

Abstract
Motivation: Cryo-electron tomography allows the imaging of macromolecular complexes in near living conditions. To enhance the nominal resolution of a structure it is necessary to align and average individual subtomograms each containing identical complexes. However, if the sample of complexes is heterogeneous, it is necessary to first classify subtomograms into groups of identical complexes. This task becomes challenging when tomograms contain mixtures of unknown complexes extracted from a crowded environment. Two main challenges must be overcomed: First, classification of subtomograms must be performed without knowledge of template structures. However, most alignment methods are too slow to perform reference-free classification of a large number of (e.g. tens of thousands) of subtomograms. Second, subtomograms extracted from crowded cellular environments, contain often fragments of other structures besides the target complex. However, alignment methods generally assume that each subtomogram only contains one complex. Automatic methods are needed to identify the target complexes in a subtomogram even when its shape is unknown.
Results: In this article, we propose an automatic and systematic method for the isolation and masking of target complexes in subtomograms extracted from crowded environments. Moreover, we also propose a fast alignment method using fast rotational matching in real space. Our experiments show that, compared with our previously proposed fast alignment method in reciprocal space, our new method significantly improves the alignment accuracy for highly distorted and especially crowded subtomograms. Such improvements are important for achieving successful and unbiased high-throughput reference-free structural classification of complexes inside whole-cell tomograms.
Contact: alber@usc.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Cryo-electron tomography enables the 3D imaging of macromolecular complexes at nanometer-scale resolution in near native conditions (Best et al., 2007; Lucic et al., 2005; Medalia et al., 2002). Tomograms of individual cells are essentially 3D representations of the entire proteome providing a snapshot of the distributions of protein complexes (Beck et al., 2011). However, comprehensive detection of individual complexes in cell tomograms is challenging because of the inherent low signal-to-noise ratio (SNR), missing data, nonisotropic resolution and the fact that individual macromolecules are difficult to recognize in a highly crowded environment (Beck et al., 2009; Best et al., 2007; Böhm et al., 2000; Frangakis et al., 2002; Medalia et al., 2002; Nickell et al., 2006).
Most methods for detecting complexes in cell tomograms rely on a template structure, which serves as a reference in searching for a similar pattern in the tomogram (e.g. Beck et al., 2009). However, for an unbiased detection and classification of all the different complexes in a cellular tomogram, template-free methods are needed (Xu et al., 2011). Such an analysis is challenging and relies on three main steps: First, the locations of potential complexes are detected by using particle-picking methods and a subregion surrounding the potential complex is extracted (i.e. the subtomogram). Second, the subtomogram regions corresponding only to the target complex must be detected, which allows masking out all background regions, which in turn contain noise and in case of crowded subtomograms also fragments of surrounding structures. Third, reference-free structural classification of the masked subtomograms is performed, which is typically based on an iterative process of subtomogram alignments, classifications and averaging (e.g. Amat et al., 2010; Bartesaghi et al., 2008; Chen et al., 2013; Förster et al., 2008; Volkmann, 2010; Xu et al., 2012). Finally, the averaging of aligned subtomograms of the same complexes will enhance the nominal resolution of the resulting density maps.
In this article, we address two main challenges in reference-free subtomogram classifications. First, we propose an automatic method for adaptive masking of target regions in crowded subtomograms without the knowledge of the shape of the complex. Such a method is particularly important for subtomograms extracted from crowded environments, such as the cell cytoplasm. In such a case, subtomograms will also contain fragmental regions of other complexes owing to the high particle density in the tomogram. Masking out these regions is of great importance in the subsequent classification process. Unlike automatic masking methods based on voxel weighting (Xu et al., 2012) or dimension reduction methods such as Principle Component Analysis (Heumann et al., 2011), our method is highly scalable because it is independent of the classification process and does not involve iterative processing of a large number of subtomograms. Second, we propose a new method for fast rotational matching of subtomograms in real space. The 3D subtomogram alignment is computationally the most intensive step in the classification process. Currently, most alignment methods are based on maximizing the overlap similarity of two subtomograms through exhaustive search over all rigid transforms (rotation and translation) of one subtomogram with respect to the other (e.g. Amat et al., 2010; Förster et al., 2008). Such methods scan exhaustively through each rotational angle, and find the best corresponding translation using Fast Fourier Transform (FFT) (e.g. Amat et al., 2010; Förster et al., 2008). These methods are computationally intensive, which limits their applicability in reference-free classifications. To increase alignment accuracy, local search methods have been developed to refine initial alignments (e.g. Bartesaghi et al., 2008; Hrabe et al., 2012; Xu and Alber, 2012). To increase computational efficiencies, two approximate alignment methods have been developed, which separate the translational from the rotational subtomogram alignments (Bartesaghi et al., 2008; Xu et al., 2012). These methods are based on similarity scores defined in the reciprocal space (i.e. Fourier space). To separate translational and rotational search, these methods use an approximate score that introduces translational invariance by eliminating the phase information of the complex coefficients in the reciprocal space and use only the magnitude of the Fourier coefficients (Bartesaghi et al., 2008; Xu et al., 2012). By expressing the structural information in Spherical Harmonics (SH) Expansion, it is possible to formulate the rotational alignment as a fast rotational matching, which simultaneously calculates alignment scores of all rotations using FFT. This procedure significantly increases the alignment speed. For example, our previously published method (Xu et al., 2012) achieved between hundreds to thousands fold speedup (depending on subtomograms size and rotational angle interval) compared with the standard scanning-based alignment method (Förster et al., 2008) by using a translational invariance approximation of a similarity score equivalent to a popular score proposed by (Förster et al., 2008).
However, formulating the fast rotational matching in reciprocal space has some limitations. In reciprocal space, the majority of informative signals of a complex is usually contained in a relatively small amount of high magnitude Fourier coefficients (Amat et al., 2010). These coefficients are often occupying relatively few voxels in the reciprocal space located within a small region centered at the origin. Therefore, the SH expansion in the fast rotational matching may be hampered by interpolation errors, which reduces the accuracy in the alignment process. In contrast, in real space, the signal covers a wider area within a subtomogram, and the SH expansion is expected to be more accurate. It is therefore beneficial to formulate fast rotational matching in real space. In addition, real space fast rotational matching uses the full constrained alignment score instead of a translation invariant approximation when the method is expressed in reciprocal space (Bartesaghi et al., 2008; Xu et al., 2012).
Here, we devise an improved alignment method, which formulates the fast rotational matching of subtomograms in real space. To achieve this goal, we first identify a keypoint in the target complex whose relative location is invariant to rigid transformations and serves as the center of rotation in the fast rotational matching of the two subtomograms. A natural choice is the center of mass of a complex. However, detection of the center of mass is not trivial and cannot be approximated by the geometrical or mass density center of the subtomogram. The reason for this complication is that tomograms are images of the crowded cellular environment (Beck et al., 2009), and a subtomogram contains not necessarily mass density of only one single complex. When a potential complex is detected and its subtomogram extracted as a rectangular cube then this subtomogram typically contains also fragments of other surrounding structures that occupy parts of the subtomogram. Then, the center of mass estimation and subtomogram alignment are affected by the existence of the additional mass density in the subtomogram. It is necessary to focus only on the regions that contain the actual target complex. Even when a subtomogram does not contain any surrounding structures, its mean intensity is often close to background intensity owing to the suppression of low frequency signal owing to the Contrast Transfer Function (CTF) effect, which also makes the estimation of the center of mass of the complex inaccurate.
