

Abstract
The clinical management of glaucoma and optic neuropathies has traditionally focused on stages of the diseases at which there are congruent losses of visual function and optic nerve tissue. Increasing clinical and experimental evidence suggests that the electrical activity of retinal ganglion cells, as measured by pattern electroretinogram (PERG), may be altered long before measurable changes in the thickness of the retinal nerve fiber layer. In addition, PERG alterations in early glaucoma may be either reversed by lowering the intraocular pressure or induced with head-down body posture. Here we apply the well-known concept of neural plasticity to model the reversible/inducible changes of retinal ganglion cell electrical activity during a critical period of dysfunction preceding death. Identification and characterization of this stage of modifiable retinal ganglion cell function represents both a rationale and a target for treatment to change the natural history of the disease.
Retinal ganglion cell (RGC) death is the final common pathway that leads to loss of vision in glaucoma and most optic neuropathies. Lethal insult to RGCs and their axons originates from stressful changes of the cellular and molecular environment that exceed their survival capacity. The clinical management of glaucoma and optic neuropathies has traditionally focused on stages of the diseases at which losses of visual function of optic nerve tissue become manifest. Relatively less attention has been given to the fundamental issue of whether visual loss precedes, accompanies, or follows cell death. In the former case, visual dysfunction can be used as a marker to predict future nerve tissue loss and to initiate therapeutic strategies to prevent cell death and possibly reverse visual loss. In the latter cases, the only available therapeutic strategy is to slow down further cell death and visual loss. While this problem has been dealt with before (1), formal theoretical concepts leading to a testable model are lacking. In our view, this represents a limitation to progress in the field of degenerative diseases of the optic nerve. Here we provide a simple framework and a unified model to aid in understanding glaucoma and optic nerve diseases and for hypothesis testing. This model is supported by an increasing number of clinical and experimental results.
CENTRAL HYPOTHESIS
A reasonable hypothesis is that the early stages of optic neuropathies are characterized by failure of autoregulatory mechanisms to sustain normal RGC function under prolonged exposure to a stressful environment. Autoregulatory failure sets in motion adaptive mechanisms to prolong RGC survival. Surviving RGCs have altered function, which may be reversible under less stressful conditions. The duration of the stage of RGC dysfunction preceding death may be relatively long in glaucoma compared with Leber hereditary optic neuropathy (LHON). In these different diseases, however, RGC may share the same fundamental stress-dependent adaptive response to use fully the residual intracellular and extracellular resources and extend the time window during which RGC dysfunction preceding death may be still reversible under less stressful environmental conditions. Independent of the mechanisms or RGC/axon insult, an understanding of the time window of reversible RGC dysfunction would be applicable to most optic neuropathies and would represent key information to develop therapeutic approaches to prevent RGC death and restore RGC function.
CONCEPTUAL MODEL
Here we apply the well-known concept of neural plasticity to describe the ability of RGCs to change their function over time in response to an environmental challenge that exceeds their autoregulatory capacity. If an intervention is applied during the plastic period, the natural history of the disease will be substantially modified. While neuroplasticity is known to be active in the early postnatal period, some degree of plasticity is retained in the adult. This is particularly evident in acquired brain injury, where plasticity represents the fundamental issue that supports a scientific basis for treatment (2,3). Synapse elimination is one of the earliest events in neurodegenerative diseases (4), and this also occurs in RGC dendrites in early stages of glaucoma in the DBA/2J mouse model through reactivation of developmental molecular mechanisms (5).
DEFINITIONS
We define RGC plasticity as the ability of RGCs to modify their electrical activity upon an environmental challenge that exceeds their autoregulatory capacity. We define critical period as the time during which changes in RGC electrical activity are possible. If RGC function is modifiable by specific stressors during the critical period of RGC plasticity, then this represents both a rationale for treatment and a target to change the natural history of the disease.
