

SUMMARY
Proteolytic fragments of the pigment cell-specific glycoprotein, PMEL, form the amyloid fibrillar matrix underlying melanins in melanosomes. The fibrils form within multivesicular endosomes to which PMEL is selectively sorted and that serve as melanosome precursors. GPNMB is a tissue-restricted glycoprotein with substantial sequence homology to PMEL but no known function, and was proposed to localize to non-fibrillar domains of distinct melanosome subcompartments in melanocytes. Here we confirm that GPNMB localizes to compartments distinct from the PMEL-containing multivesicular premelanosomes or late endosomes in melanocytes and HeLa cells, respectively, and is largely absent from fibrils. Using domain swapping, the unique PMEL localization is ascribed to its PKD domain, whereas the homologous PKD domain of GPNMB lacks apparent sorting function. The difference likely reflects extensive modification of the GPNMB PKD domain by N-glycosylation, nullifying its sorting function. These results reveal the molecular basis for the distinct trafficking and morphogenetic properties of PMEL and GPNMB, and support a deterministic function of the PMEL PKD domain in both protein sorting and amyloidogenesis.
Keywords: Pmel17, gp100, Nmb, osteoactivin, melanosome, amyloid, endosome, Polycystic kidney disease-1 repeat domain, N-Glycosylation, Multivesicular body
INTRODUCTION
Melanosomes are unique intracellular membranous organelles of epidermal melanocytes and ocular pigment cells within which melanin pigments are synthesized and stored (Hearing, 2005; Raposo and Marks, 2007). Within pigment cells, melanosomes develop through a series of morphologically defined stages, starting with non-pigmented precursors harboring an underlying fibrillar matrix and ending with mature pigmented melanosomes in which polymerized melanins fill the entire lumen (Seiji et al., 1963). Maturation occurs by the progressive accumulation of distinct protein contents, many of which are uniquely expressed in pigment cells. For example, early stages are enriched in proteolytic fragments of the pigment cell-specific protein PMEL (also called Pmel17, gp100 or Silver) (Raposo et al., 2001). These fragments are incorporated into the fibrillar matrix and appear to be the main constituent of the fibrils (Berson et al., 2001; Berson et al., 2003; Harper et al., 2008; Kushimoto et al., 2001). By contrast, later stages are enriched in pigment cell-specific enzymes that generate melanin intermediates, such as tyrosinase, tyrosinase related protein-1 (TYRP1), and dopachrome tautomerase (Kushimoto et al., 2001; Novikoff et al., 1968; Raposo et al., 2001; Seiji and Iwashia, 1965; Theos et al., 2005a), and in transporters that regulate the activity of these enzymes, such as the pigment cell-specific OCA2 (Donatien and Orlow, 1995; Rosemblat et al., 1994; Sitaram et al., 2009) and the more broadly expressed copper transporter, ATP7A (Setty et al., 2008). These proteins become incorporated into maturing melanosomes by mechanisms that are only beginning to be understood (Raposo and Marks, 2007; Sitaram and Marks, 2012).
Whereas the contents of the later stage melanosomes appear to be delivered to non-pigmented precursors by vesicular transport mechanisms — some of which are confounded in genetic diseases such as Hermansky-Pudlak syndrome (Wei and Li, 2013; Wei, 2006) — PMEL, the best known component of early stage melanosomes, appears to be incorporated by distinct mechanisms (reviewed in (Watt et al., 2010; Watt et al., 2013)). PMEL is synthesized as a type I integral membrane glycoprotein (Adema et al., 1994; Kwon et al., 1987; Maresh et al., 1994a) with a short signal sequence that is cleaved upon translocation into the endoplasmic reticulum (ER) (Maresh et al., 1994b), a large lumenal domain that becomes modified by four N-linked and multiple O-linked oligosaccharides (Harper et al., 2008; Maresh et al., 1994b; Valencia et al., 2007), a single membrane spanning domain and a 45-residue cytoplasmic domain.
Following modification of oligosaccharides in the Golgi complex (Berson et al., 2001; Harper et al., 2008), PMEL is targeted to early endosomes, most likely following transient appearance at the plasma membrane and subsequent endocytosis (Chen et al., 2012; Lepage and Lapointe, 2006; Robila et al., 2008; Theos et al., 2006a). Within vacuolar domains of early endosomes, PMEL associates with invaginating membranes that form intralumenal vesicles (ILVs) (Raposo et al., 2001). Prior to or concomitant with endosomal sorting, PMEL is cleaved by two proteases: a proprotein convertase generates a large lumenal fibrillogenic Mα fragment that remains disulfide bonded to the integral membrane Mβ fragment (Berson et al., 2003; Leonhardt et al., 2011), and an as yet undefined site 2 protease cleaves Mβ at a juxtamembrane site liberating the entire lumenal domain (Kummer et al., 2009; van Niel et al., 2011). These cleavages, together with other factors that have not yet been identified, trigger a conformational change that favors the ordered oligomerization of Mα fragments into detergent insoluble fibrils (Berson et al., 2003; Kummer et al., 2009; Zhou et al., 1994) that form and grow in association with the intralumenal membranes of vacuolar endosomes/ early stage melanosomes (Berson et al., 2001; Hurbain et al., 2008). Interference with either of the two proteolytic cleavages or with the association of PMEL with ILVs inhibits fibril formation (Berson et al., 2003; Hoashi et al., 2006; Kummer et al., 2009; Theos et al., 2006b). As the melanosomes mature, PMEL is further proteolytically processed (Harper et al., 2008; Kushimoto et al., 2001; Watt et al., 2009) and the fibrils laterally assemble into concentric sheets (Hurbain et al., 2008). The C-terminal fragments are degraded in lysosomes (van Niel et al., 2011).
Unlike the later stages of melanosome maturation into a pigment granule, which require cell type-specific transport processes and enzymes, the events leading to PMEL sorting into early endosomes, association with forming ILVs, subsequent cleavages and even nascent fibril formation are recapitulated in non-pigment cells such as HeLa by ectopic expression of PMEL (Berson et al., 2001; Berson et al., 2003; Hoashi et al., 2006; Theos et al., 2006b). This suggests that these events are intrinsic to structural determinants within PMEL. Indeed, by expressing mutagenized forms of PMEL in non-pigment cells, distinct structural domains have been shown to be necessary for its transport and fibrillogenic properties. Determinants within the cytoplasmic domain regulate association with coat proteins that facilitate ER export and endocytosis, permitting enrichment of PMEL in early endosomes (Theos et al., 2006a). Within endosomes, enrichment on intralumenal membranes appears to require two consecutive lumenal sub-domains, a ~200 residue N-terminal region (NTR) and a ~90 residue PKD (polycystic kidney disease-1) domain with homology to a repeat found in polycystin-1, such that deletion of either domain results in a failure to enrich PMEL within ILVs of HeLa cells (Theos et al., 2006b); the NTR appears to be dispensable for ILV sorting in melanoma cells (Leonhardt et al., 2013). A third lumenal subdomain, RPT, which consists of a series of imperfect direct repeats (Kwon et al., 1987; Theos et al., 2005b) and is highly modified by O-linked glycosylation (Harper et al., 2008; Valencia et al., 2007), is required in vivo for fibril formation in HeLa cells (Hoashi et al., 2006; Theos et al., 2006b) and for normal fibril assembly and morphology in melanoma cells (Leonhardt et al., 2013). Interestingly, the amyloidogenic properties of the Mα fragment in vitro are mimicked by fragments consisting of either the NTR or PKD alone (Watt et al., 2009); the unglycosylated RPT domain can also form amyloid in vitro under acidic conditions (McGlinchey et al., 2011; McGlinchey et al., 2009), but at a much slower rate than full-length Mα or NTR or PKD domains (Watt et al., 2009). Together, these data suggest that the NTR and PKD domains confer both the trafficking and amyloidogenic properties of PMEL to allow for fibril formation in early stage melanosomes. Consistently, a region of the NTR prior to the PKD homology domain is necessary for PMEL folding and downstream trafficking steps (Leonhardt et al., 2010), and mutagenesis of residues within several regions of the NTR interfere with fibril assembly (Leonhardt et al., 2013).
If the unique properties of PMEL are indeed conferred by the NTR and PKD domains, it is thus surprising that a close PMEL homologue, GPNMB, has distinct properties. GPNMB (also called Nmb, osteoactivin, hematopoietic growth factor inducible neurokinin-1 type, and DC-HIL) is a tissue-restricted glycoprotein that was first identified as the product of a mRNA expressed preferentially in non-metastatic relative to metastatic melanoma (Weterman et al., 1995), but is overexpressed in both cutaneous and uveal melanoma as well as a number of other malignancies (Kuan et al., 2006; Rich et al., 2003; Rose and Siegel, 2010; Tse et al., 2006; Williams et al., 2010). GPNMB (and its homologue from quail, QNR-71) is expressed in pigment cells (Bachner et al., 2002; Turque et al., 1996; Weterman et al., 1995), and like other melanosomal proteins its expression is regulated transcriptionally by Microphthalmia transcription factor family members (Loftus et al., 2009; Ripoll et al., 2008). A recent report suggests that in melanocytes, GPNMB expression is required for the MITF-independent expression of melanosomal proteins and for generation of melanosomes (Zhang et al., 2012). Two recent reports suggested that unlike PMEL, GPNMB localized predominantly to mature pigmented melanosomes in melanocytic cell lines (Hoashi et al., 2010a; Tomihari et al., 2009), consistent with the partial codistribution of QNR-71 with melanosomes in ammonium chloride-treated retinal pigment epithelial cells (Le Borgne et al., 2001). This differential pattern of localization for PMEL and GPNMB is surprising, given that the amino acid sequences of the NTR and PKD domains from the two proteins exhibit 35–37% identity and 58–60% similarity (Theos et al., 2005b).
