

Abstract
Motivation: Ancient DNA (aDNA) molecules in fossilized bones and teeth, coprolites, sediments, mummified specimens and museum collections represent fantastic sources of information for evolutionary biologists, revealing the agents of past epidemics and the dynamics of past populations. However, the analysis of aDNA generally faces two major issues. Firstly, sequences consist of a mixture of endogenous and various exogenous backgrounds, mostly microbial. Secondly, high nucleotide misincorporation rates can be observed as a result of severe post-mortem DNA damage. Such misincorporation patterns are instrumental to authenticate ancient sequences versus modern contaminants. We recently developed the user-friendly mapDamage package that identifies such patterns from next-generation sequencing (NGS) sequence datasets. The absence of formal statistical modeling of the DNA damage process, however, precluded rigorous quantitative comparisons across samples.
Results: Here, we describe mapDamage 2.0 that extends the original features of mapDamage by incorporating a statistical model of DNA damage. Assuming that damage events depend only on sequencing position and post-mortem deamination, our Bayesian statistical framework provides estimates of four key features of aDNA molecules: the average length of overhangs (λ), nick frequency (ν) and cytosine deamination rates in both double-stranded regions (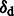 ) and overhangs (
) and overhangs (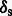 ). Our model enables rescaling base quality scores according to their probability of being damaged. mapDamage 2.0 handles NGS datasets with ease and is compatible with a wide range of DNA library protocols.
). Our model enables rescaling base quality scores according to their probability of being damaged. mapDamage 2.0 handles NGS datasets with ease and is compatible with a wide range of DNA library protocols.
Availability: mapDamage 2.0 is available at ginolhac.github.io/mapDamage/ as a Python package and documentation is maintained at the Centre for GeoGenetics Web site (geogenetics.ku.dk/publications/mapdamage2.0/).
Contact: jonsson.hakon@gmail.com
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
DNA in historical samples is subject to a plethora of environmental conditions and degradation reactions (Sawyer et al., 2012). Abasic sites, strand breaks, interstrand cross-links and a wide diversity of atypic nucleotidic bases are formed following oxidative and hydrolytic degradation (Lindahl, 1993; Pääbo et al., 2004), even in the most favorable preservation conditions.
Post-mortem DNA damage limits our ability to access ancient DNA (aDNA) sequences and increases the risk of exogenous modern contamination, as undamaged DNA molecules are more prone to enzymatic manipulation. Nucleotide misincorporation patterns, which are mostly driven by deaminated forms of cytosines (uracils), have been suggested as a powerful approach to authenticate aDNA sequences generated on next-generation sequencing (NGS) platforms (Briggs et al., 2007) and motivated the creation of the mapDamage package (Ginolhac et al., 2011). Such patterns could vary according to the specific molecular approach used for constructing (Meyer et al., 2012) and/or amplifying (Ginolhac et al., 2011) second-generation DNA libraries. For instance, for one of the most popular protocols (Meyer and Kircher, 2010), we observe inflated cytosine deamination rates at 5′-overhangs, an increase in 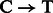 substitution rates toward sequencing starts and complementary increase in
substitution rates toward sequencing starts and complementary increase in 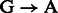 rates toward reads ends (Briggs et al., 2007). Conversely, a novel procedure targeting single-stranded templates has shown elevated
rates toward reads ends (Briggs et al., 2007). Conversely, a novel procedure targeting single-stranded templates has shown elevated 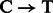 substitution rates at both ends (Meyer et al., 2012).
substitution rates at both ends (Meyer et al., 2012).
Statistical modeling of such patterns has been developed by Briggs et al., 2007 with strand break, overhangs and cytosine deamination as key factors. Using read alignment to reference genomes and maximum likelihood optimization, this approach has delivered the first quantitative estimates of damage parameters. However, the likelihood framework originally implemented scales poorly with the size of NGS datasets, and extensive running times have prevented common usage. Here, we present an extension of mapDamage, which implements a fast approximation of the DNA damage model using a Bayesian framework. mapDamage 2.0 opens the possibility of comparing DNA damage levels across temporal and environmental gradients. Posterior distributions of damage parameters also enable penalizing the quality score of likely damaged bases, reducing noise in downstream single-nucleotide polymorphism (SNP) calling procedures.
2 APPROACH
Here we build on the DNA damage model described in Briggs et al., 2007. We make the simplifying assumption that mutations and post-mortem DNA damage are independent within a fragment, with occurrences depending only on the relative position from the sequence ends.
3 METHODS
The general idea is to mutate bases following an Hasegawa, Kishino and Yano (HKY) transition matrix (Hasegawa et al., 1985) and then independently add post-mortem damage on top of mutated bases. In this framework, we have multinomial distributions describing the position-specific substitutions for any given base (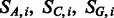 and
and 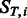 ).
).
Θ is the HKY transition matrix, and 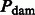 is defined as the DNA damage transition matrix. We assume post-mortem cytosine deamination is the main driver of nucleotide misincorporations in agreement with experimental evidence (Briggs et al., 2007), providing
is defined as the DNA damage transition matrix. We assume post-mortem cytosine deamination is the main driver of nucleotide misincorporations in agreement with experimental evidence (Briggs et al., 2007), providing
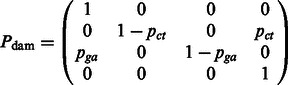 |
Where the base-specific damage probabilities are defined as
The motivation for the base-specific damage probabilities 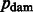 is best explained by the Markov chain in Figure 1 where the first jump decides if the position is before or after a nick; then a
is best explained by the Markov chain in Figure 1 where the first jump decides if the position is before or after a nick; then a 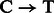 substitution could be observed following deamination in overhang or double-stranded DNA regions. A similar Markov chain could be drawn for
substitution could be observed following deamination in overhang or double-stranded DNA regions. A similar Markov chain could be drawn for 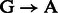 substitutions (Supplementary Section 1).
substitutions (Supplementary Section 1).
