

Abstract
The intestinal mucus layer protects the epithelium from noxious agents, viruses, and pathogenic bacteria present in the gastrointestinal tract. It is composed of mucins, predominantly mucin-2 (Muc2), secreted by goblet cells of the intestine. Experimental alcoholic liver disease requires translocation of bacterial products across the intestinal barrier into the systemic circulation, which induces an inflammatory response in the liver and contributes to steatohepatitis. We investigated the roles of the intestinal mucus layer, and in particular Muc2, in development of experimental alcohol-associated liver disease in mice. We studied experimental alcohol-induced liver disease, induced by the Tsukamoto-French method (which involves continuous intragastric feeding of an isocaloric diet or alcohol) in wild-type and Muc2−/− mice. Muc2−/− mice showed less alcohol-induced liver injury and steatosis that developed in wild-type mice. Most notably, Muc2−/− mice had significantly lower plasma levels of lipopolysaccharide than wild-type mice after alcohol feeding. In contrast to wild-type mice, Muc2−/− mice were protected from alcohol-associated microbiome changes that are dependent on intestinal mucins. The anti-microbial proteins Reg3b and Reg3g were expressed at significantly higher levels in the jejunum of Muc2−/− mice fed the isocaloric diet or alcohol, compared with wild-type mice. Consequently, Muc2−/− mice showed increased killing of commensal bacteria and prevented intestinal bacterial overgrowth. Conclusion: Muc2−/− mice are protected from intestinal bacterial overgrowth and dysbiosis in response to alcohol feeding. Subsequently, lower amounts of bacterial products such as endotoxin translocate into the systemic circulation, decreasing liver disease.
Keywords: microbiome, intestinal bacterial overgrowth, bacterial translocation, endotoxin, Reg3
Introduction
Liver cirrhosis is the 12th leading cause of death in the United States, and 48% of all cirrhotic deaths are alcohol-related (1). Alcoholic liver disease comprises hepatic steatosis, which may progress to alcoholic hepatitis, fibrosis, and cirrhosis (2). There is strong evidence for a gut-liver axis that is causatively related to alcohol-induced liver disease, both in patients and experimental animal models. Gastrointestinal permeability is greater in alcoholics compared to normal subjects (3, 4). Several animal studies demonstrated that ethanol disrupts the intestinal epithelial barrier function via a direct effect of ethanol and/or its metabolite acetaldehyde (5). Ethanol-induced gut leakiness results in elevated plasma levels of lipopolysaccharide (LPS) or endotoxin, a major component of the Gram-negative bacterial outer-membrane, and subsequent liver injury (6–8). Endotoxemia is more prevalent in patients with alcoholic liver disease as compared to normal subjects, and plasma endotoxin levels correlate with the severity of liver damage in patients with alcoholic hepatitis (9–12). The most convincing evidence for a role of gut-derived endotoxin comes from mice harboring a genetic deletion in the LPS signaling pathway. Mice deficient in Toll-like receptor 4 (TLR4) as the cellular LPS receptor, CD14 as the cellular co-receptor for LPS, or intracellular signaling molecules downstream of the LPS receptor are resistant to alcohol-induced liver injury (13–15). In addition, selective intestinal decontamination with non-absorbable antibiotics reduces plasma endotoxin levels and prevents experimental alcoholic liver disease (16–18). Although not an established therapy, treatment with antibiotics also improves liver function in patients with alcoholic cirrhosis (19).
The intestinal mucus layer forms a physical barrier between the underlying epithelium and the lumen of the gastrointestinal tract, and protects the epithelium against noxious agents, viruses, and pathogenic bacteria. It consists of two separate sublayers: the inner layer is attached to the epithelial cell layer and is devoid of bacteria; the outer layer can be washed off easily and is colonized by bacteria (20, 21). The intestinal mucus layer is composed of mucins that are synthesized and secreted by intestinal goblet cells (22). Two different types of mucins exist: secreted, or gel-forming mucins, and membrane-bound mucins. There are three gastrointestinal secreted mucins (Muc2, Muc5AC and Muc6) which are characteristically large, heavily O-glycosylated glycoproteins assembled into oligomers that contribute to the viscous properties of intestinal mucus layer (23). The intestinal membrane-bound mucins (Muc1, Muc3-4, Muc12-13 and Muc17) protect against pathogens that penetrate the inner mucus layer (24). The major and most abundant secreted mucin in the small and large intestine is mucin-2 (25). Mice deficient in Muc2 are prone to colorectal cancer and appear to have a disrupted epithelial homeostasis (25). Specific clinical symptoms such as spontaneous colitis depend on their genetic mouse strain background (26).
It has been reported that the intestinal mucus increases after alcohol feeding in rats (27). However, there are currently no patient data or experimental studies assessing the functional contribution of the intestinal mucus layer in alcoholic liver disease. We therefore took an unbiased approach to study the role of the intestinal mucus layer, and in particular of mucin-2 using a mouse model of alcoholic liver injury and steatosis.