Applying our automatic target complex segmentation method, it is possible to estimate a center of mass based only on the target complex regions in the subtomogram (i.e. the constrained center of mass). Once the constrained center of mass is detected, we design a fast subtomogram alignment method that uses real space signals and takes into account missing wedge corrections using reciprocal space signals. Moreover, the alignment focuses exclusively on the target complex regions in each subtomogram, which significantly decreases the influence of background noise and surrounding structures to the alignment process.
Our experiments show that our new approach significantly increases the alignment accuracy compared with our previous proposed fast alignment method (Xu et al., 2012) for highly distorted subtomograms. Most importantly, the new method is highly robust to the influence of surrounding structures inside crowded subtomograms.
2 METHODS
This section begins by describing the automatic segmentation of the target complex and then focuses on the fast rotational alignment in real space.
2.1 Automatic segmentation of the target complex in a subtomogram
The automatic target complex segmentation method consists of several steps (Figs 1, 2 and 7): (i) automatic scale selection is performed to maximally enhance the detection of structural boundaries in a subtomogram; (ii) segments are defined that describe the boundaries between structural elements; and (iii) a classification of the detected structural segments into candidate regions is performed by using a statistical model based clustering.
Fig. 1.
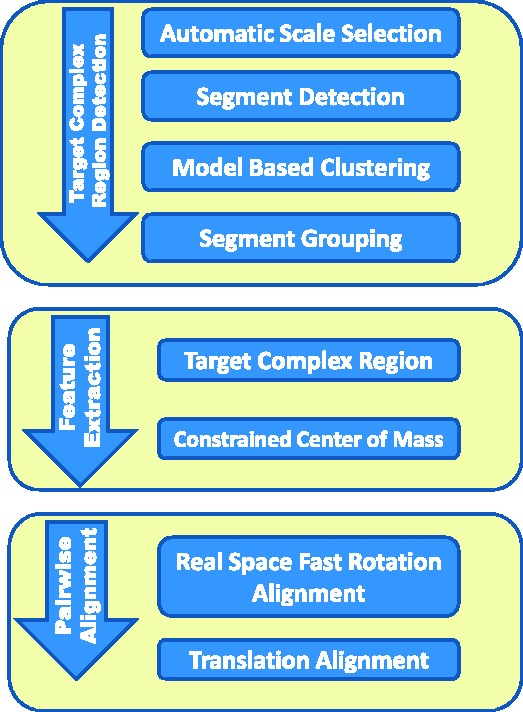
Flow chart for segmentation of target complex regions and the real space alignment of subtomogram using only target complex regions
Fig. 2.
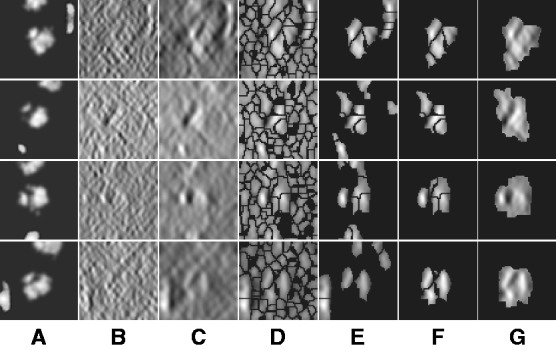
Four typical examples for target complex segmentation based on simulated data. Each row is an example, whereas each subfigure is a slice through the x-z plane in the 3D image. (A) Ground truth. (B) Simulated subtomograms at SNR 0.005 and tilt angle range 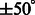 (Section 3.1). (C) After Gaussian smoothing with automatic scale selection. (D) After Watershed segmentation. (E) After boundary segment detection. (F) Selected segment cluster from model based clustering. (G) Detected target complex regions
(Section 3.1). (C) After Gaussian smoothing with automatic scale selection. (D) After Watershed segmentation. (E) After boundary segment detection. (F) Selected segment cluster from model based clustering. (G) Detected target complex regions
Fig. 7.

Segmentation of target complex regions in Leptospira interrogans subtomograms. Each row shows the segmentation process for a ribosome instance. Each subfigure in a row consists of the consecutive slices through the y-axis of the 3D image. (A) Original extracted subtomograms. (B) Images after Gaussian smoothing with automatic scale selection. (C): Images after Watershed segmentation. (D) Images after structural boundary segment detection. (E) Images of the selected segment clusters resulting from the model-based clustering. (F) Images of the final segmented target complexes. Because the contrast is low, for the detection of significant boundary segments, we relaxed significance level parameters by setting 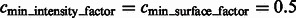 . When determining the target complex region, we set
. When determining the target complex region, we set 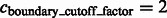
2.1.1 Automatic scale selection for optimal smoothing and boundary enhancement
In this article, we assume that in a subtomogram high image intensity corresponds to high electron density. When a subtomogram is renormalized so that its mean intensity is zero, the boundary of a complex tends to have a negative intensity surface (Fig. 3, yellow arrow). This property is due to the CTF effects in the electron tomography imaging process, which suppresses low frequency signals. We use this negative intensity surface as a main characteristic when identifying the segments at the boundary of structural elements, such as the target complex and those structures captured at the outer regions of the subtomogram. To reduce the influence of noise, smoothing of the subtmograms is needed. However, too much smoothing will weaken the negative intensity boundary. To find the optimal degree of smoothing, we use the scale-space representation method and propose an automatic scale selection method to find the optimal degree of smoothing that maximally enhances the boundary surface while minimizing the noise. Scale-space (Witkin, 1983) is a framework for multi-scale signal representation. An image is represented as a one-parameter family of smoothed images, the scale-space representation, parameterized by the size of the Gaussian smoothing kernel (Lindeberg, 1994):
Given a subtomogram, f, a Gaussian smoothing can be expressed as the convolution between f and 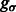 :
:
In the smoothed subtomogram 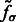 , all features with a size smaller than σ are filtered out, whereas others are preserved. Our objective is to determine the optimal scale σ to enhance the formation of a negative intensity surface while minimizing the influence of noise. To do so, we scan a range of feasible σ values. For a given smoothed subtomogram with scale σ, we find all local minima with negative intensities in
, all features with a size smaller than σ are filtered out, whereas others are preserved. Our objective is to determine the optimal scale σ to enhance the formation of a negative intensity surface while minimizing the influence of noise. To do so, we scan a range of feasible σ values. For a given smoothed subtomogram with scale σ, we find all local minima with negative intensities in 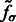 and denote this set as
and denote this set as 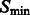 . We then define a local surface score for each minima in
. We then define a local surface score for each minima in 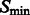 , to identify those minima that are likely part of a negative intensity surface. The surface score is calculated as follows: we calculate the Hessian matrix
, to identify those minima that are likely part of a negative intensity surface. The surface score is calculated as follows: we calculate the Hessian matrix 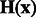 , whose elements are second-order derivatives of
, whose elements are second-order derivatives of 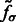 evaluated at
evaluated at  ,
,
where 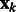 is the corresponding location of a voxel k.
is the corresponding location of a voxel k.
