

Highlights.
– Gut-derived serotonin (GDS) synthesis is increased by fasting
– GDS enhances lipolysis by signaling in adipocytes through Htr2b receptor
– GDS favors gluconeogenesis, suppresses glucose uptake in liver by acting on Htr2b
– Inhibition of GDS synthesis ameliorates hyperglycemia in type 2 diabetes
Energy release from cellular storage is mandatory for survival during fasting. This is achieved through lipolysis and liver gluconeogenesis. We show here that in the mouse gut-derived serotonin (GDS) is up-regulated during fasting and that it favors both mechanisms. In adipocytes GDS signals through the Htr2b receptor to favor lipolysis by increasing phosphorylation and activity of hormone sensitive lipase. In hepatocytes GDS signaling through Htr2b promotes gluconeogenesis by enhancing activity of two rate-limiting gluconeocenic enzymes, FBPase and G6Pase. In addition, GDS signaling in hepatocytes prevents glucose uptake in a Glut2-dependent manner thereby further favoring maintenance of blood glucose levels. As a result inhibition of GDS synthesis can improve glucose intolerance caused by high fat diet. Hence, GDS opposes deleterious consequences of food deprivation by favoring lipolysis and liver gluconeogenesis while preventing glucose uptake by hepatocytes. As a result pharmacological inhibition of its synthesis may contribute to improve type 2 diabetes.
Introduction
The ability to survive during food deprivation has been a constant necessity for living organisms throughout evolution. In vertebrates most of the energy stores are located in adipose tissue and liver. Hence, these 2 organs are the main providers of energy to the rest of the body during fasting. Adipose tissue releases FFAs and glycerol in the process of lipolysis (Rosen and Spiegelman, 2000; Zechner et al., 2012), while liver produces ketone bodies, releases triglycerides and maintains glucose levels mainly through gluconeogenesis (Lin and Accili, 2011; Rosen and Spiegelman, 2000). Given the prime importance of lipolysis and gluconeogenesis for adaptation to fasting these processes need to be tightly regulated.
The best established regulation of lipolysis is exerted on the one hand by insulin that inhibits it (Saltiel and Kahn, 2001) and on the other hand by glucocorticoids and the sympathetic nervous system that favor it (Vegiopoulos and Herzig, 2007; Zechner et al., 2012). In addition, FGF21, glucagon and ghrelin have been identified as potential regulators of lipolysis (Inagaki et al., 2007; Perea et al., 1995; Vestergaard et al., 2008). However, both the physiological importance of lipolysis and the identification in recent years through mouse genetics of novel regulators of this process suggest that additional, yet to be identified, hormones regulating positively or negatively this survival function, may exist.
A second mechanism implicated in the adaptation to food deprivation is liver de novo glucose synthesis, or gluconeogenesis. This process can be initiated from multiple substrates such as pyruvate, glycerol, amino acids or lactate (Lin and Accili, 2011). Like lipolysis, this physiological process is tightly regulated by hormonal inputs with insulin inhibiting it and glucagon, glucocorticoids, catecholamines and FGF21 favoring it (Lin and Accili, 2011; Potthoff et al., 2009; Vegiopoulos and Herzig, 2007). Just as it is the case for lipolysis, it is likely that a systematic analysis of available mutant mouse strains lacking a given circulating molecule or receptor may identify novel regulators of this process.