Here, we have reevaluated the relative distribution of PMEL and GPNMB in melanocytic and non-melanocytic cells, and the respective role of individual homologous subdomains as determinants of localization and fibril formation. Our data suggest that the PKD domain is the main determinant of both properties in PMEL, and that the corresponding domain in GPNMB acts as a “null” determinant, likely due to extensive modification and “shielding” by N-linked oligosaccharides. These data suggest that despite substantial sequence homology, PMEL and GPNMB have distinct structures and functions within pigment cells.
RESULTS
GPNMB and PMEL are differentially localized in melanocytic cells
Whereas PMEL is highly enriched in non-pigmented stage I and II melanosome precursors (premelanosomes; (Raposo et al., 2001)), a recent report suggested that GPNMB was localized predominantly to mature stage III/ IV melanosomes (Hoashi et al., 2010a). To attempt to validate this finding, we used immunofluorescence microscopy and volume deconvolution (IFM-D) to assess GPNMB localization relative to PMEL, melanosomes, and markers of other compartments in MNT-1, a pigmented human melanoma cell line with many of the morphological and melanogenic properties of eumelanogenic melanocytes (Hoek et al., 2004; Raposo et al., 2001). To detect GPNMB, we used two antibodies: AF2550, a goat polyclonal antibody that detects the GPNMB lumenal domain and that has been previously used to characterize GPNMB in melanocytic cells (Hoashi et al., 2010a), and hNMB-C, an affinity-purified antibody generated against the C-terminal peptide of the GPNMB cytoplasmic domain. Surprisingly, labeling by these two antibodies—excluding the perinuclear area, in which many organelles accumulate—showed very little overlap with each other in the periphery of MNT-1 cells (Figure 1a–c). Peripheral labeling by hNMB-C overlapped substantially, but not completely, with the ER marker calnexin (Figure 1d–f), suggesting that much of the full-length protein was confined to the ER at steady state with additional accumulation in perinuclear compartments such as the ER-Golgi intermediate compartment and perhaps the Golgi. This is similar to the situation for PMEL, which folds and exits the ER slowly and becomes proteolytically processed in post-Golgi compartments. Quantification of the overlap was not possible due to the weak GPNMB signal and the pervasive distribution of the ER. By contrast to labeling by hNMB-C, labeling for the lumenal domain by AF2550 was more punctate, and present throughout the cell. However, at high resolution provided by deconvolution microscopy, the label showed only minimal overlap either with pigment granules visualized by bright field microscopy (Figure 1a–c), or with labeling for mature forms of PMEL (detected with antibody HMB45; Figure 1g–i) or the lysosomal marker LAMP1 (Figure 1j–l), and only minimal overlap with calnexin (Figure 1m–o). Minimal overlap was also observed with markers of early sorting and recycling endosomes (Suppl. Figure S1). Identical results were obtained when cells were fixed with either methanol or 2–6% formaldehyde (not shown), ruling out fixation artifacts that might hinder detection of GPNMB on melanosomes or other structures (whereas varying concentrations of formaldehyde alters the distribution of the PMEL cytoplasmic domain relative to ER markers; ref. (Harper et al., 2008)). Moreover, epitope-tagged forms of GPNMB transiently overexpressed in other pigmented melanoma and melanocyte cell lines failed to colocalize with PMEL (Suppl. Figure S2 and data not shown). From these results, we infer that: (1) GPNMB is present in two forms in MNT-1 cells, an immature full-length form in the ER that is detected by hNMB-C, and a likely more mature form that is detected in post-ER structures by AF2550; (2) while labeling for “mature” GPNMB might partially overlap several compartments, it is not enriched in melanosomes; and, importantly, (3) GPNMB does not accumulate in the same early stage melanosomes as PMEL.
Figure 1. Localization of GPNMB within melanocytic MNT-1 cells by IFM.

MNT-1 cells were fixed with methanol, labeled with the indicated primary antibodies and fluorochrome-conjugated species-specific secondary antibodies, and analyzed by IFM-D (a, b, d, e, g, h, j–o) and by bright field microscopy (c, f, i). a–c, anti-GPNMB antibodies AF2550 and hNMB-C; d–f, hNMB-C and anti-calnexin (CLNX); g–i, AF2550 and anti-PMEL antibody HMB45; j–l, AF2550 and anti-LAMP1 antibody (H4A3); m–o AF2550 and CLNX. Boxed regions are magnified 4X in the insets at bottom right of each panel; merged images are shown in the insets of panels c, f, i, l and o, and in a–f, melanosomes (from bright field images) are pseudocolored blue. Bar, 10 μm.
GPNMB is cleaved by a site 2 protease but the lumenal domain does not preferentially cofractionate with premelanosome fibrils
To determine whether GPNMB undergoes proteolytic processing in melanocytic cells and whether the lumenal domain becomes incorporated into premelanosome fibrils, crude subcellular fractions of MNT-1 cells, obtained by differential solubility of cell components in TX-100, were analyzed by immunoblotting with AF2550 and hNMB-C. Detergent-soluble fractions (S) contain most non-membrane anchored and integral membrane proteins, whereas detergent-insoluble fractions (I) are enriched in melanin and premelanosome fibrils (Berson et al., 2003; Orlow et al., 1993). In detergent soluble fractions, AF2550 detected two bands: a ~ 100 kDa P1 (P1-N) form that likely represents the immature core-glycosylated precursor and a ~110 kDa form that likely represents the mature (M) form with Golgi-processed N-linked glycans (Figure 2a, left), consistent with observations from a previous report (Hoashi et al., 2010a). hNMB-C also detects these bands, albeit weakly. Additionally, hNMB-C occasionally detects a ~7 kDa band present in soluble fractions but not in the insoluble fractions (Figure 2a, right) or in the medium (data not shown). Since this band is recognized by hNMB-C but not AF2550, it must represent a C-terminal fragment (CTF) derived by proteolysis of full-length GPNMB, perhaps related to the larger CTFs observed in breast tumor cell lines and NIH 3T3 fibroblasts that overexpress GPNMB transgenes (Furochi et al., 2007; Rose et al., 2010). The size of the CTF in MNT-1 cells is consistent with that expected for a peptide consisting of the transmembrane and cytoplasmic domains of GPNMB, such that proteolytic cleavage to form the CTF would liberate the GPNMB lumenal domain from its membrane anchor as has been observed in other cell types (Furochi et al., 2007; Rose et al., 2010). Additional intermediate bands detected by the hNMB-C antibody are non-specific, as determined by blotting lysates from untransfected HeLa cells which lack GPNMB expression (data not shown).
Figure 2. Immunoblotting analysis of crude subcellular fractions from MNT-1 cells.
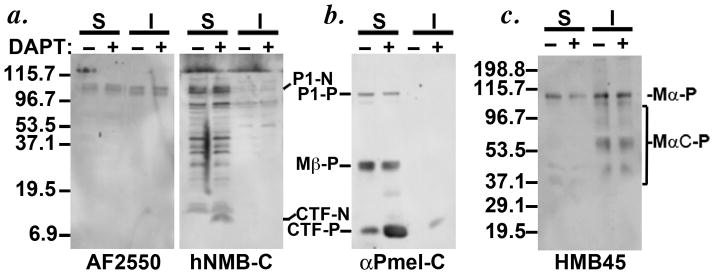
MNT-1 cells were treated with vehicle (−) or DAPT (+) for 4 h, harvested, and lysed with buffer containing Triton X-100 to obtain detergent soluble (S) and melanosome matrix-enriched insoluble (I) fractions. Lysates were fractionated further by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies for GPNMB (a) or PMEL (b, c). Arrows denote the stabilized CTFs of GPNMB (CTF-N) and PMEL (CTF-P) upon DAPT treatment, detected mainly in the soluble fractions. P1-N, GPNMB P1 precursor; P1-P, PMEL P1 precursor; Mβ-P, PMEL Mβ fragment; Mα-P, PMEL Mα fragment; MαC-P, PMEL Mα proteolytic fragments. Molecular weight markers are shown to the left of each panel.
PMEL, which is highly homologous to GPNMB, is cleaved in the juxtamembrane region by a “site 2 protease” to liberate its lumenal domain for incorporation into amyloid fibrils and a CTF that remains membrane embedded (Kummer et al., 2009). The PMEL CTF is subsequently eliminated by lysosomal proteolysis following intramembrane cleavage by the γ-secretase complex (Kummer et al., 2009; van Niel et al., 2011); hence, treatment with γ-secretase inhibitors stabilizes the CTF (Kummer et al., 2009). Consistently, treatment of MNT-1 cells with the γ-secretase inhibitor DAPT for 4 h stabilized the GPNMB CTF (CTF-N), as well as the PMEL CTF (CTF-P) (Figure 2a and b). These data suggest that GPNMB is targeted by two proteases: a site 2-like protease to release its lumenal domain, and γ-secretase to degrade the remaining CTF. These data also indicate that in MNT-1 cells, the CTF is degraded rapidly, as the CTF is only consistently detected upon γ-secretase inhibition.
To determine whether the released lumenal fragment of GPNMB, like that of PMEL, becomes incorporated into melanosome fibrils, we also analyzed detergent insoluble fractions. As shown previously (Berson et al., 2003; Kushimoto et al., 2001), PMEL lumenal fragments were enriched in the detergent-insoluble fraction (MαC-P, Figure 2c); in contrast, the GPNMB lumenal fragment detected by AF2550 showed no such enrichment and instead was detected at similar levels in both the detergent-soluble and insoluble fractions (Figure 2a), similar to results previously reported (Hoashi et al., 2010a). These data and our IFM-D analysis (Figure 1) indicate that GPNMB, unlike PMEL, is not enriched in the premelanosome fibrillar matrix. Interestingly, however, the GPNMB doublet that accumulated in the detergent-insoluble fraction migrated faster than the doublet in the detergent-soluble fraction and was not recognized by hNMB-C (Figure 2a, b). This suggests that the detergent-insoluble form of GPNMB might consist solely of the lumenal domain that has been liberated from the CTF.
