

Abstract
Background and Purpose
Corticosteroid insensitivity is a major therapeutic problem for some inflammatory diseases including chronic obstructive pulmonary disease (COPD), and it is known to be induced by reduced histone deacetylase (HDAC)-2 activities via activation of the phosphoinositide 3-kinase (PI3K) pathway. The aim of this study is to evaluate effects of a novel macrolide/fluoroketolide, solithromycin (SOL, CEM-101), on corticosteroid sensitivity induced by oxidative stress.
Experimental Approach
Corticosteroid sensitivity was determined by IC50/EC50 of dexamethasone (Dex) on TNF-α-induced CXCL8 production in U937 monocytic cell line and peripheral blood mononuclear cells (PBMC) from COPD patients. Activities of HDAC and protein phosphatase 2A (PP2A) were measured by fluorescence-based assay in cells exposed to hydrogen peroxide (H2O2). We also investigated steroid insensitive airway neutrophilia in cigarette smoke exposed mice in vivo.
Key Results
SOL (10 μM) restored Dex sensitivity in PBMC from COPD patients, H2O2-treated U937 cells and phorbol 12-myristate 13-acetate-differentiated U937 cells. In addition, SOL restored HDAC activity with concomitant inhibition of Akt phosphorylation as surrogate marker of PI3K activation. The inhibition of Akt phosphorylation by SOL was due to increased PP2A phosphatase activity, which was reduced in COPD and oxidative stress model. Other known macrolides, such as eryhthromycin, clarithromycin and azithromycin, were significantly less effective in these responses. In cigarette smoke-exposed mice, SOL (100 mg kg−1, po) showed significant but weak inhibition of neutrophilia, whereas Dex (10 mg kg−1, p.o.) showed no such effect. However, a combination of SOL and Dex inhibited neutrophilia by over 50%.
Conclusions and Implications
SOL has potential as novel therapy for corticosteroid-insensitive diseases such as COPD.
Keywords: chronic obstructive pulmonary disease, corticosteroid insensitivity, histone deacetylase 2, macrolide, oxidative stress, phosphaoinositide 3-kinase, protein phosphatase 2A, solithromycin
Introduction
Chronic obstructive pulmonary disease (COPD) is a major global health problem which is due to chronic inflammation of peripheral airways and lung parenchyma. There are no safe and effective anti-inflammatory therapies for COPD and discovery of new agents is held back by a fundamental lack of knowledge about the cellular, molecular and genetic mechanisms of the disease (Rabe et al., 2007; Ito and Barnes, 2009). Although the mechanisms of airway inflammation in COPD are complex, there is increased expression of multiple inflammatory genes, including CXC chemokines (CXCL1, CXCL8), TNF-α and MMP-9 (Nakamaru et al., 2009; Barnes, 2009a). In sharp contrast to asthma, corticosteroid treatment is poorly effective in COPD patients because the inflammation seen in COPD is corticosteroid insensitive (Barnes and Adcock, 2009).
Histone deacetylases (HDAC) are recruited by co-repressor proteins to switch off gene transcription. In particular, HDAC2 is a prerequisite molecule for corticosteroid-dependent anti-inflammatory action, and HDAC2 is associated with pro-inflammatory transcription factors bound to promoter region of pro-inflammatory genes together with activated corticosteroid receptor (GR) (Barnes, 2009b). HDAC2 activity and expression are markedly reduced in COPD lungs airways and alveolar macrophages (Cosio et al., 2004; Ito et al., 2005). This may explain the poor response to corticosteroids in COPD. Oxidative stress, which is a central driving mechanism in COPD, activates the enzyme phosphoinositide-3-kinase-δ (PI3Kδ). Phosphatidylinositol (3,4,5)-triphosphate produced via PI3Kδ activation phosphorylates downstream kinase Akt via PDK1 or PDK2 activation (Alessi and Downes, 1998), resulting in the phosphorylation and inactivation of HDAC2 (Barnes and Adcock, 2009; Barnes, 2009b). Activated Akt is regulated by dephosphorylation at Thr308 and Ser473 as well as by degradation of Akt proteins. Protein phosphatase 2A (PP2A) is a serine/threonine phosphatase that dephosphorylates Akt at these sites (Desaki et al., 2004; Parnham, 2005).
Since the discovery of erythromycin (EM) therapy for diffuse panbronchiolitis, macrolides have been postulated to reduce airway inflammation. Clinical and experimental data indicate that macrolides have anti-inflammatory properties independently of their antibacterial effects (Takizawa et al., 1995; Takizawa et al., 1997; Desaki et al., 2004; Parnham, 2005; Crosbie and Woodhead, 2009; ). Recently, a number of reports showed beneficial effects of macrolides on exacerbation of COPD (Milstone, 2008; He et al., 2010; Albert et al., 2011; Pomares et al., 2011). In addition, clarithromycin (CAM) is reported to reduce the requirement for corticosteroids in patients with corticosteroid-dependent asthma significantly (Garey et al., 2000; Gotfried et al., 2004). However the molecular mechanism of anti-inflammatory effects or corticosteroid-sparing effects has not yet been elucidated.
Solithromycin (SOL; CEM-101; Cempra Pharmaceuticals, Inc., Chapel Hill, NC, USA) is a novel fluoroketolide antibacterial agent related to 14-member-ring macrolides that has activity against many bacteria causing pneumonia (Farrell et al., 2010; McGhee et al., 2010) and is currently in clinical development for the treatment of bacterial pneumonia. In this manuscript, we investigated the effects of SOL in corticosteroid sensitivity in corticosteroid insensitive model and its molecular mechanism.
Methods
Further details have been described in web-depository supporting information.
Cells
Peripheral blood mononuclear cells (PBMCs), U937 cells, were cultured as previously described (Charron et al., 2009; Kobayashi et al., 2011). U937 were differentiated into an adherent macrophage-like morphology by exposure to phorbol 12-myristate 13-acetate (PMA; 50 or 100 ng ml−1) for 48 or 72 h.
Corticosteroid sensitivity
PBMCs were treated with dexamethasone (Dex; 10−11 to 10−6 M) for 45 min, followed by the TNF-α (10 ng mL−1) stimulation overnight. U937 cells were incubated with H2O2 4 h before, treated with macrolides 1 h before and treated with Dex 45 min before the TNF-α (10 ng mL−1) stimulation, and cells were incubated for 4 h further. PMA-differentiated U937 cells were treated with macrolides for 1 h, and then stimulated with H2O2 (1 mM) 1 hr before and treated with Dex 45 min before the TNF-α (10 ng mL−1) stimulation for 4 h. CXCL8 in supernatant was evaluated by ELISA according to the manufacturer's instructions (R&D Systems Europe, Abingdon, UK). As markers of steroid sensitivity, IC50 values for Dex (Dex-IC50) on CXCL8 production for PBMCs, Dex-IC50, EC50 values for Dex (Dex-EC50) and Emax for U937 cells were calculated using Prism 4.0 (GraphPad Software Inc., San Diego, CA, USA).