Serotonin is a bioamine derived from tryptophan that is highly conserved throughout evolution (Berger et al., 2009). In vertebrates there are two pools of serotonin, one made in neurons of the brainstem and one made in the periphery, mainly, but not only, in enterochromaffin cells of the gut. In those two compartments serotonin biosynthesis is initiated by a different rate-limiting enzyme, tryptophan hydroxylase 1 (Tph1) in the periphery and tryptophan hydroxylase 2 (Tph2) in the brain. Since serotonin does not cross the blood-brain barrier it is believed that each pool of serotonin has a discrete set of functions (Berger et al., 2009) although some neurons of the hypothalamus may be accessible to peripheral molecules that otherwise do not cross the blood-brain barrier. While brain-derived serotonin is a multifunctional neurotransmitter, gut-derived serotonin (GDS) has emerged recently as a hormone able to regulate bone formation, erythropoiesis and regenerative processes (Amireault et al., 2011; Dees et al., 2011; Fligny et al., 2008; Lesurtel et al., 2006; Yadav et al., 2008). These novel functions of GDS raise the possibility that it may have additional endocrine roles. In particular, considering the regulation of bone formation by GDS, it is important to test if GDS regulates any aspect of energy metabolism as several other hormones affecting bone mass do (Ducy et al., 1996; Lee et al., 2007; Wei et al., 2012).
In this study we show that food deprivation promotes synthesis of GDS, which in turn favors both lipolysis and liver gluconeogenesis by signaling, in adipocytes and hepatocytes, through the same receptor. Furthermore, GDS prevents glucose uptake by hepatocytes thereby further contributing to maintaining normal blood glucose levels. Taking advantage of the availability of a small molecule inhibitor of GDS synthesis we also provide pharmacological evidence that decreasing its synthesis has beneficial effects in type 2 diabetic mice.
Results
GDS promotes lipid mobilization upon fasting
To expand our knowledge about GDS biology we asked how its synthesis is regulated. Given the role of food intake during bone formation and the regulation of bone formation by GDS (Black et al., 2001; Yadav et al., 2008) we asked whether fasting affects GDS synthesis.
As shown in Figure 1A and B Tph1 expression in the gut and plasma serotonin levels were both significantly increased in fasted WT mice at all time points analyzed. Plasma serotonin levels reached 24 μM upon 48h of fasting. Although the increase in Tph1 expression in the duodenum was modest upon fasting it was specific since its expression in adipose tissue was nearly 300-fold lower than in duodenum. Likewise, expression of Tph2, the rate-limiting enzyme in brain serotonin synthesis was 200-fold lower in adipose tissue than in the brainstem (Figure S1I). The increase in circulating serotonin levels occurred before and was not of lesser amplitude than the one of glucagon (Figure 1C), a major determinant of the adaptation to fasting (Unger and Cherrington, 2012). Since metabolic adaptation to fasting relies, in part, on lipolysis, we tested whether GDS is a physiological regulator of lipolysis by analyzing mice lacking tryptophan hydroxylase 1 (Tph1), the initial and rate-limiting enzyme in GDS biosynthesis, only in the gut (Tph1gut∆/∆ mice) (Figures S1A and B) (Yadav et al., 2008).