Materials and Methods
Animal models of alcohol feeding
Male wild-type mice (C57BL/6J) were purchased from Jackson Laboratory or bred in the vivarium associated with our laboratory. Male Muc2−/− mice (back crossed to C57BL/6J for more than 10 generations) were kindly provided by Dr. Anna Velcich (Albert Einstein College of Medicine, Yeshiva University, New York). Age matched mice were used for this study. All animals received humane care in compliance with institutional guidelines. The intragastric feeding model of continuous ethanol infusion in mice has been described (28).
The Lieber DeCarli diet model of alcohol feeding for two weeks was used to determine intestinal permeability and for an in vivo luminal killing assay. We opted to assess intestinal permeability in a complementary and non-invasive mouse model of alcoholic steatohepatitis using the Lieber DeCarli diet, as prior surgery and the implanted gastrostomy catheter could affect accurate assessment of intestinal permeability. To avoid two surgeries in the same mouse and to comply with institutional IACUC requirements, we also chose to assess in vivo luminal killing of bacteria in mice that were fed the Lieber DeCarli diet.
Other materials and methods are described in the Supplementary Materials and Methods section.
Results
Alcohol abuse increases the thickness of the intestinal mucus layer in humans
It has been reported that chronic alcohol feeding increases the total mucus content in the small intestine in rats (27). We have confirmed these data in humans. Alcoholics show a significant increase in the thickness of the mucus layer on duodenal biopsies as compared to healthy humans (Fig. 1A,B).
Figure 1. Ethanol increases the intestinal mucus layer.
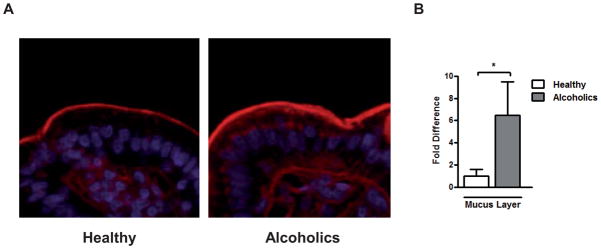
The small intestinal mucus layer was determined on duodenal biopsies obtained from healthy controls (n=12) and patients with chronic alcohol abuse (n=8) using wheat germ agglutinin staining. (A) Representative intestinal sections are shown (magnification x200). (B) Densitometry of sections was performed. *p < 0.05.
Mucin-2 deficient mice have decreased alcoholic steatohepatitis
To investigate the role of the intestinal mucus layer in experimental alcoholic liver disease, we used mice harboring a genetic deletion in the mucin-2 gene (25). Mucin-2 is the most abundant secreted mucin in the gastrointestinal tract (25) and its absence results in a significantly thinner mucus layer in mice as shown by Periodic Acid/Schiff (PAS) staining of the small intestine (Fig. 4A). To confirm that Muc2 expression is largely restricted to the intestine, we measured Muc2 mRNA levels in several organs from wild type mice. Muc2 gene expression was highest in the small and large intestine, but it was undetectable in the liver or bone marrow-derived cells (Suppl. Fig. 1A). These findings were confirmed by immunofluorescent staining. Muc2 protein was abundantly expressed in the small intestine (Suppl. Fig. 1B, left panel), but undetectable in the liver of wild type mice (Suppl. Fig. 1B, right panel). Small intestine from Muc2 deficient mice served as negative staining control (Suppl. Fig. 1B, middle panel).
Figure 4. Expression of intestinal mucins.

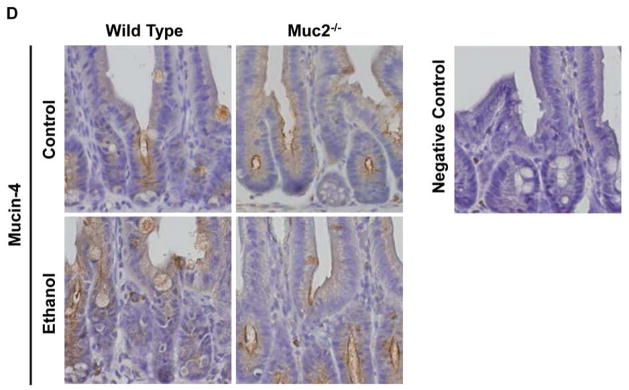
Wild type or Muc2−/− mice were fed an intragastric isocaloric diet (n=4–6) or alcohol (n=9–10) for 1 week. (A) PAS staining of jejunal sections. Representative sections are displayed. (B) Gene expression of membrane-bound mucins Muc1 and Muc4, and of secreted mucin Muc6 in jejunum of mice. (C) Immunohistochemical detection of mucin-1 in the jejunum of mice following alcohol or control feeding for 1 week. The negative control was performed using isotype IgG instead of the primary antibody. The positive control is small intestine from a 2 week old male C57BL/6J mouse (47). Representative sections are shown. (D) Immunohistochemical detection of mucin-4 in the jejunum of mice following alcohol or control feeding for 1 week. The negative control was performed without primary antibody. Representative sections are shown.