Cell lysis and Western blotting
Protein preparation from cells and Western blotting analysis were performed as previously described (Kobayashi et al., 2011). Phosphatase inhibitor (Active Motif, Rixensart, Belgium) was used as needed.
In cell HDAC assay and HDAC2 activity
For ‘In cell HDAC assay’, cells were incubated with Fluor de Lys™ substrate (40 μM) for 1 h before cell lysis. To measure HDAC2 activity, HDAC2 was immunoprecipitated (IP) with anti-HDAC2 antibody in cell lysate. HDAC2-IP samples with assay buffer were incubated with Fluor de Lys substrate for 1 h. HDAC activity was expressed as μM of fluorescence standard provided in the kit.
Protein phosphatase activity
Phosphatase activities in cell lysates and immunoprecipitates with the rabbit anti-PP2A antibody (Bethyl Laboratories Inc., Montgomery, TX, USA) were determined using the SensoLyte™ MFP Protein Phosphatase Assay system (AnaSpec, San Jose, CA, USA) as previously reported (Kobayashi et al., 2011).
Membrane protein extraction
Membrane protein fraction was isolated using ProteoExtract® Transmembrane Protein Extraction Kit (Novagen, Madison, WI, USA).
Cigarette smoke exposure model in vivo
C57BL/6J mice (male, 4 weeks) were purchased from CLEA Japan, Inc, (Tokyo, Japan) and adapted for 1 week. Mice were exposed to cigarette smoke (4% cigarette smoke diluted with compressed air) for 30 min day−1 for 12 days as previously reported (Nakamaru et al., 2009), and five mice were used for each group. SOL (100 mg kg−1, p.o.) and Dex (10 mg kg−1, p.o.) were resuspended into 0.5% carboxylmethylcysteine and administered orally 1 h before 5th–12th tobacco smoke exposure. Bronchoalveolar lavage was performed at 24 h after the last cigarette smoke exposure, and the number of alveolar macrophages and neutrophils was determined from Diff-Quick stained specimens. This animal study was approved by The Experimental Animal Ethics Committee in Kyorin University.
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Statistical analysis
Comparisons of two groups of data were performed using the Mann–Whitney U-test, a paired t-test or the Wilcoxon signed rank test. Correlation coefficients were calculated with the use of Spearman's rank method. Other data were analysed by one-way anova with post hoc test, as appropriate. The difference was considered significant at P < 0.05. The results were expressed as the mean ± SEM.
Results
SOL restored corticosteroid sensitivity in PBMCs from COPD patients and oxidative stress exposed U937cells
PBMC from six COPD patients and four healthy subjects were stimulated with TNF-α in the presence or absence of Dex, and the IC50 value on CXCL8 release was calculated as the index of Dex sensitivity (Dex-IC50). Corticosteroid sensitivity was determined as the IC50 value of Dex. The Dex-IC50 value in COPD patients was 15.0 ± 4.6 nM, which was 4.1 higher than that of healthy subjects (Dex-IC50 (3.7 ± 0.38 nM), suggesting that PBMCs from COPD patients were fourfold less sensitive to Dex than healthy subjects. SOL (10 μM) significantly improved Dex-IC50 (15.0 ± 4.6 nM in vehicle control, 6.5 ± 1.4 nM in SOL, P < 0.01) (Figure 1A). EM and CAM demonstrated a tendency to decrease Dex-IC50 values at higher concentrations (100 μM), whereas azithromycin (AZM) had no effect (Figure 1B, Supporting Information Fig. S1 for individual plots).
Figure 1.

Effects of macrolides on corticosteroid sensitivity in PBMCs from COPD patients and H2O2-treated U937 cells. (A,B) Effects of SOL, (10 μM) (A) EM, CAM and AZM at 100 μM on Dex sensitivity in PBMCs from COPD patients. PBMCs were incubated with macrolides for 30 min. The sensitivity to Dex was evaluated on TNF-α-induced CXCL8 production. (C–F) U937 cells were stimulated with H2O2 (200 μM) at 4 h before and then treated with Dex (10−11 to 10−6 M) at 45 min before TNF-α stimulation for overnight. SOL (10, 100 μM) (C), EM (10, 100 μM) (D), CAM (10, 100 μM) (E) and AZM (100 μM) (F) were added 1 h before TNF-α stimulation. Data in C–F were expressed as mean ± SEM of three experiments.
We also used an H2O2-dependent steroid-insensitive model in U937 cells. H2O2 (200 μM for 4 h) shifted Dex-inhibition curve to the right (Figure 1C) and the Dex-IC50 value of H2O2-treated cells was 16-fold higher than that of intact cells (Dex-IC50: 0.73 ± 0.065 nM in NT, 11.6 ± 1.2 nM in H2O2), suggesting 16-fold less sensitive to Dex treatment (Figure 1C, Table 1). When the macrolides were treated at 3 h after H2O2 stimulation and then cells were stimulated with TNF-α at 1 h after the macrolide treatment. EM did not restore the corticosteroid sensitivity at 10 μM, but significantly improved at 100 μM H2O2 (Dex-IC50: 11.6 nM in H2O2, 8.5 nM in H2O2 with EM at 10 μM, 2.6 nM in H2O2 with EM at 100 μM) (Figure 1D, Table 1). Similarly, CAM did not restore the corticosteroid sensitivity at 10 μM, but improved it at 100 μM H2O2 (Dex-IC50: 11.6 nM in H2O2, 13.8 nM in H2O2 with CAM at 10 μM, 4.8 nM in H2O2 with CAM at 100 μM) (Figure 1E, Table 1). In contrast, AZM did not show any significant effect on Dex sensitivity at 100 μM (Figure 1F, Table 1). Pharmacological parameter EC50 analysis also showed similar trend, and Emax was significantly improved only in SOL, 100 μM, treated cells (Table 1). Furthermore, we also evaluated Dex sensitivity in TNF-α-induced IL-1β production and also IL-6 production (Supporting Information Table S2). The level of IL-1β and IL-6 were much lower than CXCL8, but H2O2 showed reduction of Dex sensitivity. Also, SOL restored Dex sensitivity as shown above.
Table 1.