Fig. 1. GDS is required for fasting induced lipolysis.
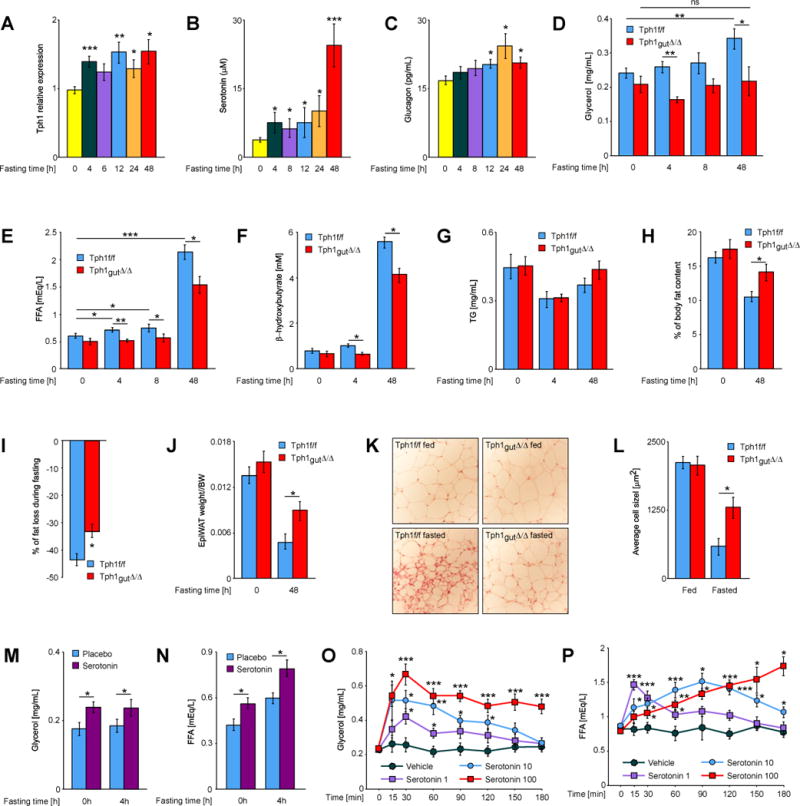
(A) Relative expression levels of Tph1 in duodenum from mice fasted for indicated times (n ≥ 5). (B) Plasma levels of serotonin and (C) glucagon in mice fasted for indicated times (n ≥ 5). Plasma levels of (D) glycerol, (E) FFAs, (F) β-hydroxybutyrate and (G) triglycerides in mice of indicated genotypes fasted for indicated times (n ≥ 7). (H) % of fat content in fed and fasted mice of indicated genotypes measured by magnetic resonance imaging (MRI) (n ≥ 5). (I) % of fat content loss during 48h fasting in relation to initial fat content of each animal, average of 5 mice of Tph1f/f and Tph1gut∆/∆ genotype. (J) Epigonadal fat pad weight to body weight ratio in fed and fasted Tph1f/f and Tph1gut∆/∆ mice (n ≥ 6). (K) Histology of fat pad from fed and 48h fasted Tph1f/f and Tph1gut∆/∆ mice (n ≥ 5). (L) Average size of adipocytes from fat pad of fed and 48h fasted Tph1f/f and Tph1gut∆/∆ mice (n ≥ 5). Plasma levels of (M) glycerol and (N) FFAs in mice with implanted placebo or serotonin-releasing pellets fasted for indicated time (n ≥ 8). Plasma levels of (O) glycerol and (P) FFAs at indicated time points after i.p. injection of serotonin (doses in mg/kg BW) (n ≥ 5). Data represented as mean +/− SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
Body, organ and fad pad weights as well as food intake were similar in Tph1gut∆/∆ and Tph1fl/fl mice fed ad libitum a normal diet (Figure S1C and D). Likewise, circulating triglycerides levels and expression of the crucial regulators of fat absorption, FIAF and LPL (Backhed et al., 2004), in intestine and adipose tissue were not changed in fed and fasted Tph1gut∆/∆ mice (Figure 1G and S1F and S2C). In contrast, fasted Tph1gut∆/∆ mice could not increase their circulating levels of glycerol as Tph1 fl/fl littermates did (Figure 1D). The lack of increase of circulating glycerol levels in Tph1 fl/fl mice after 4h of fasting compared to what they are after 48h of fasting reflects the fact that the rate of lipolysis is much higher at that latter time point. We verified that the food content in stomach, duodenum, jejunum and ileum was significantly decreased after a 4h fast (Figure S1G). Tph1gut∆/∆ mice did increase their circulating levels of FFAs during a prolonged fasting but this increase remained significantly weaker than the one seen in Tph1 fl/fl littermate controls (Figure 1E). Likewise, GDS deficiency blunted the increase of plasma ketone bodies such as β-hydroxybutyrate that normally occurs during fasting (Figure 1F). The rate of glycerol and FFAs clearance in Tph1gut∆/∆ and Tph1 fl/fl mice was the same (Figure S1J and K) and the VillinCre transgene did not alter glycerol, FFAs and β-hydroxybutyrate circulating levels in fed or fasted mice (Figure S3A–C). Hence, in absence of GDS the levels of three main energetic substrates during fasting are blunted.
