

Key Points
Human iPSCs differentiate into CD34+ HPCs.
iPSC-derived HPCs induce T-cell anergy.
Abstract
Human induced pluripotent stem cells (iPSCs) have emerged as an alternative source of pluripotent stem cells that can be used for tissue regeneration in place of the controversial human embryonic stem cells. However, immunologic knowledge about iPSC derivatives remains enigmatic. Here, we characterized human iPS-derived CD34+ hematopoietic progenitor cells (HPCs). These HPCs poorly express major histocompatibility complex (MHC) I antigens and are MHC-II negative. Interestingly, they moderately express nonclassical HLA-G and HLA-E molecules. Consequently, alloreactive HLA-A2–specific cytotoxic T cells failed to recognize HLA-A2–expressing HPCs but became anergic. Subsequent upregulation of MHC-I using interferon-γ stimulation and provision of CD28 cosignaling led to T-cell activation, confirming that poor delivery of signals 1 and 2 by the HPCs mediated T-cell anergy. These data indicate for the first time that HPCs induce T-cell anergy, a unique characteristic of iPSC-derived cells that confers immunologic advantage for allogenic transplantation. Although iPSCs are ideal for patient-tailored treatments with the anticipation that no immunosuppression will be required, in cases of gene defects, their derivatives could be used to treat diseases in nonhistocompatible recipients.
Introduction
Hematopoietic stem cells (HSCs) that are used in clinical transplantation are derived from bone marrow, peripheral blood, or umbilical cord blood (UCB).1 Unfortunately, harsh preconditioning regimens, drug toxicity, and the requirement for immunosuppression preclude routine application of these HSCs in the treatment of devastating hematopoietic malignancies. In addition, approximately two-thirds of transplantation patients lack suitable HLA-matched donors. Those patients who find donors face the burden of nonspecific immunosuppression, increased risk of opportunistic infections, and the potential development of secondary malignancies.2,3
However, pluripotent stem cells have recently emerged as an alternative source of cells that can be used in regenerative medicine.4-6 Moreover, several groups have reported that embryonic stem cells (ESCs) are poorly immunogenic due to their low expression of classical major histocompatibility complex (MHC) I and lack of MHC-II antigens.7,8 Our group recently successfully established mixed chimerism in mice transplanted with mouse ESC-derived hematopoietic progenitor cells (HPCs)7 and for the first time showed that HPC-established mixed chimerism induced transplantation tolerance to cardiac allografts.9 Moreover, unlike adult stem cells, human ESCs (hESCs) and their derivatives are not susceptible to immunologic rejection.8 However, the use of hESCs for the treatment of diseases is complicated by the limited number of available hES cell lines. Furthermore, hESCs remain ethically and morally controversial. Thus, an alternative source of pluripotent stem cells is most desirable.
Recently, Yamanaka and colleagues established induced pluripotent stem cells (iPSCs) by reprogramming fibroblasts into a pluripotent state by means of retroviral transduction of 4 factors: Oct 3/4, Sox2, Klf4, and c-Myc.10 Even though iPSCs are similar to ESCs in their morphology, expression of pluripotent stem cell genes, and ability to form embryoid bodies (EBs), and in possessing the unique potential to differentiate into lineage-committed cells, recent molecular studies show genetic and molecular differences between both forms of pluripotent stem cells,11 which might affect their differentiation into lineage-committed cells. One caveat that remains to be resolved is avoidance of viral vectors during the reprogramming process. These retroviral vectors can induce epigenetic changes, which can lead to tumor formation but also affect their potential to differentiate. Interestingly, several alternative methods for the generation of iPSCs have now been reported, including the use of only 2 reprogramming factors or the use of plasmids, recombinant proteins, and messenger RNA and micro RNA–mediated reprogramming.12-18 These new procedures, however, remain very inefficient. The use of small molecules in combination with reprogramming transcription factors is a further alternative approach in generating human iPSCs.19 Lastly, in addition to fibroblasts, many other cell types have been successfully used to generate iPSCs,20-23 broadening the alternative sources of iPSCs.
Despite these advances, little is known about the immunologic characteristics of iPSC derivatives, an important determinant of their potential clinical application. For example, in the first studied disease model of iPSCs, Hanna et al24 deleted natural killer (NK) cells in recipient syngeneic mice before transplanting iPS-HPCs, suggesting that NK cells can be a limiting factor on the engraftment and therapeutic use of iPSC-derived progenitor cells. This observation supports our own studies on ESC-HPCs in which we showed HPCs to be highly susceptible to NK cells in vivo but not in vitro.25 More recently, it was reported that mouse iPSCs were rejected in syngeneic mice, whereas ESCs were not, suggesting that iPSCs are potentially immunogenic.26 This clearly demonstrates the importance of defining the immunologic properties of iPSC derivatives to allow determination of their potential clinical application.
In this study, we show that iPSC-derived CD34+ iPS-HPCs poorly express classical MHC antigens, lack CD80 and CD86, and highly express the T-cell inhibitory ligand PD-L1. Our data show that these HPC characteristics induce T-cell anergy in alloreactive T cells, which can be exploited for allogenic transplantation of iPSC-derived progenitor cells.
Methods
Cell lines
Human iPSCs reprogrammed from fibroblasts of patients with mucopolysaccharidosis type VI (CHOPWT3.1) and from fibroblasts of apparently healthy nonfetal tissue (CHOPWT2.2) were purchased from the Children’s Hospital of Philadelphia, Center for Cellular and Molecular Therapeutics, hESC/iPSC Core Facility. Other iPSCs, GM23226 and GM23262, were purchased from Coriell Institute for Medical Research. We also generated iPSCs from MRC5 (Fibroblasts, ATCC) (supplemental Figure 1; see the Blood Web site). Human iPSCs and ESCs (H13, from Wicell) were cultured in Dulbecco’s modified Eagle medium/F12 medium supplemented with 20% Knockout Serum (GIBCO/BRL), 10 ng/mL Fibroblast Growth Factor-basic, 1 mM GlutaMax, 50 U/mL penicillin and 50 μg/mL streptomycin, 1× nonessential amino acids, and 100 µM 2-mercaptoethanol (Invitrogen). Cytotoxic T lymphocytes (CTLs), immortalized B cells, and peripheral blood mononuclear cell (PBMCs) were cultured in complete RPMI medium.