MODEL FOR SINGLE RGCS
The model is summarized in Figure 1. After a long period of a normal existence (Stage 1), the cumulative stress exposure becomes higher than the RGC can tolerate, thus setting the cell in survival mode (Stage 2). At Stage 2, the RGC is dysfunctional. However, RGC dysfunction is modifiable with stress modulation: it will worsen after a further increase in stress (e.g., by increasing intraocular pressure [IOP] in glaucoma) and will improve by reducing the level of stress. At a level of dysfunction incompatible with survival, the RGC will start the one-way process of apoptosis (Stage 3) and will be removed from the neuronal pool (Stage 4). No residual RGC function is expected at Stages 3 and 4, but the RGC body and axon would still be morphologically visible and quantifiable. This model predicts that there will be a time lag between loss of function and loss of structure, which represents the lifespan of a surviving RGC. The transition between Stage 1 and Stage 2 represents the onset of disease that is crucially important to start a timely treatment to prevent further dysfunction and neural loss.
FIG. 1.
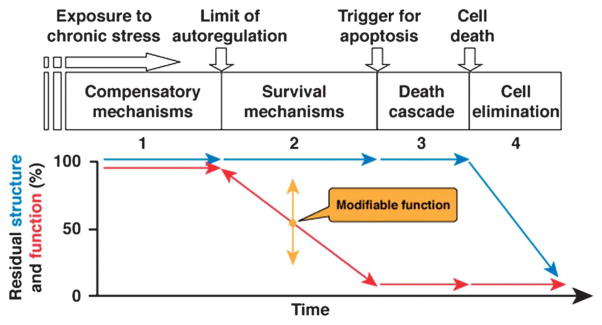
Conceptual model of structure–function relationship for 1 retinal ganglion cell undergoing several stages of progressive dysfunction and death.
MODEL FOR RGC POPULATIONS
In a large population of RGCs, it is expected that cells at different levels of dysfunction coexist with normal cells. During progression of disease, each RGC will follow structure–function time courses similar to those depicted in Figure 1 but shifted over time by different amounts. This will result in a family of functions whose average represents the time course of the functional–structural decay of the neuronal ensemble. The profiles of these time courses will be sigmoidal like those depicted in Figure 2. This figure shows 2 different conditions simulating different rates of progression of disease. One (Fig. 2A) is relatively fast as in LHON. The other (Fig. 2B) is relatively slow as in glaucoma. Except for the time scale, functions A and B are identical. The model is conceived on the assumption that RGC function/number can be specifically and quantitatively measured in vivo over time. These requirements are very difficult to achieve under ordinary circumstances. However, there are available noninvasive tools that have been shown to directly reflect RGC electrical activity, such as the pattern electroretinogram (PERG) (6–11) and RGC number, such as optical coherence tomography (OCT) (12). PERG and OCT have received extensive validation and can be used in both human patients and experimental models. The conceptual model depicted in Figure 2 is likely to apply using PERG and OCT as approximate measures of RGC function and number, respectively. Standard automated perimetry (SAP) seems less specific than PERG as a measure of RGC function, as it integrates functional changes occurring at RGC level and in the optic nerve, as well as secondary changes occurring in relay neurons of the lateral geniculate nucleus and primary visual cortex (13–15). Postretinal functional changes may either exacerbate those occurring at RGC level or even mitigate them as result of cortical compensatory mechanisms (16).
FIG. 2.
