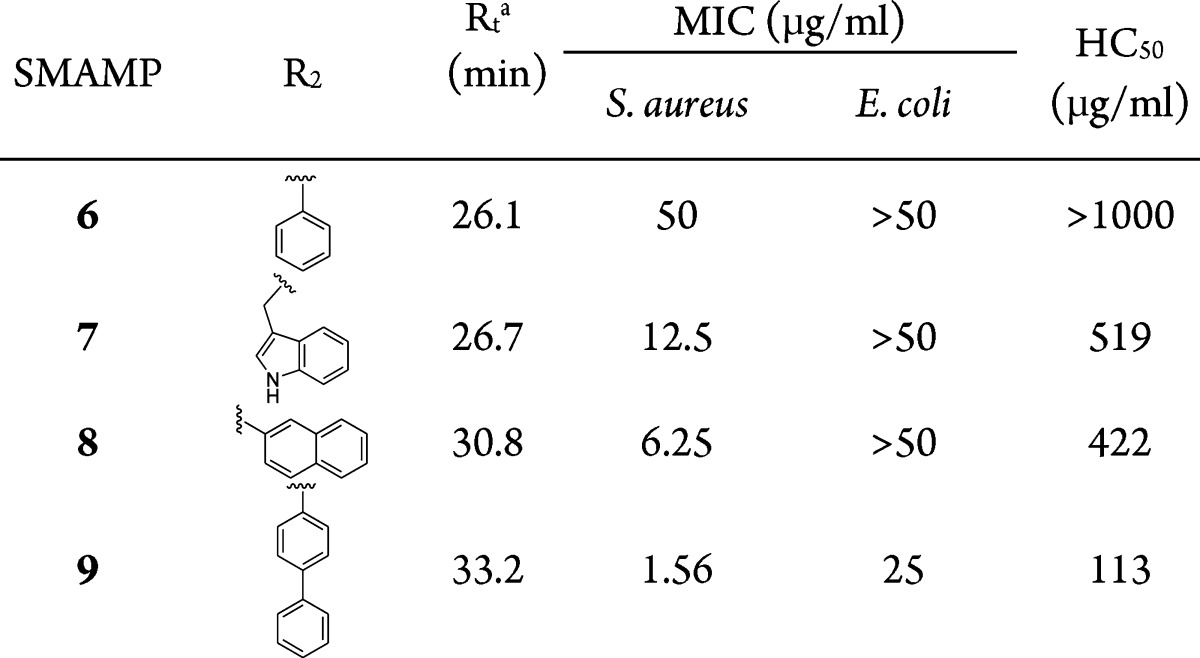
Measured by HPLC using a C8 column with a gradient of 1% acetonitrile/min starting with 100% water.
The antimicrobial and the hemolytic activities of the FA SMAMPs are summarized in Table 1. In general, all of the compounds had antimicrobial activities higher than the well-known AMP MSI-78, which is currently in phase III clinical trials as a topical antibiotic.23 All of the FA SMAMPs 2–5 showed improved antimicrobial activity against S. aureus as compared to SMAMP 1. The substitution of the pendant phenyl group with a methyl-phenyl group (SMAMP 2) resulted in a 16-fold increase in activity against S. aureus with an MIC of 0.78 μg/mL. While this modification also led to a 2-fold decrease in the HC50, it still resulted in a very high selectivity toward S. aureus (selectivity ∼350). The incorporation of a pendant indole ring in SMAMP 3 yielded the highest selectivity in the FA series against S. aureus, with selectivity values of ∼400 for S. aureus and ∼100 for E. coli, along with very low toxicity (HC50 = 634 μg/mL). This may be due to the unique membrane affinity of the indole ring, as observed with tryptophan.24 Further increases in hydrophobicity with the tert-butyl phenyl and bis(trifluoromethyl)phenyl pendant groups (SMAMPs 4 and 5) did not alter the antimicrobial activity but led to increased hemolytic potency, indicating that the SMAMPs had become less selective for both S. aureus and E. coli. This suggests that increasing the hydrophobicity beyond a particular threshold only increases hemolytic activity, without any further improvement in the antimicrobial activity.