GPNMB and PMEL are differentially localized when expressed in non-pigment cells
Melanosomes are only generated in pigment cells, but some of the early steps in premelanosome biogenesis can be partially recapitulated by ectopic expression of PMEL in non-pigment cells such as HeLa (Berson et al., 2001). In particular, when expressed in HeLa cells, PMEL accumulates on the ILVs of multivesicular late endosomes, from where it can initiate fibril formation (Berson et al., 2001; Berson et al., 2003; Hoashi et al., 2006; Theos et al., 2006b). PMEL localization to late endosomes in transfected HeLa can be visualized by IFM-D by its labeling in puncta that are surrounded by the late endosome/ lysosome marker, LAMP1 (Berson et al., 2001) or by overlap with CD63 (Figure 3a–c), which also accumulates on the internal vesicles of late endosomal ILVs (Escola et al., 1998). When expressed transiently in transfected HeLa cells and detected by AF2550, GPNMB also was present in punctate structures with an accumulation of label in the perinuclear area (Figure 3d). However, AF2550 labeling did not overlap at all with the late endosomal marker CD63 (Figure 3d–f). Similar results were obtained with hNMB-C, although substantial labeling in the ER and Golgi were also detected, consistent with results in MNT-1 cells (data not shown). To ensure that a subset of GPNMB not detected by AF2550 might be present in late endosomes, we expressed epitope-tagged forms of GPNMB in HeLa cells and assessed their localization by IFM-D. GPNMB with a myc epitope tag inserted within the proline-rich “hinge” region following the PKD domain also localized to punctate structures in HeLa cells that did not overlap with the late endosome marker, LAMP1 (Figure 3g–i; note, the anti-LAMP1 antibody was used in this case for compatibility with the anti-Myc antibody, and labels the limiting membrane of late endosomes rather than the internal vesicles). A small fraction of puncta harboring epitope-tagged GPNMB overlapped with the early sorting endosomal marker, EEA1 (Figure 3j–l), but most did not. Similarly, a small fraction of these puncta overlapped with transferrin that had been internalized for 3 h, thus marking early sorting and recycling endosomes (Figure 3m–o); overlap was more substantial in cells that overexpressed GPNMB (e.g. cell in upper right). Further efforts to identify the majority of the GPNMB-containing structures were unsuccessful. Together, these data indicate that GPNMB, when expressed in HeLa cells, accumulates partially in early endosomes and does not accumulate in late endosomes, and thus is sorted to distinct compartments from PMEL.
Figure 3. GPNMB does not accumulate in late endosomes/ lysosomes in transfected HeLa cells.

HeLa cells were transiently transfected with tagged and non-tagged forms of GPNMB or untagged wild-type PMEL, fixed with 2% formaldehyde, stained with the indicated primary and fluorochrome conjugated secondary antibodies, and analyzed by IFM-D. Wild-type PMEL (a–c), but not GPNMB (d–f), colocalizes with the late endocytic marker CD63. Myc-tagged GPNMB (g–i) does not co-localize with the lysosomal marker LAMP1. C-terminally HA-tagged GPNMB shows minimal co-localization with EEA1 (j–l) and Transferrin (Tfn) that has been internalized for 3 h to label recycling endosomes (m–o). Boxed regions are magnified 6X in the insets at bottom right of each panel; merged images are shown on the right. Bar, 20 μm.
The PKD domains distinguish the distribution of PMEL and GPNMB
PMEL and GPNMB share broad homology throughout their coding region, with 31–42% amino acid sequence identity and 56–67% similarity in the NTR, PKD, “GAP2” and KLD regions (Figure 4a). We (Theos et al., 2006b) and others (Hoashi et al., 2006) had previously shown that the PMEL PKD domain, and possibly the NTR, were required for the selective incorporation of PMEL into ILVs of late endosomes/ lysosomes in HeLa cells. To determine which domain(s) were responsible for the respective trafficking properties of GPNMB and PMEL in HeLa cells, we generated chimeric proteins in which distinct homology domains were swapped between the two proteins (Figure 4b). The chimeric proteins are named with six characters — either P for PMEL or N for GPNMB — for each of the six subdomains within the PMEL protein coding sequence (“-” is used to denote the absent RPT domain in GPNMB). The chimeric proteins were expressed by transient transfection in HeLa cells, and their steady state localization analyzed by IFM-D relative to the late endosome/ lysosome markers, LAMP1 or CD63.
Figure 4. The lumenal NTR and PKD domains of PMEL and GPNMB confer distinct trafficking properties to chimeric constructs.

a. Schematic diagram depicting the indicated homologous subdomains within PMEL and GPNMB. GPNMB lacks a RPT domain, but contains NTR (N-terminal region), PKD (Polycystic Kidney Disease homology domain), KLD (Kringle-like domain), TMD (Transmembrane domain) and Cyt (cytoplasmic domain) regions homologous to those found in PMEL. Arrows indicate cleavage sites for a proprotein convertase (PPC) and site 2 protease (S2P) in PMEL. SS, signal sequence. Numbers in the middle correspond to the sequence identity/ similarity between human PMEL and human GPNMB within the indicated regions. Regions indicated in light green and light blue have been referred to as “GAP2” by Hearing and colleagues (Hoashi et al., 2006); for chimeric proteins, these regions were appended to the RPT and KLD domains, respectively. b. Schematic diagram of PMEL, GPNMB and chimeric proteins analyzed in c–k. The 6 subdomains in PMEL are indicated in black and named with a letter P (i.e., PPPPPP for PMEL), and 5 corresponding domains in GPNMB are indicated in gray and denoted with the letter N (e.g. NN-NNN for GPNMB; the missing RPT domain in GPNMB is denoted with a −). Arrow indicates presence of a PPC cleavage site; where indicated, a myc epitope tag has been inserted into GPNMB where the RPT domain would be in PMEL.. c–k. HeLa cells were transiently transfected with the indicated chimeric proteins, processed as in Figure 3, and analyzed by IFM-D for the transgene (red) and the lysosomal protein LAMP1 (green); a merged image is shown on the right, and insets show the boxed regions that are magnified 4X. Note that substitution of the NTR and PKD domains of PMEL with those of GPNMB (NNmycPPP) causes mislocalization of PMEL, as shown by loss of colocalization with the lysosomal marker LAMP1 (c–e). Conversely, the NTR and PKD domains of PMEL (PP-NNN, f–h), but not the RPT (NNPNNN, i–k), are sufficient to confer late endosome/ lysosome targeting to GPNMB, as shown by co-labeling with LAMP1. Bars, 20 μm.
A myc-epitope-tagged chimeric protein containing the N-terminal NTR and PKD domains of GPNMB and the C-terminal KLD, TMD and CytD of PMEL (NNmycPPP) was detected with anti-myc antibody primarily on the plasma membrane and in punctate structures that largely did not overlap with LAMP-1 or CD63 (Figure 4c–e), much like GPNMB. By contrast, the anti-PMEL PKD-specific antibody NKI-beteb detected the inverse chimeric protein, containing the N-terminal NTR and PKD domains of PMEL and the C-terminal KLD, TMD and CytD of GPNMB (PP-NNN), in puncta that overlapped nearly completely with CD63 (data not shown) and LAMP1 (Figure 4f–h). This indicated that the N-terminal domains of PMEL are both necessary and sufficient to confer localization to late endosomes/ lysosomes. Consistently, insertion of the PMEL RPT domain in place of the myc epitope tag within the hinge region of GPNMB (NNPNNN) was, like intact GPNMB, detected by the PMEL RPT-specific antibody HMB45 in puncta that largely did not overlap with LAMP1 (Figure 4i–k; note that when NNPNNN is overexpressed as in the cell on the left, a portion is detected in lysosomes, but not when it is modestly expressed as in the cell on the right). This confirms that the PMEL RPT domain, which is largely resistant to degradation by virtue of its extensive glycosylation, does not confer localization to late endosomes/ lysosomes, and shows that the lack of detection of intact GPNMB or the myc-tagged chimeras in these compartments does not reflect epitope degradation.
To distinguish whether the NTR or PKD was responsible for late endosomal targeting, we expressed PMEL or GPNMB forms with single domain substitutions in HeLa cells (Figure 5a). A chimera consisting of PMEL with the NTR derived from GPNMB (NPPPPP) and detected with HMB45 was detected in puncta that overlapped completely with a subset of CD63- or LAMP1-containing compartments (Figure 5b–d), whereas a chimera with the PKD derived from GPNMB (PNPPPP) was detected in puncta that did not overlap at all with CD63 (Figure 5e–g). These data suggest that the PKD domain of PMEL, but not the NTR, confers late endosome localization, a conclusion that was confirmed by analysis of the inverse chimeras. GPNMB in which the NTR was replaced by that of PMEL (PN-NNN) was detected by AF2550 in puncta distinct from CD63 (Figure 5h–j), and the myc-tagged GPNMB with the same substitution for the NTR (PNmycNNN) was detected by anti-myc in puncta distinct from LAMP1 (Figure 5k–m). A form of GPNMB in which the PKD was replaced by that of PMEL (NP-NNN) was inefficiently expressed and largely trapped in the ER (data not shown). However, a fraction of cells expressing the chimera showed a punctate pattern; in these cells, the puncta overlapped completely with a subset of LAMP1-containing compartments (Figure 5n–p). This indicates that (1) the PMEL PKD domain is sufficient to confer localization to late endosomes/ lysosomes, and (2) optimal folding of the PMEL PKD domain requires specific interaction with the PMEL NTR.