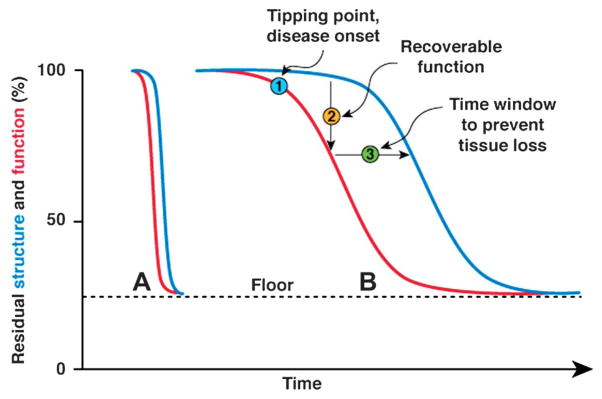
Conceptual model of structure–function relationship for a large population of retinal ganglion cells, in which different levels of cell dysfunction coexist. Condition A corresponds to a fast-progressing disease, such as Leber hereditary optic neuropathy. Condition B corresponds to a slow progressing disease such as glaucoma. The curves representing function and structure have identical form, although shifted by a time lag. Note that the residual function and structure measurable in advanced stages of the diseases with pattern electroretinogram or optical coherence tomography has a nonzero value (floor).
Predictions of the Model
Disease Onset
Given the sigmoidal nature of the curves, the model anticipates that the tipping point (disease onset) may be difficult to detect. The earliest discernable functional or structural loss must have a magnitude that exceeds the confidence intervals of the normal population (in a cross-sectional study) or the test-retest variability of individual subjects (in a longitudinal study).
Susceptibility to Provocation
A way to detect the tipping point is to use a provocative stressful event to exacerbate latent RGC dysfunction. Provocative conditions have successfully been used to disclose latent RGC dysfunction in glaucoma suspects by means of head-down (−10°) body posture. Postural change causes intraocular pressure (IOP) elevation in both normal subjects and patients with suspicion of glaucoma or early glaucoma. Head-down posture does not cause PERG alterations in normal subjects but reversibly alters the PERG of a subset of glaucoma patients (17), implying that RGC function is susceptible to IOP stress in these patients. A similar approach has been used in the DBA/2J mouse model of glaucoma. DBA/2J mice develop a pigment-liberating iris disease that causes elevated IOP and glaucoma (18). While RGC axon loss is first detectable at 8 months of age, the PERG signal can be altered approximately 3 months earlier (19,20). The time courses of RGC axon loss and PERG signal loss have profiles very similar to the theoretical model depicted in Figure 2B. In addition, the PERG signal is susceptible in an age-dependent manner to temporary IOP elevation induced by head-down (60°) body posture (21). That is, while preglaucomatous young (2- to 4-month-old) DBA/2J mice have normal PERG and are resistant to posture-induced IOP elevation, the PERG of older mice (5–6 months old) becomes abnormal during head-down body posture. At 7–8 months of age, the PERG is severely abnormal, and the loss of signal is exacerbated during head-down body posture (21).
Reversibility of RGC Dysfunction
The model (Fig. 2) predicts that there will be an excess of RGC dysfunction compared with that expected from loss of tissue. This has been shown by comparing PERG and OCT in human glaucoma (22) and LHON (23) and by comparing PERG and axon counts in DBA/2J glaucomatous mice (19,20). This excess dysfunction is potentially recoverable. A number of studies show that the losses of PERG signal in human and DBA/2J experimental glaucoma are, at least in part, recoverable (21,24–28).
Critical Period of RGC Plasticity
The model predicts that there will be a time lag between RGC dysfunction and death. A number of PERG studies in human and experimental models of glaucoma, multiple sclerosis, and LHON show that RGC dysfunction may be identifiable before structural loss within the optic nerve is evident with OCT, magnetic resonance imaging, or histology (19,20,22,23,29–32). This time lag represents the time window for therapeutic intervention to prevent cell death (33,34). Under ordinary circumstances, the time lag between dysfunction and death can only be established a posteriori by comparing the time courses of RGC dysfunction and death. Having an approximate prediction of the natural history of the disease with or without therapeutic intervention, one may study how to modify RGC electrical responsiveness under altered environmental conditions. High susceptibility of the PERG signal to provocative stress (e.g., IOP elevation (17,21), increased metabolic challenge (35,36)) would suggest a fast progressing disease. Reversibility of PERG signal after reduced stress (e.g., IOP lowering) or drug intake would suggest recovery of RGC function preceding death, and potentially that the disease can be prevented.
