

Abstract
Motivation: Genome-wide haplotype reconstruction from sequence data, or haplotype assembly, is at the center of major challenges in molecular biology and life sciences. For complex eukaryotic organisms like humans, the genome is vast and the population samples are growing so rapidly that algorithms processing high-throughput sequencing data must scale favorably in terms of both accuracy and computational efficiency. Furthermore, current models and methodologies for haplotype assembly (i) do not consider individuals sharing haplotypes jointly, which reduces the size and accuracy of assembled haplotypes, and (ii) are unable to model genomes having more than two sets of homologous chromosomes (polyploidy). Polyploid organisms are increasingly becoming the target of many research groups interested in the genomics of disease, phylogenetics, botany and evolution but there is an absence of theory and methods for polyploid haplotype reconstruction.
Results: In this work, we present a number of results, extensions and generalizations of compass graphs and our HapCompass framework. We prove the theoretical complexity of two haplotype assembly optimizations, thereby motivating the use of heuristics. Furthermore, we present graph theory–based algorithms for the problem of haplotype assembly using our previously developed HapCompass framework for (i) novel implementations of haplotype assembly optimizations (minimum error correction), (ii) assembly of a pair of individuals sharing a haplotype tract identical by descent and (iii) assembly of polyploid genomes. We evaluate our methods on 1000 Genomes Project, Pacific Biosciences and simulated sequence data.
Availability and Implementation: HapCompass is available for download at http://www.brown.edu/Research/Istrail_Lab/.
Contact: Sorin_Istrail@brown.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The genome sequence of a human individual can be modeled as 23 pairs of sequences of four nucleotide bases, A, C, G and T, representing the 22 pairs of autosomes and the sex chromosomes. However, ∼99.5% of any two individuals’ genome sequences is shared within a population. The ∼0.5% of the nucleotide bases varying within a population range from single-nucleotide polymorphisms (SNPs) to more complex structural changes, for example, deletions or insertions of genomic material. A sequence of genomic variants, typically SNPs, with the non-varying DNA removed is referred to as a haplotype.
Standard genome sequencing workflows produce contiguous DNA segments of an unknown chromosomal origin. De novo assemblies for genomes with two sets of chromosomes (diploid) or more (polyploid) produce consensus sequences in which the relative haplotype phase between variants is undetermined. The set of sequencing reads can be mapped to the phase-ambiguous reference genome and the diploid chromosome origin can be determined but, without knowledge of the haplotype sequences, reads cannot be mapped to the particular haploid chromosome sequence. As a result, reference-based genome assembly algorithms also produce unphased assemblies. However, sequence reads are derived from a single haploid fragment and thus provide valuable phase information when they contain two or more variants. The haplotype assembly problem aims to compute the haplotype sequences for each chromosome given a set of aligned sequence reads to the genome and variant information. The haplotype phase of variants is inferred from assembling overlapping sequence reads [Browning and Browning (2011); Halldórsson et al. (2003); Schwartz (2010)].
The input to the haplotype assembly problem is a matrix M whose rows correspond to aligned read fragments and columns correspond to SNPs (Fig. 1). The quality of M’s construction depends on the parameters of the sequencing workflow and the accuracy of the read alignment algorithms. Misaligned read fragments can introduce erroneous base calls or sampling biases so the careful alignment of sequence reads is necessary for high-quality haplotype assemblies. Without read alignment or sequencing errors, the haplotype assembly problem can be solved in time linear in the size of M by partitioning the fragments in two sets whereby no fragments internal to a set share an SNP and differ in the allele called. To address erroneous base calls or misplaced alignments, three primary haplotype assembly optimizations have been developed: minimum error correction (MEC), minimum SNP removal (MSR) and minimum fragment removal (MFR). The goal is to convert M into a state such that the fragments (rows of M) can be distributed into two sets corresponding to the two haplotypes. All fragments in a set must agree on the allele at each SNP site and this is accomplished using the minimum number of SNP allele flips (0 to 1 or vice versa - MEC), SNP (columns of M) removals (MSR) or fragment (rows of M) removals (MFR).
Fig. 1.
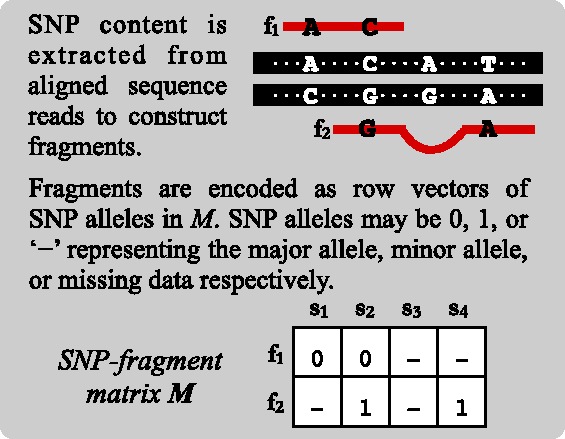
Construction of the input to the haplotype assembly problem
Lancia et al. (2001) and Rizzi et al. (2002) provide a theoretical foundation for the MFR and MSR optimizations and describe the fundamental SNP and fragment conflict graph structures. The first widely available haplotype assembly software package was presented in Panconesi and Sozio (2004) in which the authors describe the Fast Hare algorithm, which optimizes the ‘Min Element Removal’ problem. Bansal et al. (2008) describe a Markov chain model with Metropolis updating rules to sample a set of likely haplotypes under the MEC optimization. In a follow-up, the authors present a much faster algorithm on a related graph model that relates maximum cuts to SNP allele flips (in the MEC model) [Bansal and Bafna (2008)]. Still other authors have suggested reductions to the well-known maximum satisfiability problem [He et al. (2010); Mousavi et al. (2011)] The Levy et al. (2007) algorithm is a well-known heuristic that was used to haplotype assemble the HuRef genome; it assigns fragments to haplotypes in a greedy fashion and iteratively refines the solution by comparing the set of fragments to the assembled haplotypes using majority rule phasings. In a recent survey, Geraci (2010) describes the Levy et al. (2007) algorithm as, arguably, the best performing algorithm tested.
The first extension of the haplotype assembly problem that addressed the simultaneous assembly of multiple diploid chromosomes was presented in Li et al. (2006); however, the benefits of multi-haplotype assembly are not clear for a set of unrelated individuals. Halldorsson et al. (2011) continued development of this theory by describing methods for assembling individuals who share a haplotype identical by descent (IBD) using relationships among the reads.
Aguiar and Istrail (2012) introduced a new graph data structure, algorithmic framework and the minimum weighted edge removal (MWER) optimization, which together have several advantages over existing methods. Recall that the rows of M correspond to sequence read fragments with the non-polymorphic bases removed such that only SNPs remain. The HapCompass model defined in Aguiar and Istrail (2012) is composed of the compass graph GC core data structure, which summarizes the rows of M using edges weights and the MWER optimization that aims to remove a minimum weighted set of edges from GC such that a unique phasing may be constructed. The algorithm operates on the spanning-tree cycle basis of GC to iteratively remove errors that are manifested through a particular type of simple cycle [Deo et al. (1982); Mac Lane (1937)].