Consistent with their failure to induce appropriate lipolysis, Tph1gut∆/∆ mice fasted for 48h lost significantly less body weight, less fat and displayed larger adipocytes than Tph1 fl/fl mice (Figure 1I–L, S2A). Respiratory exchange rate was increased in fasted Tph1gut∆/∆ compared to littermate Tph1 fl/fl mice while energy expenditure was similar (Figure S2B). Circulating levels of insulin and leptin were normal in fed and fasted Tph1gut∆/∆ mice (Figure S2D and E) and there was no evidence of transformation of white fat into brown fat in Tph1gut∆/∆ mice as reported for FGF21-deficient animals (Fisher et al., 2012) (Figure S2C).
To assess the importance GDS regulation of lipolysis we next asked whether enhancing peripheral serotonin signaling in vivo in WT mice would promote lipolysis. Implanting subcutaneously serotonin-releasing pellets in WT mice (350 ng/h/animal for 60 days) led to an increase of circulating levels of serotonin of approximately 2.5-fold (Figure S2F) resulting in a significant increase in circulating levels of both glycerol and FFAs, an increase that was further enhanced following a 4h-long fasting period (Figure 1M and N). Similar results were obtained with single shot injections of serotonin (Figure 1O and P). Taken together these loss- and gain-of-function experiments concur to indicate that GDS is a positive regulator of lipolysis. That abrogating GDS synthesis inhibited almost completely the increase of glycerol release during lipolysis but only partially FFAs release is fully consistent with the notion that GDS is by no means the only regulator of lipolysis.
We are aware that in the conditions of this experiment, serotonin might have evoked other responses that may confound interpretation. This is why this experiment was repeated this experiment once the appropriate serotonin receptor was identified and deleted in a cell-specific manner (see below).
GDS promotes lipolysis by acting through Htr2b receptor on adipocytes
We next asked whether GDS promotes lipolysis by acting directly on adipcytes. First we treated epididymal and subcutaneous fat pads with increasing amounts of serotonin. This treatment increased significantly and in dose dependent manner release of glycerol and, to an even larger extent, the one of FFAs suggesting that serotonin acts directly on adipose tissue to favor lipolysis (Figure 2A, B and S4A, B). Next we isolated epigonadal adipocytes and treated them with serotonin. That serotonin increased the release of glycerol and FFAs in isolated adipocytes indicated that the adipocyte is a cell type on which serotonin acts to promote lipolysis (Figure S4C and D).
Fig. 2. GDS favors lipolysis through Htr2b receptor expressed in adipocytes.
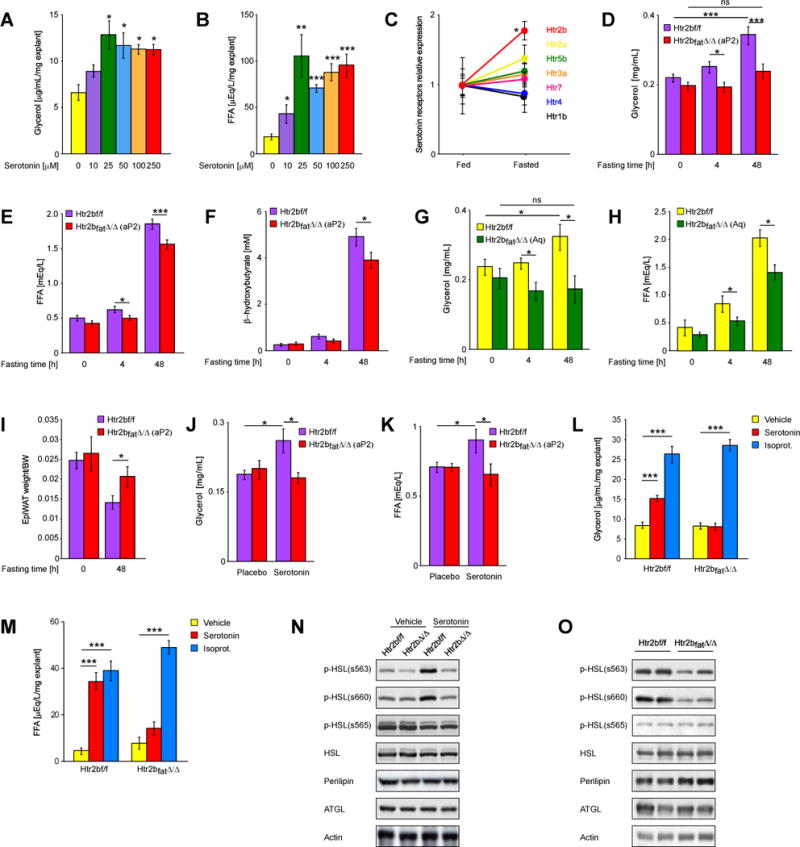