Figure 5. The PKD domain of PMEL is necessary and sufficient to confer late endosome/ lysosome localization.

Chimeric constructs with individual domain substitutions between PMEL and GPNMB (schematically indicated in panel a) were transiently expressed in HeLa cells, processed as in Figure 3, and analyzed by IFM-D for the transgene (red) and either CD63 or LAMP1 (green) as indicated; merged images are shown on the right, and insets show a 4X magnification of the boxed region. Note that substitution of the PKD domain of PMEL (PNPPPP, e–g), but not of the NTR (NPPPPP, b–d), with that of GPNMB causes loss of PMEL accumulation in late endosomes/ lysosomes. Conversely, substitution of the GPNMB PKD with that of PMEL (NP-NNN, n-p), but not of the NTR (PN-NNN, h–j or PNmycNNN, k–m), confers PMEL-like localization to GPNMB, as determined by colocalization with LAMP1 or CD63. Bars, 20 μm.
The chimeric proteins also provided information regarding epitope reactivity of the PMEL- and GPNMB-specific antibodies. As predicted from earlier work (Harper et al., 2008; Hoashi et al., 2006; Theos et al., 2006b; Yasumoto et al., 2004), the PMEL-specific antibody HMB45 reacted only with chimeras containing the PMEL RPT domain. The PMEL-specific antibodies NKI-beteb and HMB50 (the latter not shown) had been assigned either to the KLD (Hoashi et al.; Yasumoto et al., 2004) or to the PKD domain (Harper et al., 2008; Theos et al., 2006b); its immunoreactivity with NP-NNN but not with PNPPPP proves definitively that it is specific for the PKD domain. The polyclonal anti-GPNMB antibody AF2550 reacted with the chimeras NPPPPP (containing the GPNMB NTR), PN-NNN (containing all of GPNMB except the NTR), and NP-NNN (containing the PMEL PKD), but not with PNPPPP (containing the GPNMB PKD), as detected by immunoblotting of HeLa cells transfected with the respective chimeras (Figure 5 and data not shown). This indicates that AF2550 lacks antibodies that bind to the PKD domain, but likely reacts with multiple other GPNMB subdomains.
The GPNMB PKD domain is heavily modified by glycosylation and functions as a “gnull” PKD for endosomal targeting
While the data above indicate that the PKD domains of PMEL and GPNMB are functionally distinct, it was not clear whether the GPNMB PKD domain had functional targeting information itself. To test this possibility, we compared the localization of the PNPPPP chimera–containing the GPNMB PKD domain—with that of a PKD deletion mutant of PMEL (PMEL ΔPKD) expressed in HeLa cells. As previously shown (Theos et al., 2006b), whereas PMEL localizes to LAMP1-containing compartments that are distinct from recycling endosomes marked by transferrin that had been internalized for 30 min (Figure 6a–c), PMEL ΔPKD is detected in compartments that overlap extensively with internalized transferrin but not LAMP1 (Figure 6g–i). Similarly, PNPPPP also overlaps extensively with internalized transferrin (Figure 6j–l), even though epitope-tagged full-length GPNMB shows only restricted colocalization with internalized transferrin (Figure 6d–f). These data suggest that GPNMB has distinct targeting information elsewhere and that the GPNMB PKD domain does not provide targeting information in the context of PMEL, thus functioning as a “null” in terms of trafficking.
Figure 6. The PKD domain of GPNMB acts as a “gnull” PKD in terms of trafficking information.

HeLa cells transiently transfected with either wild-type PMEL (wt, a–c), GPNMB (d–f), PMEL lacking the PKD (PMEL ΔPKD, g–i), or the PMEL chimera containing the GPNMB PKD (PNPPPP, j–l) were allowed to internalize fluorochrome conjugated transferrin (Tfn) for 30 min to label recycling endosomes, processed as in Figure 3, and analyzed by IFM-D. Individual labels are shown in black and white; the boxed regions are magnified 6X in the insets and merged such that the transgene is in red, Tfn in green and LAMP1 in blue. Note that both PMELΔPKD and PNPPPP, but not wild-type PMEL, partially co-localize with internalized Tfn, indicating redistribution of PMEL to recycling compartments upon loss of the wild-type PMEL PKD. Bar, 20 μm.
The inability of the GPNMB PKD domain to confer targeting information was surprising, given that its sequence is 31% identical and 56% similar to that of the PMEL PKD. Careful inspection of the sequence, however, identifies six consensus N-linked glycosylation sites within the GPNMB PKD (Figure 7a); by contrast, the PMEL PKD domain is devoid of consensus glycosylation sites. To determine whether the GPNMB PKD domain glycosylation sites are used, we compared the mobility of newly synthesized PMEL with that of the PNPPPP chimera before and after treatment with protein N-glycanase F (PNGase F). HeLa cells transiently transfected with PMEL, PMEL ΔPKD, or PNPPPP were pulse labeled for 15 min, and then cell lysates were immunoprecipitated with an antibody to the PMEL cytoplasmic domain (αPmel-C). Immunoprecipitates were mock treated or treated with PNGase F and then fractionated by SDS-PAGE and analyzed by phosphorimaging. As shown in Figure 7b, wild-type PMEL and PMEL ΔPKD migrate with a similar Mr, with PMEL ΔPKD showing a slightly faster mobility due to the deletion of the PKD domain. In contrast, PNPPPP migrated with Mr approximately 15 kDa higher than that of wild-type PMEL, reflecting increased mass of the GPNMB PKD domain corresponding to the equivalent of 5–6 N-glycans. After treatment with PNGase F, the Mr of wild-type PMEL and PNPPPP was reduced such that they were now approximately the same, as expected since the non-glycosylated PKD domains of PMEL and GPNMB are nearly the same size; importantly, whereas the downwards shift in Mr of wild-type PMEL and PMELΔPKD after PNGase F treatment was exactly the same, PNPPPP showed a much larger shift in Mr after treatment, consistent with use of at least five of the six N-glycosylation sites. This indicated that the added mass of the GPNMB PKD domain was entirely due to N-glycosylation and corroborated that the PMEL PKD is not glycosylated. Together, these data suggest that glycosylation of the GPNMB domain renders it incapable of functioning as a late endosome targeting determinant.
Figure 7. Loss of PMEL-like subcellular distribution correlates with glycosylation of the PKD domain.

a. Primary amino acid sequence and alignment of the PKD domains of PMEL (residues 217–295) and GPNMB (residues 270–348). The potential N-glycosylation consensus sites within the PKD domain of GPNMB have been underlined; the PMEL PKD domain has none. b. HeLa cells were transiently transfected with the indicated constructs, metabolically labeled with [35]S-methionine/cysteine for 15 min, and lysed. Lysates were either mock-treated (−) or treated with PNGaseF (+) for 4 h to remove N-linked glycans. Note that in contrast to the small downwards shift in Mr of wild-type PMEL and PMEL-ΔPKD upon PNGaseF treatment, PNPPPP shows a larger (~15 kDa) shift in Mr after treatment, due to the N-glycosylation of the GPNMB-derived PKD.
The GPNMB NTR-PKD domain region has the intrinsic capacity to form amyloid
If N-glycosylation were responsible for inactivating the GPNMB PKD domain, then one would predict that the deglycosylated domain might have similar properties to the PMEL PKD domain. To test this prediction, we exploited the ability of the recombinant PMEL lumenal Mα fragment to generate amyloid fibrils in vitro (Watt et al., 2009). A “Mα” fragment corresponding to the NTR and PKD domains of GPNMB was expressed as a C-terminally His6-tagged fusion protein in E. coli, which cannot N-glycosylate target proteins; the His6-tagged PMEL Mα fragment previously described (Watt et al., 2009) was produced as a control (Figure 8a). Like the corresponding PMEL domain, the GPNMB Mα fragment was recovered completely from the inclusion body fraction of expressing bacteria (data not shown). After solubilization in 6M guanidine and purification via the His6 tag, the recombinant GPNMB-Mα fragment was diluted into renaturing buffer for 24 h, and then assayed for (1) sedimentation at 100,000 x G and (2) binding to the amyloidophilic dye thioflavin T (ThioT). The recombinant de-glycosylated fragment was completely recovered in the pellet fraction after sedimentation, indicating that it had become insoluble in aqueous solution (Figure 8b), and bound ThioT to a similar degree as the PMEL Mα fragment (Figure 8c). These data show that in the absence of glycosylation, the GPNMB NTR-PKD fragment retains the intrinsic capacity to form amyloid in vitro. Given the absence of amyloid-like fibrils in cells expressing GPNMB (data not shown), these data support the conclusion that N-glycosylation of the PKD domain nullifies its function both in protein sorting and amyloid formation.
Figure 8. The unglycosylated recombinant lumenal domain of GPNMB shows amyloid-like properties in vitro.

a. Schematic diagram of the His-tagged PMEL lumenal Mα fragment and the homologous fragment of GPNMB. b–c. Recombinant His-tagged lumenal GPNMB was expressed as in E. coli, isolated and affinity-purified under denaturing conditions from inclusion body fractions, and allowed to refold by dilution into non-denaturing buffer. Refolded protein was subjected to high speed sedimentation (b) or Thioflavin T binding and fluorescence analysis (c). Note that lumenal GPNMB, lacking any glycosylation modifications due to its production in bacteria, shows amyloid-like properties upon refolding in non-denaturing buffer, similar to what is observed for PMEL under similar conditions (Watt et al., 2009).