The DA SMAMPs 6–9 were synthesized using four different pendant groups: benzene, indole, naphthalene, and phenylbenzene. Table 2 summarizes the antimicrobial and hemolytic activities of the SMAMPs with DA topologies. The SMAMPs 6 and 7 containing benzene and indole rings, respectively, were synthesized as a direct comparison to their FA analogues, SMAMPs 1 and 3. In the case of SMAMP 6, the use of a polar amide linker to connect the two benzene rings significantly reduced the overall hydrophobicity (Rt = 26.1 min) in comparison to its FA analogue, SMAMP 1 (Rt = 28.8 min). This resulted in a complete loss of activity of SMAMP 6 against both S. aureus and E. coli. Similarly, SMAMP 7 was more hydrophilic and had a lower activity as compared to FA SMAMP 3. The use of a pendant indole ring in the DA SMAMP 7 improved the activity against S. aureus by 4-fold in comparison to SMAMP 6, but no change in activity was observed against E. coli.
Increasing the hydrophobicity of the SMAMPs in the DA series to match the hydrophobicity range of the FA SMAMPs required the use of more bulky and hydrophobic aromatic residues. For this purpose, naphthalene and phenylbenzene groups were used. SMAMP 8, with an amide linker and a naphthalene pendant ring, had a retention time of 30.8 min, which is comparable to the FA SMAMPs 2 and 3. Despite identical hydrophobicities by HPLC, SMAMP 8 showed no activity against E. coli, while SMAMP 3 had an MIC of 6.25 μg/mL. The hydrophobicity was further increased by the addition of a phenylbenzene group (SMAMP 9 with Rt = 33.2 min), which improved the MIC against S. aureus (1.56 μg/mL) while remaining inactive against E. coli. Plots of antimicrobial potency (1/MIC) of the FA and DA SMAMPs against both E. coli and S. aureus as a function of the HPLC retention time (see Figure S9 in the Supporting Information) suggest that in the case of S. aureus while amphiphilicity is important, it is the overall hydrophobicity that determines their activity. However, for Gram-negative E. coli, antimicrobial activity was more sensitive to changes in amphiphilicity than S. aureus, since none of the DA SMAMPs were active against E. coli.
Further assessment of a correlation between the antimicrobial activity and the amphiphilicity of these SMAMPs required the quantification of their amphiphilicity. The amphiphilicity of natural as well as synthetic AMPs is usually measured in terms of the hydrophobic moment, but this is not possible in the case of small molecule SMAMPs. For this purpose, VolSurf was used, which converts the 3D molecular interaction fields of a molecule into a set of simple descriptors.25 One of the descriptors is the integy moment (IW), which is a vector pointing from the center of mass of the molecule to the center of the hydrophilic region. A higher IW value is indicative of higher segregation of the hydrophilic and hydrophobic regions. Thus, IW is a convenient parameter for quantifying the amphiphilicity of a molecule. The IW values for both the FA and DA series of SMAMPs (in their charged state) were determined using the VolSurf software (Table S3 in the Supporting Information).
A plot of antimicrobial potency against E. coli versus IW is shown in Figure 2. In the DA SMAMPs (red dots), the presence of the amide linker was enough to disrupt the amphiphilicity, as evidenced by the lower IW values (less than 0.24) that remained constant even upon the addition of bulky hydrophobic groups. On the other hand, the more potent FA SMAMPs (black squares) all had IW values greater than 0.24. These results indicate that a threshold value of amphiphilicity exists, above which activity against Gram-negative E. coli can be achieved.
Figure 2.
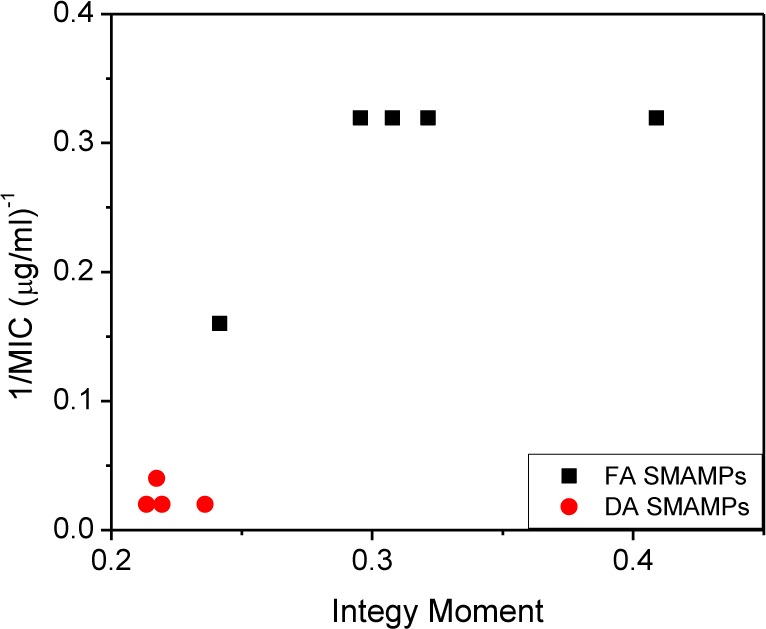
Plot of the E. coli antimicrobial potency vs the IW for the SMAMPs in the DA series (red dots) and the FA series (black squares).