Fig. 3.

A subtomogram’s image features. The boundary feature is a surface that has strong negative intensity (yellow arrow). A local maxima in 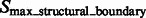 is highlighted by a red cross, which is inside the macromolecular complex. Its corresponding structural boundary segment is highlighted by a red circle. A local maxima in
is highlighted by a red cross, which is inside the macromolecular complex. Its corresponding structural boundary segment is highlighted by a red circle. A local maxima in 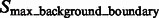 is highlighted by a green cross, which is inside a background region. Its corresponding background boundary segment is highlighted by green circle. See Section 2.1.2 for the details of these two types of local maxima
is highlighted by a green cross, which is inside a background region. Its corresponding background boundary segment is highlighted by green circle. See Section 2.1.2 for the details of these two types of local maxima
For any 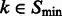 , let
, let 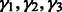 be the ordered eigenvalues of
be the ordered eigenvalues of 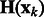 such that
such that 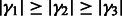 . Then, we can construct a local surface score
. Then, we can construct a local surface score 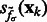 similar to (Martinez-Sanchez et al., 2011) as follows:
similar to (Martinez-Sanchez et al., 2011) as follows:
We find the σ value that maximally enhances the surface scores of all the minima so that the most negative minima tends to have a strong surface score. To do so, we calculate an accumulative surface score from all the minima. First, the minima are ordered in ascending order in 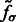 such that
such that
where 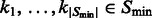 . Then the accumulative surface score
. Then the accumulative surface score 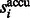 is defined as
is defined as
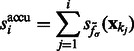 |
Because 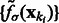 is in ascending order with respect to i, this score
is in ascending order with respect to i, this score 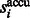 forms a monotonically increasing function of
forms a monotonically increasing function of 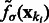 . We can measure the Area Under Curve,
. We can measure the Area Under Curve, 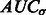 , using
, using 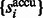 and
and 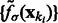 .
. 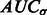 tends to be large when the most negative minima have large surface scores. To measure how much the AUC reflects true surface signals rather than random noise, we also randomly permutate all voxels in f and repeat the aforementioned procedure to calculate a permutated AUC,
tends to be large when the most negative minima have large surface scores. To measure how much the AUC reflects true surface signals rather than random noise, we also randomly permutate all voxels in f and repeat the aforementioned procedure to calculate a permutated AUC, 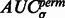 .
.
To determine the optimal Gaussian filtering value σ, we scan through a range of different σs to find the 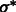 value that maximizes
value that maximizes
For simplicity, in the following sections, we denote 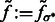 (Figs 2C and 7B) and also denote
(Figs 2C and 7B) and also denote 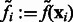 for any voxel i.
for any voxel i.
2.1.2 Detecting structural, boundary and background segments in the subtomogram
After identifying the optimal scale 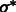 , we now determine those local minima that have strong surface scores and are likely to separate different structural elements (i.e. the target complex and surrounding structures). We define them as those local minima with both high surface scores and low intensity values. To do so, first we select the minima whose intensity is significantly smaller than the average intensity, i.e.
, we now determine those local minima that have strong surface scores and are likely to separate different structural elements (i.e. the target complex and surrounding structures). We define them as those local minima with both high surface scores and low intensity values. To do so, first we select the minima whose intensity is significantly smaller than the average intensity, i.e.
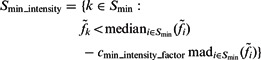 |
where 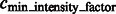 is a constant parameter to control the significance level, and
is a constant parameter to control the significance level, and 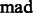 is the Median Absolute Deviation, which is a robust measure of variability.
is the Median Absolute Deviation, which is a robust measure of variability.
In addition, we also collect those minima whose surface score is significantly larger than the average scores of all minima, i.e.
 |
where 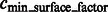 is a constant parameter.
is a constant parameter.
All minima with strong surface scores are defined as the intersection of both groups.
We denote these minima as the boundary surface minima.
We then determine all local intensity maxima 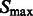 in
in 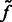 , which are likely to be inside structural elements. We first perform Watershed segmentation (Beucher and Lantuéjoul, 1979; Volkmann, 2002) using
, which are likely to be inside structural elements. We first perform Watershed segmentation (Beucher and Lantuéjoul, 1979; Volkmann, 2002) using 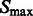 as seeds (Figs 2D and 7C). Each segment corresponds to one local maximum, and therefore the terms local maximum and segment are used interchangeably. We then select all the segments that border with local minima defined in
as seeds (Figs 2D and 7C). Each segment corresponds to one local maximum, and therefore the terms local maximum and segment are used interchangeably. We then select all the segments that border with local minima defined in  and denote the corresponding set of maxima as
and denote the corresponding set of maxima as  . The actual separations between structural elements and background regions are always at the boundary between segments in
. The actual separations between structural elements and background regions are always at the boundary between segments in 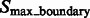 . The segments in
. The segments in  can be divided into two types: a structural boundary segment is located in the target complex or the surrounding structures (Fig. 3, inside red circle), and it contains a maximum with high intensity value (Fig. 3, highlighted by red cross); a background boundary segment is located in the regions that do not contain structural elements (Fig. 3, inside green circle), and which contain a maximum with low intensity value (Fig. 3, highlighted by green cross).
can be divided into two types: a structural boundary segment is located in the target complex or the surrounding structures (Fig. 3, inside red circle), and it contains a maximum with high intensity value (Fig. 3, highlighted by red cross); a background boundary segment is located in the regions that do not contain structural elements (Fig. 3, inside green circle), and which contain a maximum with low intensity value (Fig. 3, highlighted by green cross).
To separate these two types of boundary segments, we perform k-means clustering on 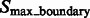 (with cluster number
(with cluster number 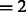 ), resulting in the two groups
), resulting in the two groups 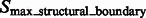 and
and 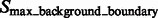 .
. 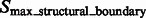 are segments containing local maxima with higher intensity values and are defined as structural boundary segments (Figs 2E and 7D), whereas
are segments containing local maxima with higher intensity values and are defined as structural boundary segments (Figs 2E and 7D), whereas 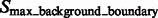 are defined as background boundary segments.
are defined as background boundary segments.
After focusing on boundary segments (i.e. those that border with surface minima), we now characterize the remaining segments as either part of structural elements or background regions. To do this, we first determine a cutoff intensity value 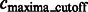 as the largest intensity value in all the background boundary segments
as the largest intensity value in all the background boundary segments 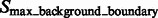 . All remaining segments that contain a local maxima with an intensity value smaller that
. All remaining segments that contain a local maxima with an intensity value smaller that 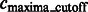 are assigned as background regions.
are assigned as background regions.
Finally, the remaining segments whose maxima have intensity values larger than 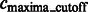 are defined as structural segments and are therefore assumed to be either part of the target complex or other structural elements, i.e.
are defined as structural segments and are therefore assumed to be either part of the target complex or other structural elements, i.e. 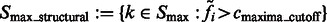 .
.