We therefore subjected wild type and Muc2−/− mice to the intragastric feeding model of continuous ethanol infusion for one week. Mice fed an isocaloric diet served as controls. Administration of ethanol lead to a comparable increase of liver weight to body weight ratio (Suppl. Fig. 2A). Plasma Alanine Aminotransferase (ALT) levels as measures for liver injury were significantly lower in alcohol-fed Muc2−/− mice as compared to wild type mice (Fig. 2A). Micro- and macrovesicular steatosis occurred after one week following alcohol administration as compared to wild type mice receiving an isocaloric diet. Hepatic fat accumulation was markedly lower in Muc2−/− mice as compared to wild type mice following one week of continuous intragastric ethanol feeding (Fig. 2B). This was confirmed by lower hepatic triglycerides in Muc2−/− mice after alcohol administration (Fig. 2C). Plasma triglyceride levels were similar between wild type and Muc2−/− mice fed an isocaloric and alcohol diet intragastrically for one week (Suppl. Fig. 2B) suggesting no difference in intestinal lipid absorption. Hepatic oxidative stress was also significantly lower in Muc2−/− mice compared to wild type mice following one week of intragastric alcohol feeding, as supported by Thiobarbituric Acid Reactive Substances (TBARS) assay (Fig. 2D) and by staining for 4-hydroxynonenal (4-HNE) (Fig. 2E). Thus, mucin-2 deficiency, and hence a thinner intestinal mucus layer, ameliorates experimental alcohol-induced steatohepatitis.
Figure 2. Mucin-2 deficiency ameliorates experimental alcoholic liver injury and steatosis.

Wild type or Muc2−/− mice were fed an intragastric isocaloric diet (n=5–6) or alcohol (n=9–14) for 1 week. (A) Plasma ALT levels. (B) Representative liver sections after hematoxylin-eosin staining. (C) Hepatic triglyceride levels. (D) Hepatic oxidative stress levels. (E) Staining for 4-hydroxynonenal. *p < 0.05.
Alcohol metabolism and expression of intestinal mucins after ethanol feeding in mucin-2 deficient mice
To explain the different hepatic phenotype, we investigated whether mucin-2 deficiency affects the intestinal absorption or hepatic metabolism of alcohol. Plasma alcohol levels were found to be comparable in wild type and Muc2−/− mice following one week of intragastric alcohol feeding (Fig. 3A). Alcohol dehydrogenase (Adh) and cytochrome p450 enzyme 2E1 (Cyp2E1) are the two main hepatic enzymes to metabolize alcohol and to convert alcohol to acetaldehyde (29). Microsomal Cyp2E1 protein was similarly upregulated in the ethanol-treated groups (Fig. 3B). Despite higher hepatic Adh activity in Muc2−/− mice as compared to wild type mice after intragastric administration of an isocaloric diet that was not observed after ethanol administration (Fig. 3C), plasma acetaldehyde levels were not different following one week of intragastric alcohol feeding (Fig. 3D). To investigate whether the absence of mucin-2 results in a compensatory upregulation of other intestinal mucins after ethanol administration, intestinal gene and protein expression of several mucins was assessed. Deficiency in Muc2 did not result in a compensatory increase in the thickness of the intestinal mucus layer following intragastric alcohol feeding (Fig. 4A). There was no significant difference in the gene expression of secreted mucin Muc6 or of membrane-bound mucins (such as Muc1 and Muc4) in Muc2−/− mice relative to wild type mice after one week of intragastric feeding of ethanol (Fig. 4B). These findings were confirmed using immunohistochemistry for mucin-1 and mucin-4 in small intestinal sections of wild type and Muc2−/− mice fed an intragastric isocaloric or alcohol diet (Fig. 4C and D).
Figure 3. Mucin-2 deficient mice exhibit a similar alcohol metabolism as compared to wild type mice after ethanol feeding.

Wild type or Muc2−/− mice were fed an intragastric isocaloric diet (n=2–5) or alcohol (n=7–12) for 1 week. (A) Plasma ethanol levels. (B) Western blot for Cyp2E1 protein in liver microsomes; VDAC1 is shown as loading control for liver microsomes. (C) Hepatic Adh activity. (D) Plasma acetaldehyde levels. Adh, alcohol dehydrogenase; Cyp2E1, cytochrome p450 enzyme 2E1; VDAC1, Voltage-dependent anion-selective channel protein 1. *p < 0.05.
Mucin-2 deficient mice exhibit lower plasma LPS levels and are protected from microbiome changes after alcohol feeding
Alcoholic steatohepatitis is dependent on endotoxin derived from intestinal bacteria (2, 30). Since mucin-2 is expressed in the intestine but not the liver, we next investigated whether translocation of bacterial products from the intestine to the systemic circulation is affected by the absence of mucin-2. Indeed, systemic endotoxin levels were significantly lower in Muc2−/− mice that were fed an isocaloric diet and alcohol intragastrically for one week as compared to wild type mice (Fig. 5A). Altered intestinal permeability or a quantitative decrease of the intestinal microflora might allow less endotoxin to escape from the gut into the systemic circulation. We therefore assessed intestinal permeability by measuring fecal albumin following a Lieber DeCarli diet for two weeks (31). Fecal albumin was higher in Muc2 deficient mice at baseline and after alcohol feeding indicative of increased intestinal permeability (Fig. 5B). To confirm our findings and to directly assess intestinal permeability we used an in vivo method by measuring recovery of ingested dextran labeled with fluorescein isothiocyanate (FITC). Isocaloric Lieber DeCarli diet or alcohol feeding for two weeks resulted in a significant increase of fluorescence in the plasma of Muc2−/− mice as compared to wild type mice indicative of increased intestinal permeability (Fig. 5C). Thus, despite a leakier gut barrier, Muc2−/− mice showed lower translocation of bacterial products.