DISCUSSION
GPNMB and PMEL are highly homologous at the amino acid sequence level, yet their function and subcellular distribution appear to be distinct. Here, we provide evidence that the distinct properties of these two proteins are at least in part mediated by their PKD homology domains. Despite the high sequence similarity between them, the PKD domain of GPNMB fulfills a distinct structural and functional role. Whereas the PMEL PKD domain mediates protein sorting within endosomes to ILVs and accumulates in premelanosome amyloid fibrils, the GPNMB PKD domain lacks protein sorting function and does not lead to accumulation in fibrils, despite an intrinsic capacity to form amyloid fibrils in vitro. The failure of the GPNMB PKD domain to mediate these functions is likely due to extensive modification by N-glycosylation. These results, and the lack of specific accumulation of GPNMB in melanosomes, have important implications for GPNMB function. Moreover, they point to the unique character of the PMEL PKD domain in PMEL function.
GPNMB clearly plays a role in iris pigmentation (Anderson et al., 2006; Anderson et al., 2002; Haraszti et al., 2011) and melanosome biogenesis in skin melanocytes (Zhang et al., 2012), has been identified in melanosome-enriched fractions of MNT-1 cells by proteomics analysis (Chi et al., 2006), and has been interpreted from low resolution IFM analyses and subcellular fractionation of MNT-1 and B16 melanoma to be localized to melanosomes (Hoashi et al., 2010a; Tomihari et al., 2009). Here, using the high resolution afforded by IFM-D, we failed to observe a selective enrichment of the GPNMB lumenal domain within either pigmented stage III/ IV melanosomes or non-pigmented stage II melanosomes in pigmented MNT-1; similar results were observed in a second pigmented melanoma cell line (data not shown). This supports earlier work showing that the quail homologue of GPNMB, QNR-71, did not localize at steady state to melanosomes in pigmented quail neuretinal cells (Le Borgne et al., 2001). Similarly, neither intact GPNMB nor epitope-tagged fusion proteins localized effectively to late endosomes/ lysosomes—the target of most melanosomal proteins–upon ectopic expression in HeLa cells. While a small fraction of the structures to which lumenal GPNMB localized in both cell types overlapped with markers of early endosomes, the majority did not overlap with markers of the ER, ER exit sites, Golgi, early or late stage melanosomes (in MNT-1 cells), late endosomes, or lysosomes, and could not be definitively identified. As discussed below, the majority of the intracellular signal for endogenous GPNMB in MNT-1 cells detected by the lumenal domain-directed AF2550 antibody likely corresponds to the released, soluble lumenal domain, a significant fraction of which is secreted into the medium (Hoashi et al., 2010a) (and our unpublished data). Its distribution is thus likely to be regulated by lumenal protein: protein or protein: lipid interactions, which might be promiscuous and dependent on culture conditions, perhaps explaining the apparent codistribution with melanosomes in prior analyses. Since GPNMB harbors an RGD motif that likely functions in extracellular matrix binding and cell adhesion (Tomihari et al., 2009), it is also possible that GPNMB impacts melanosome integrity in iris pigmented epithelium through cell signaling properties rather than as a melanosome resident itself. Indeed, this is supported by the effects of loss of GPNMB expression on the accumulation of mRNA for melanosomal constituents in melanocytes (Zhang et al., 2012).
Importantly, our data suggest that GPNMB largely accumulates in cells as a lumenal fragment that is not selectively associated with premelanosome fibrils. Antibodies to the GPNMB cytoplasmic domain largely labeled the ER and perinuclear region (likely Golgi), whereas the AF2550 antibody to the lumenal domain labeled largely distinct punctate structures. Like for PMEL (Harper et al., 2008; Kummer et al., 2009; Raposo et al., 2001), this distinction in labeling by the lumenal and cytoplasmic domains likely represents the separation of the GPNMB lumenal domain from the integral membrane CTF by juxtamembrane proteolysis in a post-Golgi compartment, and subsequent γ-secretase-dependent degradation of the remaining integral membrane CTF. This interpretation is supported by the detection of the CTF in cell lysates and its stabilization by γ-secretase inhibitors, analogous to data for PMEL (Kummer et al., 2009) (and our unpublished data). We thus propose that soon after exit from the Golgi, the majority of GPNMB is cleaved by a juxtamembrane “site 2” protease, releasing the large lumenal GPNMB fragment as a soluble protein. Importantly, crude subcellular fractionation studies in melanocytes show that unlike the PMEL lumenal domain, the GPNMB lumenal fragment does not preferentially associate with detergent-insoluble melanosome fibrils, showing similar levels in both detergent soluble and insoluble fractions. Moreover, GPNMB does not associate at all with insoluble fractions in transfected HeLa cells, indicating that the protein does not have intrinsic aggregation properties like PMEL. Thus, despite the high sequence homology, GPNMB has a distinct subcellular and molecular fate from PMEL. The homology likely reflects divergent evolution of a similar structural foundation for a distinct functional purpose.
Using a chimeric protein approach, we show that the distinct localization patterns for PMEL and GPNMB are not related to the PMEL-specific RPT region, but rather to differences between the highly homologous PKD domains of the two proteins. Placement of the PMEL PKD domain within the homologous region of GPNMB resulted in targeting to late endosomes/ lysosomes, whereas placement of the GPNMB PKD domain within the homologous region of PMEL resulted in a loss of selective sorting to lysosomes from early endosomal intermediates and resultant accumulation in recycling compartments. This distinct behavior correlated with the use of at least five of six potential N-linked glycosylation sites within the GPNMB PKD domain, whereas the PMEL PKD domain is devoid of glycosylation. Indeed, the PMEL PKD throughout vertebrate evolution lacks any consensus N-glycosylation sites, whereas N-glycosylation sites are well conserved among GPNMB orthologue PKD domains, with at least three in the amphibian Xenopus tropicalis orthologue (NCBI accession #AAI68052), four each in the Xenopus laevis orthologue (NCBI accession #NP_001089087) and quail orthologue QNR-71 (NCBI accession # CAA63859), and six in all known mammalian orthologues. N-glycosylation of the GPNMB PKD domain might be important for proper protein folding in the endoplasmic reticulum, since prolonged treatment of MNT-1 cells with the N-glycosylation inhibitor, tunicamycin, resulted in accumulation of GPNMB within the ER, whereas under the same conditions PMEL progresses to later stages of the secretory pathway (data not shown). Without N-glycosylation, the recombinant GPNMB NTR-PKD region generated in bacteria—like the homologous region of PMEL (Watt et al., 2009)—can be effectively incorporated into amyloid fibrils, suggesting that the polypeptide sequence of the GPNMB PKD domain per se can function in some ways similar to that of the PMEL PKD domain. However, the recombinant glycosylated GPNMB lumenal domain generated in insect cells is highly soluble (Kuan et al., 2006), suggesting that amyloid formation is not a property of the appropriately modified lumenal domain. Those features of the PMEL PKD domain that are critical for the amyloidogenic and protein sorting properties still need to be identified, but likely lack of glycosylation is critical.
The most distinctive feature of PMEL is its incorporation into functional amyloid for melanin deposition (Fowler et al., 2006). The data shown here suggest that although highly homologous, GPNMB does not subserve a similar function. This is likely a consequence of: (a) a failure to be selectively sorted onto endosomal ILVs, which appear to seed fibril formation (Hurbain et al., 2008); (b) the lack of a RPT domain, which is required for PMEL fibril formation in vivo in HeLa cells (Hoashi et al., 2006; Theos et al., 2006b); (c) the lack of a specific proteolytic cleavage site to separate the potential fibrillogenic fragment from the remaining region of the lumenal domain, which has been shown to be required for PMEL fibril formation (Berson et al., 2003); and (d) the distinct properties of the PKD domains shown here. Whereas we have presented evidence that the PMEL PKD domain is the core of the melanosome functional amyloid fibrils (Watt et al., 2009), others have argued that the RPT domain is the amyloid core based on the acid-dependent formation of fibrils by the purified human PMEL RPT domain over long time periods in vitro (McGlinchey et al., 2009; Pfefferkorn et al., 2011), the conservation of similar amyloidogenic properties by sequence divergent RPT homologues in other vertebrates (McGlinchey et al., 2011), and the lack of evidence for fibril formation by the highly homologous PKD domain of GPNMB. Our evidence here nullifies the latter argument by showing that despite sequence homology, the GPNMB PKD domain cannot form fibrils, likely due to glycosylation. Moreover, the effect of glycosylation on nullifying the intrinsic fibrillogenic properties of this protein provide further evidence against a role for the RPT domain in amyloid core formation, given that this domain is highly modified by O-linked glycans (Harper et al., 2008; Valencia et al., 2007). Leonhardt and colleagues have recently provided supporting evidence that the RPT domain is dispensable for fibril formation in melanocytes and that the PKD domain likely contains the fibril core (Leonhardt et al., 2013). Resolution of this issue will require detailed analyses of the fibrils formed in melanocytic cells and/or reconstitution of fibril formation by properly modified and folded forms of PMEL.
MATERIALS AND METHODS
Antibodies and reagents
Unless otherwise specified, chemicals were obtained from Sigma-Aldrich (St. Louis, MO) and tissue culture reagents were from Invitrogen (Carlsbad, CA). PCR primers were purchased from Thermo Fisher Scientific (Fremont, CA), GoTaq DNA polymerase was from Promega Corp. (Madison, WI), and restriction enzymes and ligase from New England Biolabs (Ipswich, MA). The mouse monoclonal antibodies used, their targets and sources were as follows: HMB45 and NKI-Beteb to PMEL were from Lab Vision (Freemont, CA); H4A3 to LAMP1 was from Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA); anti-calnexin was from Chemicon (Temecula, CA); anti-CD63 was a gift from Meenhard Herlyn (Wistar Inst., Philadelphia, PA); and anti-myc (clone 9E10) was generated from the hybridoma from American Type Culture Collection (Manassas, VA). Goat polyclonal antibody AF2550 to human GPNMB was from R&D Systems (Minneapolis, MN). Rabbit polyclonal antibodies were αPep13h to the C-terminal peptide of hPMEL (Berson et al., 2001) and α-LAMP1 from Affinity BioReagents (Golden, CO). The hNMB-C rabbit antiserum was raised by Genemed Synthesis (San Antonio, TX) against a peptide (residues 543 – 560) mapping to the C-terminus of human GPNMB and conjugated to Keyhole Limpet Hemocyanin, and the antiserum was affinity purified using SulfoLink beads (Pierce, Rockford, IL) coupled to the peptide as described (Berson et al., 2001).