While SMAMPs likely kill bacteria via multiple mechanisms, similar to AMPs, membrane interactions seem to be critical.14 This increased importance of the FA topology in killing Gram-negative bacteria is consistent with the requirement for the generation of negative Gaussian curvature leading to pore formation in bacterial membranes.26 The ability to induce negative Gaussian curvature requires both positive and negative mean curvature components.27 The SMAMPs contribute both the negative-inducing components via cationic amines and the positive-inducing component via insertion of hydrophobic residues.14 The Gram-negative E. coli lipid membrane is richer in negative intrinsic curvature lipid phosphatidylethanolamine (PE ∼ 80%) as compared to the Gram-positive S. aureus lipid membrane. Thus, it is probable that disruption of Gram-negative bacteria requires more efficient insertion of hydrophobic components. Our aryl SMAMPs 2 and 8, for example, have four amines and similar overall hydrophobicities as measured by HPLC (Rt = 30.2 and 30.8 min, respectively); however, the hydrophobicity of the DA SMAMP 8 is disrupted by the presence of the amide linker. The high energy penalty of 2 kcal/mol required for the insertion of a polar amide moiety into the hydrophobic core limits the penetration ability of the DA SMAMPs.28 This reduces the volume of the hydrophobic groups that is inserted into the membrane resulting in less positive curvature generation, which is needed for pore formation and a broad range of barrier disruption events. This is reflected in the difference in activities of the DA and FA SMAMPs 8 and 2 against E. coli (MIC > 50 vs 3.13 μg/mL, respectively).
In summary, the SAR studies of the FA and DA SMAMPs described here reveal the significance of topologically homogeneous amphiphilicity for attaining antimicrobial activity, especially against Gram-negative E. coli. The FA SMAMPs had broad spectrum activity against both S. aureus and E. coli, whereas the insertion of a disruptive amide linker in the DA SMAMPs led to a complete loss of activity against E. coli. The IW values of the SMAMPs provided an efficient approach to quantify the amphiphilicity of these small molecules, and this method can aid in the design of potent and selective SMAMPs with broad spectrum activity that are potentially useful for therapeutic applications.
Acknowledgments
We acknowledge Melissa Lackey and Michael Lis for their invaluable comments on the early drafts.
Supporting Information Available
Experimental details, HPLC purity data, broad spectrum antimicrobial activity, and additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
This work has been supported by the NIH (AI-074866 and U01 AI-082192). Mass spectral data were obtained at the University of Massachusetts Mass Spectrometry Facility, which is supported, in part, by the National Science Foundation. This work also used shared facilities supported from the MRSEC on Polymers at UMass (DMR-0820506).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Taubes G. The bacteria fight back. Science 2008, 321, 356–361. [DOI] [PubMed] [Google Scholar]
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. W.; Sahl H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [DOI] [PubMed] [Google Scholar]
- Jenssen H.; Hamill P.; Hancock R. E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr A.; Gooderham W.; Hancock R. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [DOI] [PubMed] [Google Scholar]
- Rotem S.; Mor A. Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 1582–1592. [DOI] [PubMed] [Google Scholar]
- Som A.; Vemparala S.; Ivanov I.; Tew G. N. Synthetic mimics of antimicrobial peptides. Biopolymers 2008, 90, 83–93. [DOI] [PubMed] [Google Scholar]
- Porter E. A.; Weisblum B.; Gellman S. H. Mimicry of host-defense peptides by unnatural oligomers: Antimicrobial β-peptides. J. Am. Chem. Soc. 2002, 124, 7324–7330. [DOI] [PubMed] [Google Scholar]
- Andreu D.; Ubach J.; Boman A.; Wahlin B.; Wade D.; Merrifield R. B.; Boman H. G. Shortened cecropin A-melittin hybrids. Significant size reduction retains potent antibiotic activity. FEBS Lett. 1992, 296, 190–194. [DOI] [PubMed] [Google Scholar]