(A) Glycerol and (B) FF As release from mouse epigonadal fat explants stimulated with indicated doses of serotonin (n > 5). (C) Relative expression of indicated serotonin receptors in adipose tissue of fed and fasted mice. Plasma levels of (D) glycerol, (E) FF As and (F) β-hydroxybutyrate in mice lacking Htr2b in adipose tissue (deletion by aP2-Cre – indicated on graphs Htr2bfatΔ/Δ (aP2)) fasted for indicated times (n ≥ 20). Plasma levels of (G) glycerol and (H) FFAs in mice lacking Htr2b in adipose tissue (deletion by Adiponectin-Cre – indicated on graphs Htr2bfatΔ/Δ (Aq) fasted for indicated times (n > 6). (I) Epigonadal fat pad weight to body weight ratio in fed and fasted Htr2bf/f and Htr2bfatΔ/Δ(aP2) mice (n > 12). (J) Glycerol and (K) FFAs levels in Htr2bf/f and Htr2bfatΔ/Δ(aP2) mice implanted with serotonin-releasing pellets. (L) Glycerol and (M) FFAs release from epigonadal fat explants isolated from Htr2bf/f and Htr2bfatA/A(aP2) mice and stimulated with 50μM serotonin or 1μM isoproterenol (n = 5). (N) Western blot analysis of expression and activation of indicated proteins in fat explants isolated from Htr2bf/f and Htr2bfatΔ/Δ(aP2) mice and stimulated with 50μM serotonin. (O) Western blot analysis of expression and activation of indicated proteins in fat pads isolated from Htr2bf/f and Htr2bfatΔ/Δ(aP2) mice fasted for 48h. Data represented as mean +/− SEM, *P < 0.05, ***P < 0.001.
In view of these results we sought to identify serotonin receptor expressed in adipocytes. Among the fourteen known serotonin receptors (Berger et al., 2009) Htr2b was by far the most highly expressed in mouse adipocytes and its expression in white adipose tissue markedly increased upon fasting (Figure S4E and F and Figure 2C). Given this pattern of expression we tested whether Htr2b mediates serotonin regulation of lipolysis in vivo by analyzing mice lacking this receptor only in adipocytes (Htr2bfat∆/∆ mice). Prior to studying these mutant mice we verified that Htr2b had been deleted in adipocytes but not in macrophages (Figure S4G).
Like Tph1gut∆/∆ mice, Htr2bfat∆/∆ mice displayed normal body and organ weight when fed a normal diet ad libitum. Likewise, their circulating levels of insulin, leptin and triglycerides where normal in fed and fasted conditions (Figure S4I–M). Upon fasting, however, we did not observe in Htr2bfat∆/∆ mice the increase in circulating glycerol levels seen in Htr2b fl/fl control mice and the increase in FFAs and β-hydroxybutyrate circulating levels was significantly weaker in Htr2bfat∆/∆ than in Htr2b fl/fl mice (Figure 2D–F). Moreover, Htr2bfat∆/∆ mice maintained bigger fat pads and larger adipocytes after a 48h-long fasting than Htr2b fl/fl mice did (Figure 2I, Figure S4N and O). Hence, the metabolic profile of Htr2bfat∆/∆ mice is similar to the one observed in Tph1gut∆/∆ mice. The aP2-Cre transgene did not affect glycerol, FFAs and β-hydroxybutyrate levels when compared to littermate WT mice, whether in fed or fasted conditions (Figure S3H–J).