Structure–Function Correlations
The model (Fig. 2) predicts that structure–function correlations will be poor around the tipping point, whereas they will be relatively good at later stages, when both function and structure progressively deteriorate. Good correspondence between structural loss in the optic nerve head (as determined by either optic disc photos or OCT) and functional deficits in the visual field is a hallmark for establishing a clinical diagnosis of glaucoma and other optic nerve diseases. Some recent structure–function models based on the comparison between SAP and estimates of RGC number show that there is a precise causal relationship between loss of structure and loss of function in patients with glaucoma (37–41), ischemic optic neuropathy (42), and in nonhuman primates with experimental glaucoma (43). Other models would suggest that both RGC death and RGC dysfunction are necessary to account for empiric findings about relationships between perimetric and structural measures of glaucomatous damage (44). Our conceptual model includes these deterministic factors, which are based on parallelism between structure and function at stages of disease characterized by progressive RGC loss (37,43). Also, the model includes a key factor—the time lag between RGC dysfunction and death—that provides the window of opportunity to rescue ailing RGCs. The most sought after hallmark of glaucoma and optic neuropathies—correlation between structure and function —should be considered typical of a relatively late stage of disease. In contrast, lack of correlation between structure and function should characterize early potentially reversible stages of disease.
CONCLUSIONS
The concepts presented here provide a comprehensive unifying model of an identifiable stage of plasticity of RGC electrical responsiveness preceding death. Identification of this stage represents both a rationale and a target for treatment. The model does not make assumptions about mechanisms resulting in RGC dysfunction and death or does it make assumptions about genetic susceptibility to chronic stress exposure—and about the nature of stress—in different optic neuropathies. The model allows a set of testable predictions that are supported by an increasing number of clinical and experimental findings. It may be used as a framework for better understanding of how glaucoma and optic nerve disorders develop as well as the appropriate time window for treatment. The model has intrinsic limitations. First, both PERG and OCT have a limited dynamic range; the magnitudes of both PERG and OCT signals do not go to zero in advanced stages of optic neuropathy (floor effect) (22,40,45), limiting the use of the model to early-to-moderate stages of disease. Second, the OCT signal may spuriously increase as a result of either inflammation or gliosis of optic nerve axons (46). These factors need to be considered to reconcile the model with experimental findings.
Acknowledgments
Supported by NIH-NEI RO1 EY019077 (VP), R01EY014957 (VP), P30-EY014801 (VP), and an unrestricted grant to Bascom Palmer Eye Institute from Research to Prevent Blindness, Inc.
Footnotes
The authors report no conflict of interest.