Cells, culture and transfection conditions
The highly pigmented MNT-1 human melanoma cell line was cultured as described (Raposo et al., 2001); HeLa cells were cultured and transiently transfected using Fugene 6 (Roche Diagnostics, Indianapolis, IN) as described (Berson et al., 2001), and analyzed 48 h post-transfection. To inhibit γ-secretase cleavage, cells were treated for 4 h at 37 C with either vehicle (DMSO) or 1μM DAPT (EMD Biosciences, San Diego, CA), harvested with 5mM EDTA in PBS, and frozen prior to analysis.
DNA constructs and cloning
pCI-hGPNMB encodes the cDNA of full-length hGPNMB from residues 1–560 (isoform b, accession number NP_002501), which was obtained by RT-PCR from RNA purified from MNT-1 cells. RNA was isolated using RNeasy Mini (Qiagen, Valencia, CA) and RT-PCR performed according to manufacturers instructions (Promega Corp., Madison, WI). The PMEL-GPNMB chimeras were constructed by site directed mutagenesis of pCI-hPMEL and pCI-GPNMB using PCR to create novel restriction sites, followed by restriction endonuclease digestion and ligation to swap homologous domains. The respective residues included in the chimeras are as follows: NNmycPPP, residues M1-K334 of GPNMB, a myc epitope, and residues T426–V668 of PMEL; PP-NNN, residues M1-T314 of PMEL and residues T336-S560 of GPNMB; NNPPPP, residues M1-K334 of GPNMB and residues A294-V668 of PMEL; NNPNNN, residues M1-P320 of GPNMB followed by residues L298-D474 of PMEL and residues C357-S560 of GPNMB; PN-NNN, residues M1-V213 of PMEL and residues P237-S560 of GPNMB; NP-NNN, residues M1-A224 of GPNMB, residues H202-P297 of PMEL, a myc epitope, and residues G321-S560 of GPNMB; NPPPPP, residues M1-Q235 of GPNMB and residues V213-V668 of PMEL; PNPPPP, residues M1-H202 of PMEL, residues Y226-P320 of GPNMB, and residues L298-V668 of PMEL; PNmycNNN, residues M1-V213 of PMEL and residues P237-S560 of GPNMB with a myc epitope inserted at the junction of residues K334 and T336. All sequences were verified by automated dideoxy sequencing (University of Pennsylvania DNA sequencing facility, Philadelphia, PA), and relevant sequences and restriction maps are available upon request. pCI-GPNMB with an internal myc epitope tag was created by ligation of annealed oligonucleotides corresponding to the myc epitope into a GPNMB cDNA engineered to contain a unique BsiWI restriction site within the proline-rich linker region between the PKD domain and KLD; pCI-GPNMB with a C-terminal HA tag was created by PCR using a mutagenic oligonucleotide primer on the pCI-GPNMB plasmid template. For recombinant His-tagged fusion protein production, the lumenal NTR and PKD domains of GPNMB (residues A22-L349) were subcloned into pET28a(+) (Novagen-EMD Biosciences, Gibbstown, NJ) for C-terminal hexahistidine (His6) tag fusions lacking the N-terminal His6 or T7 tags; the GPNMB sequence is followed by a gly-gly-leu-glu linker upstream of the His6-tag. The PMEL Mα-His6 fragment was constructed similarly, as previously described (Watt et al., 2009).
Immunofluorescence microscopy and image deconvolution
MNT-1 cells were fixed with either ice-cold 100% methanol for 2 min at −20 C or with 2–6% formaldehyde in PBS at room temperature for 20–30 min. HeLa cells were fixed with 2% formaldehyde in PBS at room temperature for 20–30 min. Fixed cells were incubated with primary and fluorochrome-conjugated secondary antibodies, as described (Raposo et al., 2001) and analyzed in a DM IRBE microscope (Leica Microsystems, Wetzlar, Germany) using a 63X Plan Apo lens, 1.4 NA. Digital images in consecutive z-planes were captured at 0.2 μm intervals using an Orca (Hamamatsu, Bridgewater, NJ) or Retiga-SRV (QImaging, Surrey, BC, Canada) digital camera and OpenLab software (Perkin-Elmer, Waltham, MA). Images were deconvolved and manipulated with OpenLab and Adobe Photoshop software. To label recycling compartments with Alexa-dye conjugated human transferrin, cells were starved for 30 min at 37°C with serum free media containing 0.5% BSA, 15mM HEPES, followed by a 30 min or 3 h incubation at 37°C with starvation media containing 7.5 μg/ml Alexa-dye transferrin (Molecular Probes, Eugene, OR), after which cells were fixed as described above.
Immunoblotting
Frozen cell pellets were lysed with TX-100 lysis buffer (150 mM NaCl, 1% (w/v) Triton X-100, 10 mM Tris pH 8.0) containing a protease inhibitor mix (Roche Diagnostics) for 30 min on ice, and centrifuged 20 min at 4°C to obtain detergent-soluble and -insoluble fractions. Detergent insoluble fractions were solubilized with 0.5% SDS, 1% 2-mercaptoethanol and boiled for 10 min, after which cooled lysates were incubated with 0.2 mg/ml DNAse I for 15 min at room temperature to digest nuclear DNA. Detergent-soluble and -insoluble lysates were combined with SDS sample buffer under reducing conditions, boiled, and further fractionated by SDS-PAGE. Proteins were transferred to Immobilon-P membranes (Millipore, Billerica, MA), probed with the indicated primary antibodies, and detected with alkaline phosphatase-conjugated secondary antibodies, enhanced chemifluorescence and imaging using phosphorimaging analysis with a Molecular Dynamics STORM 860 and ImageQuant software (GE Healthcare, Piscataway, NJ).
Metabolic Labeling, Immunoprecipitation, and Endoglycosidase Treatment
Transfected cells were starved, metabolically labeled for 15 min with [35S]-methionine/cysteine, lysed, and immunoprecipitated as described previously (Berson et al., 2000). Immunoprecipitated protein was subjected to treatment with either protein N-glycanase F (PNGaseF; New England Biolabs, Ipswich, MA) or mock treatment for 4 h as described (Harper et al., 2008), and analyzed by SDS-PAGE and Phosphorimaging.
Recombinant Protein Production and analysis
Recombinant protein production, purification, refolding into native buffer, and analysis was performed as described previously (Watt et al., 2009). Briefly, recombinant His6-tagged proteins were solubilized from purified inclusion bodies in 6M guanidine HCl and purified by affinity chromatography in 6M guanidine HCl using HisSelect resin (Sigma). Protein was “refolded” by dilution into buffer lacking chaotropic agent. For sedimentation analysis, refolded protein was subjected to centrifugation at 100,000 × g at 4°C to fractionate soluble and insoluble material, combined with SDS-sample buffer, boiled and separated by SDS-PAGE, followed by Coomassie Blue R-250 staining. For Thioflavin T (ThioT) binding and fluorescence analysis, 2 μM refolded protein was combined with 200 μM ThioT, and fluorescence emission at 490 nm (excitation, 440 nm; cutoff, 475 nm), was measured directly after mixing with a SpectraMax Gemini fluorometer and SoftMax Pro 4.0 software (Molecular Devices, Sunnyvale, CA).
Supplementary Material
SIGNFICANCE.
PMEL and GPNMB are highly homologous proteins that play essential but distinct roles in melanosome biogenesis. This work is important in dissecting the molecular basis for their distinct functions in two ways. First, our data challenge a prevailing view of GPNMB, suggesting that it functions from a subcellular compartment distinct from mature melanosomes. Second, we ascribe the distinct cellular properties of PMEL and GPNMB largely to critical differences in a single subdomain that shares high sequence similarity between PMEL and GPNMB but is extensively post-translationally modified only in GPNMB. These data explain how two homologous melanocyte proteins can have disparate functions.
Acknowledgments
We thank Graça Raposo and Guillaume van Neil for valuable discussions and critical reading of the manuscript. This work was supported by National Institutes of Health grant R01 AR048155 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (to MSM), National Institutes of Health Training Grant T32 GN997229 and Fellowship F31 GM08917 from the NIGMS (to BW), and National Institutes of Health Training Grant 5 T32 CA09140 from the NCI, a Shaffer grant from the Glaucoma Research Foundation and American Health Assistance Foundation grant G2012025 (to ACT).