2.1.3 Combining structural segments into complexes using model based clustering
We now cluster structural segments in 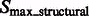 according to the intensity values and location of their local maxima so that each cluster of segments will approximately correspond to one consecutive structural region. We choose a model-based clustering approach because it allows an automatic determination of the optimal set of clusters. To perform the clustering, we assume that the location and intensity of local maxima from clusters follows a probabilistic mixture model, where each component probability distribution corresponds to a cluster (Fraley and Raftery, 2002). We model the probability of independently observing both location and intensity of a local maxima
according to the intensity values and location of their local maxima so that each cluster of segments will approximately correspond to one consecutive structural region. We choose a model-based clustering approach because it allows an automatic determination of the optimal set of clusters. To perform the clustering, we assume that the location and intensity of local maxima from clusters follows a probabilistic mixture model, where each component probability distribution corresponds to a cluster (Fraley and Raftery, 2002). We model the probability of independently observing both location and intensity of a local maxima 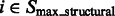 from a cluster k as:
from a cluster k as:
| (1) |
where 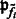 is a probability proportional to the intensity
is a probability proportional to the intensity 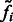 , and
, and 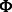 is a probability density function in the form of a multivariate normal distribution with mean
is a probability density function in the form of a multivariate normal distribution with mean 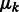 and covariance
and covariance 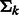 . For simplification, we assume that the clusters are spherical with different sizes, i.e.
. For simplification, we assume that the clusters are spherical with different sizes, i.e. 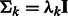 . The main difference between our model and the standard model-based clustering (Fraley and Raftery, 2002) is the inclusion of the image intensity related term
. The main difference between our model and the standard model-based clustering (Fraley and Raftery, 2002) is the inclusion of the image intensity related term 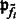 in Equation (1). Our simulation experiments show that the inclusion of image intensity improves the clustering performance (Supplementary Document).
in Equation (1). Our simulation experiments show that the inclusion of image intensity improves the clustering performance (Supplementary Document).
The likelihood for the mixture model with K clusters is:
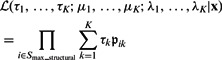 |
where x represents the collection of observed local maxima, and 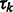 is the probability of cluster k (i.e. cluster mixing probability). A model-based clustering determines the parameters
is the probability of cluster k (i.e. cluster mixing probability). A model-based clustering determines the parameters 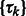 ,
, 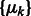 ,
, 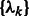 that maximizes
that maximizes 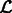 .
.
We use a scale-space representation to estimate cluster centers 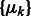 . Given a scale
. Given a scale 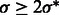 , we obtain the set
, we obtain the set 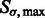 of local maxima with positive intensities in
of local maxima with positive intensities in 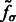 . The local maxima
. The local maxima 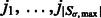 are ordered with decreasing intensity values, i.e.
are ordered with decreasing intensity values, i.e. 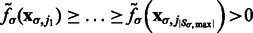 , where
, where 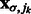 is the corresponding location of local maxima jk on
is the corresponding location of local maxima jk on 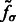 . We choose cluster centers as the first K local maxima, i.e
. We choose cluster centers as the first K local maxima, i.e 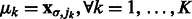
Similar to the standard model based clustering (Celeux and Govaert, 1995; Fraley and Raftery, 2002), an Expectation Maximization algorithm is used to estimate 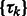 and
and 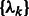 . The expectation maximization is defined by two iterative steps:
. The expectation maximization is defined by two iterative steps:
E-step: update an estimated posterior probability 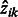 that the local maximum i belongs to cluster k:
that the local maximum i belongs to cluster k:
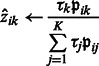 |
M-step: update the cluster mixing probability 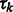 , and variances
, and variances 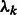 , given the new parameters in the previous step:
, given the new parameters in the previous step:
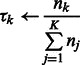 |
where
and
where
The Expectation Maximization algorithm iterates until there is no significant change in the likelihood 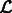 . The clustering of local maxima leads to clusters of the corresponding segments (Figs 2F and 7E). Using statistical modeling for clustering enables the determination of an optimal set of clusters through model selection. By varying the scale σ and cluster number K, we obtain the optimal values for scale and cluster set using the Bayesian Information Criterion (Fraley and Raftery, 2002):
. The clustering of local maxima leads to clusters of the corresponding segments (Figs 2F and 7E). Using statistical modeling for clustering enables the determination of an optimal set of clusters through model selection. By varying the scale σ and cluster number K, we obtain the optimal values for scale and cluster set using the Bayesian Information Criterion (Fraley and Raftery, 2002):
The final target complex is then represented by combining multiple segments that fulfill the following criteria: (i) they belong to same cluster in the model based clustering; (ii) they are in consecutive contact to each other; and (iii) the maximum intensity values 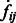 of the voxels between two consecutive segments i and j is larger than a threshold. The threshold is computed as
of the voxels between two consecutive segments i and j is larger than a threshold. The threshold is computed as 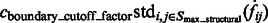 , where
, where 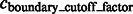 is a constant parameter.
is a constant parameter.
The target complex region is then selected as the combined segments that contain at least one structural boundary segment (defined by 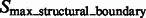 ) and are located closest to the center of the subtomogram. To prevent that small features of the target complex are excluded, a dilation operation is performed to define the target complex region A (Figs 2G and 7F).
) and are located closest to the center of the subtomogram. To prevent that small features of the target complex are excluded, a dilation operation is performed to define the target complex region A (Figs 2G and 7F).
2.2 Real space fast subtomogram alignment using the target complex region and its center of mass
Fast rotational matching in real space relies on an initial alignment of the center of masses of the two subtomograms. Therefore, we describe first how the center of mass of each target complex is calculated before we describe the fast rotational matching approach.
2.2.1 Calculation of center of mass for target complex region
The determination of the center of mass in subtomograms is not trivial, primarily owing to high noise levels, the suppression of low frequency signals in the reciprocal space and the existence of surrounding structures inside a subtomogram.
Here, the center of mass is calculated only based on the regions in the target complex A. We set a thresholded region 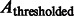 on the filtered subtomogram
on the filtered subtomogram 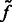 , i.e.
, i.e. 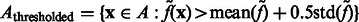 . Then, we use
. Then, we use 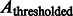 to calculate the center of mass. We define another intensity function
to calculate the center of mass. We define another intensity function
We then calculate the constrained center of mass 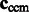 of
of 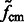 .
.
2.2.2 Fast rotation alignment
We represent two subtomograms f and g as two integrable functions 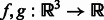 . They have been transformed such that (i) their mean intensities are adjusted to zero, (ii) the voxel intensities outside the target complex region A is set to zero, and (iii) they are translated according to
. They have been transformed such that (i) their mean intensities are adjusted to zero, (ii) the voxel intensities outside the target complex region A is set to zero, and (iii) they are translated according to 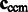 so that their constrained centers of mass are aligned. The constrained center of mass is used as rotational centers in our fast rotational alignment. To correct for the missing wedge distortions, we only keep the Fourier coefficients within the overlap of nonmissing wedge regions of both subtomograms. Following Bartesaghi et al. (2008), Förster et al. (2008) and Xu et al. (2012), we use a binary missing wedge mask function as
so that their constrained centers of mass are aligned. The constrained center of mass is used as rotational centers in our fast rotational alignment. To correct for the missing wedge distortions, we only keep the Fourier coefficients within the overlap of nonmissing wedge regions of both subtomograms. Following Bartesaghi et al. (2008), Förster et al. (2008) and Xu et al. (2012), we use a binary missing wedge mask function as 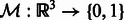 , which defines valid and missing Fourier coefficients in reciprocal space. For example, with a tilt angle range
, which defines valid and missing Fourier coefficients in reciprocal space. For example, with a tilt angle range 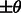 , the missing wedge mask function can be defined as
, the missing wedge mask function can be defined as 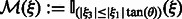 , where
, where  is the indicator function. Given
is the indicator function. Given 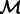 , the real space subtomogram that excludes any coefficients inside of any of the two missing wedge regions is defined as,
, the real space subtomogram that excludes any coefficients inside of any of the two missing wedge regions is defined as,
where 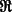 denotes the real part of a complex function;
denotes the real part of a complex function; 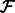 is the Fourier transform operator; and
is the Fourier transform operator; and 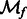 and
and 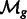 are missing wedge masks for f and g, respectively.
are missing wedge masks for f and g, respectively.