In this work, we prove a number of theoretical results for the previously described MWER optimization on compass graphs. The main result proves MWER is NP-hard and motivates the use of our heuristic algorithms. Further, we demonstrate how extensions to the generalized diploid HapCompass model can enable (i) usage of different optimizations, for example, MEC and MWER, to be used in the local optimization step, (ii) simultaneous assembly of two individuals sharing a haplotype tract IBD and (iii) haplotype assembly of a single polyploid organism. Finally, we evaluate our methods on 1000 Genomes Project, Pacific Biosciences and simulated data.
2 METHODS
Let a fragment f be a sequence read with the non-polymorphic bases removed such that only SNPs remain. Fragments may be either a single contiguous region of DNA or contain any number of gaps between contiguous regions (for example, one gap between two contiguous regions in paired-end sequencing). Each SNP must be heterozygous and each row must cover at least two SNPs to be able to extract useful haplotype phase information from sequence reads. An SNP allele is encoded as 0 or 1 corresponding to the major or minor allele. The kth base of the ith fragment is referred to as 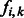 . If fi does not include the base k in the sequence read (within the gap of a paired-read, for instance), then
. If fi does not include the base k in the sequence read (within the gap of a paired-read, for instance), then  ‘–'. Let M be the
‘–'. Let M be the  SNP-fragment matrix with m rows corresponding to the m fragments and n columns corresponding to n SNPs. Two fragments fi and fj are in fragment conflict if
SNP-fragment matrix with m rows corresponding to the m fragments and n columns corresponding to n SNPs. Two fragments fi and fj are in fragment conflict if
| (1) |
Informally, fragment conflict represents two fragments that include the same SNP but differ in the allele. The fragment conflict graph GF has a vertex for each fragment in M and an edge between two fragments if they are in fragment conflict. M is feasible if a bipartition exists in GF or, equivalently, the fragments of M can be partitioned in two sets such that no two fragments within each set are in fragment conflict.
2.1 The MWER optimization and HapCompass models
The HapCompass model defined in Aguiar and Istrail (2012) is composed of the compass graph GC data structure and optimizations on the spanning-tree cycle basis of this graph. GC is a graph with a vertex for each SNP and an edge between two SNPs if at least one read contains both SNPs (Fig. 2). The weight on the edge, defined by the function fMWER, is the difference between the number of fragments that suggest a 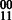 phasing and the number of fragments that suggest
phasing and the number of fragments that suggest 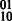 .
.
Fig. 2.
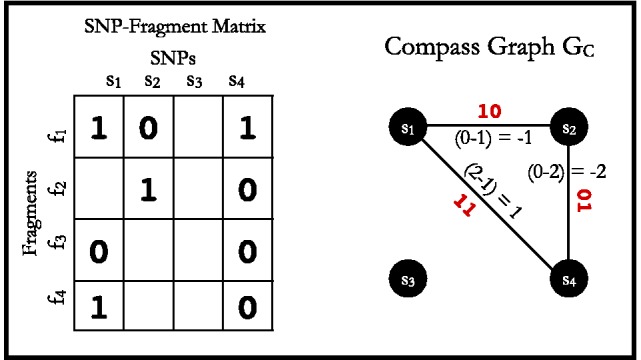
Construction of the compass graph from SNP-fragment matrix M. The SNP-fragment matrix M (left) contains four fragments and four SNPs. Each SNP’s pairwise phasing relationship defined by the fragments is represented on the edges of the compass graph (right). The majority rule phasing for one of the haplotypes is shown in red on the compass graph edges
A path in GC corresponds to a phasing of the SNP vertices by concatenating the phasings on the edges. For example, the 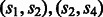 path in Figure 2 corresponds to the concatenation of the
path in Figure 2 corresponds to the concatenation of the 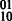 phasing with the
phasing with the 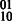 phasing, yielding the
phasing, yielding the 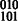 phasing. A number of subtle combinatorial properties of the diploid read information define the contiguity of the assembly; in haplotype assembly of polyploid genomes where more than two haplotypes exist, these combinatorial properties will be made explicit and generalized as a basis for the development of polyploid haplotype assembly algorithms.
phasing. A number of subtle combinatorial properties of the diploid read information define the contiguity of the assembly; in haplotype assembly of polyploid genomes where more than two haplotypes exist, these combinatorial properties will be made explicit and generalized as a basis for the development of polyploid haplotype assembly algorithms.
A spanning tree in GC corresponds to a valid phasing of the SNPs of GC. Simple cycles in GC have the property of being non-conflicting, whereby every path in the cycle including the same set of vertices corresponds to the same phasing, or conflicting, whereby there is no unique phasing. Aguiar and Istrail (2012) show that a simple cycle is conflicting if and only if there is a 0-weight edge or an odd number of negative weight edges and non-conflicting otherwise. For the MWER optimization, the HapCompass algorithm constructs a spanning-tree cycle basis of GC and removes edges of small weight (in absolute value) from conflicting cycles until GC is void of conflicts.
The generalized HapCompass model described in this work supports multiple optimizations on compass graphs, joint haplotype assembly of individuals sharing a haplotype tract IBD and haplotype assembly of polyploid organisms. To support these algorithmic extensions, we examine key concepts of the HapCompass model and describe their generalizations.
The core of the HapCompass framework constructs the compass graph GC, a spanning-tree cycle basis of GC, and then corrects conflicting cycles. One such method for correcting conflicting cycles was presented in Aguiar and Istrail (2012) where edge weights are used to compute a set of edges whose removal would eliminate conflicting cycles (the MWER optimization). In principle, other methods may be used to remove edges, or entirely new optimizations may be employed, for example, MEC. Specifically, we implement an algorithm for the MEC optimization on compass graphs. However, before an implementation of an MEC algorithm on compass graphs can be realized, the HapCompass framework must be generalized to allow for corrections to fragments.
Concept 1: edge weights. The HapCompass framework proposed in Aguiar and Istrail (2012) defines edge weights as the difference between the number of reads indicating the 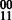 and
and 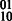 phasings. The generalized model includes a vector for edge e, ve, consisting of four integers corresponding to the four possible haplotypes between two SNPs: 00, 01, 10, 11. A function, f(e), maps the vector to a meaningful value interpreted by the HapCompass algorithm. For example, in the MWER HapCompass algorithm,
phasings. The generalized model includes a vector for edge e, ve, consisting of four integers corresponding to the four possible haplotypes between two SNPs: 00, 01, 10, 11. A function, f(e), maps the vector to a meaningful value interpreted by the HapCompass algorithm. For example, in the MWER HapCompass algorithm, 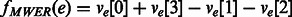 where
where 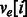 is the count of the phasings 00, 01, 10, 11 for
is the count of the phasings 00, 01, 10, 11 for 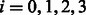 , respectively.
, respectively.