References
- Adema GJ, de Boer AJ, Vogel AM, Loenen WAM, Figdor CG. Molecular characterization of the melanocyte lineage-specific antigen gp100. J Biol Chem. 1994;269:20126–20133. [PubMed] [Google Scholar]
- Anderson MG, Haraszti T, Petersen GE, Wirick S, Jacobsen C, John SW, Grunze M. Scanning transmission X-ray microscopic analysis of purified melanosomes of the mouse iris. Micron. 2006;37:689–698. doi: 10.1016/j.micron.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Anderson MG, Smith RS, Hawes NL, Zabaleta A, Chang B, Wiggs JL, John SWM. Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nature Genet. 2002;30:81–85. doi: 10.1038/ng794. [DOI] [PubMed] [Google Scholar]
- Bachner D, Schroder D, Gross G. mRNA expression of the murine glycoprotein (transmembrane) nmb (Gpnmb) gene is linked to the developing retinal pigment epithelium and iris. Brain Res Gene Expr Patterns. 2002;1:159–165. doi: 10.1016/s1567-133x(02)00012-1. [DOI] [PubMed] [Google Scholar]
- Berson JF, Frank DW, Calvo PA, Bieler BM, Marks MS. A common temperature-sensitive allelic form of human tyrosinase is retained in the endoplasmic reticulum at the nonpermissive temperature. J Biol Chem. 2000;275:12281–12289. doi: 10.1074/jbc.275.16.12281. [DOI] [PubMed] [Google Scholar]
- Berson JF, Harper D, Tenza D, Raposo G, Marks MS. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol Biol Cell. 2001;12:3451–3464. doi: 10.1091/mbc.12.11.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson JF, Theos AC, Harper DC, Tenza D, Raposo G, Marks MS. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J Cell Biol. 2003;161:521–533. doi: 10.1083/jcb.200302072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chalouni C, Tan C, Clark R, Venook R, Ohri R, Raab H, Firestein R, Mallet W, Polakis P. The melanosomal protein PMEL17 as a target for antibody drug conjugate therapy in melanoma. J Biol Chem. 2012;287:24082–24091. doi: 10.1074/jbc.M112.361485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi A, Valencia JC, Hu ZZ, Watabe H, Yamaguchi H, Mangini NJ, Huang H, Canfield VA, Cheng KC, Yang F, et al. Proteomic and bioinformatic characterization of the biogenesis and function of melanosomes. J Proteome Res. 2006;5:3135–3144. doi: 10.1021/pr060363j. [DOI] [PubMed] [Google Scholar]
- Donatien PD, Orlow SJ. Interaction of melanosomal proteins with melanin. Eur J Biochem. 1995;232:159–164. doi: 10.1111/j.1432-1033.1995.tb20794.x. [DOI] [PubMed] [Google Scholar]
- Escola JM, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, Geuze HJ. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem. 1998;273:20121–20127. doi: 10.1074/jbc.273.32.20121. [DOI] [PubMed] [Google Scholar]
- Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006;4:e6. doi: 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furochi H, Tamura S, Mameoka M, Yamada C, Ogawa T, Hirasaka K, Okumura Y, Imagawa T, Oguri S, Ishidoh K, et al. Osteoactivin fragments produced by ectodomain shedding induce MMP-3 expression via ERK pathway in mouse NIH-3T3 fibroblasts. FEBS Lett. 2007;581:5743–5750. doi: 10.1016/j.febslet.2007.11.036. [DOI] [PubMed] [Google Scholar]
- Haraszti T, Trantow CM, Hedberg-Buenz A, Grunze M, Anderson MG. Spectral analysis by XANES reveals that GPNMB influences the chemical composition of intact melanosomes. Pigment Cell Melanoma Res. 2011;24:187–196. doi: 10.1111/j.1755-148X.2010.00788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper DC, Theos AC, Herman KE, Tenza D, Raposo G, Marks MS. Premelanosome amyloid-like fibrils are composed of only golgi-processed forms of pmel17 that have been proteolytically processed in endosomes. J Biol Chem. 2008;283:2307–2322. doi: 10.1074/jbc.M708007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearing VJ. Biogenesis of pigment granules: a sensitive way to regulate melanocyte function. J Dermatol Sci. 2005;37:3–14. doi: 10.1016/j.jdermsci.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Hoashi T, Muller J, Vieira WD, Rouzaud F, Kikuchi K, Tamaki K, Hearing VJ. The repeat domain of the melanosomal matrix protein Pmel17/gp100 is required for the formation of organellar fibers. J Biol Chem. 2006;281:21198–22208. doi: 10.1074/jbc.M601643200. [DOI] [PubMed] [Google Scholar]
- Hoashi T, Sato S, Yamaguchi Y, Passeron T, Tamaki K, Hearing VJ. Glycoprotein nonmetastatic melanoma protein b, a melanocytic cell marker, is a melanosome-specific and proteolytically released protein. FASEB J. 2010a;24:1616–1629. doi: 10.1096/fj.09-151019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoashi T, Tamaki K, Hearing VJ. The secreted form of a melanocyte membrane-bound glycoprotein (Pmel17/gp100) is released by ectodomain shedding. FASEB J. 2010b;24:916–930. doi: 10.1096/fj.09-140921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek K, Rimm DL, Williams KR, Zhao H, Ariyan S, Lin A, Kluger HM, Berger AJ, Cheng E, Trombetta ES, et al. Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res. 2004;64:5270–5282. doi: 10.1158/0008-5472.CAN-04-0731. [DOI] [PubMed] [Google Scholar]
- Hurbain I, Geerts WJC, Boudier T, Marco S, Verkleij A, Marks MS, Raposo G. Electron tomography of early melanosomes: implications for melanogenesis and the generation of fibrillar amyloid sheets. Proc Natl Acad Sci USA. 2008;105:19726–19731. doi: 10.1073/pnas.0803488105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CT, Wakiya K, Dowell JM, Herndon JE, 2nd, Reardon DA, Graner MW, Riggins GJ, Wikstrand CJ, Bigner DD. Glycoprotein nonmetastatic melanoma protein B, a potential molecular therapeutic target in patients with glioblastoma multiforme. Clin Cancer Res. 2006;12:1970–1982. doi: 10.1158/1078-0432.CCR-05-2797. [DOI] [PubMed] [Google Scholar]
- Kummer MP, Maruyama H, Huelsmann C, Baches S, Weggen S, Koo EH. Formation of Pmel17 amyloid is regulated by juxtamembrane metalloproteinase cleavage, and the resulting C-terminal fragment is a substrate for gamma-secretase. J Biol Chem. 2009;284:2296–2306. doi: 10.1074/jbc.M808904200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushimoto T, Basrur V, Valencia J, Matsunaga J, Vieira WD, Ferrans VJ, Muller J, Appella E, Hearing VJ. A model for melanosome biogenesis based on the purification and analysis of early melanosomes. Proc Natl Acad Sci USA. 2001;98:10698–10703. doi: 10.1073/pnas.191184798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon BS, Halaban R, Kim GS, Usack L, Pomerantz S, Haq AK. A melanocyte-specific complementary DNA clone whose expression is inducible by melanotropin and isobutylmethyl xanthine. Mol Biol Med. 1987;4:339–355. [PubMed] [Google Scholar]
- Le Borgne R, Planque N, Martin P, Dewitte F, Saule S, Hoflack B. The AP-3-dependent targeting of the melanosomal glycoprotein QNR-71 requires a di-leucine-based sorting signal. J Cell Sci. 2001;114:2831–2841. doi: 10.1242/jcs.114.15.2831. [DOI] [PubMed] [Google Scholar]
- Leonhardt RM, Vigneron N, Hee JS, Graham M, Cresswell P. Critical residues in the PMEL/Pmel17 N-terminus direct the hierarchical assembly of melanosomal fibrils. Mol Biol Cell. 2013 doi: 10.1091/mbc.E12-10-0742. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt RM, Vigneron N, Rahner C, Cresswell P. Proprotein convertases process Pmel17 during secretion. J Biol Chem. 2011;286:9321–9337. doi: 10.1074/jbc.M110.168088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt RM, Vigneron N, Rahner C, Van den Eynde BJ, Cresswell P. Endoplasmic reticulum (ER)-export, subcellular distribution and fibril formation by PMEL17 requires an intact N-terminal domain junction. J Biol Chem. 2010;285:16166–16683. doi: 10.1074/jbc.M109.097725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepage S, Lapointe R. Melanosomal targeting sequences from gp100 are essential for MHC class II–restricted endogenous epitope presentation and mobilization to endosomal compartments. Cancer Res. 2006;66:2423–2432. doi: 10.1158/0008-5472.CAN-05-2516. [DOI] [PubMed] [Google Scholar]
- Loftus SK, Antonellis A, Matera I, Renaud G, Baxter LL, Reid D, Wolfsberg TG, Chen Y, Wang C, Program NCS, et al. Gpnmb is a melanoblast-expressed, MITF-dependent gene. Pigment Cell Melanoma Res. 2009;22:99–110. doi: 10.1111/j.1755-148X.2008.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresh GA, Marken JS, Neubauer M, Aruffo A, Hellström I, Hellström KE, Marquardt H. Cloning and expression of the gene for the melanoma-associated ME20 antigen. DNA and Cell Biology. 1994a;13:87–95. doi: 10.1089/dna.1994.13.87. [DOI] [PubMed] [Google Scholar]
- Maresh GA, Wang WC, Beam KS, Malacko AR, Hellström I, Hellström KE, Marquardt H. Differential processing and secretion of the melanoma-associated ME20 antigen. Arch Biochem Biophys. 1994b;311:95–102. doi: 10.1006/abbi.1994.1213. [DOI] [PubMed] [Google Scholar]
- McGlinchey RP, Shewmaker F, Hu KN, McPhie P, Tycko R, Wickner RB. The repeat domains of melanosome matrix protein Pmel17 orthologs form amyloid fibrils at the acidic melanosomal pH. J Biol Chem. 2011;286:8385–8393. doi: 10.1074/jbc.M110.197152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlinchey RP, Shewmaker F, McPhie P, Monterroso B, Thurber K, Wickner RB. The repeat domain of the melanosome fibril protein Pmel17 forms the amyloid core promoting melanin synthesis. Proc Natl Acad Sci USA. 2009;106:13731–13736. doi: 10.1073/pnas.0906509106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikoff AB, Albala A, Biempica L. Ultrastructural and cytochemical observations on B-16 and Harding-Passey mouse melanomas. The origin of premelanosomes and compound melanosomes. J Histochem Cytochem. 1968;16:299–319. doi: 10.1177/16.5.299. [DOI] [PubMed] [Google Scholar]