Effect of macrolides on Dex-concentration response on TNF-α-induced CXCL8 production in U937 cells
| Treatment | Concentration | Intact U937 (H2O2, 200 μM, 4 h) | PMA-U937 (H2O2, 1 mM, 30 min) | ||||
|---|---|---|---|---|---|---|---|
| IC50-Dex | EC50-Dex | Emax | IC50-Dex | EC50-Dex | Emax | ||
| (μM) | (nM) | (nM) | (%) | (nM) | (nM) | (%) | |
| NT | 0.73 ± 0.065 | 0.56 ± 0.12 | 90 ± 3.2 | 8.5 ± 0.59 | 4.9 ± 0.56 | 76 ± 1.6 | |
| H2O2 | 11.6 ± 1.2 | 4.5 ± 0.60 | 68 ± 0.76 | 38.5 ± 3.4 | 21.3 ± 2.6 | 76 ± 0.41 | |
| +SOL | 10 | 2.5 ± 0.25 | 1.1 ± 0.18 | 74 ± 0.93 | |||
| 100 | 1.4 ± 0.15 | 0.84 ± 0.12 | 88 ± 0.36 | 5.0 ± 0.69 | 3.2 ± 1.6 | 74 ± 0.27 | |
| +EM | 10 | 8.5 ± 0.40 | 3.1 ± 0.31 | 68 ± 0.74 | |||
| 100 | 2.6 ± 0.28 | 1.1 ± 0.13 | 76 ± 0.54 | 54.4 ± 2.4 | 17.3 ± 0.98 | 71 ± 2.9 | |
| +CAM | 10 | 13.8 ± 1.5 | 5.1 ± 1.5 | 70 ± 4.0 | |||
| 100 | 4.8 ± 0.41 | 2.4 ± 0.25 | 82 ± 0.97 | 56.2 ± 8.4 | 17.9 ± 5.0 | 71 ± 1.4 | |
| +AZM | 100 | 9.4 ± 1.6 | 3.1 ± 0.27 | 70 ± 2.6 | 83.5 ± 16.0 | 46.5 ± 21.8 | 71 ± 1.1 |
We then evaluated Dex sensitivity in PMA-differentiated macrophage like U937 cells. The Dex-IC50 value in intact U937 cells was 0.73 ± 0.065 nM as shown before, but the Dex-IC50 value in PMA-treated U937 cells was 8.5 ± 0.59 nM, suggesting that macrophage-like cells were 12-fold less sensitive to Dex. These PMA-treated cells became less sensitive by H2O2 exposure (1 mM for 1 h) (Dex-IC50: 8.5 ± 0.59 nM in NT, 38.5 ± 3.4 nM in H2O2) (Table 1). When the macrolides were added 1 h before H2O2 stimulation and 2 h before TNF-α stimulation, SOL (10 μM) restored Dex sensitivity reduced by exposure to H2O2 (Dex-IC50: 38.5 nM in H2O2, 5.0 nM in H2O2 with SOL at 100 μM) (Table 1). Neither EM, CAM nor AZM restored the corticosteroid sensitivity at 100 μM (Table 1). Analysis using pharmacological parameter EC50 also showed similar trend, and none of the macrolides increased Emax (Table 1).
Reversal of corticosteroid resistance in cigarette smoke-exposed mice in vivo by SOL
C57BL/6J mice were exposed to cigarette smoke for 10 days, and treated with SOL at 100 mg kg−1, p.o. and/or Dex (10 mg kg−1, p.o.) once daily in the last 3 days of cigarette smoke exposure. Cigarette smoke exposure significantly increased alveolar macrophages (air: 4.3 ± 0.53 × 104 cells mL−1, smoke + vehicle: 16.6 ± 0.81 × 104) and neutrophils (air: 2.2 ± 0.39 × 104 cells mL−1, smoke + vehicle: 12.9 ± 1.2 × 104). As shown in Figure 2A,B, Dex did not inhibit alveolar macrophage and neutrophil accumulation. Although SOL showed only 17% inhibition on alveolar macrophage accumulation (smoke + vehicle: 16.6 ± 0.81 × 104 cells mL−1, smoke + SOL: 14.5 ± 1.6 × 104), it significantly inhibited neutrophil accumulation by 38% (smoke + vehicle: 12.9 ± 1.2 × 104 cells mL−1, smoke + SOL: 8.8 ± 0.8 × 104). When SOL was administered with Dex, the combination strongly inhibited both alveolar macrophage (11.2 ± 8.3 × 104 cells, 43% inhibition) and neutrophil accumulation (6.4 ± 0.95 × 104 cells, 61% inhibition) (Figure 2A,B).
Figure 2.
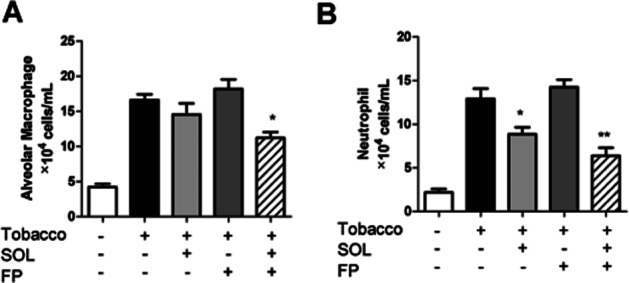
Effects of SOL with or without Dex on alveolar macrophage and neutrophil accumulation in cigarette smoke-exposed mice. C57BL/6J mice (n = 5 per group) were exposed to cigarette smoke (4%) for 30 min day−1 for 12 days. SOL (100 mg kg−1 orally), Dex (10 mg kg−1 orally), a combination of these treatments were administered once daily at 1 h before 5th−12th smoke exposure. BAL was collected 24 h after the final smoke exposure. The number of alveolar macrophages (A) and neutrophils (B) in BAL fluid was calculated; *P < 0.05, **P < 0.01 (vs. smoke control).
SOL restores HDAC activity and expression in parallel with inhibition of Akt phosphorylation under oxidative stress
PMA-dependent differentiation to macrophage-type cells caused reduction of HDAC activity in U937 cells as seen in Figure 3A [HDAC activity (μM of standard): 0.40 ± 0.020 in intact U937 cells: 0.17 ± 0.015 in PMA differentiated U937 cells]. This reduction was completely inhibited by 100 μM of N-acetyl cysteine, an antioxidant, suggesting that the reduction is due to endogenous oxidative stress produced during differentiation. The total HDAC activity in PMA-differentiated cells was further reduced by 36% after H2O2 exposure to 0.11 ± 0.022 μM of standard (Figure 3A). SOL concentration-dependently increased total HDAC activity, and the level at 100 μM was close to the level seen in intact U937 cells rather than that of PMA-differentiated U937 cells (Figure 3B). All standard macrolides (EM, CAM and AZM) also restored H2O2-dependent reduction of HDAC activity (33–333 μM), but the efficacies were not significant. The concentrations to increase total HDAC activity by 300% of that of H2O2-exposed PMA U937 cells (EnC300) were 6.7, 101, 128 and 169 μM in SOL, EM, CAM and AZM respectively. Thus, SOL was 10-fold more potent on an increase in HDAC activity.