2.2 An MEC HapCompass optimization
The MEC optimization on GC aims to flip the minimum number of alleles such that all of the cycles are non-conflicting. The MEC algorithm proceeds by building a spanning tree cycle basis of the compass graph. The following steps are repeated until each edge is non-conflicting. (i) For each edge e in the set of conflicting cycles: let v1 and v2 be the two vertices adjacent to e. (ii) If 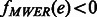 , we check the fragments that include both v1 and v2, and temporarily flip the fragment alleles of v1 (v2 in following iteration) to indicate
, we check the fragments that include both v1 and v2, and temporarily flip the fragment alleles of v1 (v2 in following iteration) to indicate 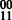 phasings. The other alleles in the fragments cause edges adjacent to v1 (v2) to change weight as well. We record the number of conflicting cycles resolved and created by checking each cycle in the cycle basis including an edge that was modified by the flipping of a fragment allele. (iii) The case of
phasings. The other alleles in the fragments cause edges adjacent to v1 (v2) to change weight as well. We record the number of conflicting cycles resolved and created by checking each cycle in the cycle basis including an edge that was modified by the flipping of a fragment allele. (iii) The case of 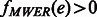 is handled analogously with the exception of flipping the alleles to indicate
is handled analogously with the exception of flipping the alleles to indicate 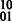 phasings. (iv) Let the number of conflicting cycles resolved by processing e be
phasings. (iv) Let the number of conflicting cycles resolved by processing e be 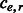 and the number of conflicting cycles created be
and the number of conflicting cycles created be 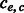 . If
. If 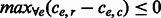 , then there does not exist a favorable switching of fragment alleles and an edge is removed following the MWER algorithm. Otherwise, the fragment changes giving
, then there does not exist a favorable switching of fragment alleles and an edge is removed following the MWER algorithm. Otherwise, the fragment changes giving 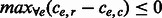 are introduced in GC. (v) When all cycles are non-conflicting, we output the phasing defined by any spanning tree.
are introduced in GC. (v) When all cycles are non-conflicting, we output the phasing defined by any spanning tree.
The primary data structure change in GC introduces a mapping of edges to fragments. The primary addition to the HapCompass framework is a definition of optimization function to remove conflicting cycles from GC.
2.3 IBD tracts and haplotype assembly
Thus far, the HapCompass framework has only been defined for a single diploid individual. The generalization of haplotype assembly to multiple genomes must be selective for which individuals to assemble jointly. For example, if two individuals do not share a haplotype by descent, one individual’s set of reads does not provide any information for the other. However, when two individuals do share a haplotype by descent, the shared haplotype provides phasing information across homozygous sites as long as one individual remains heterozygous (Fig. 3). Regions of homozygosity in an individual, which would otherwise disconnect SNPs and partition haplotype solutions, can be phased together as long as the jointly assembled genotype has heterozygous SNPs within the interval.
Fig. 3.
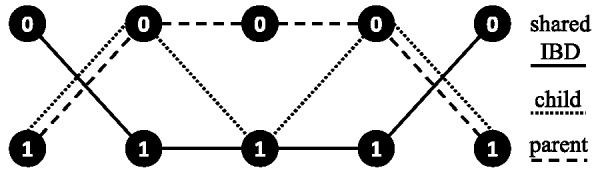
A graph of the haplotype transitions defined by the majority rule phasings of a compass graph. SNPs 1, 2, 3, 4 and 5 (left to right) are shown with both alleles (vertices), and edge transitions are encoded by a specific type of line depending on whether the haplotype is shared IBD or unique to the child or parent. The genotype of the parent and child are 22 222 and 22 122, respectively (where the two corresponds to the heterozygote and 0 and 1 correspond to homozygous for the major and minor alleles, respectively)
Concept 2: multiple genotypes. The problem of joint assembly of two individuals who share a haplotype IBD (hereafter referred to as a pair) is different from jointly assembling two individuals who do not share a haplotype. In the compass graph, two unrelated genotypes have the effect that both individuals can be heterozygous but have completely different phasings. However, if they share a haplotype, a transition from a doubly heterozygous SNP to another doubly heterozygous SNP forces exactly two phasings, namely 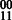 or
or 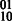 (for example, SNP transitions (1,2) and (4,5) in Fig. 3). For the doubly heterozygous to singly heterozygous transitions, we may have exactly three of the four possible 2-SNP haplotypes. In Figure 3, the child’s genotype is 22 122 and to phase this block using the child’s data alone, we require a read to cover at least one of the first two SNPs and at least one of the last two SNPs, which may be impossible depending on the distance between the SNPs and sequence read insert length. However, if we assemble the parent with the child, we can use the shared haplotype to decode the parent’s phase across SNPs 2, 3 and 4 to be
(for example, SNP transitions (1,2) and (4,5) in Fig. 3). For the doubly heterozygous to singly heterozygous transitions, we may have exactly three of the four possible 2-SNP haplotypes. In Figure 3, the child’s genotype is 22 122 and to phase this block using the child’s data alone, we require a read to cover at least one of the first two SNPs and at least one of the last two SNPs, which may be impossible depending on the distance between the SNPs and sequence read insert length. However, if we assemble the parent with the child, we can use the shared haplotype to decode the parent’s phase across SNPs 2, 3 and 4 to be 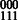 . Because they share a haplotype, the 111 haplotype must be the shared haplotype and it can be inferred that the child’s phased haplotypes are
. Because they share a haplotype, the 111 haplotype must be the shared haplotype and it can be inferred that the child’s phased haplotypes are 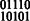 .
.
Joint haplotype assembly in HapCompass is thus encoded as follows. Each edge now has two sets of vectors corresponding to the 2-SNP haplotype transitions of the parent and child. For a doubly heterozygote to doubly heterozygote transition, the weight function can be computed as before using the coverage from both individuals (because there are exactly two disjoint phasings). For a singly heterozygote to doubly heterozygote transition (or vice-versa), the weight function can solely use the heterozygous–heterozygous transmission data from a single individual.
2.4 Haplotype assembly of polyploid genomes
The research literature concerning polyploid haplotype assembly is essentially non-existent. The analysis of k-ploid genomes (k sets of chromosomes) has been hindered by the complexity of sequencing and assembling k chromosomes concurrently. With high-throughput sequencing technologies, genotype inference in polyploid organisms is manageable; sequence reads are mapped to a reference genome, and the relative quantities of alleles at an SNP can be inferred from sequence coverages. However, the basic assumption that there exists exactly two phasing between two SNPs no longer holds. We note that the polyploidy assembly problem is similar to a number of problems in other areas of haplotype reconstruction (when the number of haplotypes is known or unknown) such as modeling metagenomics (organism identification), HIV (viral quasispecies identification in the ‘metagenome’ of patients), cancer (tumor and plasma) and epigenetics (regulatory region methylation reconstruction similar to ‘probabilistic haplotype’ inference).