- Wieprecht T.; Dathe M.; Krause E.; Beyermann M.; Maloy W. L.; MacDonald D. L.; Bienert M. Modulation of membrane activity of amphipathic, antibacterial peptides by slight modifications of the hydrophobic moment. FEBS Lett. 1997, 417, 135–140. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Mant C. T.; Farmer S. W.; Hancock R. E.; Vasil M. L.; Hodges R. S. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Vidal M.; Jayasinghe S.; Ladokhin A. S.; White S. H. Folding amphipathic helices into membranes: amphiphilicity trumps hydrophobicity. J. Mol. Biol. 2007, 370, 459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley W. C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Gordon V. D.; Mishra A.; Som A.; Purdy K. R.; Davis M. A.; Tew G. N.; Wong G. C. Synthetic antimicrobial oligomers induce a composition-dependent topological transition in membranes. J. Am. Chem. Soc. 2007, 129, 12141–12147. [DOI] [PubMed] [Google Scholar]
- Mihajlovic M.; Lazaridis T. Charge distribution and imperfect amphipathicity affect pore formation by antimicrobial peptides. Biochim. Biophys. Acta 2012, 1818, 1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z.; Vasil A. I.; Gera L.; Vasil M. L.; Hodges R. S. Rational design of alpha-helical antimicrobial peptides to target Gram-negative pathogens, Acinetobacter baumannii and Pseudomonas aeruginosa: Utilization of charge, “specificity determinants,” total hydrophobicity, hydrophobe type and location as design parameters to improve the therapeutic ratio. Chem. Biol. Drug Des. 2011, 77, 225–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H.; Doerksen R.; Jones T.; Klein M.; Tew G. Biomimetic Facially Amphiphilic Antibacterial Oligomers with Conformationally Stiff Backbones. Chem. Biol. 2006, 13, 427–435. [DOI] [PubMed] [Google Scholar]
- Tew G. N.; Liu D.; Chen B.; Doerksen R. J.; Kaplan J.; Carroll P. J.; Klein M. L.; DeGrado W. F. De novo design of biomimetic antimicrobial polymers. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 5110–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H.; Doerksen R. J.; Tew G. N. Synthesis of urea oligomers and their antibacterial activity. Chem. Commun. 2005, 1537. [DOI] [PubMed] [Google Scholar]
- Arnt L.; Rennie J. R.; Linser S.; Willumeit R.; Tew G. N. Membrane activity of biomimetic facially amphiphilic antibiotics. J. Phys. Chem. B 2006, 110, 3527–3532. [DOI] [PubMed] [Google Scholar]
- Arnt L.; Tew G. N. Cationic facially amphiphilic poly(phenylene ethynylene)s studied at the air-water interface. Langmuir 2003, 19, 2404–2408. [Google Scholar]
- Thaker H. D.; Som A.; Ayaz F.; Lui D.; Pan W.; Scott R. W.; Anguita J.; Tew G. N. Synthetic mimics of antimicrobial peptides with immunomodulatory responses. J. Am. Chem. Soc. 2012, 134, 11088–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell C. D.; Hiss J. A.; Hancock R. E. W.; Schneider G. Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discovery 2012, 11, 37–51. [DOI] [PubMed] [Google Scholar]
- Chan D. I.; Prenner E. J.; Vogel H. J. Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim. Biophys. Acta 2006, 1758, 1184–1202. [DOI] [PubMed] [Google Scholar]
- Cruciani G.; Crivori P.; Carrupt P. A.; Testa B. Molecular fields in quantitative structure–permeation relationships: The VolSurf approach. J. Mol. Struct.: THEOCHEM 2000, 503, 17–30. [Google Scholar]
- Zimmerberg J.; Kozlov M. M. How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 2006, 7, 9–19. [DOI] [PubMed] [Google Scholar]
- Schmidt N. W.; Mishra A.; Lai G. H.; Davis M.; Sanders L. K.; Tran D.; Garcia A.; Tai K. P.; McCray P. B.; Ouellette A. J.; Selsted M. E.; Wong G. C. Criterion for amino acid composition of defensins and antimicrobial peptides based on geometry of membrane destabilization. J. Am. Chem. Soc. 2011, 133, 6720–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley W. C. Energetics of peptide and protein binding to lipid membranes. Adv. Exp. Med. Biol. 2010, 677, 14–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.