Given a 3D rotation R, we can calculate a correlation
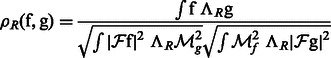 |
(2) |
where 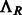 is the rotation operator such that
is the rotation operator such that 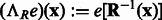 for any function
for any function 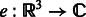 , and
, and  is the rotation matrix corresponding to 3D rotation R. As in Xu et al. (2012), the correlation measure
is the rotation matrix corresponding to 3D rotation R. As in Xu et al. (2012), the correlation measure 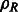 in Equation (2) is equivalent (up to a constant factor.) to a popular constrained correlation score with missing wedge correction (Förster et al., 2008).
in Equation (2) is equivalent (up to a constant factor.) to a popular constrained correlation score with missing wedge correction (Förster et al., 2008).
The fast rotational alignment is based on sampling of 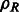 simultaneously over all rotations R. To do so, we apply a fast 3D volumetric rotational matching (Kovacs and Wriggers, 2002; Xu et al., 2012). It can be seen that Equation (2) can be formulated as being composed of three rotational correlation functions of the form
simultaneously over all rotations R. To do so, we apply a fast 3D volumetric rotational matching (Kovacs and Wriggers, 2002; Xu et al., 2012). It can be seen that Equation (2) can be formulated as being composed of three rotational correlation functions of the form 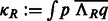 , where p and q are component functions;
, where p and q are component functions; 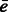 denotes the complex conjugate (when e is a real valued function, the complex conjugate
denotes the complex conjugate (when e is a real valued function, the complex conjugate 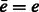 .) of a function e. Specifically, we can represent
.) of a function e. Specifically, we can represent 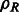 as
as
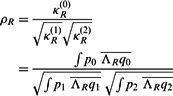 |
(3) |
where 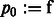 ,
, 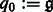 ,
, 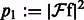 ,
, 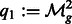 ,
, 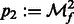 , and
, and 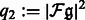 .
.
When represented in spherical coordinates, the p and q components of these three functions 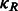 can be approximated by an SH expansion. Following Garzón et al. (2007), Kovacs and Wriggers (2002) and Xu et al., (2012), the p and q components in Equation (3) can be expressed as:
can be approximated by an SH expansion. Following Garzón et al. (2007), Kovacs and Wriggers (2002) and Xu et al., (2012), the p and q components in Equation (3) can be expressed as:
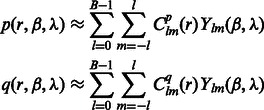 |
where B is the bandwidth, and 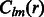 are the coefficients associated with the complex-valued spherical harmonic function
are the coefficients associated with the complex-valued spherical harmonic function 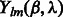 with degree l and order m, where r, β and λ are the radial distance, co-latitude and longitude, respectively, which define the position in spherical coordinate.
with degree l and order m, where r, β and λ are the radial distance, co-latitude and longitude, respectively, which define the position in spherical coordinate.
When a suitable parameterization of the 3D rotational group is achieved, the rotational correlation function 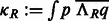 of all sampled rotations R can be represented as an inverse FFT of a 3D array of the sum of integrals as follows (Garzón et al., 2007; Kovacs and Wriggers, 2002; Xu et al., 2012):
of all sampled rotations R can be represented as an inverse FFT of a 3D array of the sum of integrals as follows (Garzón et al., 2007; Kovacs and Wriggers, 2002; Xu et al., 2012):
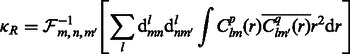 |
where 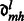 are real coefficients defining the elements of the Wigner small d-matrix evaluated at
are real coefficients defining the elements of the Wigner small d-matrix evaluated at  .
.
As a consequence of the aforementioned formulation, the cross-correlation functions can be efficiently sampled by using FFT simultaneously over all sampled rotations (Kovacs and Wriggers, 2002; Xu et al., 2012). The sampling is given as twice the bandwidth B used in the SH transformations. Therefore, 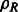 can be efficiently computed over all sampled rotations R. Similar to Bartesaghi et al. (2008) and Xu et al. (2012), the set of candidate rotations are then obtained by identifying the local maxima of ρ with respect to the rotational degrees of freedom R. To obtain the optimal translation, a fast translational search is performed for each candidate rotation over the full correlation function
can be efficiently computed over all sampled rotations R. Similar to Bartesaghi et al. (2008) and Xu et al. (2012), the set of candidate rotations are then obtained by identifying the local maxima of ρ with respect to the rotational degrees of freedom R. To obtain the optimal translation, a fast translational search is performed for each candidate rotation over the full correlation function 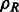 by using FFT. Finally, the best combination of rotation and translation is chosen.
by using FFT. Finally, the best combination of rotation and translation is chosen.
3 RESULTS
We assess the performance of our method by using simulated subtomograms of a phantom model. In addition, we test our method by experimentally determined structures of five complexes as well as experimental ribosome subtomograms extracted from a whole-cell tomogram.
3.1 Generation of simulated tomograms from phantom and experimental structures
We follow a previously applied methodology for generating subtomograms from initial structures by simulating the tomographic imaging process as realistically as possible, allowing for the inclusion of noise, tomographic distortions due to missing wedge and electron optical factors such as CTF and Modulation Transfer Function (MTF) (Beck et al., 2009; Förster et al., 2008; Nickell et al., 2005; Xu and Alber, 2012; Xu et al., 2011, 2012).
A density map of the complexes (Fig. 4) are generated by applying a low pass filter at 4 nm resolution to the atomic structures using the PDB2VOL program of the Situs 2.0 package (Wriggers et al., 1999) with voxel length of 1 nm. In addition, a density map of a phantom model is generated (Fig. 4A). These density maps are used as base maps, both of size 643 voxels, and they are randomly rotated and translated to generate density map instances in uncrowded conditions. To generate a crowded environment, two additional structures (randomly chosen from the phantom model or the ribosome complex) are randomly oriented and placed into the density map ∼32 voxels away from the center of the map.
Fig. 4.