Since aP2-Cre deleted Htr2b in hypothalamus (Figure S4G) we repeated this experiment using a different Cre driver, the Adiponectin-Cre that did not remove Htr2b in other cell types than the adipocytes and in particular not in hypothalamic neurons (Eguchi et al., 2011) (Figure S4H). Glycerol levels did not increase either upon fasting and the increase in FFAs levels was also blunted in Adiponectin-Cre; Htr2bfl/fl mice (Figure 2G and H).
In agreement with these loss-of-function results, implantation of serotonin-releasing pellets did not increase glycerol and FFAs circulating levels in Htr2bfat∆/∆ mice as it did in Htr2b fl/fl littermates (Figure 2J and K). Moreover, in cell culture studies, while isoproterenol, a positive control, promoted the release of FFAs and glycerol equally well in WT and Htr2b-deficient adipose tissue explants, serotonin increased the release of FFAs and glycerol in WT but not in Htr2b-deficient explants (Figure 2L and M). Taken together this set of experiments indicates that GDS favors lipolysis by signaling through the Htr2b receptor expressed in adipocytes.
To begin deciphering the molecular mode of action of serotonin in adipocytes downstream of Htr2b we analyzed expression, accumulation and phosphorylation of key lipolytic enzymes (Haemmerle et al., 2006; Martinez-Botas et al., 2000; Osuga et al., 2000). While serotonin did not alter expression or accumulation of enzymes involved in lipolysis, it favored, ex vivo, phosphorylation of hormone sensitive lipase (HSL) on serine 563 and 660 in an Htr2b-dependent manner (Figure 2N and S4P). These two phosphorylation events have been shown to enhance HSL activity (Anthonsen et al., 1998; Greenberg et al., 2001). To determine if this regulation of phosphorylation of HSL by serotonin occurs also in vivo we analyzed Htr2bfat∆/∆ and Htr2b fl/fl mice that had been fasted for 48h. As shown in Figure 2O phosphorylation of HSL was decreased in Htr2bfat∆/∆ adipose tissue indicating that a signaling event elicited by serotonin in adipocytes is to favor HSL phosphorylation, an event that should increase HSL activity. Since both HSL and ATGL promote lipolysis we tested whether serotonin affects ATGL localization but failed to observe any effect of serotonin on its translocation to lipid droplets (Figure S4Q). In view of these data we conclude that one mechanism whereby serotonin favors lipolysis is phosphorylation of HSL on residues s563 and s660, two events viewed as activating HSL (Djouder et al., 2010; Iwata et al., 2012). We cannot exclude, however, that serotonin may also increase activation of ATGL and perilipin, two aspects of lipolysis that could not been tested due to the lack of reliable reagents, or other component of lipolytic machinery.
GDS promotes liver gluconeogenesis
Glycerol is a substrate for liver gluconeogenesis upon fasting (Lin and Accili, 2011) thus the results presented above implied that GDS should promote gluconeogenesis because of its ability to increase glycerol levels.
Glycerol injections, however, did not increase circulating glucose levels to the same extent in Tph1gut∆/∆ than in Tph1 fl/fl control mice (Figure 3A). Moreover, injections of pyruvate, the initial substrate in liver gluconeogenesis, whose circulating levels are not influenced by lipolysis, also failed to increase blood glucose levels in Tph1gut∆/∆ mice as they did in Tph1 fl/fl control mice (Figure 3B). Villin-Cre transgenic mice did not show any alteration of gluconeogenesis compared to littermate controls after glycerol or pyruvate injections (Figure S3D and E). These results implied that the favorable influence of GDS on liver gluconeogenesis is not a mere consequence of its up-regulation of lipolysis. Indeed, in WT mice implanted with serotonin-releasing pellets, which increased plasma serotonin levels 2.5-times (Figure S2F), glycerol injection increased blood glucose to higher levels than in control mice (Figure 3C). Similar results were obtained following a single injection of serotonin (10μg/g BW) in both Tph1 fl/fl and Tph1gut∆/∆ mice (Figure 3D).