Concept 3: uniqueness and disjoint phasings. One difficulty of polyploid haplotype assembly emerges from the non-disjointness of phasing solutions between SNPs. With the assumption that SNPs are biallelic, at least one haplotype will be shared by two or more phasings between two SNPs. In the diploid case, a read suggesting the 00 phasing could be interpreted as evidence for 11 on the other haplotype (uniqueness of phasing) and also evidence contradicting the 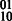 phasing (disjointness of haplotypes in phasing solutions). In the tetraploid case, for example, if the genotype for each of 2 SNPs is
phasing (disjointness of haplotypes in phasing solutions). In the tetraploid case, for example, if the genotype for each of 2 SNPs is 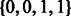 then there exists three possible haplotype phasings: (00,00,11,11), (01,01,10,10), (00,01,10,11).
then there exists three possible haplotype phasings: (00,00,11,11), (01,01,10,10), (00,01,10,11).
In general, the number of haplotype phasings on an edge is a function of the ploidy of the organism and the alleles at each SNP. As in the diploid case, each SNP must have at least one of each allele or else the SNP is homozygous and sequence observations of an allele do not provide any phasing information. As a result, every 2-SNP haplotype includes either 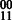 or
or 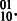
However, unlike in the diploid case, the extension from one edge in GC to the next may not be deterministic. For example, in diploid assembly, if a reads suggest a 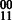 phasing for SNPs 1 and 2, and a
phasing for SNPs 1 and 2, and a 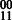 phasing for SNPs 2 and 3, the extension would give us a phasing of
phasing for SNPs 2 and 3, the extension would give us a phasing of 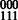 . A conflicting cycle in GC could then be generated if reads connecting SNPs 1 and 3 disagreed with this phasing. For the polyploid case, if the genotypes for each of SNP 1 and 2 are (0,0,1), then both the (00,00,11) phasing and (00,01,10) phasing are valid. Assume that we can compute the phasings between SNPs 1 and 2 and SNPs 2 and 3 to be (00,00,11); we can extend as we did in the diploid case to create the phasing (000,000,111). Then, if a read suggests a 01 phasing between SNPs 1 and 3, we again generate a conflicting cycle. However, if the SNPs were phased using (00,01,10) for SNPs 1,2 and 2,3, then either phasing (000,010,101) or (001,010,100) is possible. Both are completely valid phasings consistent with the genotype and read data but fragments connecting SNPs 1 and 3 may constrain the phasing solution to be unique.
. A conflicting cycle in GC could then be generated if reads connecting SNPs 1 and 3 disagreed with this phasing. For the polyploid case, if the genotypes for each of SNP 1 and 2 are (0,0,1), then both the (00,00,11) phasing and (00,01,10) phasing are valid. Assume that we can compute the phasings between SNPs 1 and 2 and SNPs 2 and 3 to be (00,00,11); we can extend as we did in the diploid case to create the phasing (000,000,111). Then, if a read suggests a 01 phasing between SNPs 1 and 3, we again generate a conflicting cycle. However, if the SNPs were phased using (00,01,10) for SNPs 1,2 and 2,3, then either phasing (000,010,101) or (001,010,100) is possible. Both are completely valid phasings consistent with the genotype and read data but fragments connecting SNPs 1 and 3 may constrain the phasing solution to be unique.
Concept 4: polyploid edge decidability. The polyploid HapCompass model retains the axiom that each edge is decidable; that is, each edge has a unique and computable phasing as defined by the reads. The compass graph and spanning tree cycle basis is built from the input genotypes and reads as before. The distribution of haplotype configurations between two SNPs are defined by the genotypes, and a singular configuration is computed using the available read data. The first approach attempts to assign reads into haplotype bins that represent the haplotype distribution for a valid phasing between two SNPs. Given a 2-SNP genotype, a binning is an assignment of reads to haplotypes. For example, if two SNPs both had two 0 alleles and two 1 alleles, there would exist three haplotype phasings: (00,00,11,11), (01,10,11,00), (01,10,01,10), each with 4 bins. The phasing (00,00,11,11), for instance, would contain two 00 bins and two 11 bins.
Greedy binning algorithm. Input: a maximum distance d between any two bins, a set of haplotype phasings P and a set of reads R. Output: the haplotype phasing most supported by the reads.
For each haplotype phasing
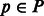
For each haplotype bin
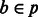 , do steps (3–5).
, do steps (3–5).Loop through steps (4–5) until all read fragments have been assigned.
Select a read
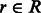 such that the edit distance between r and an available haplotype bin
such that the edit distance between r and an available haplotype bin 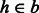 is minimal.
is minimal.Place r in the selected bin h and remove this read from the read set.
Report the haplotype phasing with the binning of minimum total edit distance as the optimal phasing.
We enforce that the difference of haplotypes in each bin must be at most d haplotypes to avoid always preferring diverse haplotype phasings [e.g. (10,10,01,01) versus (00,11,10,01)]. This condition defines which haplotype bin is available during each iteration.
Probabilistic binning algorithm. Alternatively, probabilities of each phasing given the set of reads can be computed and uncertainty can be accounted for when extending phase to adjacent edges. In particular, we wish to compute the likelihood of a phasing given the set of input sequence reads. Let pi be the ith phasing for two adjacent SNPs, P the set of all possible phasings for the two SNPs, rj be the jth read and se the probability of a sequencing error. Then, the likelihood of a particular phasing pp is
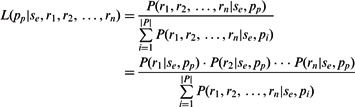 |
which may be computed using the assumption that sequence reads are independent. The probability of a read ri given sequencing error se and phasing p can be computed by marginalizing over all possible haplotypes h sampled for phasing p:
| (2) |
Thus, the edge is decisive for the haplotype phasing with the maximum likelihood for all reads that span the two SNPs. The original diploid scoring scheme can be recreated with a manipulation of the unnormalized phasing likelihoods: 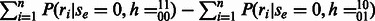 .
.
Concept 5: conflicting cycles and phase extensions. Both the greedy and probabilistic binning algorithms decide the haplotype phase of edges. In the diploid case, the extension of phasings from edges to paths was unambiguous because for each of the two phasings, exactly one haplotype begins with 0 (or 1) and exactly one haplotype ends with 0 (or 1). Therefore, the computation of phasings for paths and conflicting cycles was easily determined given the decided edges. In polyploid genomes, each SNP variant in GC is still assumed to have only two possible alleles but each edge has three or more haplotypes. When extending phase from one edge to an adjacent edge, the haplotypes on different edges that share an allele can be used for extending phase. If this allele is present in k haplotypes, then there are 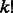 possible extensions.
possible extensions.
Phase extension algorithm. We introduce the chain graph Gh, which is defined on a path or cycle in GC for a k-ploid genome. Let 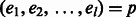 denote a path of edges in GC of length l. Each edge ei is phased (by the greedy or probabilistic method) and each haplotype in the phasing introduces a vertex in Gh at level i. Thus, Gh contains k vertices for each
denote a path of edges in GC of length l. Each edge ei is phased (by the greedy or probabilistic method) and each haplotype in the phasing introduces a vertex in Gh at level i. Thus, Gh contains k vertices for each 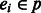 and a total of
and a total of 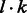 vertices in total. Two haplotype vertices are connected by an edge if and only if they share an SNP position and allele. Because haplotypes at adjacent levels uniquely share an SNP position in Gh, edges only exist between adjacent levels and a path through the chain graph corresponds to a joining (or extension) of haplotypes. Therefore, there is always a valid phasing for a Gh defined on a path of GC.
vertices in total. Two haplotype vertices are connected by an edge if and only if they share an SNP position and allele. Because haplotypes at adjacent levels uniquely share an SNP position in Gh, edges only exist between adjacent levels and a path through the chain graph corresponds to a joining (or extension) of haplotypes. Therefore, there is always a valid phasing for a Gh defined on a path of GC.