Figure 5. Lower plasma LPS levels and no intestinal bacterial overgrowth are found in mucin-2 deficient mice after alcohol feeding.

Wild type or Muc2−/− mice were fed an intragastric isocaloric diet (n=4–7) or alcohol (n=9–10) for 1 week (A, D, E, F – middle panel) or 3 weeks (F –left panel; n=3 for isocaloric diet and n=5 for alcohol). Mice were orally fed a Lieber DeCarli isocaloric diet (n=3–8) or alcohol (n=4–6) for 2 weeks (B and C). (A) Plasma endotoxin levels. (B) Fecal albumin content. (C) FITC-dextran in plasma 4 hrs after gavage. (D, E, F – middle panel) Total intestinal bacteria, Lactobacillus and Akkermansia muciniphila in cecum of mice. (F – left panel) Akkermansia muciniphila in the small intestine of mice. (F – right panel) Akkermansia muciniphila was cultured in vitro in Brain Heart Infusion (BHI) medium with or without mucus in the absence or presence of 10 or 100mM ethanol for 24 hrs. OD600 was measured. Shown is the mean of three independent experiments (n=4–6). *p < 0.05.
Only a minority of the enteric bacteria can be cultured by conventional culture techniques (32). To assess quantitative changes in the intestinal microbiome, the total bacterial load was measured by qPCR using universal 16S rRNA bacterial primer sets. As reported by us (28), intragastric ethanol feeding induced intestinal bacterial overgrowth in wild type mice as compared to isocaloric diet fed wild type mice (Fig. 5D). Interestingly, Muc2−/− mice are protected from intestinal bacterial overgrowth after alcohol feeding (Fig. 5D).
We have also shown that alcohol-associated changes in the enteric microbiome are characterized by a significant suppression of the commensal probiotic microflora including Lactobacillus (28). We have confirmed a significant reduction of Lactobacillus in wild type mice following intragastric ethanol feeding for one week as compared to control animals (Fig. 5E). Muc2−/− mice are not only protected from a suppression of Lactobacillus, they actually demonstrate higher numbers of Lactobacillus after alcohol feeding as compared to control fed Muc2−/− mice (Fig. 5E). In addition, we have previously shown and confirmed that chronic intragastric alcohol feeding for 3 weeks results in an increase of Gram-negative (33) Akkermansia muciniphila (Fig. 5F – left panel) (28). Although no significant change was observed in wild type mice following one week of intragastric alcohol feeding as compared to isocaloric diet feeding, Akkermansia muciniphila was significantly lower in Muc2−/− mice as compared to wild type mice after alcohol feeding (Fig. 5F – middle panel). Growth of Akkermansia muciniphila is dependent on the presence of mucus in vitro, but not ethanol (Fig. 5F – right panel). Thus, the absence of Muc2 results in dysbiosis characterized by a decrease in Gram-negative Akkermansia muciniphila that likely contributes to lower systemic levels of endotoxin. Littermate and non-littermate wild type mice did not show significant differences at baseline in ALT, intestinal permeability, intestinal bacterial burden, the quantity of the two major intestinal bacterial phyla, Bacteroidetes and Firmicutes, and Lactobacillus (Suppl. Fig. 3). Taken together, Muc2−/− mice are protected from alcohol-associated quantitative and qualitative changes in the microbiome and have lower plasma levels of LPS.
Antimicrobial protein expression and activity are enhanced in the intestine of mucin-2 deficient mice
Several factors control the bacterial load of intestine including host antimicrobial molecules that are secreted by epithelial cells and Paneth cells. We have previously reported that the expression of regenerating islet-derived 3 beta (Reg3b) and gamma (Reg3g) are reduced in the small intestine of mice fed alcohol as compared to control mice (28). The inhibition was pronounced in the proximal small intestine, the site with the largest relative increase in luminal bacteria and with highest intraluminal alcohol concentrations (28). We confirmed alcohol-induced inhibition of Reg3b and Reg3g protein expression in the jejunum of wild type mice (Fig. 6A and C). Strikingly, Reg3b and Reg3g expression was much higher in Muc2−/− mice receiving an isocaloric diet or alcohol via an intragastric feeding tube for one week as compared to wild type mice (Fig. 6A and C). Other antimicrobial molecules such as cathelicidin antimicrobial peptide (Camp) or defensin beta 1 (Defb1) show similar responses to intragastric alcohol in wild type and Muc2−/− mice (Fig. 6B). IL-22 is required for the induction of intestinal Reg3b and Reg3g expression (34). IL-22 gene expression showed a trend to be higher expressed in the small intestine of isocaloric and ethanol-fed Muc2−/− mice as compared to wild type mice (Suppl. Fig. 4). These results suggest that mucin-2 deficiency results in a strong induction of antimicrobial factors that restrict survival or replication of the commensal microflora.
Figure 6. Mucin-2 deficient mice display an upregulated intestinal antimicrobial protein expression.