Fig. 3. GDS promotes gluconeogenesis in liver.
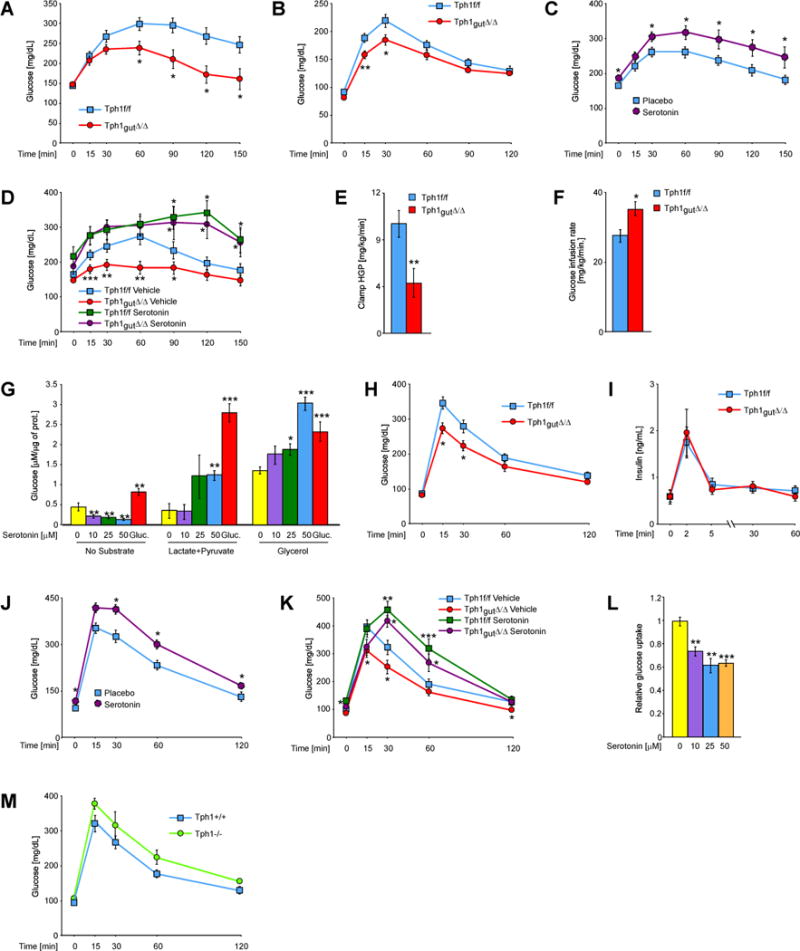
(A) Glycerol-evoked glucose production in Tph1f/f and Tph1gut∆/∆ mice (n ≥ 8). (B) Pyruvate tolerance test in Tph1f/f and Tph1gut∆/∆ mice (n ≥ 8). (C) Glycerol tolerance test in mice with implanted placebo or serotonin-releasing pellets (n ≥ 8). (D) Glycerol-evoked glucose production in Tph1f/f and Tph1gut∆/∆ mice injected with serotonin (10 mg/kg BW) 90min. before the test (n ≥ 7). Hepatic glucose production (E) and (F) glucose infusion rate during hyperinsulinemic-euglycemic clamp in Tph1f/f and Tph1gut∆/∆ mice (n ≥ 7). (G) Production of glucose from indicated substrates by primary hepatocytes stimulated with indicated doses of serotonin or 100nM glucagon (n = 5). (H) Glucose tolerance test in Tph1f/f and Tph1gut∆/∆ mice (n ≥ 7). (I) Plasma insulin levels during glucose tolerance test in Tph1f/f and Tph1gut∆/∆ mice (n ≥ 7). (J) Glucose tolerance test in mice with implanted placebo or serotonin-releasing pellets (n ≥ 8). (K) Glucose tolerance test in Tph1f/f and Tph1gut∆/∆ mice injected with serotonin (10 mg/kg BW) 90min. before the test (n ≥ 7). (L) Glucose uptake in primary hepatocytes stimulated with indicated doses of serotonin. (M) Glucose tolerance test in Tph1+/+ and Tph1−/− mice (n ≥ 3). Data represented as mean +/− SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
To further test if GDS can induce gluconeogenesis from endogenous substrates we measured hepatic glucose production (HGP) in Tph1gut∆/∆ and Tph1 fl/fl mice in conditions of hyperinsulinemic-euglycemic clamps and observed that hepatic glucose production was reduced more than 2-fold in Tph1gut∆/∆ mice fasted for 4h (Figure 3E and F). Although the effect that insulin infusion might have on serotonin levels was not measured in this assay, this result supports the contention that GDS is a positive regulator of liver gluconeogenesis. We next tested if serotonin acts directly on hepatocytes for that purpose. When added to hepatocytes cultures serotonin (10–50μM) stimulated conversion of glycerol as well as lactate/pyruvate to glucose like the positive control glucagon did (Figure 3G). That serotonin decreased the rate of glucose production in the absence of substrate suggests that it specifically promotes gluconeogenesis. Taken together our results indicate that GDS is necessary but not sufficient for proper induction of gluconeogenesis upon fasting. That Tph1 deletion does not result in a decrease of fasting glucose levels is consistent with the fact that other hormones also regulate gluconeogenesis during fasting (Lin and Accili, 2011; Vegiopoulos and Herzig, 2007).