Structures used for simulating the tomographic image process. (A) Top: a phantom model that consists of four elliptical Gaussian functions as branches and one spherical Gaussian function to connect the elliptical functions. Bottom: Ribosome complex (PDB ID: 2AW7, 2AWB). Left: isosurface of the two structures. Right: Slices of the corresponding x-z plane in the simulated tomograms with different degree of distortions, i.e. different SNRs and tilt angle ranges. (B) The isosurfaces of four additional benchmark complexes (Xu et al., 2012)
The density maps of the target complex and neighboring structures are used as input for simulating electron micrograph images at different tilt angles. Our simulated subtomograms therefore contain a wedge-shaped region in reciprocal space for which no structure factors have been measured (i.e. the missing wedge effect), resulting in distortions in the density map as observed in the experimental measurements. To generate realistic micrographs, noise is added to the images according to a given SNR level (ranging between 0.05 and 0.001), defined as the ratio between the variances of the signal and noise (Förster et al., 2008) (Fig. 4A).
The resulting image is convoluted with a CTF, which describes the imaging in the transmission electron microscope in a linear approximation (Frank, 2006; Nickell et al., 2005). We also convolute the density map with the corresponding MTF of a typical detector used in tomography. Typical experimental acquisition parameters (Beck et al., 2009) were used: voxel grid length  nm, the spherical aberration
nm, the spherical aberration 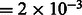 m, the defocus value
m, the defocus value 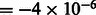 m, the MTF corresponded to a realistic electron detector (McMullan et al., 2009), defined as
m, the MTF corresponded to a realistic electron detector (McMullan et al., 2009), defined as 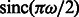 where ω is the fraction of the Nyquist frequency. Finally, we use a backprojection algorithm (Nickell et al., 2005) to generate a tomogram from the individual 2D micrographs generated at the various tilt angles (Beck et al., 2009; Xu et al., 2011, 2012; Xu and Alber, 2012).
where ω is the fraction of the Nyquist frequency. Finally, we use a backprojection algorithm (Nickell et al., 2005) to generate a tomogram from the individual 2D micrographs generated at the various tilt angles (Beck et al., 2009; Xu et al., 2011, 2012; Xu and Alber, 2012).
3.2 Segmentation of the target complex in simulated subtomograms
To determine the optimal scale for surface enhancement, we vary σ from 1.0 nm to 3.0 nm. At high noise levels (e.g. 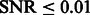 ), the optimal
), the optimal 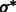 is usually obtained at ∼2.0 nm.
is usually obtained at ∼2.0 nm.
For the detection of boundary segments, we set the significance levels 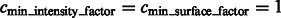 . For determining the target complex, we choose
. For determining the target complex, we choose 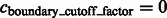 , ignoring all boundary segment maxima with negative intensities in
, ignoring all boundary segment maxima with negative intensities in 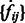 . For combining segments, we scan σ from
. For combining segments, we scan σ from  to 10 nm and scan the cluster number K from 1 to 5.
to 10 nm and scan the cluster number K from 1 to 5.
To assess the target complex segmentation, we calculate the overlap between the determined target complex A and the ground truth 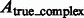 . We also calculate the overlap between A and regions occupied by the surrounding structures in the crowded ground truth density map
. We also calculate the overlap between A and regions occupied by the surrounding structures in the crowded ground truth density map 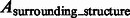 . Then, we calculate the median of the following three overlap scores across 100 simulated subtomograms at each distortion level and for each of the benchmark structures:
. Then, we calculate the median of the following three overlap scores across 100 simulated subtomograms at each distortion level and for each of the benchmark structures:
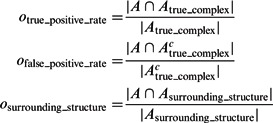 |
where Ac denotes the complementary set of A. Our results show that for all distortion levels, the median of 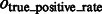 is at least 0.9, and
is at least 0.9, and 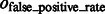 is at most 0.004, demonstrating a good performance in segmenting the areas of the corresponding target structures. In contrast, the median of
is at most 0.004, demonstrating a good performance in segmenting the areas of the corresponding target structures. In contrast, the median of 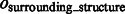 is close to zero for most of the distortion levels, indicating that our target complex segmentation method can successfully exclude surrounding structures in the subtomograms. Only at high distortion levels, the median of
is close to zero for most of the distortion levels, indicating that our target complex segmentation method can successfully exclude surrounding structures in the subtomograms. Only at high distortion levels, the median of  starts to deviate from zero (Supplementary Tables S7–S12).
starts to deviate from zero (Supplementary Tables S7–S12).
3.3 Assessment of constrained center of mass detection
The constrained center of mass is expected to be invariant to rigid transforms of the corresponding structures. The assessment of the rotational invariance of our constrained center of mass is performed as follows. We randomly select 100 pairs of subtomograms for each of the benchmark structures. For each pair of subtomograms i and j, we calculate
as a measure of the degree of invariance, where 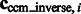 and
and 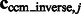 are the corresponding locations of the constrained center of masses in the original complex that is used to generate the instances by random rigid transforms. The smaller
are the corresponding locations of the constrained center of masses in the original complex that is used to generate the instances by random rigid transforms. The smaller 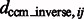 , the higher is the degree of invariance, indicating a good performance.
, the higher is the degree of invariance, indicating a good performance.
The performance is expressed as the median of 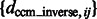 , which is listed in Figure 5 and Supplementary Tables S1, S4, S13 and S16. We can see that the estimation error is generally smaller than 3, indicating high accuracy even at high distortion levels. As can be seen in the following section, the degree of invariance is sufficiently small to allow successful pairwise fast rotational alignments in real space.
, which is listed in Figure 5 and Supplementary Tables S1, S4, S13 and S16. We can see that the estimation error is generally smaller than 3, indicating high accuracy even at high distortion levels. As can be seen in the following section, the degree of invariance is sufficiently small to allow successful pairwise fast rotational alignments in real space.
Fig. 5.
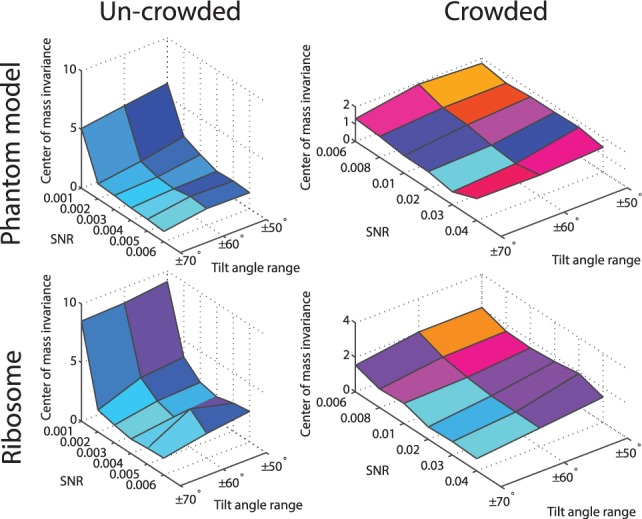
The performance of the center of mass invariance, expressed as the median of 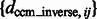 across 100 pairwise comparisons of subtomograms simulated at different distortion levels. The surface region color is rescaled hue saturation value (HSV) color and proportional to the surface height
across 100 pairwise comparisons of subtomograms simulated at different distortion levels. The surface region color is rescaled hue saturation value (HSV) color and proportional to the surface height
3.4 Fast rotational alignment of subtomograms
Our fast alignment method relies on fast rotational matching. In the following two subsections, we calculate the rotational alignment errors for the pairs of complexes, both for the crowded as well as uncrowded cases. The rotational alignment error is calculated as follows: Suppose 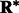 is the rotation matrix calculated for an alignment, and
is the rotation matrix calculated for an alignment, and 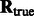 is the true rotation matrix. The rotation alignment error can then be measured as
is the true rotation matrix. The rotation alignment error can then be measured as
where 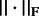 is Frobenius norm.
is Frobenius norm.