Cytotoxicity assay
HLA-A2–specific CTLs were generated as we previously described.27 Briefly, we cocultured PBMCs with irradiated immortalized B cells (HLA*A2/3, B7, Cw7) for 7 days. Consequently, CTLs were restimulated every 7 to 10 days 2 times and then restimulated with HSB-2 (HLA*A1/2, B44/w57, Cw1/3) for 7 days. After this restimulation, HLA-A2–specific CTLs were restimulated with immortalized B cells every 7 days in complete RPMI-1640 medium supplemented with recombinant interleukin 2 (IL-2). These CTLs were used as effector cells, whereas iPS-HPCs, M23e immortalized B cells, and M28e cells were used as target cells in 4-hour 51Cr-release assay, as described previously.25
IFN-γ treatment
To upregulate expression of MHC-I, iPS-HPCs were treated with recombinant human interferon (rhIFN)-γ (50 ng/mL; PeproTech) for 48 hours. MHC-I upregulation was confirmed through flow cytometry with a phycoerythrin-conjugated HLA-ABC antibody. Cytotoxicity and enzyme-linked immunospot (ELISPOT) assays using IFN-γ–treated iPSC-HPCs were performed as described previously.
Statistical analysis
All of the experiments were performed at least 3 times in order to achieve statistically significant results (P < .05). GraphPad Prism 5 Statistics Software was used for statistical evaluation.
Results
Human iPSCs differentiate into HPCs
iPSCs were generated from commercially available fibroblasts using the Yamanaka factors Oct4, Sox2, Klf4, and c-Myc cloned into pMIG-GFP retroviral vectors (supplemental Figure 1A). Our generated iPSCs had normal karyotypes and were positive for human pluripotent stem cell markers (supplemental Figure 1B-C). More importantly, the cells formed EBs that expressed genes of all 3 germ layers (supplemental Figure 1D). The cell line 4F-2 generated in our laboratory is representative of our iPS cell lines and was used in the following experiments. Supplemental Table 1 shows that the DNA short tandem repeats profile for the iPSCs was identical to that of the parental fibroblasts. Lastly, to more definitively establish iPSC pluripotency, cells were injected into NOD-Scid IL2Rγnull mice to determine teratoma formation. These mice developed teratomas after 9 to 12 weeks. Hematoxylin and eosin staining of the teratomas revealed tissues of all 3 germ layers (supplemental Figure 1E). These data altogether show that we successfully generated pluripotent iPSCs from fibroblasts.
Next, we differentiated iPSCs into CD34+ HPCs and studied their immunologic characteristics. iPSCs were differentiated using a modified protocol.28,29 To determine the phenotype of iPS-HPCs, the differentiated cells were isolated using 0.05% trypsin and stained with various antibodies directed against hematopoietic cell markers (Figure 1A). On day 9 of the differentiation process, HPCs expressed CD34 (17%), the erythroid cell marker CD235a (8.61%), CD34/CD43 (5.9%), and CD34/CD45 (3.3%). These results are summarized in Figure 1B. In addition, HPCs were generated from 4 additional iPS cell lines either purchased or kindly provided by other investigators. As depicted in Figure 1C, the yield of CD43+/CD34+ HPCs varied among these 5 different iPS cell lines, suggesting possible epigenetic and molecular differences between the iPS cell lines. Furthermore, to confirm that the iPS-HPCs were indeed hematopoietic cells, reverse transcription–polymerase chain reaction (RT-PCR) was performed for the hematopoietic genes HoxA9, GATA-1, CD41, and CD45 (Figure 1D). Undifferentiated iPSCs and CD34+ cells from UCB were used as controls. Additionally, iPS-HPCs uniformly expressed the HSC marker CD90, similar to UCB-CD34+ cells (supplemental Figure 2). CD33, which is expressed by lineage-committed cells,30 was not expressed by iPS-HPCs but was found on the majority of UCB-CD34+ cells (supplemental Figure 2). Finally, unlike UCB-CD34+ cells, iPS-HPCs do not express CD38, which is a marker expressed by a variety of mature hematopoietic cell types.30 Altogether, these data indicate that iPS-HPCs, as compared with UCB-CD34+ cells, are more primitive HSCs.
Figure 1.
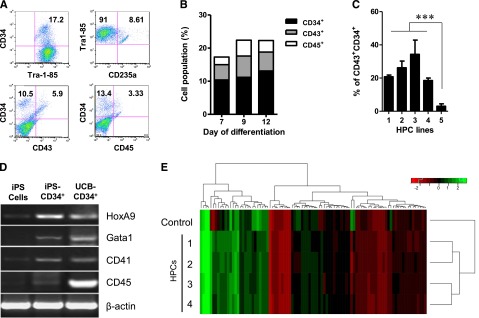
Human iPS-HPCs. (A) To determine the phenotype of iPS-HPCs, iPSCs cultivated on OP9 cells were harvested on differentiation day 9 and phenotypically characterized for various hematopoietic cell markers. iPS-HPCs expressed the hematopoietic cell surface markers CD34, CD43, CD45, and CD235a. We used the Tra-1-85 antibody to discriminate human cells. (B) The relative percentages of CD34+, CD43+, and CD45+ cells during the differentiation days 7, 9, and 12. (C) Five different human iPS cell lines were used to generate HPCs, respectively. The yield of CD43+/CD34+ HPCs was compared among these cell types. The data show that the efficiency of HPC derivation varied greatly among the iPSCs studied (mean ± standard deviation [SD]). ***P < .001. In particular, cell line 5 yielded the fewest number of CD34+ cells. (D) Total RNA was isolated from iPSCs, iPS-HPCs, and PBMCs enriched for CD34, respectively. RT-PCR of the hematopoietic-related genes HoxA9, Gata1, CD41, and CD45 was performed. β-Actin was used as a loading control. The results show that PBMC CD34+ cells and iPS-HPCs were identical in their gene expression patterns. (E) Microarray analysis of our 4 different iPS-HPC cell lines and UCB-CD34+ cells demonstrated that these cell lines have nearly identical hematopoietic gene expression profiles. These data suggest that the iPS-HPCs are similar to one another and confirm the authentic hematopoietic quality of these cells.