Figure 3.

Effects of macrolides on total HDAC activity and Akt phosphorylation under oxidative stress. (A) Reduction of HDAC activity in PMA-differentiated U937 cells, and reversal by N-acetyl cysteine (NAC). Cells were treated with PMA (50 ng mL−1) for 48 h, and NAC 100 μM was treated 20 min before and 24 h after PMA treatment. (B) Effects of macrolides on total HDAC activity in PMA-differentiated U937 cells. Cells were pretreated with macrolides (10 to 330 μM) for 20 min and then treated with H2O2 (1 mM) for 30 min. Total HDAC activity was determined by in-cell HDAC activity assay. (C) Effects of macrolides on H2O2-induced phosphorylation of Akt in PMA-differentiated U937 cells. Cells were pretreated with macrolides (33 to 330 μM) for 20 min, followed by H2O2 (1 mM) stimulation for 30 min. Phosphorylation levels of Akt were measured by Western blot (C) and calculated relative to total Akt protein (D). Values represent means of three experiments ± SEM. #P < 0.05 (vs. vehicle control), *P < 0.05, **P < 0.01 (vs. treatment with H2O2).
H2O2 (1 mM) also clearly increased the level of Akt phosphorylation (Figure 3C,D). SOL inhibited the H2O2-induced Akt phosphorylation in a concentration-dependent manner, and the IC50 value was 21 μM, and the level was similar to the EnC300 for HDAC activity by SOL (6.7 μM, Table 2). EM also concentration-dependently inhibited Akt phosphorylation and showed 46% inhibition at 100 μM. However, the inhibitory activity was not increased at 333 μM. Both CAM and AZM also weakly inhibited it with IC50 values of 201 and 247 μM respectively (Figure 3C,D) (Table 2).
Table 2.
Effect of macrolides on H2O2-induced reduction of HDAC activity and Akt phosphorylation
| HDAC activity | pAkt | |
|---|---|---|
| (H2O2, 1 mM, 30 min) | (H2O2, 1 mM, 30 min) | |
| EnC300 (μM) | IC50(μM) | |
| SOL | 6.7 | 21 |
| EM | 101 | 46% at 100 μM |
| CAM | 128 | 201 |
| AZM | 169 | 247 |
EnC300: concentrations to achieve to 300% of the level of PMA-H2O2 control.
Although effects of macrolides on PI3K enzyme activity were evaluated as the molecular mechanism of inhibition of Akt phosphorylation, none of macrolides inhibited PI3Kα, β, γ and δ enzyme activity up to 333 μM (Supporting Information Table S3).
Protein phosphatase PP2A is involved in SOL-mediated effects
As pAkt is reported to be dephosphorylated by phosphatase PP2A, we investigated the effects of okadaic acid (OA) (10−9 M), a selective PP2A inhibitor, on restoration of corticosteroid sensitivity by SOL. Although pretreatment with SOL (10 μM) completely restored Dex sensitivity reduced under H2O2 (500 μM, 1 h) in PMA-differentiated U937 cells (Dex-IC50: 8.5 ± 0.59 nM in vehicle control, 38.5 ± 3.4 nM in H2O2, 5.0 ± 0.69 nM μM in H2O2 with SOL, n = 3). However, OA treatment completely reversed SOL effects (45.8 ± 7.4 nM in H2O2 with SOL plus OA, n = 3) (Figure 4A).
Figure 4.

Effects of protein phosphatase PP2A inhibitor on SOL-mediated actions under oxidative stress and PP2A activity in PBMCs from COPD patients. Cells preincubated with a selective PP2A inhibitor, OA (10−9 M) for 1 h (A and B) or overnight (C and D) were treated with SOL (100 μM) for 20 min, followed by exposure to H2O2 (500 μM for 30 min). (A) Effect of OA on SOL-mediated restoration of Dex sensitivity in PMA-differentiated U937 cells. (B) Effect of OA on SOL-mediated HDAC2 activation in PMA-differentiated U937 cells. HDAC2 was immunopurified and HDAC2 activity was determined. (C,D) Effect of OA on SOL-mediated dephosphorylation of HDAC2 and Akt in PMA-differentiated U937 cells. Phosphorylation levels of HDAC2 (C) and Akt (D) were measured by Western blot and normalized to the expression of total HDAC2 or Akt protein. (E) PP2A activity in PBMCs from four healthy volunteers (HV), four patients with mild and moderate COPD (GOLD stage I and II) and five patients with severe and very severe COPD (GOLD stage III and IV). (F) Effect of SOL on PP2A activity in PBMCs from COPD patients. Cells were treated with SOL (10 μM) for 20 min before lysation. Values represent means of three experiments ± SEM. Individual values and means of subjects are shown in E. Six individual values are shown in F. #P < 0.05 (vs. HV or NT), *P < 0.05, **P < 0.01 (as shown between two groups).
As shown in Figure 4B, H2O2 exposure (500 μM, 1 h) significantly reduced activity of IP HDAC2 by 35% [HDAC2 activity (μM of standard): 0.12 ± 0.010 of non-treatment control, 0.078 ± 0.014 of H2O2 treated, n = 3] and SOL (10 μM) clearly restored H2O2-induced reduction of HDAC2 activity [HDAC2 activity (μM of standard): 0.16 ± 0.014 of H2O2 plus SOL treated, n = 3]. However, OA almost completely reversed the effect of SOL [HDAC2 activity (μM of standard); 0.12 ± 0.010 in NT, 0.078 ± 0.014 in H2O2, 0.16 ± 0.014 in H2O2 with SOL, 0.079 ± 0.0081 in H2O2 with SOL plus OA, n = 3] (Figure 4B). Furthermore, H2O2 (500 μM, 30 min) enhanced HDAC2 phosphorylation with concomitant induction of Akt phosphorylation, and OA abrogated SOL-mediated dephosphorylation of HDAC2 and Akt (Figure 4C,D).
As shown in Figure 4E, PP2A activity was significantly reduced in PBMCs from patients with COPD compared with healthy volunteers. PBMCs from COPD GOLD stages III–IV showed further reduction of PP2A activity compared with patients with COPD stages I–II, and there was a positive correlation between PP2A activity in PBMCs and lung functions [FEV1 (% predicted) and FEV1/FVC] (Supporting Information Fig. S2). Interestingly, SOL increased PP2A activity in PBMC from patients with COPD [PP2A activity (AFU μg−1 min−2); 0.94 ± 0.13 in NT, 1.51 ± 0.20 in SOL, n = 6] (Figure 4F). In addition, although H2O2 decreased PP2A activity, SOL restored H2O2-induced reduction of PP2A activity in PMA-differentiated U937 cells [PP2A activity (AFU μg−1 min−2); 0.77 ± 0.01 in NT, 0.68 ± 0.02 in H2O2, 0.92 ± 0.06 in H2O2 with SOL, n = 3]. SOL also activated PP2A in cell membrane fraction within 3 min after treatment and this effect lasted more than 20 min (Supporting Information Fig. S3). In addition, immunopurified PP2A from U937 cells was activated by SOL (10 μM), and the effect was more potent or similar to that of EM (100 μM), at 10 times higher concentration than SOL [PP2A activity (AFU/μg min−1); 0.63 ± 0.05 in NT, 1.14 ± 0.10 in SOL, 0.97 ± 0.06 in EM, n = 4] (Supporting Information Fig. S3C).