Cycles introduce complexity in Gh. Gh defined on a cycle retains the characteristics of the path chain graph, but also includes source and sink nodes:  and
and  , respectively. Let
, respectively. Let 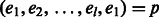 denote any path of edges in GC of length l with the addition of the
denote any path of edges in GC of length l with the addition of the 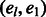 edge. Source nodes are connected arbitrarily to haplotypes on level 1 but haplotypes on level l are only connected to sink nodes if the shared variant position agrees with the haplotype the source was connected to; for example, in Figure 5 Top, t2 is connected to both 00 haplotypes at level l because s2 is connected to a haplotype starting with 0. The sources and sinks represent the
edge. Source nodes are connected arbitrarily to haplotypes on level 1 but haplotypes on level l are only connected to sink nodes if the shared variant position agrees with the haplotype the source was connected to; for example, in Figure 5 Top, t2 is connected to both 00 haplotypes at level l because s2 is connected to a haplotype starting with 0. The sources and sinks represent the 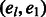 edge and a path from si to ti represents one valid haplotype. This intuition enables the formulation of the k vertex disjoint paths problem on chain graphs. If there exists k vertex disjoint, si to ti paths for
edge and a path from si to ti represents one valid haplotype. This intuition enables the formulation of the k vertex disjoint paths problem on chain graphs. If there exists k vertex disjoint, si to ti paths for 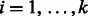 , we have k valid phasings for the cycle; otherwise, the cycle is conflicting and there is no valid phasing.
, we have k valid phasings for the cycle; otherwise, the cycle is conflicting and there is no valid phasing.
Fig. 5.

The chain graphs (top) 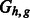 and (bottom)
and (bottom) 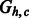 corresponding to
corresponding to 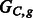 and
and 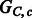 , respectively
, respectively
To further build intuition, consider a conflicting cycle of GC and Gh in the diploid case. A cycle was conflicting if the number of negative weighted edges in GC was odd. Relating this to the chain graph Gh, an si node would be connected to a 0 (or 1) and each negative edge would flip the next bit. So, a conflicting cycle has an odd number of negative edges, which translates into an odd number of bit flips resulting in no si to ti path for  . Figure 4 gives an example of non-conflicting and conflicting cycles in polyploid compass graphs and Figure 5 their chain graphs.
. Figure 4 gives an example of non-conflicting and conflicting cycles in polyploid compass graphs and Figure 5 their chain graphs.
Fig. 4.
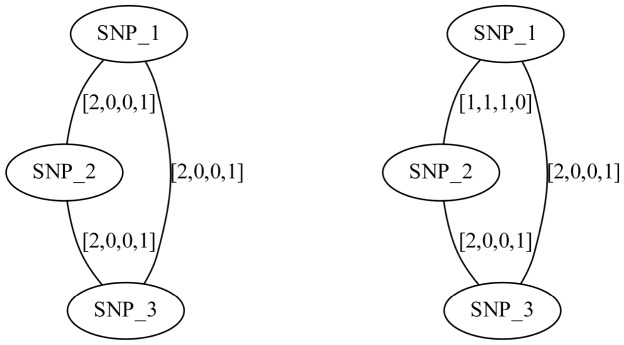
Compass graphs 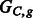 , a non-conflicting polyploid cycle (left), and
, a non-conflicting polyploid cycle (left), and 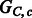 , a conflicting polyploid cycle (right). The vector on the edge corresponds to the haplotype counts for an edge in the format [00,01,10,11]. In both compass graphs, the haplotypes are 000, 000 and 111, while the reads in
, a conflicting polyploid cycle (right). The vector on the edge corresponds to the haplotype counts for an edge in the format [00,01,10,11]. In both compass graphs, the haplotypes are 000, 000 and 111, while the reads in 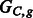 are 000, 000 and 111, and the reads in
are 000, 000 and 111, and the reads in 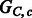 are 00–, 01–, 10–, –00, –00, –11, 0–0, 0–0 and 1–1
are 00–, 01–, 10–, –00, –00, –11, 0–0, 0–0 and 1–1
Gh enables the (i) determination of conflicting cycles and (ii) computation of the phased haplotypes for a path or cycle using disjoint paths. The k-disjoint paths problem is a well-studied optimization in the field of discrete mathematics [Robertson and Seymour (1995)]. A polynomial-time solution is known to exist for the node disjoint paths problem when k is known as part of the input [Robertson and Seymour (1995); Kawarabayashi et al. (2012)], but these algorithms require manipulation of enormous constants rendering them difficult to implement in practical settings.
Fortunately, the structure of Gh enables a much more efficient solution to the problem. All paths from si to ti can be computed by a modified depth first search algorithm. A depth first search is started from each source si and the path from source to the current node is stored. Each node contains a list of integers initially empty. When the algorithm either encounters the sink node ti, or a node already labeled with i, all nodes on the current path have i added to their list. After each source–sink pair is processed, each node contains a label i if there is an si to ti path that includes the node. The runtime of this algorithm is O(kve) where k is the ploidy, and v and e are the number of vertices and edges in Gh, respectively.
After all nodes are labeled, we iterate through each level of Gh and create an auxiliary flow graph 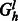 where l is the level.
where l is the level. 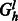 defines a bipartite graph where one set of vertices corresponds to the source haplotype paths, which are connected to a set of vertices corresponding to the haplotypes of the phasing level l. A flow in
defines a bipartite graph where one set of vertices corresponds to the source haplotype paths, which are connected to a set of vertices corresponding to the haplotypes of the phasing level l. A flow in 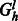 of total value k where each edge has capacity 1 corresponds to a maximum matching and thus a valid assignment of haplotype paths to haplotypes of the phasing at level l. This flow can be found in time linear in the size of the edge set of
of total value k where each edge has capacity 1 corresponds to a maximum matching and thus a valid assignment of haplotype paths to haplotypes of the phasing at level l. This flow can be found in time linear in the size of the edge set of 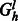 . If every level of the chain graph has a valid bijection, then the cycle is non-conflicting and the path given by the matchings define a valid phasing. Figure 6 give an example of the auxiliary flow graphs for level 1 of the chain graphs defined in Figure 5.
. If every level of the chain graph has a valid bijection, then the cycle is non-conflicting and the path given by the matchings define a valid phasing. Figure 6 give an example of the auxiliary flow graphs for level 1 of the chain graphs defined in Figure 5.
Fig. 6.