References
- 1.Osborne NN, Wood JP, Chidlow G, Bae JH, Melena J, Nash MS. Ganglion cell death in glaucoma: what do we really know? Br J Ophthalmol. 1999;83:980–986. doi: 10.1136/bjo.83.8.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jenkins WM, Merzenich MM. Reorganization of neocortical representations after brain injury: a neurophysiological model of the bases of recovery from stroke. Prog Brain Res. 1987;71:249–266. doi: 10.1016/s0079-6123(08)61829-4. [DOI] [PubMed] [Google Scholar]
- 3.Kolb B, Whishaw IQ. Plasticity in the neocortex: mechanisms underlying recovery from early brain damage. Prog Neurobiol. 1989;32:235–276. doi: 10.1016/0301-0082(89)90023-3. [DOI] [PubMed] [Google Scholar]
- 4.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 5.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson K, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 6.Mafei L, Fiorentini A. Electroretinographic responses to alternating gratings before and after section of the optic nerve. Science. 1981;211:953–955. doi: 10.1126/science.7466369. [DOI] [PubMed] [Google Scholar]
- 7.Porciatti V, Pizzorusso T, Cenni MC, Maffei L. The visual response of retinal ganglion cells is not altered by optic nerve transection in transgenic mice overexpressing Bcl-2. Proc Natl Acad Sci U S A. 1996;93:14955–14959. doi: 10.1073/pnas.93.25.14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zrenner E. The physiological basis of the pattern electroretinogram. In: Osborne N, Chader G, editors. Progress in Retinal Research. Oxford: Pergamon Press; 1990. pp. 427–464. [Google Scholar]
- 9.Holder GE. The pattern electroretinogram in anterior visual pathway dysfunction and its relationship to the pattern visual evoked potential: a personal clinical review of 743 eyes. Eye. 1997;11:924–934. doi: 10.1038/eye.1997.231. [DOI] [PubMed] [Google Scholar]
- 10.Porciatti V. The mouse pattern electroretinogram. Doc Ophthalmol. 2007;115:145–153. doi: 10.1007/s10633-007-9059-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bach M, Hoffmann MB. Update on the pattern electroretinogram in glaucoma. Optom Vis Sci. 2008;85:386–395. doi: 10.1097/OPX.0b013e318177ebf3. [DOI] [PubMed] [Google Scholar]
- 12.Harwerth RS, Wheat JL. Modeling the effects of aging on retinal ganglion cell density and nerve fiber layer thickness. Graefes Arch Clin Exp Ophthalmol. 2008;246:305–314. doi: 10.1007/s00417-007-0691-5. [DOI] [PubMed] [Google Scholar]
- 13.Yucel YH, Zhang Q, Weinreb RN, Kaufman PL, Gupta N. Effects of retinal ganglion cell loss on magno-, parvo-, koniocellular pathways in the lateral geniculate nucleus and visual cortex in glaucoma. Prog Retin Eye Res. 2003;22:465–481. doi: 10.1016/s1350-9462(03)00026-0. [DOI] [PubMed] [Google Scholar]
- 14.Gupta N, Yucel YH. Glaucoma as a neurodegenerative disease. Curr Opin Ophthalmol. 2007;18:110–114. doi: 10.1097/ICU.0b013e3280895aea. [DOI] [PubMed] [Google Scholar]
- 15.Duncan RO, Sample PA, Weinreb RN, Bowd C, Zangwill LM. Retinotopic organization of primary visual cortex in glaucoma: Comparing fMRI measurements of cortical function with visual field loss. Prog Retin Eye Res. 2007;26:38–56. doi: 10.1016/j.preteyeres.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lam DY, Kaufman PL, Gabelt BT, To EC, Matsubara JA. Neurochemical correlates of cortical plasticity after unilateral elevated intraocular pressure in a primate model of glaucoma. Invest Ophthalmol Vis Sci. 2003;44:2573–2581. doi: 10.1167/iovs.02-0779. [DOI] [PubMed] [Google Scholar]