Lastly, to determine whether the HPCs derived from the 4 iPS cell lines are similar to each other, we performed human gene 1.0 ST microarrays (Gene Expression Omnibus accession number: GSE46888). UCB-CD34+ cells were used for comparison. Heat map and hierarchical clustering of 84 hematopoietic-related genes showed that the 4 different iPS-HPCs exhibit near identical gene expression patterns that are also similar to that of UCB-CD34+ cells. This result was not expected, because all HPCs were derived from iPSCs generated in different labs using different reprogramming techniques. Because iPS cell line 5 only yielded very few CD34+ cells, it was excluded from the DNA array analysis. The list of hematopoietic-related genes is presented in supplemental Table 2.
To further determine the hematopoietic potential of the iPS-HPCs, colony forming units (CFUs) were studied. Human iPS-HPCs were able to generate multilineage hematopoietic cells, namely, CFU-E (erythroid), -GEMM (granulocyte, erythrocyte, macrophage, megakaryocyte), -M (macrophage), -GM (granulocyte, macrophage), and -G (granulocyte) (Figure 2A). The relative percentages of these lineage-committed cells are summarized in Figure 2B, showing that the iPS-HPCs were capable of multilineage differentiation.
Figure 2.
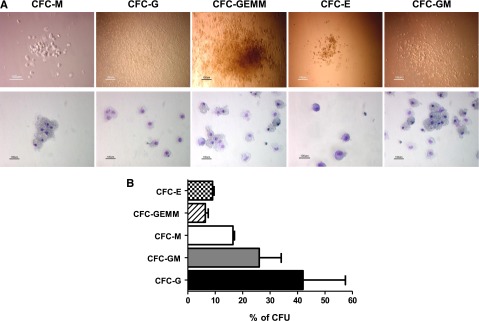
iPS-HPCs differentiate into multilineage progenitors. (A) To determine the multilineage potential of iPS-HPCs, HPCs were used to form CFUs. The upper panel shows the morphology of the CFUs, and the lower panel shows the CFUs after staining with Giemsa-Wright solution. These CFUs are similar to those formed by human bone marrow stem cells. (B) Relative percentages of CFU-E, -GEMM, -M, -GM, and -G. CFU-G shows the highest frequency. CFU-E and CFU-GEMM showed the lowest frequency (mean ± SD).
iPS-HPCs poorly express HLA molecules
Our goal here was to determine the immunologic characteristics of iPS-HPCs, in particular the level of MHC expression. Although transplantation of iPSC-derived cells into the somatic cell donor may not require T cell–directed immunosuppression to avoid rejection, NK cells are a potential barrier to engraftment. For example, in the published sickle cell anemia therapeutic model using iPSCs,24 deletion of NK cells was a requirement for the HSCs to engraft in syngeneic recipients. This suggested to us that the iPSCs derivatives in the study were possibly susceptible to NK cells, which may have played a significant role in regulating the engraftment of iPSC derivatives. Indeed, our studies recently showed that NK cells regulated the engraftment of mouse ESC-HPCs.25
Here, we studied the MHC expression by iPS-HPCs, iPSCs, and the parental fibroblasts. ESCs, ESC-HPCs, and UCB-CD34+ cells were included as controls. Interestingly, MHC expression by iPSCs was less than that by fibroblasts and by UCB-CD34+ cells (Figure 3). This is consistent with our own studies on rat embryonic-like stem cells, which poorly expressed MHC,31 now a well-established characteristic of ESC-HPCs.7,32 Further, iPS-HPCs expressed the nonclassical MHC molecules HLA-G and HLA-E (Figure 3 and supplemental Table 3), albeit at modest levels. Additionally, HLA-G expression varied among the iPS cell lines used (supplemental Table 3). Interestingly, treatment of HPCs with IFN-γ enhanced their expression of HLA-E but not HLA-G (supplemental Figure 4). Lastly, iPS-HPCs slightly expressed MHC class I–related chain (MIC)-A/B, ligands for NK cell–activating receptors (supplemental Table 3). In addition, iPS-HPCs remained negative for MHC-II molecules, an observation that is consistent with our prior studies on mouse ESC-HPCs. In contrast, UCB-CD34+ cells highly expressed MHC-I but had poor expression of MHC-II antigens.
Figure 3.

MHC expression profiles of human iPSCs and iPS-HPCs. Parental fibroblasts, iPSCs, iPS-HPCs, hESCs, ESC-HPCs, and UCB-CD34+ cells (control) were stained with anti–HLA-ABC, HLA-DR, HLA-G, and HLA-E antibodies. The gray histograms indicate the isotype control. These data show that reprogrammed human iPSCs exhibit low expression of HLA-ABC compared with parental fibroblasts, whereas the expression of other molecules was not significantly different. The expression of the MHC molecules on iPSCs is very similar to that of hESCs. This pattern is also seen in iPS-HPCs and hESC-HPCs. However, the nonclassical MHC molecule HLA-G is upregulated after differentiation. All cell types expressed HLA-E. These data are representative of 7 experiments.
iPS-HPCs do not stimulate HLA-A2–specific CTLs
We and others previously reported that mouse ESCs and ESC derivatives are poorly immunogenic.32,33 To determine whether low MHC-I expression influenced the susceptibility of iPS-HPCs toward alloreactive T-cell killing, we established HLA-A2–specific CTLs as we previously described.27 After 5 weeks in culture, 85% of the CTLs were CD8+ (Figure 4A). CTL cytotoxicity was tested using a 4-hour 51Cr-release assay. The CTLs strongly killed HLA-A2 target cells in a 4-hour 51Cr- release assay but surprisingly failed to kill iPS-HPCs (Figure 4B). Similarly, third-party target cells that did not express HLA-A2 were not lysed. To determine whether iPS-HPCs activate CTLs, we used the ELISPOT assay to measure IL-2 secretion by CTLs in response to stimulator cells at different time points. We observed robust IL-2 secretion by CTLs cultured with immortalized B cells (HLA-A2) but not by CTLs cultured with either HPCs or with a third-party control (Figure 4C). Altogether, our data demonstrate that iPS-HPCs failed to stimulate the HLA-A2–specific CTLs despite their expression of HLA-A2, suggesting that these cells poorly engage with HLA-A2–specific T-cell receptors (TCRs).