Discussion and conclusions
In this study, we demonstrated that a novel macrolide/fluoroketolide, SOL (CEM-101), restored corticosteroid sensitivity in PBMC from COPD patients. SOL up-regulated HDAC2 activity under conditions of oxidative stress via inhibition of Akt phosphorylation, resulting in restoration of corticosteroid sensitivity. Protein phosphatase PP2A activation by SOL is likely a key molecular mechanism in PI3K signalling inhibition, thus demonstrating a novel mechanism of action.
A major problem in the therapy of COPD is its poor response to corticosteroids. In our data, the Dex-IC50 value was fourfold higher in PBMCs of COPD patients than that of healthy subjects. We used TNF-α-induced CXCL8 release in PBMCs as readout to determine corticosteroid sensitivity. CXCL8 is abundant in bronchoalveolar lavage fluid and sputum obtained from COPD patients (Hollander et al., 2007; Paone et al., 2011). This is a chemotactic factor of neutrophils, and activated neutrophils cause lung injury and emphysema (Hollander et al., 2007). Therefore, CXCL8 is believed to be involved in pathogenesis of COPD. Even more importantly, CXCL8 is relatively corticosteroid insensitive in intact cells and also more resistant to corticosteroid treatment in COPD in vitro and in vivo (Ford et al., 2010; Knobloch et al., 2011). In addition, practically the induction level of CXCL8 is robust and the CXCL8 production was not often interfered with test agents. To determine corticosteroid sensitivity of patients, we found that TNF-α-induced CXCL8 in PBMCs is better readout than other cytokines and stimulation. For example, LPS-induced cytokine production is corticosteroid sensitive and the levels of cytokines are very variable, possibly due to different expression of TLR4 in patients. Therefore, we have established this system for evaluation of agents on corticosteroid sensitivity, and we already tested theophylline, formoterol, salmeterol, nortriptyline, PI3K inhibitors and p38MAPK inhibitors (To et al., 2010; Mercado et al., 2011a,b; 2012; Kobayashi et al., 2012). Thus, this system using TNF-α-induced CXCL8 system is robust and reproducible, and we are able to detect efficacy using PBMCs from six donors based on power calculations ‘if the agent induces more than 2 fold reduction of Dex-IC50’. Therefore, we recruited six COPD patients initially. This Dex insensitivity seen in PBMCs of COPD on TNF-α-induced CXCL8 production was also replicated well in U937 monocytic cells exposed to H2O2 (Rossios et al., 2012). As shown in Figure 1C, H2O2-treated cells were 10-fold less sensitive to Dex treatment than intact cells. In addition, the Dex insensitivity by H2O2 was also seen in TNFα-induced IL-1β and IL-6 production, and SOL reversed the corticosteroid insensitivity in U937 cells (Supporting Information Table S2).
The main aim of this study was to evaluate SOL in these corticosteroid-insensitive models, and our data for the first time showed that SOL restored corticosteroid sensitivity in PBMC from COPD patients and human monocytic culture cells under conditions of oxidative stress (Figure 1A,B,C). Macrolides currently used in the clinic, EM and CAM, partially (but not significantly) restored corticosteroid sensitivity in PBMCs from COPD patients at 10-fold higher concentrations compared with SOL (Figure 1B, Supporting Information Fig. S1A–C). These findings were supported by previous reports showing that CAM significantly reduced the requirement for corticosteroids in patients with corticosteroid-dependent asthma (Garey et al., 2000; Gotfried et al., 2004). If the clinical effect of CAM is real, SOL could have the potential to improve corticosteroid sensitivity further. As resident macrophage is known to be less sensitive to corticosteroid treatment, we also prepared macrophage-like cells by incubation with PMA. As seen in Figure 4A and Table 1, PMA-differentiated macrophage like U937 cells was less sensitive to Dex and H2O2 treatment further reduced corticosteroid sensitivity. In this system, only SOL restored Dex sensitivity at 10 μM.
This enhancement of corticosteroid effect by SOL was also confirmed in corticosteroid-insensitive inflammation in cigarette smoke-exposed mice in vivo. Eleven-day smoke exposure-induced airway neutrophilia was not inhibited by Dex (10 mg kg−1, p.o.), which is able to inhibit LPS-induced neutrophilia by 70% (data not shown). Neutrophil accumulation was more sensitive to SOL treatment than alveolar macrophage accumulation. As we reported that SOL is able to inhibit CXCL8 production induced by LPS in U937 cells with IC50 of 78 μM, which is more than 10-fold superior to any other macrolides (Kobayashi et al., 2013), SOL affected alveolar macrophages to inhibit CXCL8, leading inhibition of neutrophil chemotaxis.