Wild type or Muc2−/− mice were fed an intragastric isocaloric diet (n=4–6) or alcohol (n=9–10) for 1 week. (A, B) Gene expression of Reg3b, Reg3g, Camp and Defb1 in jejunum of mice. (C) Western blot for Reg3b and Reg3g; β-Actin was used as loading control. Representative Western blots are shown, which were reproduced (n=6 in each group). Densitometry of Western blot images was performed. Camp, cathelicidin antimicrobial peptide; Defb1, defensin beta 1; Reg3, regenerating islet-derived 3. *p < 0.05.
To investigate whether these findings directly translate into quantitative alterations of the commensal microflora, we used an in vivo luminal killing assay of non-pathogenic Escherichia coli (E.coli) in the gut of wild type and mucin-2 deficient mice as described by us (35, 36). A 4-cm loop of the proximal jejunum was ligated (without interrupting the blood supply) in anesthetized mice and injected with bioluminescent, non-pathogenic E.coli. To analyze luminal survival and killing, IVIS imaging of bioluminescent E.coli was performed at 0 min and 3.5 hrs after injection of bacteria into ligated jejunal loops. Whereas loops of Muc2−/− mice after feeding a Lieber DeCarli isocaloric diet or alcohol for two weeks were essentially devoid of luminescent bacteria, bioluminescent bacteria were found in alcohol and control fed wild type mice at a significantly higher percentage after 3.5 hrs (Fig. 7A,B). This result suggests that commensal bacteria are killed more effectively in jejunal loops of Muc2−/− mice than in wild type mice (Fig. 7C), thereby limiting intestinal bacterial overgrowth after alcohol feeding.
Figure 7. Mucin-2 deficient mice exhibit an enhanced intestinal antimicrobial activity.

Wild type or Muc2−/− mice were orally fed a Lieber DeCarli isocaloric diet (n=3–7) or alcohol (n=4–6) for 2 weeks. (A) Bioluminescent imaging after injection of luciferase-expressing Escherichia coli into jejunal loop at 0min and 3.5 hrs. (B, C) Survival in percentage (B) and effective killing (C) of injected E.coli after 3.5 hrs. *p < 0.05.
Enteral LPS administration increases experimental alcoholic liver disease in Muc2−/− mice
To demonstrate that Muc2−/− mice are protected due to intestinal changes, but not secondary to hepatic adaptations, we have chosen to administer LPS enterally. When mice were given LPS through the intragastric feeding tube daily for one week in addition to ethanol, increased bacterial products from Gram-negative E.coli were found in the liver of Muc2−/− mice comparable to levels seen in wild type mice (Suppl. Fig. 5A). This restoration of hepatic endotoxemia exacerbated alcoholic steatohepatitis in Muc2−/− mice fed ethanol and LPS (Suppl. Fig. 5B and C). This supports our finding that a decreased endotoxemia contributes to the protection of Muc2−/− mice form experimental alcoholic liver disease despite a leakier gut.
Discussion
The first, and arguably best, opportunity for the body to limit toxic effects of orally administered alcohol is the gastrointestinal tract. In this study, we investigate the role of mucins and in particular intestinal mucin-2 in alcoholic steatohepatitis. Alcohol increases the thickness of the intestinal mucus layer in patients with alcohol abuse. Alcoholic steatohepatitis was ameliorated in mice deficient in mucin-2, which could not be explained by altered ethanol metabolism or a compensatory upregulation of other intestinal mucins. We provide evidence that mucin-2 deficiency results in altered microbiome composition and an increased expression of antimicrobial molecules. This is associated with enhanced intraluminal killing of bacteria and a decrease in the intestinal bacterial burden in mucin-2 deficient mice. Less bacterial products such as LPS translocate from the intestine to the systemic circulation and cause less liver injury and steatosis (Fig. 8).
Figure 8.
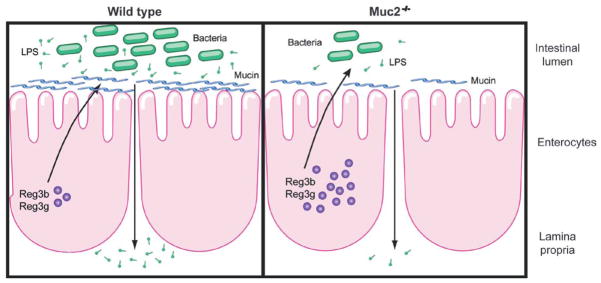
Model depicting the role of mucin-2 in experimental alcoholic liver disease.
Experimental alcoholic liver disease is dependent on gut-derived bacterial products that drive liver injury and steatosis (2). There is an evolving concept that changes in the gut microflora and microbiome affect bacterial translocation, both in patients and experimental models of alcoholic steatohepatitis. Increased plasma endotoxin and bacterial DNA have been associated with small intestinal bacterial overgrowth in cirrhotic patients. Furthermore, small intestinal bacterial overgrowth was an independent and major risk factor for the presence of bacterial DNA in the systemic circulation in cirrhotic patients (37, 38). Interestingly, selective intestinal decontamination decreased translocation to the mesenteric lymph nodes to the level of non-cirrhotic patients, and although not an established therapy, it also benefits patients with alcoholic liver cirrhosis by improving their liver function (19, 39). Thus, intestinal bacterial overgrowth predisposes patients with liver disease to bacterial translocation.