- 17.Ventura LM, Golubev I, Lee W, Nose I, Parel JM, Feuer WJ, Porciatti V. Head-down posture induces PERG alterations in early glaucoma. J Glaucoma. doi: 10.1097/IJG.0b013e318232973b. [published online ahead of print December 1, 2011] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Libby RT, Anderson MG, Pang IH, Robinson ZH, Savinova OV, Cosma IM, Snow A, Wilson LA, Smith RS, Clark AF, John SW. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005;22:637–648. doi: 10.1017/S0952523805225130. [DOI] [PubMed] [Google Scholar]
- 19.Saleh M, Nagaraju M, Porciatti V. Longitudinal evaluation of retinal ganglion cell function and IOP in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2007;48:4564–4572. doi: 10.1167/iovs.07-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barta JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SW. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523–1537. doi: 10.1083/jcb.200706181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagaraju M, Saleh M, Porciatti V. IOP-dependent retinal ganglion cell dysfunction in glaucomatous DBA/2J mice. Invest Ophthalmol Vis Sci. 2007;48:4573–4579. doi: 10.1167/iovs.07-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ventura LM, Sorokac N, De Los Santos R, Feuer WJ, Porciatti V. The relationship between retinal ganglion cell function and retinal nerve fiber thickness in early glaucoma. Invest Ophthalmol Vis Sci. 2006;47:3904–3911. doi: 10.1167/iovs.06-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lam BL, Feuer WJ, Porciatti V, Abukhalil F, Morante A, Guy JR. Leber hereditary optic neuropathy gene therapy clinical trial: preparatory phase year one. ARVO Meeting Abstracts. 2011;51:994. [Google Scholar]
- 24.Ventura LM, Porciatti V. Restoration of retinal ganglion cell function in early glaucoma after intraocular pressure reduction: a pilot study. Ophthalmology. 2005;112:20–27. doi: 10.1016/j.ophtha.2004.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salgarello T, Falsini B, Stifano G, Montrone L, Iarossi G, Balestrazzi E, Colotto A. Morpho-functional follow-up of the optic nerve in treated ocular hypertension: disc morphometry and steady-state pattern electroretinogram. Curr Eye Res. 2008;33:709–721. doi: 10.1080/02713680802277692. [DOI] [PubMed] [Google Scholar]
- 26.Sehi M, Grewal DS, Goodkin ML, Greenfield DS. Reversal of retinal ganglion cell dysfunction after surgical reduction of intraocular pressure. Ophthalmology. 2010;117:2329–2336. doi: 10.1016/j.ophtha.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 27.Ventura LM, Feuer WJ, Porciatti V. Progressive loss of retinal ganglion cell function is hindered with IOP-lowering treatment in early glaucoma. Invest Ophthalmol Vis Sci. 2012;53:659–663. doi: 10.1167/iovs.11-8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porciatti V, Nagaraju M. Head-up tilt lowers IOP and improves RGC dysfunction in glaucomatous DBA/2J mice. Exp Eye Res. 2010;90:452–460. doi: 10.1016/j.exer.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enriquez-Algeciras M, Ding D, Chou TH, Want J, Padgett KR, Pociatti V, Bhattacharya SK. Evaluation of a transgenic mice model of multiple sclerosis with non invasive methods. Invest Ophthalmol Vis Sci. 2011;52:2405–2411. doi: 10.1167/iovs.10-6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porciatti V, Sartucci F. Retinal and cortical evoked responses to chromatic contrast stimuli. Specific losses in both eyes of patients with multiple sclerosis and unilateral optic neuritis. Brain. 1996;119:723–740. doi: 10.1093/brain/119.3.723. [DOI] [PubMed] [Google Scholar]
- 31.Falsini B, Marangoni D, Salgarello T, Stifano G, Montrone L, Campagna F, Aliberti S, Balestrazzi E, Colotto A. Structure–function relationship in ocular hypertension and glaucoma: interindividual and interocular analysis by OCT and pattern ERG. Graefes Arch Clin Exp Ophthalmol. 2008;246:1153–1162. doi: 10.1007/s00417-008-0808-5. [DOI] [PubMed] [Google Scholar]