The major cause of corticosteroid insensitivity in COPD is reported to be down-regulation of HDAC2 activity and expression (Cosio et al., 2004; Ito et al., 2005). HDAC2 is known to be phosphorylated mainly at Ser349, Ser422 and Ser424 (Tsai and Seto, 2002). Activated PI3Kδ, which is up-regulated/activated in COPD and by oxidative stress, and consequently phosphorylated Akt, phosphorylates HDAC2, leading to enhancement of ubiquitination and degradation of HDAC2 by the proteasome pathway (Ito et al., 2007; Adenuga et al., 2009; Barnes and Adcock, 2009; Osoata et al., 2009). Recent studies showed that inhibition of PI3Kδ restores corticosteroid function in cells from COPD patients and in an oxidative stress-induced corticosteroid insensitive model (Marwick et al., 2009; 2010). We demonstrated that PMA treatment reduced HDAC activity by production of endogenous oxidative stress (Figure 3A) and H2O2 further reduced HDAC activity. In this system, SOL remarkably reverses oxidative stress-induced reduction of HDAC2 activity, and increased further to the level of intact (non-PMA treated) U937 cells (Figure 3B). The increase in HDAC activity was in parallel with inhibition of Akt phosphorylation (Figure 3C,D) and dephosphorylation of HDAC2 (Figure 4C). Other macrolides, such as EM and CAM, also restored HDAC activity and inhibited Akt phosphorylation, but the effects were more than 100-fold weaker than SOL, which is consistent in all systems through this manuscript. It is not clear why SOL is better than other known macrolides. EM is the first macrolide from which acid-stable second-generation macrolides, CAM and AZM were derived. These antibiotics had the same antibacterial activity and anti-inflammatory properties but had better pharmacokinetics than the older EM. The antibacterial activity of the macrolides is attributed to the binding of the N-dimethyl group on the desosamine sugar which is common to all macrolides. The structure responsible for the anti-inflammatory properties is not known but is attributed to the macrocyclic ring as all macrolides appear to have some of this activity. A new generation of macrolides was developed called the ketolides, in which the cladinose sugar of CAM is displaced by a keto group, and a C11-C12 carbamate with a side chain added stability to the macrocyclic ring and adding a second binding site to the bacterial ribosome. Telithromycin was the first approved ketolide but has failed because of serious adverse events. SOL differs from telithromycin in not having a pyridine in the side chain which has been attributed to have resulted in ACh nicotinic acid receptor activity inhibition resulting in major adverse events (Bertrand et al., 2010). In addition, in SOL the keto group at the 3-position is protected by a fluorine at the 2-position and remains in the keto configuration, and unlike in the older ketolides like telithromycin which converts where the keto group converts to an enol group. The fluorine group likely contributes to better bioavailability and pharmacokinetics as well also is a third binding site on the ribosome (Llano-Sotelo et al., 2010). SOL has been demonstrated to accumulate 200 times the plasma level in the alveolar macrophages and 10 times the concentration in the epithelial lining fluid (Rodvold et al., 2012), and also accumulated more in macrophage-type THP1 cells (Lemaire et al., 2009). Thus, unique chemical structure contributes to the potent efficacy. Further structure–activity relationship analysis will be required.
PI3Kδ enzyme inhibition is reported as the mechanism of restoration of HDAC activity (Marwick et al., 2009; To et al., 2010). However, none of the macrolides showed inhibitory effects in a direct PI3Kα, β, γ and δ enzyme assay (Supporting Information Table S3) in contrast to theophylline and IC87114. This suggests that SOL inactivates PI3K-Akt pathway in a different way rather to direct kinase inhibition. PP2A, a serine/threonine phosphatase, is capable of dephosphorylating Akt in vitro, and OA, a PP2A inhibitor, has been shown to reverse PP2A-mediated Akt dephosphorylation (Andjelkovic et al., 1996). We found that PP2A inhibition by OA abrogated SOL-mediated restoration effects on HDAC2 activity and corticosteroid sensitivity which are impaired under oxidative stress (Figure 4A,B) as well as pAkt inhibition (Figure 4D). Moreover, SOL increased PP2A activity, which was reduced in COPD and in an oxidative stress model (Figure 4 and Supporting Information Fig. S3A). The effect of SOL was equivalent or superior to that of EM at 10 times higher concentration than SOL (Supporting Information Fig. S3). Another possible mechanism of PI3K-Akt inhibition would be the enhancement of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) activation (Cantley and Neel, 1999). Oxidative stress activates PI3K-dependent signalling via inactivation of PTEN (Leslie et al., 2003). Although it remains unclear whether SOL can activate PTEN, PTEN is unlikely involved in SOL's effect as U937 cells have undetectable levels of PTEN expression (Dahia et al., 1999) and we did find that SOL did not change PTEN activity in A549 cells (data not shown). It is still unclear how SOL activates PP2A. PP2A is known to activate by cAMP (Feschenko et al., 2002), but we reported that formoterol restored corticosteroid sensitivity in severe asthma via PP2A activation but in cAMP-independent manner (Kobayashi et al., 2012). Further, molecular-based analysis, including structure–activity relationship, will be required to identify the mechanism of PP2A activation by SOL.
In conclusion, our findings revealed that a novel macrolide fluoroketolide, SOL (CEM-101), restored corticosteroid sensitivity by inhibition of PI3K signalling under oxidative stress, and was more potent than any other macrolides tested. SOL has potential as a therapy for corticosteroid insensitive diseases, such as COPD.
Acknowledgments
We would like to thank Dr Prabhavathi Fernandes (Cempra Phamaceuticals) for supplying the CEM-101 and relevant information for the compound. We would also like to thank Mr Arnell Colongon and Ms Sally Meah for their support in the recruitment of patients.
Glossary
- AZM
azithromycin
- CAM
clarithromycin
- COPD
chronic obstructive pulmonary disease
- Dex
dexamethasone
- EM
erythromycin
- HDAC2
histone deacetylase 2
- PBMC
peripheral blood mononuclear cells: PI3K, phosphoinositide 3 kinase
- PMA
phorbol 12-myristate 13-acetate
- PP2A
protein phosphatase 2
- SOL
solithromycin
Conflicts of interest
This work was supported by project grant from Cempra Pharmaceuticals. The funding sources provided relevant information of compound and reviewed the manuscript before submission, but played no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. KI is currently employed by RespiVert Ltd and has honorary contract with Imperial College. PJB has served on Scientific Advisory Boards of AstraZeneca, Boehringer-Ingelheim, Chiesi, Daiichi-Sankyo, GlaxoSmithKline, Novartis, Nycomed, Pfizer, Teva and UCB, and has received research funding from Aquinox Pharmaceutiocals, AstraZeneca, Boehringer-Ingelheim, Chiesi, Daiichi-Sankyo, GlaxoSmithKline, Novartis, Nycomed, Pfizer, RespiVert and Prosonix. The other authors have declared that no competing interests exist for this research.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Effects of macrolides on steroid sensitivity in PBMCs from COPD patients. Cells were incubated with EM (100 μM) (A), CAM (100 μM) (B) and AZM (100 μM) (C) for 30 min. The cell sensitivity to Dex was evaluated. Five (A and C) and six (B) individual values are shown.
Figure S2 Correlation between PP2A activity in PBMCs and lung functions. Correlation with FEV1 (% predicted) (A) and FEV1 FVC−1 (B). Thirteen individual values and the dotted lines indicating 95% confidence interval are shown.
Figure S3 Effects of SOL on PP2A activity. (A) Effect of SOL on H2O2-induced reduction of PP2A activity in PMA-differentiated U937 cells. Cells pretreated with SOL (10 μM) for 20 min were exposed to H2O2 (200 μM) for 4 h before whole-cell extraction. (B) Time-course study of SOL effect on membrane-located PP2A activity. Activities of immunopurified PP2A from membrane protein fraction were measured with SOL (10 μM; solid triangle and 100 μM; open triangle) treatment for 3–25 min. Data are expressed as fold changes against baseline (Time 0). (C) Effects of SOL and EM on IP PP2A activity in U937 cells. SOL and EM were incubated with IP-PP2A for 20 min. Values represent means of three (A,C) or four (B) experiments ± SEM. #P < 0.05, ##P < 0.01 (vs. non-treatment control or Time 0), **P < 0.01 (vs. as shown between two groups).