We have recently demonstrated quantitative (overgrowth) changes in the enteric microbiome using a model of intragastric alcohol feeding in mice. Suppression of alcohol-induced intestinal bacterial overgrowth with non-absorbable antibiotics decreases systemic levels of LPS and ameliorates alcoholic steatohepatitis in rats (18). On the other hand, if bacterial overgrowth is induced experimentally in the small intestine, this causes liver inflammation and injury (40).
We speculate that activation of the mucosal innate immune system as demonstrated by increased levels of Reg3b and Reg3g, contributes to reduced intestinal bacterial overgrowth in Muc2−/− mice. Prebiotics restore Reg3b and Reg3g expression, limit bacterial overgrowth and ameliorate alcohol-induced steatohepatitis (28). However, other antimicrobial molecules or components of the mucosal innate immune system might work in concert with Reg3b and Reg3g, and future studies are required for further investigations. Thus, based on our study, we propose a concept in which suppression of intestinal bacterial overgrowth by host antimicrobial molecules results in a decreased availability of intraluminal bacterial products. Less of these products are able to cross the intestinal barrier into the portal circulation, which eventually limits alcoholic liver disease.
We have also recently demonstrated qualitative changes in the enteric microbiome (dysbiosis) using a model of intragastric alcohol feeding in mice. Alcohol-associated dysbiosis is characterized by a profound suppression of commensal probiotic bacteria including Lactobacillus (28). Several studies have shown that a restoration of eubiosis using supplemental probiotic Lactobacillus ameliorates alcoholic steatohepatitis in rodents (41, 42). Interestingly, Muc2−/− mice are protected from alcohol-associated changes in the microbial composition including a suppression of Lactobacillus. In addition, mucin-2 deficiency limits the proliferation of bacteria (such as Gram-negative Akkermansia muciniphila) that use mucins as carbon source. Thus, the absence of mucin-2 prevents alcohol-associated dysbiosis, restores intestinal homeostasis and inhibits experimental alcoholic liver disease.
The mucus layer has a very important role in the intestine. It largely prevents the translocation of viable bacteria from the gut lumen to extraintestinal organs such as lymph nodes and the systemic circulation (43). The absence of mucin-2 as a major component of the intestinal mucus layer has no obvious adverse effect for the gut-liver axis at baseline without challenge. The thickness of the mucus layer increases in alcoholics as shown in our study and by others in rodents (27), which could be interpreted as a defense against alcohol or more likely against intestinal epithelial cell injury. And indeed, enteric infections also increase the mucin-2 production and the mucus layer (43). A downside of this obvious good reaction of the intestine of increasing the mucus layer is that the vigorous immune defense system of enterocytes against bacteria is impaired. We currently can only speculate of how an increase in the mucus layer might affect the expression of antimicrobial molecules as part of the mucosal innate immune system. One possibility is that bacterial ligands are not as accessible to enterocytes to stimulate the expression of antimicrobials. Reg3g expression has been shown to be TLR5 and IL- 22 dependent and can be induced by flagellin (34, 44, 45), but intestinal IL-22 did not correlate with Reg3 protein expression in our study. And indeed, Reg3g expression is induced through cell-autonomous MyD88-dependent toll-like receptor (TLR) activation in intestinal Paneth cells (46). Thus, when the body is challenged with alcohol, the thickness of the intestinal mucus layer increases, and less antimicrobial molecules reach the lumen to control proliferation of intestinal bacteria. An apparently good reaction of the body to respond to alcohol-induced epithelial cell damage impairs the mucosal innate immune system and results in the intestinal homeostasis system to fail. One should note that this is not a general response in mucin-2 deficient mice upon intestinal injury or inflammation, but is rather specific for alcohol. Other studies have shown that colitis induced by the pathogen Citrobacter rodentium is exacerbated in Muc2deficient mice (43).
Our study demonstrates that deficiency of one host gene Muc2 that is not expressed in the liver or in inflammatory cells, but largely restricted to the intestine, decreases alcoholic steatohepatitis. Our findings are consistent with the large body of evidence that experimental alcoholic liver disease is driven by the gut. Alcohol-associated changes in the microbiome, and in particular intestinal bacterial overgrowth, contributes to alcohol-induced liver injury. Taken together, our study emphasizes again the importance of the gut-liver axis. Treatment targeting the mucosal innate immune system and intestinal bacterial overgrowth might contribute to the clinical management of alcohol-induced liver disease in patients.
Supplementary Material
Acknowledgments
We thank Akiko Ueno and Raul Lazaro from the Animal Core facility of the Southern California Research Center for Alcoholic Liver and Pancreatic Diseases (ALPD) and Cirrhosis, at the University of Southern California for performing animal studies described in this study. We also thank Derick Han for tissue sharing, and Yaron Niv and Anna Velcich for helpful discussion and careful reading of the manuscript. This study was supported in part by NIH grants K08 DK081830 and R01 AA020703 (to BS) and by ABMRF/The Foundation for Alcohol Research (to BS). This study was also supported by the Pilot Project Program (to BS) and the Lee Summer Fellowship Award (to PH) of the Southern California Research Center for ALPD and Cirrhosis (P50AA11999) funded by the National Institute on Alcohol Abuse and Alcoholism. The study received also support from the UCSD Digestive Diseases Research Development Center, U.S. PHS grant #DK080506.