Figure 4.
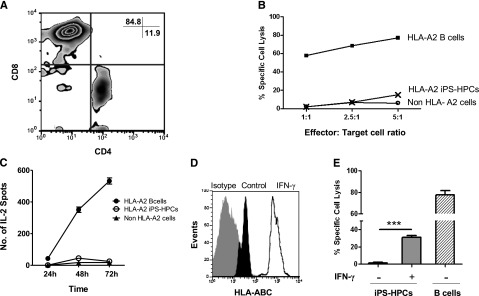
iPS-HPCs are not susceptible to allogenic CTLs in vitro. (A) In vitro–generated HLA-A2–specific CTLs were mostly CD8+. (B) To determine the susceptibility of the iPS-HPCs to CTL cytotoxicity, a CTL cytotoxicity assay was performed. HLA-A2–specific CTLs were used as effector cells. iPS-HPCs (HLA-A2), immortalized B cells (HLA-A2–expressing positive control), and non–HLA-A2 immortalized B cells served as target cells. iPS-HPCs show very low susceptibility to killing by allogenic cytotoxic T cells. ***P < .001. As expected, HLA-A2 immortalized B cells were lysed. (C) To further study the interaction of HPCs and HLA-A2–specific CTLs, IL-2 release by CTLs was measured by the ELISPOT assay following stimulation of the CTLs with iPS-HPCs (HLA-A2), immortalized B-cells (HLA-A2–expressing positive control), and non–HLA-A2 cells (negative control). iPS-HPCs failed to activate CTLs in a time-dependent manner (mean ± SD). (D) To upregulate MHC-I expression by iPS-HPCs, HPCs were stimulated with IFN-γ for 48 hours. iPS-HPCs were harvested, and the expression of MHC-I molecules was analyzed. IFN-γ (empty overlay) upregulated expression of MHC-I molecules by HPCs compared with untreated iPS-HPCs (dark). Gray represents the isotype control. (E) After IFN-γ treatment, iPS-HPCs showed significantly increased susceptibility to CTL killing compared with untreated cells (mean ± SD).
We speculated that lack of CTL activation by HPCs was due to poor MHC expression. Here, to determine whether upregulation of MHC-I molecules in the iPS-HPCs enhances their lysis by CTLs, HPCs were stimulated with IFN-γ for 48 hours. Upregulation of MHC-I antigens was confirmed by flow cytometry (Figure 4D). These cells were subsequently used as target cells of alloreactive CTLs. As expected, the iPS-HPCs were now modestly susceptible to CTL killing (Figure 4E). These results were of interest because they suggested that the iPS-HPCs were protected from CTL killing due to low MHC expression.
IFN-γ treatment and CD28 costimulation enhance HPC stimulation of CTLs
Interaction of B7 family proteins with CD28 is a requirement for T-cell stimulation in concert with TCR engagement. PD-L1 (also called B7-H1, CD274) and PD-L2 (also known as B7–dentritic cell CD273) interact with PD-1 (programmed cell death 1). PD-1 is an inhibitory receptor expressed on T cells, NK cells, B cells, and activated monocytes.34,35 Engagement of PD-1 with PD-L1 leads to the inhibition of TCR-mediated lymphocyte proliferation and cytokine secretion.36 PD-1 is significant for the induction of peripheral CD8+ T-cell tolerance by resting dendritic cells in vivo.35 Furthermore, infection with the helminthes Schistosoma mansoni or Taenia crassiceps upregulates the expression of PD-L1 on activated macrophages, which acquire immunosuppressive activity and induce T-cell anergy.35,37
The extent of costimulatory molecule expression on iPS-HPCs was determined using flow cytometry. iPS-HPCs do not express CD80, CD86, or PD-L2, but considerable levels of PD-L1, which engages PD-1, suppressing the proliferation of CD8+ T-cells38 (Figure 5A). Interestingly, expression of the costimulatory molecules was not affected by treatment with IFN-γ (Figure 5B).
Figure 5.