The auxiliary flow graphs (top) 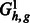 and (bottom)
and (bottom) 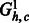 . For a k-ploid organism (in this case k = 3), a flow of k with 1 capacity on each edge corresponds to a valid assignment of haplotype paths to haplotypes of the phasing a level 1
. For a k-ploid organism (in this case k = 3), a flow of k with 1 capacity on each edge corresponds to a valid assignment of haplotype paths to haplotypes of the phasing a level 1
3 RESULTS
3.1 Theoretical
We first present results on the complexity of the MWER optimization, and related minimum weighted vertex (SNP) removal (MWVR) problems on the compass graph GC. These results motivate the usage of our heuristics for the diploid and polyploid algorithms. Let 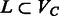 be a subset of vertices in GC, and let
be a subset of vertices in GC, and let 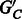 be the resulting graph created from removing L from VC. The MWVR optimization aims to compute an L such that the following conditions are satisfied:
be the resulting graph created from removing L from VC. The MWVR optimization aims to compute an L such that the following conditions are satisfied:
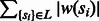 is minimal where
is minimal where 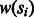 is the weight of the ith SNP (cost of removed vertices is minimal);
is the weight of the ith SNP (cost of removed vertices is minimal);All edges in
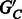 are decisive (each edge has a majority rule phasing);
are decisive (each edge has a majority rule phasing);Choosing a phasing for each edge in
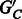 by majority rule gives a unique phasing for
by majority rule gives a unique phasing for 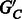 .
.
We omit the straightforward proofs that the MWVR and MWER problems are in NP. It remains to be shown that known NP-hard problems can be reduced to MWVR and MWER.
We restate the conflict graph generality lemma from Lippert et al. (2002).
Lemma 1 —
Let
be an arbitrary graph. Then there exists an SNP-fragment matrix M such that
.
Proof —
Introduce a fragment fi for each vertex
. For every two adjacent vertices
, introduce a new SNP column sk in M where
and
.
Let M be the SNP-fragment matrix constructed from Lemma 1, GF the corresponding fragment conflict graph of M and GC the compass graph of M.
Lemma 2 —
Every simple cycle of odd length in GF produces exactly one conflicting simple cycle in GC.
For the proof of Lemma 2 please see Supplementary Appendix Proof of Lemma 2.
Lemma 3 —
Every conflicting simple cycle in GC includes exactly one odd length simple cycle in GF.
Proof —
We now interpret conflicting cycles in GC as a set of vertices of GC, which define a set of edges in GF.
Because of the previous lemma, every conflicting cycle in GC can be resolved by removing an edge of GF, which corresponds to removing a vertex in GC.
Corollary 1 —
There exists no conflicting cycles in GC if and only if there are no cycles of odd length in GF.
Lemma 4 —
Given an M produced from Lemma 1, the compass graph
is the line graph of
with weights of GC as defined by the phasing relationships of the fragments of M.
Proof —
The SNPs (columns) of M contain exactly two alleles from two fragments that conflict. Therefore, in GF, each SNP uniquely defines an edge, and in GC, each SNP uniquely defines a vertex. All that remains is to show that every two adjacent edges in GF produce an edge in GC. Consider an SNP s whose conflicts involve fragments fi and fj. The edge defined by s in GF is adjacent to edges defined by the other conflicts of fi and fj. The vertex s in GC is defined exactly as the pairwise phasing relationships as defined by the SNP s and other SNP alleles in fragments fi and fj, which in turn define the adjacencies in GF.
Because GC is the line graph of GF, if k simple cycles in GC share an edge then k simple cycles in GF share a vertex.
Theorem 1 —
MWVR is NP-hard.
Proof —
See the MWVR Proof section in Supplementary Appendix.
Theorem 2 —
MWER is NP-hard.
Proof —
The reduction is from the problem of removing the minimum number of edges of a graph to make it bipartite. Let G be an arbitrary graph and M the SNP-fragment matrix as defined in Lemma 1. We modify
by adding two additional degree 2 vertices to each edge, effectively converting each edge to a length 3 path. Cycles of odd (even) length retain their odd (even) length, thus odd length cycles still create conflicting cycles in GC. All vertices of degree k produce cliques of size k in GC, which do not correspond to any cycles in
. Therefore, we label all edges of clique vertices produced from a single vertex with weight
. All paths of GF will be encoded with two edges of GC; both of which cannot be removed in an optimal solution to MWER. Given a solution to the MWER optimization, we can determine the minimum number of edges in GF to make it bipartite.
3.2 Experimental
We evaluate the HapCompass MEC, HapCompass IBD and polyploid HapCompass algorithms using 1000 Genomes Project [The 1000 Genomes Project Consortium (2010)], Pacific Biosciences and simulated data.
Metrics. To evaluate the accuracy of our diploid haplotype assembly methods, we use the following measures, which capture different aspects of haplotype assembly quality. In Aguiar and Istrail (2012), we introduce the fragment mapping phase relationship (FMPR) distance, which counts the number of pairwise phase relationships (as defined by the input read fragments) that do not exist in the solution. The related boolean fragment mapping (BFM) distance counts the number of read fragments that do not map back to the solution. The third evaluation criteria we use is the MEC measure, which counts the number of allele flips in the fragments required to produce the phased haplotype assembly solution. In all previously described measures, lower values are desired. These metrics are similar to read mapping metrics in genome and transcriptome assembly, where good-quality assemblies will allow for many reads to map back to them.
3.3 Pacific biosciences data
Single molecule sequencing has great potential to become a preferred method for haplotype assembly, but current algorithmic techniques are untested on data with high error rates. We downloaded the chromosome 20 data from individuals HG00321, HG00577, HG01101, NA18861, NA19313, NA19740, NA20296 and NA20800 [PacBio Data (2013)]. Haplotype assembly solutions were produced by HapCompass, Levy et al. (2007) and HapCUT to obtain the results in Table 1 (run times can be found in the Pacific Biosciences run times section in Supplementary Appendix). HapCompass outperforms the competition in terms of MEC using both optimizations. Interestingly, the Levy et al. (2007) algorithm is the most accurate in terms of FMPR and BFM. This is likely due to the Levy et al. (2007) algorithm processing entire read fragments each iteration while HapCompass focuses on correcting multiple fragments at adjacent SNPs. Because the Pacific Biosciences read lengths are long (several kb), more emphasis is placed on matching reads with large overlaps on the same haplotype. This result further suggests that it is important to consider the input data and the desired results when preparing data for a haplotype assembly experiment.
Table 1.
The total FMPR, BFM and MEC scores aggregated across individuals HG00321, HG00577, HG01101, NA18861, NA19313, NA19740, NA20296 and NA20800 in the Pacific Biosciences data
| HapCompass MWER | HapCompass MEC | Levy | HapCUT | |
|---|---|---|---|---|
| FMPR | 163 799 | 169 385 | 153 433 | 169 890 |
| BFM | 39 827 | 40 470 | 38 318 | 41 006 |
| MEC | 48 631 | 49 591 | 66 299 | 50 164 |
3.4 1000 genomes project data
To further evaluate the HapCompass MEC implementation, we haplotype assembled the genome of 1000 Genomes Project NA12878 CEU child using our implementation of the Levy et al. (2007) method, HapCUT (v0.5) and the HapCompass MWER and MEC algorithms. Table 2 shows that the HapCompass MWER algorithm clearly performs best overall. The full table for all chromosomes is given in Supplementary Appendix (section 1000 Genomes Project Results). Surprisingly, even though the MWER algorithm does not directly optimize the MEC measure, it produces the best haplotypes in respect to this measure for all but two chromosomes.