Abbreviations
- 4-HNE
4-hydroxynonenal
- Adh1
alcohol dehydrogenase 1
- ALT
alanine aminotransferase
- Camp
cathelicidin antimicrobial peptide
- Cyp2E1
cytochrome p450 enzyme 2E1
- Defb1
defensin beta 1
- E.coli
Escherichia coli
- FITC
fluorescein isothiocyanate
- LPS
lipopolysaccharide
- Muc
mucin
- PAS
Periodic Acid/Schiff stain
- Reg3
regenerating islet-derived 3
- TBARS
Thiobarbituric Acid Reactive Substances
- TLR
Toll-like receptor
- WGA
wheat germ agglutinin
- VDAC1
Voltage-dependent anion-selective channel protein 1
References
- 1.Kim WR, Brown RS, Jr, Terrault NA, El-Serag H. Burden of liver disease in the United States: summary of a workshop. Hepatology. 2002;36:227–242. doi: 10.1053/jhep.2002.34734. [DOI] [PubMed] [Google Scholar]
- 2.Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol. 2010;16:1321–1329. doi: 10.3748/wjg.v16.i11.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keshavarzian A, Fields JZ, Vaeth J, Holmes EW. The differing effects of acute and chronic alcohol on gastric and intestinal permeability. Am J Gastroenterol. 1994;89:2205–2211. [PubMed] [Google Scholar]
- 4.Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol. 1999;94:200–207. doi: 10.1111/j.1572-0241.1999.00797.x. [DOI] [PubMed] [Google Scholar]
- 5.Ferrier L, Berard F, Debrauwer L, Chabo C, Langella P, Bueno L, Fioramonti J. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol. 2006;168:1148–1154. doi: 10.2353/ajpath.2006.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choudhry MA, Fazal N, Goto M, Gamelli RL, Sayeed MM. Gut-associated lymphoid T cell suppression enhances bacterial translocation in alcohol and burn injury. Am J Physiol Gastrointest Liver Physiol. 2002;282:G937–947. doi: 10.1152/ajpgi.00235.2001. [DOI] [PubMed] [Google Scholar]
- 7.Keshavarzian A, Choudhary S, Holmes EW, Yong S, Banan A, Jakate S, Fields JZ. Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. J Pharmacol Exp Ther. 2001;299:442–448. [PubMed] [Google Scholar]
- 8.Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology. 2000;32:1008–1017. doi: 10.1053/jhep.2000.19621. [DOI] [PubMed] [Google Scholar]
- 9.Fujimoto M, Uemura M, Nakatani Y, Tsujita S, Hoppo K, Tamagawa T, Kitano H, et al. Plasma endotoxin and serum cytokine levels in patients with alcoholic hepatitis: relation to severity of liver disturbance. Alcohol Clin Exp Res. 2000;24:48S–54S. [PubMed] [Google Scholar]
- 10.Hanck C, Rossol S, Bocker U, Tokus M, Singer MV. Presence of plasma endotoxin is correlated with tumour necrosis factor receptor levels and disease activity in alcoholic cirrhosis. Alcohol. 1998;33:606–608. doi: 10.1093/alcalc/33.6.606. [DOI] [PubMed] [Google Scholar]
- 11.Parlesak A, Schafer C, Schutz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. 2000;32:742–747. doi: 10.1016/s0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 12.Urbaschek R, McCuskey RS, Rudi V, Becker KP, Stickel F, Urbaschek B, Seitz HK. Endotoxin, endotoxin-neutralizing-capacity, sCD14, sICAM-1, and cytokines in patients with various degrees of alcoholic liver disease. Alcohol Clin Exp Res. 2001;25:261–268. [PubMed] [Google Scholar]
- 13.Yin M, Bradford BU, Wheeler MD, Uesugi T, Froh M, Goyert SM, Thurman RG. Reduced early alcohol-induced liver injury in CD14-deficient mice. J Immunol. 2001;166:4737–4742. doi: 10.4049/jimmunol.166.7.4737. [DOI] [PubMed] [Google Scholar]
- 14.Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology. 2001;34:101–108. doi: 10.1053/jhep.2001.25350. [DOI] [PubMed] [Google Scholar]
- 15.Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt-Jones E, et al. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 2008;48:1224–1231. doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Enomoto N, Yamashina S, Kono H, Schemmer P, Rivera CA, Enomoto A, Nishiura T, et al. Development of a new, simple rat model of early alcohol-induced liver injury based on sensitization of Kupffer cells. Hepatology. 1999;29:1680–1689. doi: 10.1002/hep.510290633. [DOI] [PubMed] [Google Scholar]
- 17.Enomoto N, Ikejima K, Yamashina S, Hirose M, Shimizu H, Kitamura T, Takei Y, et al. Kupffer cell sensitization by alcohol involves increased permeability to gut-derived endotoxin. Alcohol Clin Exp Res. 2001;25:51S–54S. doi: 10.1097/00000374-200106001-00012. [DOI] [PubMed] [Google Scholar]
- 18.Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology. 1995;108:218–224. doi: 10.1016/0016-5085(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 19.Madrid AM, Hurtado C, Venegas M, Cumsille F, Defilippi C. Long-Term treatment with cisapride and antibiotics in liver cirrhosis: effect on small intestinal motility, bacterial overgrowth, and liver function. Am J Gastroenterol. 2001;96:1251–1255. doi: 10.1111/j.1572-0241.2001.03636.x. [DOI] [PubMed] [Google Scholar]