iPS-HPCs induce CD8 T-cell anergy due to poor MHC-I expression and lack of costimulatory molecules. (A) iPS-HPCs do not express the costimulatory molecules CD80 or CD86, respectively, as determined by flow cytometry, suggesting their inability to present antigen to T cells. Furthermore, iPS-HPCs express the T-cell inhibitory ligand PD-L1 but not PD-L2. (B) Expression levels of costimulatory molecules were unchanged following treatment with IFN-γ. (C) IFN-γ secretion by isotype control–treated CTLs cocultured with IFN-γ–pretreated iPS-HPCs was increased by 50-fold compared with isotype control or after treatment with an anti-CD28 antibody. IFN-γ served to upregulate the expression of MHC-I. However, anti-CD28 antibody–treated CTLs cocultured with IFN-γ–pretreated iPS-HPCs secreted 120 times more IFN-γ compared with isotype control or anti-CD28 antibody–treated CTLs cultured with untreated HPCs (mean ± SD). **P < .01; *P < .05. This result showed that delivery of costimulation through the anti-CD28 antibody rescued the stimulatory capacity of HPCs. (D) Anti-CD28 antibody–treated CTLs cocultured with IFN-γ–pretreated HPCs highly release IL-2 compared with isotype controls or anti-CD28 antibody–treated CTLs cocultured with untreated iPS-HPCs (mean ± SD). **P < .01; *P < .05. (E) Alloreactive T cells stimulated with iPS-HPCs did not appear to be stimulated, as their growth remained slow and in single-cell suspension. However, T cells stimulated with immortalized B cells formed clusters as a sign of stimulation. (F) T cells restimulated with iPS-HPCs failed to respond to stimulation with immortalized B cells in the absence of IL-2 as determined by IFN-γ secretion. After addition of IL-2, T-cell proliferation recovered suggesting that IL-2 stimulation reactivated anergic T cells (mean ± SD). **P < .01. In contrast, T cells stimulated with immortalized B cells in the first and second stimulations did not show significantly increased secretion of IFN-γ in the presence of IL-2. Thus, iPS-HPCs induce T-cell anergy that was rescued by IL-2. (G) iPS-HPCs were pretreated with a PD-L1–blocking antibody or isotype control antibody and then cocultured with CTLs for 3 days. The CTLs were then plated into an IFN-γ ELISPOT plate with irradiated M23 cells and treated with recombinant IL-2. IFN-γ production by IL-2–treated CTLs cocultured with iPS-HPCs pretreated with the PD-L1–blocking antibody was significantly higher than that of IL-2–treated CTLs cocultured with untreated iPS-HPCs (mean ± SD). *P < .05. This experiment definitively suggests that PD-L1 has a role in the induction of T-cell anergy by iPS-HPCs. These experiments were repeated 3 times.
To verify that poor MHC-I expression and lack of costimulation of iPS-HPCs induced T-cell anergy, ELISPOT assays for both IFN-γ and IL-2 were performed. As depicted in Figure 5C, IFN-γ secretion by CTLs cultured with iPS-HPCs was at a basal level when treated with an isotype control antibody or by an anti-CD28 antibody alone. In contrast, CTLs cultured with IFN-γ–treated HPCs secreted 50 times more IFN-γ than controls. This effect was further significantly enhanced when CD28 costimulation (Figure 5C) was provided. This increased CTL response is due to the enhanced MHC-I expression and signal 2 (delivered through the anti-CD28 antibody). This effect was reciprocated when we measured IL-2 release by the CTLs (Figure 5D). Thus, lack of positive costimulatory molecules on the HPCs mediated lack of T-cell activation.
iPS-HPCs induce anergy of CD8+ T cells
Human CD34+ stem cells have been reported to present antigen to T cells,39 leading to T-cell stimulation.40 It is this characteristic that forms a formidable barrier to successful bone marrow transplantation. Because we did not observe lysis of the iPS-HPCs by alloreactive T cells, we wondered about the impact of iPS-HPCs on alloreactive T cells. To address this question, HLA-A2–specific alloreactive CTLs were generated and further cultivated with either irradiated HLA-A2 iPS-HPCs or with HLA-A2 immortalized B cells for 3 days. Figure 5E depicts the aggregate morphology of activated CTLs cultured with immortalized B cells, which contrasts sharply with the appearance of CTLs stimulated with iPS-HPCs. B cell–stimulated T cells formed cell clusters in culture as a sign of stimulation. In contrast, T cells stimulated with iPS-HPCs remained in free suspension as single cells showing no sign of stimulation.
We predicted that poor stimulation of T cells due to the lack of signal 1 and signal 2 could induce T-cell anergy. The hallmark of T-cell anergy is the failure of IL-2 production by T cells upon stimulation and by defective production of inflammatory mediators such as IFN-γ and tumor necrosis factor α.41,42 An important distinguishing characteristic between T-cell anergy and T-cell exhaustion is that anergy can be broken by recombinant IL-2 treatment.43 Therefore, to determine whether iPS-HPCs induce T-cell anergy, CTLs were incubated with either iPS-HPCs or with irradiated immortalized B cells for 3 days. CTLs were subsequently harvested and restimulated with immortalized B cells, respectively, to determine IFN-γ release via the ELISPOT assay. Recombinant IL-2 was added in additional cultures to determine whether T cells responded to IL-2. As represented in Figure 5F, nontreated CTLs precultured with iPS-HPCs showed minimal secretion of IFN-γ upon stimulation with immortalized B cells. However, there was a significant increase in IFN-γ release by these same CTLs upon treatment with IL-2, suggesting that the T cells cultured with iPS-HPCs had in fact become anergic. As expected, CTLs precultured with immortalized B cells robustly secreted IFN-γ upon stimulation, and IFN-γ release did not significantly change upon IL-2 treatment (Figure 5F).
To dissect the functional contribution of PD-L1 expression by iPS-HPCs in the induction of T-cell anergy, CTL/iPS-HPC cocultures were treated with a PD-L1–blocking antibody or an isotype control. After 3 days of the coculture, CTLs were harvested and an IFN-γ ELISPOT assay was performed with immortalized B cells as stimulator cells. IFN-γ–treated HPCs were used for this assay. The data (Figure 5G) clearly demonstrated that blocking PD-L1 signaling by iPS-HPCs resulted in enhanced IFN-γ release by IL-2–treated CTLs. This experiment suggests that PD-L1 has a prominent role in the induction of T-cell anergy by iPS-HPCs.
We previously discussed that iPS-HPCs express low levels of HLA-E and HLA-G (Figure 3 and supplemental Table 3) and that IFN-γ treatment had an impact on the expression of HLA-E but not HLA-G (supplemental Figure 4). This raised the question of whether these nonclassical MHC molecules played a role in the induction of T-cell anergy by iPS-HPCs. To address this, we treated CTL-HPC cocultures with anti–HLA-G or anti–HLA-E and performed an IFN-γ ELISPOT assay with immortalized B cells as stimulator cells. The CTL-HPC cocultures consisted of IFN-γ–treated iPS-HPCs. Blocking HLA-G (supplemental Figure 5) or HLA-E (data not shown) did not impact the ability of HPCs in inducing T-cell anergy, suggesting that HLA-G and HLA-E are not decisive cues for the induction of T-cell anergy by iPS-HPCs.