3.4.1 Uncrowded subtomograms
First, we test our alignment method with uncrowded subtomograms, which contain one complex without surrounding structures. We compare the alignment errors between our new alignment method presented here and our previous method that relied on fast rotational matching in reciprocal space (Xu et al., 2012) (left column of Fig. 6 and Supplementary Tables S2 and S5). It is evident that our new method improves the alignment accuracy on highly distorted subtomograms.
Fig. 6.
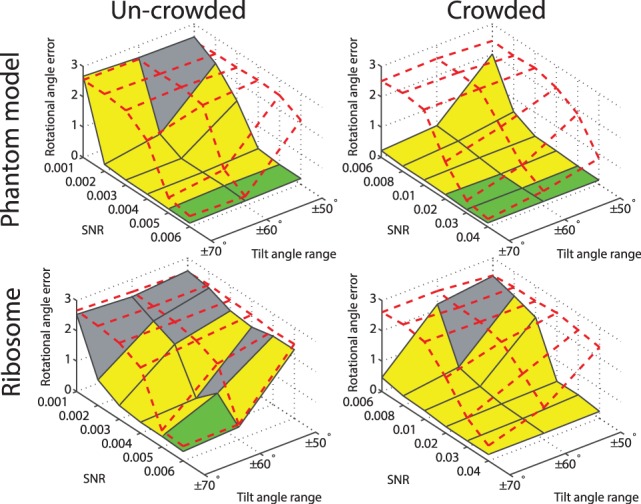
Rotational alignment error for the phantom model and the ribosome complex inside uncrowded and crowded subtomograms simulated at different distortion levels. The error is measured in terms of the median 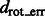 across 100 pairwise alignments. The nodes on the solid black grid correspond to the alignment performance of our new method. The nodes in the red dashed grid correspond to the alignment performance of our previous method (Xu et al., 2012). The green regions correspond to the distortion levels at which both methods can successfully perform alignments. The yellow regions correspond to the distortion levels at which our new method outperforms our previous method and correctly aligns the subtomograms, whereas our previous method (Xu et al., 2012) fails. The gray region corresponds to the distortion levels at which both methods fail. For both alignment methods, we choose a rotation angle interval
across 100 pairwise alignments. The nodes on the solid black grid correspond to the alignment performance of our new method. The nodes in the red dashed grid correspond to the alignment performance of our previous method (Xu et al., 2012). The green regions correspond to the distortion levels at which both methods can successfully perform alignments. The yellow regions correspond to the distortion levels at which our new method outperforms our previous method and correctly aligns the subtomograms, whereas our previous method (Xu et al., 2012) fails. The gray region corresponds to the distortion levels at which both methods fail. For both alignment methods, we choose a rotation angle interval 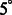 , i.e. the bandwidth B = 36
, i.e. the bandwidth B = 36
3.4.2 Crowded subtomograms
Next, we compare the two alignment methods (Xu et al., 2012) with crowded subtomograms, which also contain fragments of additional complexes (right column of Fig. 6 and Supplementary Tables S14 and S17). It can be clearly seen that when considering crowded subtomograms without excluding surrounding structures, our previous method (Xu et al., 2012) generally fails in finding the correct alignment. By contrast, our new alignment method aided by target complex segmentation is only marginally affected by crowding and the existence of the surrounding structures. Our new method clearly outperforms our previous method when dealing with subtomograms from crowded cellular environments. Similar improvement in performance is also observed by testing the crowded subtomograms of four additional benchmark complexes (Supplementary Tables S19–S22) (Fig. 4B) used in (Xu et al., 2012).
We have also measured the correlation between 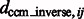 and
and 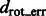 (Supplementary Tables S3, S6, S15 and S18) and tested the performance with respect to the levels of crowdedness (Supplementary Figs S1–S3). At low to median distortion levels, we observe moderate correlation between the aforementioned two quantities. At high distortion levels, the correlations are small, showing that the alignment is more affected by high levels of distortions.
(Supplementary Tables S3, S6, S15 and S18) and tested the performance with respect to the levels of crowdedness (Supplementary Figs S1–S3). At low to median distortion levels, we observe moderate correlation between the aforementioned two quantities. At high distortion levels, the correlations are small, showing that the alignment is more affected by high levels of distortions.
3.5 Segmenting target complexes in subtomograms extracted from experimental whole-cell tomogram of the human pathogen Leptospira interrogans
We tested our method also on experimental subtomograms extracted from a whole-cell tomogram of Leptospira interrogans in undisturbed conditions (Beck et al., 2009). Segmenting of complexes in such subtomograms is significantly more challenging because the macromolecular crowding is high and because the SNR levels and resolution of these subtomograms is typically low. Beck et al. (2009) have identified several complexes in the tomogram using template matching (Best et al., 2007). The templates were generated by using density maps of structures in the Protein Databank and convoluted with CTF and MTF. As a test case, we use two subtomograms with high template matching scores to the ribosome template. For each of these matches, we extracted a subtomogram of size 493 voxels that was large enough to contain an instance of the complex and certain amount of surrounding structure fragments.
The results shown in Figure 7 demonstrate that our method can successfully segment the target ribosome complex while surrounding structures are clearly excluded. The ribosome positions agree with those detected in the template matching. We further extended the study to the subtomograms corresponding to the top 20 template matching scores. Overall, 18 of them showed successful segmentation (Supplementary Fig. S4). We also analyzed the target complex segmentation performance with respect to the level of crowdedness. The crowdedness of the Leptospira interrogans subtomograms ranges from 0 to 60%. The true-positive and false-positive rates plotted in Supplementary Figure S4 shows that, in most situations, our method can correctly identify the target complex region. Only at high crowdedness level, (i.e. >50%), our method fails to correctly identify the target complex regions.
3.6 Analysis of computational costs
We implemented the methods in MATLAB and carried out our tests on a computer cluster consisting of 2.3–2.7 GHz computer nodes. On average, the target complex detection step takes 14.8 s. In this step, the major proportion of computation time was used for the scale selection (8.6 s on average). In practice, we can determine a fixed scale parameter σ from a set of training subtomograms and directly apply it to all subtomograms. Then, on average, only 6.2 s would be used for detecting the target complex. The computer memory consumption is small. Because a subtomogram is typically small (a subtomogram of size 643 corresponds to at most 2 M memory), our detection step uses no more memory than 20 times the size of a single subtomogram. The most computational intensive step in the classification process is the alignment. The computational cost of our new method is essentially the same as our previous method (Xu et al., 2012) and therefore allows high-throughput subtomogram classifications.