Table S1 Study subjects for PBMCs.
Table S2 Effect of macrolides on Dex-concentration response on TNFα-induced IL-1β and IL-6 production in U937 cells.
Table S3 Effects of SOL and EM on PI3K enzyme activity.
References
- Adenuga D, Yao H, March TH, Seagrave J, Rahman I. Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am J Respir Cell Mol Biol. 2009;40:464–473. doi: 10.1165/rcmb.2008-0255OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert RK, Connett J, Bailey WC, Casaburi R, Cooper JA, Jr, Criner GJ, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365:689–698. doi: 10.1056/NEJMoa1104623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Downes CP. The role of PI 3-kinase in insulin action. Biochim Biophys Acta. 1998;1436:151–164. doi: 10.1016/s0005-2760(98)00133-7. [DOI] [PubMed] [Google Scholar]
- Andjelkovic M, Jakubowicz T, Cron P, Ming XF, Han JW, Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc Natl Acad Sci U S A. 1996;93:5699–5704. doi: 10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. The cytokine network in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2009a;41:631–638. doi: 10.1165/rcmb.2009-0220TR. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Histone deacetylase-2 and airway disease. Ther Adv Respir Dis. 2009b;3:235–243. doi: 10.1177/1753465809348648. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373:1905–1917. doi: 10.1016/S0140-6736(09)60326-3. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Bertrand S, Neveu E, Fernandes P. Molecular characterization of off-target activities of telithromycin: a potential role for nicotinic acetylcholine receptors. Antimicrob Agents Chemother. 2010;54:5399–5402. doi: 10.1128/AAC.00840-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron CE, Chou PC, Coutts DJ, Kumar V, To M, Akashi K, et al. Hypoxia-inducible factor 1alpha induces corticosteroid-insensitive inflammation via reduction of histone deacetylase-2 transcription. J Biol Chem. 2009;284:36047–36054. doi: 10.1074/jbc.M109.025387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosio BG, Tsaprouni L, Ito K, Jazrawi E, Adcock IM, Barnes PJ. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med. 2004;200:689–695. doi: 10.1084/jem.20040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosbie PA, Woodhead MA. Long-term macrolide therapy in chronic inflammatory airway diseases. Eur Respir J. 2009;33:171–181. doi: 10.1183/09031936.00042208. [DOI] [PubMed] [Google Scholar]
- Dahia PL, Aguiar RC, Alberta J, Kum JB, Caron S, Sill H, et al. PTEN is inversely correlated with the cell survival factor Akt/PKB and is inactivated via multiple mechanisms in haematological malignancies. Hum Mol Genet. 1999;8:185–193. doi: 10.1093/hmg/8.2.185. [DOI] [PubMed] [Google Scholar]
- Desaki M, Okazaki H, Sunazuka T, Omura S, Yamamoto K, Takizawa H. Molecular mechanisms of anti-inflammatory action of erythromycin in human bronchial epithelial cells: possible role in the signaling pathway that regulates nuclear factor-kappaB activation. Antimicrob Agents Chemother. 2004;48:1581–1585. doi: 10.1128/AAC.48.5.1581-1585.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell DJ, Sader HS, Castanheira M, Biedenbach DJ, Rhomberg PR, Jones RN. Antimicrobial characterisation of CEM-101 activity against respiratory tract pathogens, including multidrug-resistant pneumococcal serogroup 19A isolates. Int J Antimicrob Agents. 2010;35:537–543. doi: 10.1016/j.ijantimicag.2010.01.026. [DOI] [PubMed] [Google Scholar]
- Feschenko MS, Stevenson E, Nairn AC, Sweadner KJ. A novel cAMP-stimulated pathway in protein phosphatase 2A activation. J Pharmacol Exp Ther. 2002;302:111–118. doi: 10.1124/jpet.302.1.111. [DOI] [PubMed] [Google Scholar]
- Ford PA, Durham AL, Russell RE, Gordon F, Adcock IM, Barnes PJ. Treatment effects of low-dose theophylline combined with an inhaled corticosteroid in COPD. Chest. 2010;137:1338–1344. doi: 10.1378/chest.09-2363. [DOI] [PubMed] [Google Scholar]
- Garey KW, Rubinstein I, Gotfried MH, Khan IJ, Varma S, Danziger LH. Long-term clarithromycin decreases prednisone requirements in elderly patients with prednisone-dependent asthma. Chest. 2000;118:1826–1827. doi: 10.1378/chest.118.6.1826. [DOI] [PubMed] [Google Scholar]
- Gotfried MH, Jung R, Messick CR, Rubinstein I, Garey KW, Rodvold KA, et al. Effects of six-week clarithromycin therapy in corticosteroid-dependent asthma: a randomized, double-blind, placebo-controlled pilot study. Curr Ther Res. 2004;65:1–12. doi: 10.1016/S0011-393X(04)90000-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He ZY, Ou LM, Zhang JQ, Bai J, Liu GN, Li MH, et al. Effect of 6 months of erythromycin treatment on inflammatory cells in induced sputum and exacerbations in chronic obstructive pulmonary disease. Respiration. 2010;80:445–452. doi: 10.1159/000321374. [DOI] [PubMed] [Google Scholar]
- Hollander C, Sitkauskiene B, Sakalauskas R, Westin U, Janciauskiene SM. Serum and bronchial lavage fluid concentrations of IL-8, SLPI, sCD14 and sICAM-1 in patients with COPD and asthma. Respir Med. 2007;101:1947–1953. doi: 10.1016/j.rmed.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135:173–180. doi: 10.1378/chest.08-1419. [DOI] [PubMed] [Google Scholar]
- Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352:1967–1976. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- Ito K, Caramori G, Adcock IM. Therapeutic potential of phosphatidylinositol 3-kinase inhibitors in inflammatory respiratory disease. J Pharmacol Exp Ther. 2007;321:1–8. doi: 10.1124/jpet.106.111674. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch J, Hag H, Jungck D, Urban K, Koch A. Resveratrol impairs the release of steroid-resistant cytokines from bacterial endotoxin-exposed alveolar macrophages in chronic obstructive pulmonary disease. Basic Clin Pharmacol Toxicol. 2011;109:138–143. doi: 10.1111/j.1742-7843.2011.00707.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Mercado N, Barnes PJ, Ito K. Defects of protein phosphatase 2A causes corticosteroid insensitivity in severe asthma. PLoS ONE. 2011;6:e27627. doi: 10.1371/journal.pone.0027627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, Mercado N, Miller-Larsson A, Barnes PJ, Ito K. Increased corticosteroid sensitivity by a long acting beta2 agonist formoterol via beta2 adrenoceptor independent protein phosphatase 2A activation. Pulm Pharmacol Ther. 2012;25:201–207. doi: 10.1016/j.pupt.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Wada H, Rossios C, Takagi D, Higaki M, Mikura S, et al. A novel macrolide solithromycin exerts superior anti-inflammatory effect via NF-κB inhibition. J Pharmacol Exp Ther. 2013;345:76–84. doi: 10.1124/jpet.112.200733. [DOI] [PubMed] [Google Scholar]