Discussion
In the past 7 years, research on iPSCs has exploded due to the potential for establishing patient-tailored therapies in regenerative medicine. In the interim, several new approaches for the derivation of lineage-committed cells have been established. Unfortunately, little is known about the immunologic characteristics of iPSC-derived progenitor cells, which represents a significant knowledge gap for the future translational application of iPSCs.
Here, we established immunologic characteristics of human iPSC-derived CD34+ HPCs. Previous reports showed that hESCs display poor expression of MHC-I molecules and do not express MHC-II or HLA-G molecules.44 Similar to these previous reports, iPSCs poorly express MHC-I molecules but not MHC-II or HLA-G molecules. This outcome is supported by Suárez-Alvarez et al,45 who reported low gene expression of MHC-I and untraceable expression of MHC-II in human iPSCs. Additionally, this group showed that the genetic expression of Transporter associated with Antigen Processing (TAP)-1, TAP-2, and tapasin molecules was weak in ESCs and iPSCs. Tapasin stabilizes the TAP-1/2 complex and is involved in peptide loading of MHC-I onto the cell surface. They suggested that the absence of tapasin expression in undifferentiated hESCs may be due to the presence of the repression marker H3K9me3.45 During the reprogramming process and conversion of fibroblasts into iPSCs, epigenetic modifications, such as hypermethylation of DNA promoter regions, and histone modifications, have been observed.46 Earlier studies by Ladhoff et al47 showed that ESCs poorly expressed MHC-II molecules due to lack of expression of the MHC-II transcription factor CIITA. Thus, poor expression of MHC antigens is an inherent characteristic of embryonic tissues because they have not yet upregulated transcription factors of the MHC complex.
After the differentiation of iPSCs into HPCs, these progenitors express HLA-G and MIC-A/B (Figure 3 and supplemental Table 3). HLA-G is a ligand for an NK cell–inhibitory receptor, whereas MIC-A/B is a ligand for NKG2D, which is an NK cell–activating receptor.48 Thus, we consider that iPS-HPCs have the potential to activate NK cells. However, an elusive balance of inhibitory and activating signals to NK cells determines whether the target cell is killed, and thus it remains to be seen whether iPS-HPCs are actually susceptible to NK-cell cytotoxicity. However, supplemental Figure 3 provides evidence that iPS-HPCs are not susceptible to NK-cell killing in vitro. This is consistent with our own data using mouse HPCs.25 Additionally, iPS-HPCs, similar to UCB-CD34+ HPCs, do not express ULBP-1,2,5,6, which are ligands of the NK cell–activating receptor NKG2D (supplemental Figure 2). This evidence is consistent with data from the in vitro cytotoxicity assay (supplemental Figure 3) indicating that iPS-HPCs are not susceptible to NK cells.
Our data show that we successfully derived CD34+ HPCs from human iPSCs and that the cells express hematopoietic genes similar to those expressed by UCB-CD34+ cells. Although we studied CD34+ cells derived from 4 iPS cell lines derived in different labs, their gene expression patterns looked very similar. This was encouraging, suggesting that our iPS cell differentiation protocol was efficient. Moreover iPS-HPCs were able to sustain hematopoiesis as confirmed by the CFU data. So far, no data are available in the literature on the long-term survival of human ES/iPS-HPCs cells or their derivatives in vivo. We believe that the current protocols only allow the derivation of primitive but not definitive HPCs that can survive and self-renew long term in vivo. Several groups have used different approaches to generate HPCs from pluripotent stem cells. For example, Lako and colleagues used AM20.1B4, UG26.1B6, EL08.1D2, and AGM cell lines for HPC differentiation from hESCs.49 Coculture of ESCs with the AGM-derived stromal cell line notably increased the efficiency of differentiation into HPCs and ultimately enhanced their reconstitution into the host system upon their engraftment into immunodeficient NOD-Scid IL2Rγnull recipient mice. However, differentiation efficiency and long-term engraftment still remain a challenge yet to be overcome. Humanized mouse models may allow for enhanced chimerism and long-term engraftment of human ES/iPS-HPCs. In the mouse, ESC-HPCs have only been able to induce long-term engraftment after transduction with the transcription factor HoxB4.32,50
Although T cell–mediated rejection of iPSC-derived progenitor cells has been proposed to be irrelevant when cells are transplanted back to the donor of the somatic cells, their lack of MHC antigens could make them targets for NK cells. For example, the use of iPSCs to cure sickle cell anemia required depletion of NK cells using an anti-NK antibody.24 Furthermore, according to a recently published report,26 mouse iPSCs induce T cell–dependent rejection in syngeneic recipients, strongly demonstrating that research on the immunogenicity of iPSCs and iPSC derivatives is of great importance and clinical relevance. Here, we show that iPS-HPCs are poor targets of alloreactive T cells. However, when MHC-I expression was upregulated by IFN-γ stimulation, the iPS-HPCs were readily lysed in vitro. Thus, these cells could potentially be rejected in vivo by T cells after upregulation of their MHC-I molecules by serum IFN-γ. In the syngeneic situation, presentation of aberrant peptides by MHC molecules could lead to significant T-cell stimulation. However, although this is a possibility, our data also show that iPS-HPCs induce T-cell anergy, which is a characteristic that supports the potential use of these cells in a clinical setting. More specifically, we demonstrated that iPS-HPCs induce T-cell anergy on the basis of their low MHC expression and low expression of the costimulatory molecules CD80, CD86, and PD-L2. Furthermore, these cells express high levels of the T cell–inhibitory ligand PD-L1. Recently published work demonstrating that iPSCs are rejected in autologous recipients seems to suggest that the induction of T-cell anergy may be beneficial in the transplantation of patient-tailored cells and tissues.26 Although the effect of iPS-HPCs on CTLs appears to be similar to the vetolike activity attributed to human HSCs and certain bone marrow–resident populations,51,52 there are several characteristic differences. iPS-HPCs do not induce lysis of allospecific CTLs but induce reversible T-cell anergy. Additionally, iPS-HPCs do not express CD8, which has been implicated in mediating the veto effect by a subset of bone marrow cells. Thus, iPS-HPCs are incapable of cross-linking with CTLs.