Table 2.
Haplotype assembly results for the 454 data from 1000 Genomes Project NA12878 individual for chromosomes 1–22 and algorithms HapCompass MWER, HapCompass MEC, Levy et al. (2007) and HapCUT
| HapCompass MWER | HapCompass MEC | Levy | HapCUT | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| FMPR | BFM | MEC | FMPR | BFM | MEC | FMPR | BFM | MEC | FMPR | BFM | MEC |
| 64 128 | 36 578 | 37 597 | 68 623 | 39 221 | 40 269 | 64 789 | 39 832 | 40 946 | 65 724 | 37 606 | 38 372 |
Note: The full table is given in the section 1000 Genomes Project Results in Supplementary Appendix.
3.5 IBD haplotype assembly
Jointly assembling the haplotypes of related individuals has considerable benefits. The first benefit comes from the extra coverage on the shared haplotype, which helps with differentiating true phasings from sequencing errors. However, the most notable advantage is being able to extend phasing past homozygous blocks. We compared the size of the phased haplotype blocks when assembling chromosome 22 of the NA12878 child in the 1000 Genomes Project data alone versus jointly with the mother. Figure 7 compares the maximum achievable haplotype block sizes of any single individual haplotype assembly algorithm to IBD haplotype assembly; it demonstrates that larger haplotype blocks are achievable by assembling two individuals with a shared haplotype together rather than separately.
Fig. 7.
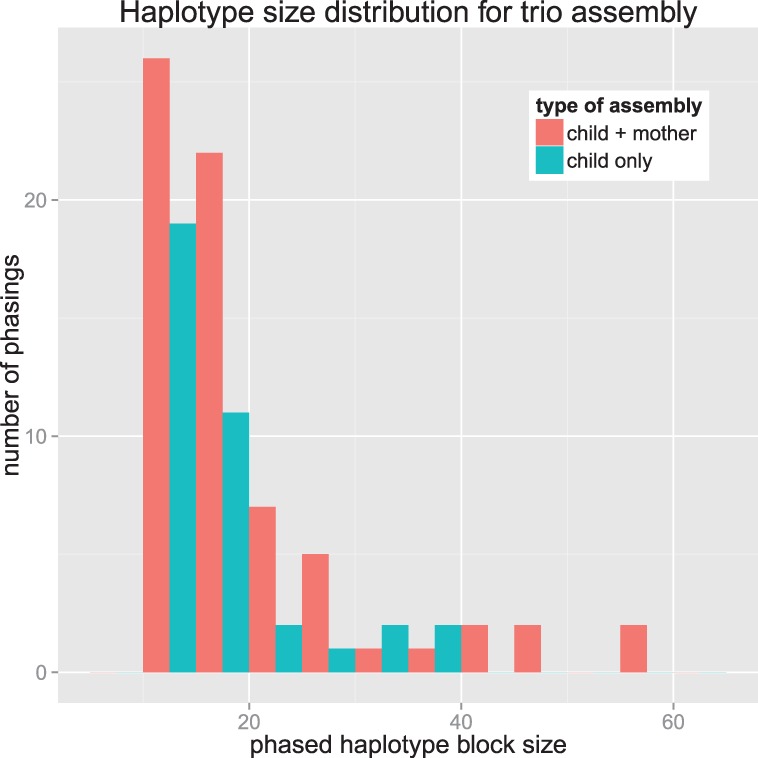
Comparison between haplotype assembling the child individually versus with a parent. The haplotype size is number of SNPs in the component of GC, which represents the maximum number of SNPs that may be phased together
3.6 Polyploid algorithm
Finally, to evaluate the polyploid algorithm, we simulated three haplotypes at random and simulated reads from these haplotypes. The simulated reads were guaranteed to contain two SNPs (assuring they are useful for haplotype assembly) and given normally distributed insert sizes. The polyploid algorithm was run using both the greedy and probabilistic binning algorithms for deciding edge phasings. Figure 8 demonstrates two interesting results: (i) for a small number of reads, the quality of haplotype phasing is independent of the choice of binning method and (ii) that the probabilistic algorithm produces a more accurate phased solution than the greedy binning method for a large range of simulated read counts.
Fig. 8.
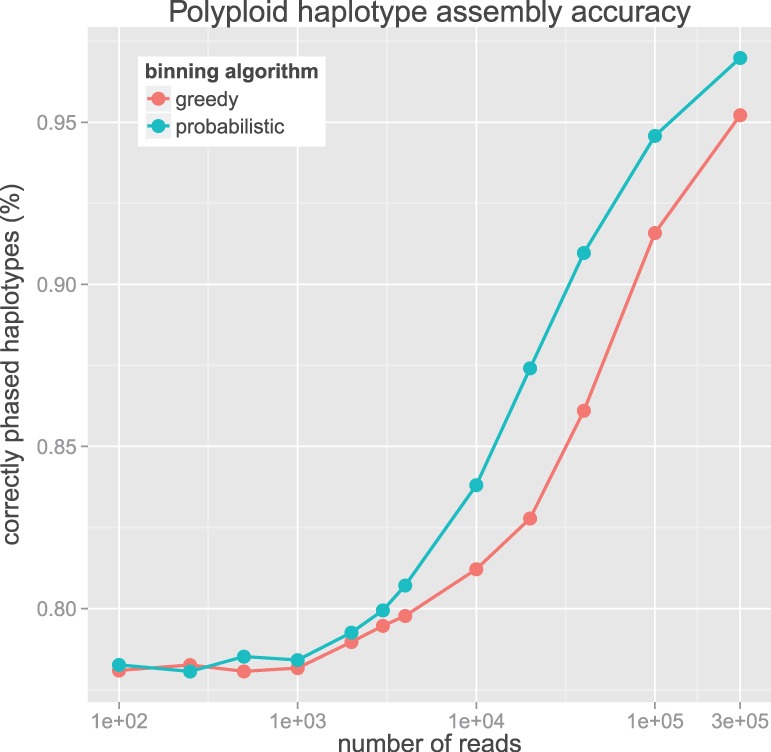
Comparison of the percentage of correctly phased polyploid SNP pairs for the greedy and probabilistic binning algorithms for varying number of input reads
4 DISCUSSION
Diploid haplotype inference is still a difficult task, in part due to the exponentially many solutions given the input genotype or sequence reads. HapCompass is a proven framework for haplotype assembly but there are a number of extensions that may improve results. For instance, we did not mention the usage of base call or read mapping quality scores in our computations. HapCompass can filter based on user-defined thresholds but a more elegant solution would be to convert the base call quality score for a particular allele call into a probability the base was called correctly. This probability can then define the contribution of weight to the edges of GC rather than the current weight contribution of 1 for each SNP allele called. Also we demonstrated in the Pacific Biosciences experiments that the choice of assembly method should be informed by the sequencing technology and desired result. The Levy et al. (2007) method mapped more fragments error free than HapCompass but contained many more single base changes in fragments required to reproduce the inferred haplotypes. Considering the Pacific Biosciences data has high error rates and generating an error-free read is unlikely, a solution with the minimum number of corrected errors is likely preferred over a solution that successfully maps more fragments without errors.