- Lemaire S, Van Bambeke F, Tulkens PM. Cellular accumulation and pharmacodynamic evaluation of the intracellular activity of CEM-101, a novel fluoroketolide, against Staphylococcus aureus, Listeria monocytogenes, and Legionella pneumophila in human THP-1 macrophages. Antimicrob Agents Chemother. 2009;53:3734–3743. doi: 10.1128/AAC.00203-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003;22:5501–5510. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano-Sotelo B, Dunkle J, Klepacki D, Zhang W, Fernandes P, Cate JH, Mankin AS. Binding and action of CEM-101, a new fluoroketolide antibiotic that inhibits protein synthesis. Antimicrob Agents Chemother. 2010;54:4961–4970. doi: 10.1128/AAC.00860-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marwick JA, Caramori G, Stevenson CS, Casolari P, Jazrawi E, Barnes PJ, et al. Inhibition of PI3Kdelta restores glucocorticoid function in smoking-induced airway inflammation in mice. Am J Respir Crit Care Med. 2009;179:542–548. doi: 10.1164/rccm.200810-1570OC. [DOI] [PubMed] [Google Scholar]
- Marwick JA, Caramori G, Casolari P, Mazzoni F, Kirkham PA, Adcock IM, et al. A role for phosphoinositol 3-kinase delta in the impairment of glucocorticoid responsiveness in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2010;125:1146–1153. doi: 10.1016/j.jaci.2010.02.003. [DOI] [PubMed] [Google Scholar]
- McGhee P, Clark C, Kosowska-Shick KM, Nagai K, Dewasse B, Beachel L, et al. In vitro activity of CEM-101 against Streptococcus pneumoniae and Streptococcus pyogenes with defined macrolide resistance mechanisms. Antimicrob Agents Chemother. 2010;54:230–238. doi: 10.1128/AAC.01123-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado N, To Y, Ito K, Barnes PJ. Nortriptyline reverses corticosteroid insensitivity by inhibition of phosphoinositide-3-kinase-delta. J Pharmacol Exp Ther. 2011a;337:465–470. doi: 10.1124/jpet.110.175950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado N, To Y, Kobayashi Y, Adcock IM, Barnes PJ, Ito K. p38 mitogen-activated protein kinase-gamma inhibition by long-acting beta2 adrenergic agonists reversed steroid insensitivity in severe asthma. Mol Pharmacol. 2011b;80:1128–1135. doi: 10.1124/mol.111.071993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado N, Hakim A, Kobayashi Y, Meah S, Usmani OS, Chung KF, et al. Restoration of corticosteroid sensitivity by p38 mitogen activated protein kinase inhibition in peripheral blood mononuclear cells from severe asthma. PLoS ONE. 2012;7:e41582. doi: 10.1371/journal.pone.0041582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstone AP. Use of azithromycin in the treatment of acute exacerbations of COPD. Int J Chron Obstruct Pulmon Dis. 2008;3:515–520. doi: 10.2147/copd.s1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamaru Y, Vuppusetty C, Wada H, Milne JC, Ito M, Rossios C, et al. A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J. 2009;23:2810–2819. doi: 10.1096/fj.08-125468. [DOI] [PubMed] [Google Scholar]
- Osoata GO, Yamamura S, Ito M, Vuppusetty C, Adcock IM, Barnes PJ, et al. Nitration of distinct tyrosine residues causes inactivation of histone deacetylase 2. Biochem Biophys Res Commun. 2009;384:366–371. doi: 10.1016/j.bbrc.2009.04.128. [DOI] [PubMed] [Google Scholar]
- Paone G, Conti V, Vestri A, Leone A, Puglisi G, Benassi F, et al. Analysis of sputum markers in the evaluation of lung inflammation and functional impairment in symptomatic smokers and COPD patients. Dis Markers. 2011;31:91–100. doi: 10.3233/DMA-2011-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnham MJ. Immunomodulatory effects of antimicrobials in the therapy of respiratory tract infections. Curr Opin Infect Dis. 2005;18:125–131. doi: 10.1097/01.qco.0000160901.71813.fe. [DOI] [PubMed] [Google Scholar]
- Pomares X, Monton C, Espasa M, Casabon J, Monso E, Gallego M. Long-term azithromycin therapy in patients with severe COPD and repeated exacerbations. Int J Chron Obstruct Pulmon Dis. 2011;6:449–456. doi: 10.2147/COPD.S23655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–555. doi: 10.1164/rccm.200703-456SO. [DOI] [PubMed] [Google Scholar]
- Rodvold KA, Gotfried MH, Still JG, Clark K, Fernandes P. Comparison of plasma, epithelial lining fluid, and alveolar macrophage concentrations of solithromycin (CEM-101) in healthy adult subjects. Antimicrob Agents Chemother. 2012;56:5076–5081. doi: 10.1128/AAC.00766-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossios C, To Y, Osoata G, Ito M, Barnes PJ, Ito K. Corticosteroid insensitivity is reversed by formoterol via phosphoinositide-3-kinase inhibition. Br J Pharmacol. 2012;167:775–786. doi: 10.1111/j.1476-5381.2012.01864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa H, Desaki M, Ohtoshi T, Kikutani T, Okazaki H, Sato M, et al. Erythromycin suppresses interleukin 6 expression by human bronchial epithelial cells: a potential mechanism of its anti-inflammatory action. Biochem Biophys Res Commun. 1995;210:781–786. doi: 10.1006/bbrc.1995.1727. [DOI] [PubMed] [Google Scholar]
- Takizawa H, Desaki M, Ohtoshi T, Kawasaki S, Kohyama T, Sato M, et al. Erythromycin modulates IL-8 expression in normal and inflamed human bronchial epithelial cells. Am J Respir Crit Care Med. 1997;156:266–271. doi: 10.1164/ajrccm.156.1.9612065. [DOI] [PubMed] [Google Scholar]
- To Y, Ito K, Kizawa Y, Failla M, Ito M, Kusama T, et al. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:897–904. doi: 10.1164/rccm.200906-0937OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SC, Seto E. Regulation of histone deacetylase 2 by protein kinase CK2. J Biol Chem. 2002;277:31826–31833. doi: 10.1074/jbc.M204149200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.