Although iPSCs may be rejected even in autologous transplants, we show that iPS-HPCs may be protected from T cell–mediated rejection due to their ability to induce T-cell anergy. We propose that poor recognition of iPS-HPCs is due to their low expression of classical MHC molecules. However, iPSCs express high levels of Oct4, which has been suggested to contribute to T-cell responses to iPSCs.53 It will therefore be beneficial to design new reprogramming approaches that reduce Oct4 expression.
Human iPS-HPCs are very similar to ESC-HPCs in many respects, including poor expression of MHC-I, lack of MHC-II expression, and no expression of the T-cell costimulatory molecules CD80 and CD86. Thus, it is likely that ESC-HPCs also induce T-cell anergy, which is especially significant because therapy using these cells involves allogenic transplantation. In this study, we conclude that human iPS-HPCs hold enormous promise due to their possible regulatory role on T cells. Our data reveal for the first time a previously unknown aspect of iPS-HPCs, which is most valuable for both autologous and allogenic transplants.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development (1I01BX001125-02) and the National Institutes of Health, National Heart, Lung, and Blood Institute (grants 5R01HL073015-08 and 3R01HL073015-04A1S1).
The content of this publication does not necessarily reflect the views or policies of the granting agencies.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: E.-M.K. designed research, generated human iPSCs, performed HPC differentiation experiments, CFU assay, RT-PCR, CTL assay, and CTL culture, and wrote the manuscript; G.S.M. performed ELISPOT assays, RT-PCR, and flow cytometry analysis; and N.Z. designed the research, analyzed data, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Nicholas Zavazava, University of Iowa & Iowa City VAMC, 200 Hawkins Dr, Iowa City, IA 52242; e-mail: nicholas-zavazava@uiowa.edu.
References
- 1.Sorrentino BP. Clinical strategies for expansion of haematopoietic stem cells. Nat Rev Immunol. 2004;4(11):878-888. [DOI] [PubMed]
- 2.Wang L, Menendez P, Cerdan C, Bhatia M. Hematopoietic development from human embryonic stem cell lines. Exp Hematol. 2005;33(9):987-996. [DOI] [PubMed]
- 3.McKinney-Freeman SL, Daley GQ. Towards hematopoietic reconstitution from embryonic stem cells: a sanguine future. Curr Opin Hematol. 2007;14(4):343-347. [DOI] [PubMed]
- 4.Bauwens CL, Peerani R, Niebruegge S, et al. Control of human embryonic stem cell colony and aggregate size heterogeneity influences differentiation trajectories. Stem Cells. 2008;26(9):2300-2310. [DOI] [PubMed]
- 5.Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol. 2000;18(4):399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- 6.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145-1147. [DOI] [PubMed]
- 7.Bonde S, Zavazava N. Immunogenicity and engraftment of mouse embryonic stem cells in allogeneic recipients. Stem Cells. 2006;24(10):2192-2201. [DOI] [PubMed]
- 8.Drukker M, Katchman H, Katz G, et al. Human embryonic stem cells and their differentiated derivatives are less susceptible to immune rejection than adult cells. Stem Cells. 2006;24(2):221–229. doi: 10.1634/stemcells.2005-0188. [DOI] [PubMed] [Google Scholar]
- 9.Bonde S, Chan KM, Zavazava N. ES-cell derived hematopoietic cells induce transplantation tolerance. PLoS One. 2008;3(9):e3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663-676. [DOI] [PubMed]
- 11.Chin MH, Mason MJ, Xie W, et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5(1):111–123. doi: 10.1016/j.stem.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huangfu D, Maehr R, Guo W, et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008;26(7):795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huangfu D, Osafune K, Maehr R, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26(11):1269-1275. [DOI] [PubMed]
- 14.Kim D, Kim CH, Moon JI, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4(6):472-476. [DOI] [PMC free article] [PubMed]
- 15.Anokye-Danso F, Trivedi CM, Juhr D, et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8(4):376-388. [DOI] [PMC free article] [PubMed]
- 16.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322(5903):945-949. [DOI] [PMC free article] [PubMed]
- 17.Yu J, Hu K, Smuga-Otto K, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324(5928):797-801. [DOI] [PMC free article] [PubMed]
- 18.Zhou H, Wu S, Joo JY, et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4(5):381-384. [DOI] [PMC free article] [PubMed]
- 19.Lin T, Ambasudhan R, Yuan X, et al. A chemical platform for improved induction of human iPSCs. Nat Methods. 2009;6(11):805–808. doi: 10.1038/nmeth.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aasen T, Raya A, Barrero MJ, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26(11):1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 21.Aoi T, Yae K, Nakagawa M, et al. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science. 2008;321(5889):699–702. doi: 10.1126/science.1154884. [DOI] [PubMed] [Google Scholar]
- 22.Brown ME, Rondon E, Rajesh D, et al. Derivation of induced pluripotent stem cells from human peripheral blood T lymphocytes. PLoS ONE. 2010;5(6):e11373. doi: 10.1371/journal.pone.0011373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JB, Sebastiano V, Wu G, et al. Oct4-induced pluripotency in adult neural stem cells. Cell. 2009;136(3):411–419. doi: 10.1016/j.cell.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 24.Hanna J, Wernig M, Markoulaki S, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318(5858):1920-1923. [DOI] [PubMed]