Fig. 1.
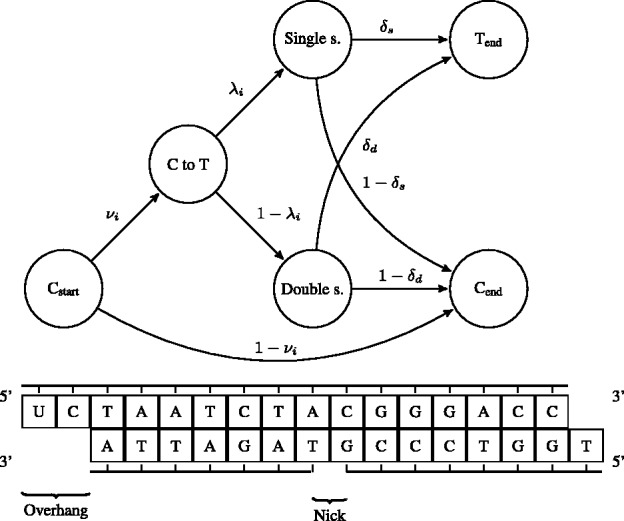
A schematic view describing the DNA damage Markov chain, which extends the DNA substitution model. The states 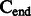 and
and 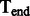 correspond to the final nucleotides in the sequences
correspond to the final nucleotides in the sequences
For rescaling base quality scores, we assume that 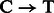 and
and 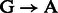 substitutions either originate from true biological differences or from damage driven misincorporations. We can derive an estimate for the probability that a
substitutions either originate from true biological differences or from damage driven misincorporations. We can derive an estimate for the probability that a 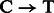 (similar for
(similar for 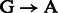 ) misincorporation at position i along the reads is due to damage using
) misincorporation at position i along the reads is due to damage using
We can now correct base quality scores provided in alignment BAM files [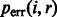 at position i for read r] using
at position i for read r] using
4 DISCUSSION
We applied mapDamage2.0 on a series of aDNA sequence datasets generated from a range of periods, source materials and environments (Supplementary Section 3). Posterior predictive intervals and empirical frequencies are in general agreement, as shown for the ancient plague dataset (Supplementary Table S2 and Supplementary Figs S4–S9) (Schuenemann et al., 2011), demonstrating the adequacy of our method. We observed a ratio of cytosine deamination rates for double- and single-stranded regions orders of magnitude greater than estimates based on in vitro experiments in aqueous solution (0.007 in Lindahl, 1993 versus 0.026–0.070 for Schuenemann et al., 2011 in Supplementary Table S1). This suggests that tissue- and sample-specific micro-environmental characteristics drive different DNA damage kinetics in situ. We also found a significant rank correlation between the posterior mean for single-stranded cytosine deamination and sample age (Supplementary Table S3) in agreement with Sawyer et al., 2012. However, remains of similar age and location showed diverse parameter estimates (Supplementary Table S2), suggesting a prominent role of micro-environmental characteristics over age in diagenesis.
We also applied our quality rescaling scheme to the sequence data of an Australian Aboriginal individual who died in 1920s (Rasmussen et al., 2011). This increased the overlap of genotype calls to dbSNP v137, suggesting that lower false-positive SNP calls were achieved (Supplementary Table S4).
5 CONCLUSION
We have developed a computational method for inferring aDNA damage parameters from NGS sequence datasets, with minimal changes to the DNA damage model presented by Briggs et al., 2007. Our model is compatible with the specificities of different sequencing and library building protocols. We believe that downscaling quality scores of likely damaged bases is the first from a long list of possible applications for damage parameter posterior distributions, limiting the impact of nucleotide misincorporations in downstream sequence analyses. The knowledge of such distributions could also be instrumental for improving mapping procedures to reference genomes (Schubert et al., 2012).
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to EUROTAST (Marie Curie FP7 Initial Training Network), a Marie Curie Career Integration Grant (293845), a Marie Curie Intra-European Fellowship (299176) and Danish Council for Independent Research; Natural Sciences (FNU) for funding. Johannes Krause, Simon Rasmussen and Stefan Prost for sharing data/scripts.
Conflict of Interest: none declared.
REFERENCES
- Briggs AW, et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl Acad. Sci. USA. 2007;104:14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginolhac A, et al. mapDamage: testing for damage patterns in ancient DNA sequences. Bioinformatics. 2011;27:2153–2155. doi: 10.1093/bioinformatics/btr347. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, et al. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010;2010 doi: 10.1101/pdb.prot5448. pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- Meyer M, et al. A high-coverage genome sequence from an archaic denisovan individual. Science. 2012;338:222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pääbo S, et al. Genetic analyses from ancient DNA. Annu. Rev. Genet. 2004;38:645–679. doi: 10.1146/annurev.genet.37.110801.143214. [DOI] [PubMed] [Google Scholar]
- Rasmussen M, et al. An aboriginal Australian genome reveals separate human dispersals into Asia. Science. 2011;334:94–98. doi: 10.1126/science.1211177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer S, et al. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PloS One. 2012;7:e34131. doi: 10.1371/journal.pone.0034131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M, et al. Improving ancient DNA read mapping against modern reference genomes. BMC Genomics. 2012;13:178. doi: 10.1186/1471-2164-13-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuenemann VJ, et al. Targeted enrichment of ancient pathogens yielding the pPCP1 plasmid of Yersinia pestis from victims of the Black Death. Proc. Natl Acad. Sci. USA. 2011;108:E746–E752. doi: 10.1073/pnas.1105107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.