- Orlow SJ, Zhou BK, Boissy RE, Pifko-Hirst S. Identification of a mammalian melanosomal matrix glycoprotein. J Invest Dermatol. 1993;101:141–144. doi: 10.1111/1523-1747.ep12363626. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn CM, McGlinchey RP, Lee JC. Effects of pH on aggregation kinetics of the repeat domain of a functional amyloid, Pmel17. Proc Natl Acad Sci USA. 2011 doi: 10.1073/pnas.1006424107. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Marks MS. Melanosomes - dark organelles enlighten endosomal membrane transport. Nat Rev Mol Cell Biol. 2007;8:786–798. doi: 10.1038/nrm2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Tenza D, Murphy DM, Berson JF, Marks MS. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J Cell Biol. 2001;152:809–823. doi: 10.1083/jcb.152.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich JN, Shi Q, Hjelmeland M, Cummings TJ, Kuan CT, Bigner DD, Counter CM, Wang XF. Bone-related genes expressed in advanced malignancies induce invasion and metastasis in a genetically defined human cancer model. J Biol Chem. 2003;278:15951–15957. doi: 10.1074/jbc.M211498200. [DOI] [PubMed] [Google Scholar]
- Ripoll VM, Meadows NA, Raggatt LJ, Chang MK, Pettit AR, Cassady AI, Hume DA. Microphthalmia transcription factor regulates the expression of the novel osteoclast factor GPNMB. Gene. 2008;413:32–41. doi: 10.1016/j.gene.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Robila V, Ostankovitch M, Altrich-Vanlith ML, Theos AC, Drover S, Marks MS, Restifo N, Engelhard VH. MHC class II presentation of gp100 epitopes in melanoma cells requires the function of conventional endosomes and is influenced by melanosomes. J Immunol. 2008;181:7843–7852. doi: 10.4049/jimmunol.181.11.7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AA, Annis MG, Dong Z, Pepin F, Hallett M, Park M, Siegel PM. ADAM10 releases a soluble form of the GPNMB/Osteoactivin extracellular domain with angiogenic properties. PLoS One. 2010;5:e12093. doi: 10.1371/journal.pone.0012093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AA, Siegel PM. Emerging therapeutic targets in breast cancer bone metastasis. Future Oncol. 2010;6:55–74. doi: 10.2217/fon.09.138. [DOI] [PubMed] [Google Scholar]
- Rosemblat S, Durham-Pierre D, Gardner JM, Nakatsu Y, Brilliant MH, Orlow SJ. Identification of a melanosomal membrane protein encoded by the pink-eyed dilution (type II oculocutaneous albinism) gene. Proc Natl Acad Sci USA. 1994;91:12071–12075. doi: 10.1073/pnas.91.25.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiji M, Fitzpatrick TM, Simpson RT, Birbeck MSC. Chemical composition and terminology of specialized organelles (melanosomes and melanin granules) in mammalian melanocytes. Nature. 1963;197:1082–1084. doi: 10.1038/1971082a0. [DOI] [PubMed] [Google Scholar]
- Seiji M, Iwashia S. Intracellular localization of tyrosinase and site of melanin formation in melanocyte. J Invest Dermatol. 1965;45:305–314. doi: 10.1038/jid.1965.135. [DOI] [PubMed] [Google Scholar]
- Setty SRG, Tenza D, Sviderskaya EV, Bennett DC, Raposo G, Marks MS. Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature. 2008;454:1142–1146. doi: 10.1038/nature07163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitaram A, Marks MS. Mechanisms of protein delivery to melanosomes in pigment cells. Physiology. 2012;27:85–99. doi: 10.1152/physiol.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitaram A, Piccirillo R, Palmisano I, Harper DC, Dell’Angelica EC, Schiaffino MV, Marks MS. Localization to mature melanosomes by virtue of cytoplasmic dileucine motifs is required for human OCA2 function. Mol Biol Cell. 2009;20:1464–1477. doi: 10.1091/mbc.E08-07-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theos AC, Berson JF, Theos SC, Herman KE, Harper DC, Tenza D, Sviderskaya EV, Lamoreux ML, Bennett DC, Raposo G, et al. Dual loss of ER export and endocytic signals with altered melanosome morphology in the silver mutation of Pmel17. Mol Biol Cell. 2006a;17:3598–3612. doi: 10.1091/mbc.E06-01-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theos AC, Tenza D, Martina JA, Hurbain I, Peden AA, Sviderskaya EV, Stewart A, Robinson MS, Bennett DC, Cutler DF, et al. Functions of AP-3 and AP-1 in tyrosinase sorting from endosomes to melanosomes. Mol Biol Cell. 2005a;16:5356–5372. doi: 10.1091/mbc.E05-07-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theos AC, Truschel ST, Raposo G, Marks MS. The Silver locus product Pmel17/ gp100/ Silv/ ME20: Controversial in name and in function. Pigment Cell Res. 2005b;18:322–336. doi: 10.1111/j.1600-0749.2005.00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theos AC, Truschel ST, Tenza D, Hurbain I, Harper DC, Berson JF, Thomas PC, Raposo G, Marks MS. A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev Cell. 2006b;10:343–354. doi: 10.1016/j.devcel.2006.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomihari M, Hwang SH, Chung JS, Cruz PD, Jr, Ariizumi K. Gpnmb is a melanosome-associated glycoprotein that contributes to melanocyte/keratinocyte adhesion in a RGD-dependent fashion. Exp Dermatol. 2009;18:586–595. doi: 10.1111/j.1600-0625.2008.00830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse KF, Jeffers M, Pollack VA, McCabe DA, Shadish ML, Khramtsov NV, Hackett CS, Shenoy SG, Kuang B, Boldog FL, et al. CR011, a fully human monoclonal antibody-auristatin E conjugate, for the treatment of melanoma. Clin Cancer Res. 2006;12:1373–1382. doi: 10.1158/1078-0432.CCR-05-2018. [DOI] [PubMed] [Google Scholar]
- Turque N, Denhez F, Martin P, Planque N, Bailly M, Begue A, Stehelin D, Saule S. Characterization of a new melanocyte-specific gene (QNR-71) expressed in v-myc-transformed quail neuroretina. EMBO J. 1996;15:3338–3350. [PMC free article] [PubMed] [Google Scholar]
- Valencia JC, Rouzaud F, Julien S, Chen KG, Passeron T, Yamaguchi Y, Abu-Asab M, Tsokos M, Costin GE, Yamaguchi H, et al. Sialylated core 1 O-glycans influence the sorting of Pmel17/gp100 and determine its capacity to form fibrils. J Biol Chem. 2007;282:11266–11280. doi: 10.1074/jbc.M608449200. [DOI] [PubMed] [Google Scholar]
- van Niel G, Charrin S, Simoes S, Romao M, Rochin L, Saftig P, Marks MS, Rubinstein E, Raposo G. The Tetraspanin CD63 integrates ESCRT-independent and dependent sorting at a common endosome. Dev Cell. 2011;21:708–721. doi: 10.1016/j.devcel.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt B, Raposo G, Marks MS. Pmel17: An amyloid determinant of organelle structure. In: Rigacci S, Bucciantini M, editors. Functional Amyloid Aggregation. Trivandrum, Kerala, India: Research Signpost; 2010. pp. 89–113. [Google Scholar]
- Watt B, van Niel G, Fowler DM, Hurbain I, Luk KC, Stayrook SE, Lemmon MA, Raposo G, Shorter J, Kelly JW, et al. N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J Biol Chem. 2009;284:35543–35555. doi: 10.1074/jbc.M109.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt B, van Niel G, Raposo G, Marks MS. PMEL: A pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res. 2013 doi: 10.1111/pcmr.12067. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei A-H, Li W. Hermansky-Pudlak syndrome: Pigmentary and non-pigmentary defects and their pathogenesis. Pigment Cell Melanoma Res. 2013 doi: 10.1111/pcmr.12051. In press. [DOI] [PubMed] [Google Scholar]
- Wei ML. Hermansky–Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- Weterman MA, Ajubi N, van Dinter IM, Degen WG, van Muijen GN, Ruitter DJ, Bloemers HP. nmb, a novel gene, is expressed in low-metastatic human melanoma cell lines and xenografts. Int J Cancer. 1995;60:73–81. doi: 10.1002/ijc.2910600111. [DOI] [PubMed] [Google Scholar]
- Williams MD, Esmaeli B, Soheili A, Simantov R, Gombos DS, Bedikian AY, Hwu P. GPNMB expression in uveal melanoma: a potential for targeted therapy. Melanoma Res. 2010;20:184–190. doi: 10.1097/CMR.0b013e3283364a08. [DOI] [PubMed] [Google Scholar]
- Yasumoto K, Watabe H, Valencia JC, Kushimoto T, Kobayashi T, Appella E, Hearing VJ. Epitope mapping of the melanosomal matrix protein gp100 (PMEL17): rapid processing in the endoplasmic reticulum and glycosylation in the early Golgi compartment. J Biol Chem. 2004;279:28330–28338. doi: 10.1074/jbc.M401269200. [DOI] [PubMed] [Google Scholar]
- Zhang P, Liu W, Zhu C, Yuan X, Li D, Gu W, Ma H, Xie X, Gao T. Silencing of GPNMB by siRNA inhibits the formation of melanosomes in melanocytes in a MITF-independent fashion. PLoS ONE. 2012;7:e42955. doi: 10.1371/journal.pone.0042955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BK, Kobayashi T, Donatien PD, Bennett DC, Hearing VJ, Orlow SJ. Identification of a melanosomal matrix protein encoded by the murine si (silver) locus using “organelle scanning”. Proc Natl Acad Sci USA. 1994;91:7076–7080. doi: 10.1073/pnas.91.15.7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.