The size of the haplotype blocks produced and, ultimately, the quality of the assembled haplotypes is a function of several factors. The primary difficulty for obtaining large haplotype blocks is the small nature and lack of diversity of insert lengths. We demonstrated a novel modeling and computational method that begins to address this difficulty by exploiting shared IBD haplotype structure. In general, assembling the haplotypes of related individuals has considerable benefits, which help overcome undesirable properties of the sequencing data. The first benefit comes from the extra coverage on the shared haplotype, which helps in differentiating actual phasings from sequencing errors. However, the most notable advantage is being able to include more SNPs into the haplotype assembly, which helps extend the assembly (past regions of low read coverage for example). But, the major advances in block sizes will likely be the result of novel experimental procedures and technologies; for instance, not only do the single molecule sequencers promise larger read lengths, they also enable the inclusion of multiple and large insert lengths.
Organisms having more than two sets of homologous chromosomes are becoming the target of many research groups interested in studying the genomics of disease, phylogenetics and evolution [Chen and Ni (2006); Leitch and Leitch (2008)]. Polyploidy occurs in human disease usually due to the duplication of a particular chromosome, for example, in Edwards, Patau and Down syndrome. While far fewer mammalian organisms are polyploid, specific mammalian cells may undergo polyploidization, for example, in human liver hepatocytes [Gentric et al. (2012)]. In addition, polyploid organisms are ubiquitous in the Plant and Fungi clades, present in crops that we ingest, convert into bioenergy and feed to livestock. Understanding the genomics of both the desirable—e.g. increased crop yield—and undesirable—e.g. susceptibility to disease—properties of plants may lead to critical advances in many research areas but requires untangling the polyploid genome and its variation. As more polyploid data becomes available, our approach may be used to infer haplotypes and begin to understand what effects haplotype variation may influence.
Funding: This work was supported by the National Science Foundation [1048831 to S.I.].
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Aguiar D, Istrail S. Hapcompass: a fast cycle basis algorithm for accurate haplotype assembly of sequence data. J. Comput. Biol. 2012;19:577–590. doi: 10.1089/cmb.2012.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal V, Bafna V. HapCUT: an efficient and accurate algorithm for the haplotype assembly problem. Bioinformatics. 2008;24:i153–i159. doi: 10.1093/bioinformatics/btn298. [DOI] [PubMed] [Google Scholar]
- Bansal V, et al. An MCMC algorithm for haplotype assembly from whole-genome sequence data. Genome Res. 2008;18:1336–1346. doi: 10.1101/gr.077065.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Browning BL. Haplotype phasing: existing methods and new developments. Nat. Rev. Genet. 2011;12:703–714. doi: 10.1038/nrg3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Ni Z. Mechanisms of genomic rearrangements and gene expression changes in plant polyploids. BioEssays. 2006;28:240–252. doi: 10.1002/bies.20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo N, et al. Algorithms for generating fundamental cycles in a graph. ACM Trans. Math. Softw. 1982;8:26–42. [Google Scholar]
- Gentric G, et al. Polyploidy and liver proliferation. Clin. Res. Hepatol. Gastroenterology. 2012;36(1):29–34. doi: 10.1016/j.clinre.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Geraci F. A comparison of several algorithms for the single individual SNP haplotyping reconstruction problem. Bioinformatics. 2010;26:2217–2225. doi: 10.1093/bioinformatics/btq411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halldórsson BV, et al. Proceedings of the 4th international conference on Discrete mathematics and theoretical computer science, DMTCS’03. Berlin, Heidelberg: Springer-Verlag; 2003. Combinatorial problems arising in snp and haplotype analysis; pp. 26–47. [Google Scholar]
- Halldorsson BV, et al. Proceedings of the Pacific Symposium on Biocomputing. Kohala Coast, Hawaii, USA: 2011. Haplotype phasing by multi-assembly of shared haplotypes: Phase-dependent interactions between rare variants; pp. 88–99. [DOI] [PubMed] [Google Scholar]
- He D, et al. Optimal algorithms for haplotype assembly from whole-genome sequence data. Bioinformatics. 2010;26:i183–i190. doi: 10.1093/bioinformatics/btq215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi K, et al. The disjoint paths problem in quadratic time. J. Comb. Theory B. 2012;102:424–435. [Google Scholar]
- Lancia G, et al. ESA ’01: Proceedings of the 9th Annual European Symposium on Algorithms. London, UK: Springer-Verlag; 2001. SNPs problems, complexity, and algorithms; pp. 182–193. [Google Scholar]
- Leitch AR, Leitch IJ. Genomic plasticity and the diversity of polyploid plants. Science. 2008;320:481–483. doi: 10.1126/science.1153585. [DOI] [PubMed] [Google Scholar]
- Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZP, et al. A dynamic programming algorithm for the k-haplotyping problem. Acta Math. Appl. Sin. (English Series) 2006;22:405–412. [Google Scholar]
- Lippert R, et al. Algorithmic strategies for the single nucleotide polymorphism haplotype assembly problem. Brief Bioinform. 2002;3:23–31. doi: 10.1093/bib/3.1.23. [DOI] [PubMed] [Google Scholar]
- Mac Lane S. A combinatorial condition for planar graphs. Fundam. Math. 1937;28:22–32. [Google Scholar]
- Mousavi SR, et al. Effective haplotype assembly via maximum Boolean satisfiability. Biochem. Biophys. Res. Commun. 2011;404:593–598. doi: 10.1016/j.bbrc.2010.12.001. [DOI] [PubMed] [Google Scholar]
- PacBio Data. 2013. Broad institute hapmap pacific biosciences data. https://github.com/PacificBiosciences/DevNet/wiki/Datasets. (15 January 2013, date last accessed) [Google Scholar]
- Panconesi A, Sozio M. Fast hare: a fast heuristic for single individual snp haplotype reconstruction. In: Jonassen I, Kim J, editors. Algorithms in Bioinformatics, volume 3240 of Lecture Notes in Computer Science. Berlin/Heidelberg: Springer; 2004. pp. 266–277. [Google Scholar]
- Rizzi R, et al. Proceedings of the Second International Workshop on Algorithms in Bioinformatics, WABI ’02. London, UK: Springer-Verlag; 2002. Practical algorithms and fixed-parameter tractability for the single individual snp haplotyping problem; pp. 29–43. [Google Scholar]
- Robertson N, Seymour P. Graph minors.xiii. the disjoint paths problem. J. Comb. Theory B. 1995;63:65–110. [Google Scholar]
- Schwartz R. Theory and algorithms for the haplotype assembly problem. Commun. Inf. Syst. 2010;10:23–38. [Google Scholar]
- The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.