- 25.Tabayoyong WB, Salas JG, Bonde S, Zavazava N. HOXB4-transduced embryonic stem cell-derived Lin-c-kit+ and Lin-Sca-1+ hematopoietic progenitors express H60 and are targeted by NK cells. J Immunol. 2009;183(9):5449–5457. doi: 10.4049/jimmunol.0901807. [DOI] [PubMed] [Google Scholar]
- 26.Zhao T, Zhang Z-N, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474(7350):212–215. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
- 27.Zavazava N, Krönke M. Soluble HLA class I molecules induce apoptosis in alloreactive cytotoxic T lymphocytes. Nat Med. 1996;2(9):1005–1010. doi: 10.1038/nm0996-1005. [DOI] [PubMed] [Google Scholar]
- 28.Eun-Mi K, Nicholas Z. Derivation of hematopoietic progenitor cells from human embryonic stem cells. 2010;2010:215-222. [Google Scholar]
- 29.Vodyanik MA, Bork JA, Thomson JA, Slukvin II. Human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood. 2005;105(2):617-626. [DOI] [PubMed]
- 30.Liu XL, Yuan JY, Zhang JW, Zhang XH, Wang RX. Differential gene expression in human hematopoietic stem cells specified toward erythroid, megakaryocytic, and granulocytic lineage. J Leukoc Biol. 2007;82(4):986–1002. doi: 10.1189/jlb.0107014. [DOI] [PubMed] [Google Scholar]
- 31.Fändrich F, Lin X, Chai GX, et al. Preimplantation-stage stem cells induce long-term allogeneic graft acceptance without supplementary host conditioning. Nat Med. 2002;8(2):171–178. doi: 10.1038/nm0202-171. [DOI] [PubMed] [Google Scholar]
- 32.Chan KM, Bonde S, Klump H, Zavazava N. Hematopoiesis and immunity of HOXB4-transduced embryonic stem cell-derived hematopoietic progenitor cells. Blood. 2008;111(6):2953-2961. [DOI] [PMC free article] [PubMed]
- 33.Bonde S, Dowden AM, Chan KM, Tabayoyong WB, Zavazava N. HOXB4 but not BMP4 confers self-renewal properties to ES-derived hematopoietic progenitor cells. Transplantation. 2008;86(12):1803-1809. [DOI] [PMC free article] [PubMed]
- 34.Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006;27(4):195–201. doi: 10.1016/j.it.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8(3):239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 36.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith P, Walsh CM, Mangan NE, et al. Schistosoma mansoni worms induce anergy of T cells via selective up-regulation of programmed death ligand 1 on macrophages. J Immunol. 2004;173(2):1240–1248. doi: 10.4049/jimmunol.173.2.1240. [DOI] [PubMed] [Google Scholar]
- 38.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173(2):945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 39.Rondelli D, Andrews RG, Hansen JA, Ryncarz R, Faerber MA, Anasetti C. Alloantigen presenting function of normal human CD34+ hematopoietic cells. Blood. 1996;88(7):2619–2675. [PubMed] [Google Scholar]
- 40.van Rhee F, Jiang YZ, Vigue F, et al. Human G-CSF-mobilized CD34-positive peripheral blood progenitor cells can stimulate allogeneic T-cell responses: implications for graft rejection in mismatched transplantation. Br J Haematol. 1999;105(4):1014–1024. doi: 10.1046/j.1365-2141.1999.01470.x. [DOI] [PubMed] [Google Scholar]
- 41.Mescher MF, Curtsinger JM, Agarwal P, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. 2006;211(1):81–92. doi: 10.1111/j.0105-2896.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 42.Wells AD. New insights into the molecular basis of T cell anergy: anergy factors, avoidance sensors, and epigenetic imprinting. J Immunol. 2009;182(12):7331–7341. doi: 10.4049/jimmunol.0803917. [DOI] [PubMed] [Google Scholar]
- 43.Schwartz RH. Models of T cell anergy: is there a common molecular mechanism? J Exp Med. 1996;184(1):1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drukker M, Katz G, Urbach A, et al. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc Natl Acad Sci USA. 2002;99(15):9864–9869. doi: 10.1073/pnas.142298299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suárez-Alvarez B, Rodriguez RM, Calvanese V, et al. Epigenetic mechanisms regulate MHC and antigen processing molecules in human embryonic and induced pluripotent stem cells. PLoS ONE. 2010;5(4):e10192. doi: 10.1371/journal.pone.0010192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hotta A, Ellis J. Retroviral vector silencing during iPS cell induction: an epigenetic beacon that signals distinct pluripotent states. J Cell Biochem. 2008;105(4):940-948. [DOI] [PubMed]
- 47.Ladhoff J, Bader M, Brösel S, et al. Low immunogenicity of endothelial derivatives from rat embryonic stem cell-like cells. Cell Res. 2009;19(4):507–518. doi: 10.1038/cr.2009.21. [DOI] [PubMed] [Google Scholar]
- 48.Seliger B, Abken H, Ferrone S. HLA-G and MIC expression in tumors and their role in anti-tumor immunity. Trends Immunol. 2003;24(2):82–87. doi: 10.1016/s1471-4906(02)00039-x. [DOI] [PubMed] [Google Scholar]
- 49.Ledran MH, Krassowska A, Armstrong L, et al. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell. 2008;3(1):85-98. [DOI] [PubMed]
- 50.Kyba M, Perlingeiro RC, Daley GQ. HoxB4 confers definitive lymphoid-myeloid engraftment potential on embryonic stem cell and yolk sac hematopoietic progenitors. Cell. 2002;109(1):29-37. [DOI] [PubMed]
- 51.Rachamim N, Gan J, Segall H, et al. Tolerance induction by “megadose” hematopoietic transplants: donor-type human CD34 stem cells induce potent specific reduction of host anti-donor cytotoxic T lymphocyte precursors in mixed lymphocyte culture. Transplantation. 1998;65(10):1386–1393. doi: 10.1097/00007890-199805270-00017. [DOI] [PubMed] [Google Scholar]
- 52.Ophir E, Reisner Y. The use of donor-derived veto cells in hematopoietic stem cell transplantation. Front Immunol. 2012;3:93. [DOI] [PMC free article] [PubMed]
- 53.Dhodapkar KM, Feldman D, Matthews P, et al. Natural immunity to pluripotency antigen OCT4 in humans. Proc Natl Acad Sci USA. 2010;107(19):8718–8723. doi: 10.1073/pnas.0915086107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.