- 20.Matsuo K, Ota H, Akamatsu T, Sugiyama A, Katsuyama T. Histochemistry of the surface mucous gel layer of the human colon. Gut. 1997;40:782–789. doi: 10.1136/gut.40.6.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105:15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Klinken BJ, Einerhand AW, Duits LA, Makkink MK, Tytgat KM, Renes IB, Verburg M, et al. Gastrointestinal expression and partial cDNA cloning of murine Muc2. Am J Physiol. 1999;276:G115–124. doi: 10.1152/ajpgi.1999.276.1.G115. [DOI] [PubMed] [Google Scholar]
- 23.McGuckin MA, Linden SK, Sutton P, Florin TH. Mucin dynamics and enteric pathogens. Nat Rev Microbiol. 2011;9:265–278. doi: 10.1038/nrmicro2538. [DOI] [PubMed] [Google Scholar]
- 24.Linden SK, Florin TH, McGuckin MA. Mucin dynamics in intestinal bacterial infection. PLoS One. 2008;3:e3952. doi: 10.1371/journal.pone.0003952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, Kucherlapati R, et al. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science. 2002;295:1726–1729. doi: 10.1126/science.1069094. [DOI] [PubMed] [Google Scholar]
- 26.Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Buller HA, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131:117–129. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 27.Grewal RK, Mahmood A. Ethanol effects on mucin glycosylation of mucins in rat intestine. Annals of Gastroenterology. 2009;22:178–183. [Google Scholar]
- 28.Yan AW, Fouts DE, Brandl J, Stärkel P, Torralba M, Schott E, Tsukamoto H, et al. Enteric Dysbiosis Associated with a Mouse Model of Alcoholic Liver Disease. Hepatology. 2011;53:96–105. doi: 10.1002/hep.24018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lieber CS. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health. 2003;27:220–231. [PMC free article] [PubMed] [Google Scholar]
- 30.Nagy LE. Recent insights into the role of the innate immune system in the development of alcoholic liver disease. Exp Biol Med (Maywood) 2003;228:882–890. doi: 10.1177/153537020322800803. [DOI] [PubMed] [Google Scholar]
- 31.Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belzer C, de Vos WM. Microbes inside--from diversity to function: the case of Akkermansia. ISME J. 2012;6:1449–1458. doi: 10.1038/ismej.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 35.Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J Exp Med. 2007;204:1891–1900. doi: 10.1084/jem.20070563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, Schnabl B, et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature. 2008;455:804–807. doi: 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bauer TM, Fernandez J, Navasa M, Vila J, Rodes J. Failure of Lactobacillus spp. to prevent bacterial translocation in a rat model of experimental cirrhosis. J Hepatol. 2002;36:501–506. doi: 10.1016/s0168-8278(02)00003-x. [DOI] [PubMed] [Google Scholar]
- 38.Jun DW, Kim KT, Lee OY, Chae JD, Son BK, Kim SH, Jo YJ, et al. Association Between Small Intestinal Bacterial Overgrowth and Peripheral Bacterial DNA in Cirrhotic Patients. Dig Dis Sci. 2010;55:1465–1471. doi: 10.1007/s10620-009-0870-9. [DOI] [PubMed] [Google Scholar]
- 39.Cirera I, Bauer TM, Navasa M, Vila J, Grande L, Taura P, Fuster J, et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J Hepatol. 2001;34:32–37. doi: 10.1016/s0168-8278(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 40.Lichtman SN, Sartor RB, Keku J, Schwab JH. Hepatic inflammation in rats with experimental small intestinal bacterial overgrowth. Gastroenterology. 1990;98:414–423. doi: 10.1016/0016-5085(90)90833-m. [DOI] [PubMed] [Google Scholar]
- 41.Nanji AA, Khettry U, Sadrzadeh SM. Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver (disease) Proc Soc Exp Biol Med. 1994;205:243–247. doi: 10.3181/00379727-205-43703. [DOI] [PubMed] [Google Scholar]
- 42.Forsyth CB, Farhadi A, Jakate SM, Tang Y, Shaikh M, Keshavarzian A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol. 2009;43:163–172. doi: 10.1016/j.alcohol.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, et al. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010;6:e1000902. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, Flavell RA, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. 2012;36:276–287. doi: 10.1016/j.immuni.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kinnebrew MA, Ubeda C, Zenewicz LA, Smith N, Flavell RA, Pamer EG. Bacterial flagellin stimulates Toll-like receptor 5-dependent defense against vancomycin-resistant Enterococcus infection. J Infect Dis. 2010;201:534–543. doi: 10.1086/650203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lacunza E, Ferretti V, Barbeito C, Segal-Eiras A, Croce MV. Immunohistochemical evidence of Muc1 expression during rat embryonic development. Eur J Histochem. 2010;54:e49. doi: 10.4081/ejh.2010.e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.