- 32.North RV, Jones A, Drasdo N, Wild JM, Morgan JE. Electrophysiological evidence for early functional damage in glaucoma and ocular hypertension. Invest Ophthalmol Vis Sci. 2010;51:1216–1222. doi: 10.1167/iovs.09-3409. [DOI] [PubMed] [Google Scholar]
- 33.Koilkonda RD, Chou T-H, Ruggeri M, Porciatti V, Howswirth WW, Chiodo V, Boye SL, Guy J. Early functional and long-term rescue of the LHON mouse with wild-type human ND4 gene delivery. ARVO Meeting Abstracts. 2011;52:1416. [Google Scholar]
- 34.Howell GR, Soto I, Zhu X, Ryan M, Macalinao DG, Sousa GL, Caddle LB, MacNicoll KH, Barbay JM, Porciatti V, Anderson MG, Smith RS, Clark AF, Libby RT, John SW. Radiation treatment inhibits monocyte entry into the optic nerve head and prevents neuronal damage in a mouse model of glaucoma. J Clin Invest. 2012;122:1246–1261. doi: 10.1172/JCI61135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parekh PK, Bosse B, Shif OA, Ventura LM, Porciatti V. Metabolic adaptation of the steady-state PERG in normal controls and early glaucoma patients. ARVO Meeting Abstracts. 2011;52:5479. [Google Scholar]
- 36.Porciatti V, Chou TH. Adaptive changes in the oattern electroretinogram (PERG) induced by fiickering light in the mouse. ARVO Meeting Abstracts. 2011;52:5477. [Google Scholar]
- 37.Hood DC, Anderson SC, Wall M, Kardon RH. Structure versus function in glaucoma: an application of a linear model. Invest Ophthalmol Vis Sci. 2007;48:3662–3668. doi: 10.1167/iovs.06-1401. [DOI] [PubMed] [Google Scholar]
- 38.Garway-Heath DF, Caprioli J, Fitzke FW, Hitchings RA. Scaling the hill of vision: the physiological relationship between light sensitivity and ganglion cell numbers. Invest Ophthalmol Vis Sci. 2000;41:1774–1782. [PubMed] [Google Scholar]
- 39.Garway-Heath DF, Poinoosawmy D, Fitzke FW, Hitchings RA. Mapping the visual field to the optic disc in normal tension glaucoma eyes. Ophthalmology. 2000;107:1809–1815. doi: 10.1016/s0161-6420(00)00284-0. [DOI] [PubMed] [Google Scholar]
- 40.Hood DC, Kardon RH. A framework for comparing structural and functional measures of glaucomatous damage. Prog Retin Eye Res. 2007;26:688–710. doi: 10.1016/j.preteyeres.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harwerth RS, Vilupuru AS, Rangaswamy NV, Smith EL., 3rd The relationship between nerve fiber layer and perimetry measurements. Invest Ophthalmol Vis Sci. 2007;48:763–773. doi: 10.1167/iovs.06-0688. [DOI] [PubMed] [Google Scholar]
- 42.Hood DC, Anderson S, Rouleau J, Wenick AS, Groves LK, Behrens MM, Odel JG, Lee AG, Karden RH. Retinal nerve fiber structure versus visual field function in patients with ischemic optic neuropathy: A test of a linear model. Ophthalmology. 2008;115:904–910. doi: 10.1016/j.ophtha.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harwerth RS, Carter-Dawson L, Smith EL, 3rd, Barnes G, Holt WF, Crawford ML. Neural losses correlated with visual losses in clinical perimetry. Invest Ophthalmol Vis Sci. 2004;45:3152–3160. doi: 10.1167/iovs.04-0227. [DOI] [PubMed] [Google Scholar]
- 44.Swanson WH, Felius J, Pan F. Perimetric defects and ganglion cell damage: interpreting linear relations using a two-stage neural model. Invest Ophthalmol Vis Sci. 2004;45:466–472. doi: 10.1167/iovs.03-0374. [DOI] [PubMed] [Google Scholar]
- 45.Hood DC, Xu L, Thienprasiddhi P, Greenstein VC, Odel JG, Grippo TM, Liebmann JM, Ritch R. The pattern electroretinogram in glaucoma patients with confirmed visual field deficits. Invest Ophthalmol Vis Sci. 2005;46:2411–2418. doi: 10.1167/iovs.05-0238. [DOI] [PubMed] [Google Scholar]
- 46.Pasol J. Neuro-ophthalmic disease and optical coherence tomography: glaucoma look-alikes. Curr Opin Ophthalmol. 2011;22:124–132. doi: 10.1097/ICU.0b013e328343c1a3. [DOI] [PubMed] [Google Scholar]