4 CONCLUSION
Cryo-electron tomograms provide useful information for simultaneously detecting the native structures and their spatial cellular locations of a large number of macromolecular complexes. However, the high level of macromolecular crowding and distortions in the extracted subtomograms makes such analysis challenging. In addition, the availability of a large number of subtomograms containing potential complexes requires high-throughput analysis techniques. In this article, we propose an automatic segmentation technique that isolates the target complex inside a crowded subtomogram. We further detect the rigid transform invariant center of mass of the target complex and propose an improved fast alignment method using fast rotational matching in real space. The new method shows good performance in segmenting target complexes in crowded subtomograms and performing fast rotational alignments using both simulated and experimental subtomograms. This performance is a necessary condition for high-throughput structural classifications of complexes in whole-cell tomograms. This work represents a step toward high-throughput and reference-free Visual Proteomics analysis of highly crowded whole-cell cryo-electron tomograms.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Martin Beck for providing the experimental tomograms for testing. They thank the anonymous reviewers for their constructive suggestions.
Funding: Human Frontier Science Program RGY0079/2009-C, the Arnold and Mabel Beckman foundation; NIH [R01GM096089 and U54RR022220] and NSF CAREER [1150287] (to F.A.). F.A. is a Pew Scholar in Biomedical Sciences, supported by the Pew Charitable Trusts.
Conflict of Interest: none declared.
REFERENCES
- Amat F, et al. Subtomogram alignment by adaptive Fourier coefficient thresholding. J. Struct. Biol. 2010;171:332–344. doi: 10.1016/j.jsb.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi A, et al. Classification and 3D averaging with missing wedge correction in biological electron tomography. J. Struct. Biol. 2008;162:436–450. doi: 10.1016/j.jsb.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, et al. Visual proteomics of the human pathogen Leptospira interrogans. Nat. Methods. 2009;6:817–823. doi: 10.1038/nmeth.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, et al. Exploring the spatial and temporal organization of a Cell’s proteome. J. Struct. Biol. 2011;173:483–496. doi: 10.1016/j.jsb.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best C, et al. Localization of protein complexes by pattern recognition. Methods Cell Biol. 2007;79:615–638. doi: 10.1016/S0091-679X(06)79025-2. [DOI] [PubMed] [Google Scholar]
- Beucher S, Lantuéjoul C. International Workshop on Image Processing, Real-Time Edge and Motion Detection/Estimation, CCETT/IRISA, Rennes, France. 1979. Use of watersheds in contour detection. [Google Scholar]
- Böhm J, et al. Toward detecting and identifying macromolecules in a cellular context: template matching applied to electron tomograms. Proc. Natl Acad. Sci. USA. 2000;97:14245–14250. doi: 10.1073/pnas.230282097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeux G, Govaert G. Gaussian parsimonious clustering models. Pattern Recognit. 1995;28:781–793. [Google Scholar]
- Chen Y, et al. Fast and accurate reference-free alignment of subtomograms. J. Struct. Biol. 2013 doi: 10.1016/j.jsb.2013.03.002. [Epub ahead of print, doi: 10.1016/j.jsb.2013.03.002, March 22, 2013] [DOI] [PubMed] [Google Scholar]
- Förster F, et al. Classification of cryo-electron sub-tomograms using constrained correlation. J. Struct. Biol. 2008;161:276–286. doi: 10.1016/j.jsb.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Fraley C, Raftery A. Model-based clustering, discriminant analysis, and density estimation. J. Am. Stat. Assoc. 2002;97:611–631. [Google Scholar]
- Frangakis A, et al. Identification of macromolecular complexes in cryoelectron tomograms of phantom cells. Proc. Natl Acad. Sci. USA. 2002;99:14153–14158. doi: 10.1073/pnas.172520299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J. Three-dimensional electron microscopy of macromolecular assemblies. New York: Oxford University Press; 2006. [Google Scholar]
- Garzón J, et al. Adp_em: fast exhaustive multi-resolution docking for high-throughput coverage. Bioinformatics. 2007;23:427–433. doi: 10.1093/bioinformatics/btl625. [DOI] [PubMed] [Google Scholar]
- Heumann J, et al. Clustering and variance maps for cryo-electron tomography using wedge-masked differences. J. Struct. Biol. 2011;175:288–299. doi: 10.1016/j.jsb.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabe T, et al. Pytom: a python-based toolbox for localization of macromolecules in cryo-electron tomograms and subtomogram analysis. J. Struct. Biol. 2012;178:177–188. doi: 10.1016/j.jsb.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Kovacs J, Wriggers W. Fast rotational matching. Acta. Crystallogr. D. Biol. Crystallogr. 2002;58:1282–1286. doi: 10.1107/s0907444902009794. [DOI] [PubMed] [Google Scholar]
- Lindeberg T. Scale-Space Theory in Computer Vision. The Kluwer International Series in Engineering and Computer Science. Dordrecht, The Netherlands: Kluwer Academic Publishers; 1994. [Google Scholar]
- Lucic V, et al. Structural studies by electron tomography: from cells to molecules. Annu. Rev. Biochem. 2005;74:833–865. doi: 10.1146/annurev.biochem.73.011303.074112. [DOI] [PubMed] [Google Scholar]
- Martinez-Sanchez A, et al. A differential structure approach to membrane segmentation in electron tomography. J. Struct. Biol. 2011;175:372–383. doi: 10.1016/j.jsb.2011.05.010. [DOI] [PubMed] [Google Scholar]
- McMullan G, et al. Detective quantum efficiency of electron area detectors in electron microscopy. Ultramicroscopy. 2009;109:1126–1143. doi: 10.1016/j.ultramic.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medalia O, et al. Macromolecular architecture in eukaryotic cells visualized by cryoelectron tomography. Science. 2002;298:1209–1213. doi: 10.1126/science.1076184. [DOI] [PubMed] [Google Scholar]
- Nickell S, et al. TOM software toolbox: acquisition and analysis for electron tomography. J. Struct. Biol. 2005;149:227–234. doi: 10.1016/j.jsb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Nickell S, et al. A visual approach to proteomics. Nat. Rev. Mol. Cell. Bio. 2006;7:225–230. doi: 10.1038/nrm1861. [DOI] [PubMed] [Google Scholar]
- Volkmann N. A novel three-dimensional variant of the watershed transform for segmentation of electron density maps. J. Struct. Biol. 2002;138:123–129. doi: 10.1016/s1047-8477(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Volkmann N. Methods for segmentation and interpretation of electron tomographic reconstructions. Methods Enzymol. 2010;483:31–46. doi: 10.1016/S0076-6879(10)83002-2. [DOI] [PubMed] [Google Scholar]
- Witkin AP. International Joint Conference on Artificial Intelligence, Karlsruhe, Germany. 1983. Scale-space filtering; pp. 1019–1022. [Google Scholar]
- Wriggers W, et al. Situs: a package for docking crystal structures into low-resolution maps from electron microscopy. J. Struct. Biol. 1999;125:185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- Xu M, Alber F. High precision alignment of cryo-electron subtomograms through gradient-based parallel optimization. BMC. Syst. Biol. 2012;6(Suppl. 1):S18. doi: 10.1186/1752-0509-6-S1-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, et al. Template-free detection of macromolecular complexes in cryo electron tomograms. Bioinformatics. 2011;27:i69–i76. doi: 10.1093/bioinformatics/btr207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, et al. High-throughput subtomogram alignment and classification by Fourier space constrained fast volumetric matching. J. Struct. Biol. 2012;178:152–164. doi: 10.1016/j.jsb.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.