GDS inhibits glucose uptake by hepatocytes
During fasting glucose uptake by hepatocytes is partially blocked (Wahren and Ekberg, 2007). To test if serotonin also modulates this aspect of adaptation to fasting we subjected fasted Tph1gut∆/∆ and control mice to glucose tolerance tests. Clearance of a glucose load was significantly improved in Tph1gut∆/∆ compared to Tph1 fl/fl mice (Figure 3H) and was not affected by expression of the VilinCre transgene (Figure S3F). Importantly, Tph1gut∆/∆ mice showed normal glucose stimulated insulin secretion at basal conditions (Figure 3I). Conversely, WT mice implanted with serotonin-releasing pellets, which increased plasma serotonin levels 2.5-times (Figure S2G), showed impaired glucose clearance during glucose tolerance test compared to placebo-implanted controls (Figure 3J). Similar results were obtained after a single injection of serotonin (10μg/g BW) in both Tph1gut∆/∆ and Tph1 fl/fl mice (Figure 3K). Moreover, stimulation of hepatocytes by serotonin (10–50μM) reduced glucose uptake by 40% (Figure 3L). These results indicate that GDS regulation of glucose uptake by hepatocytes and glucose clearance in Tph1gut∆/∆ mice occurs because of a direct action of this hormone on liver. That these results obtained through the analysis of cell-specific deletion differ from those obtained when analyzing mice lacking Tph1 in all cells (Figure 3M) (Paulmann et al., 2009) implies that Tph1 has additional functions in other cell types.
GDS promotes liver gluconeogenesis through Htr2b in hepatocytes
As it is the case in adipocytes, Htr2b is by far the most highly expressed serotonin receptor in the liver where its expression also increases upon fasting (Figure 4A and B). Therefore we tested whether Htr2b mediates serotonin regulation of hepatic gluconeogenesis through the analysis of mutant mice lacking this receptor only in hepatocytes (Htr2bliver∆/∆ mice). Prior to study these mutant mice we verified that Htr2b had been efficiently removed from hepatocytes but not from other cell types (Figure S6A).
Fig. 4. GDS promotes gluconeogenesis in liver by signaling through the Htr2b receptor.
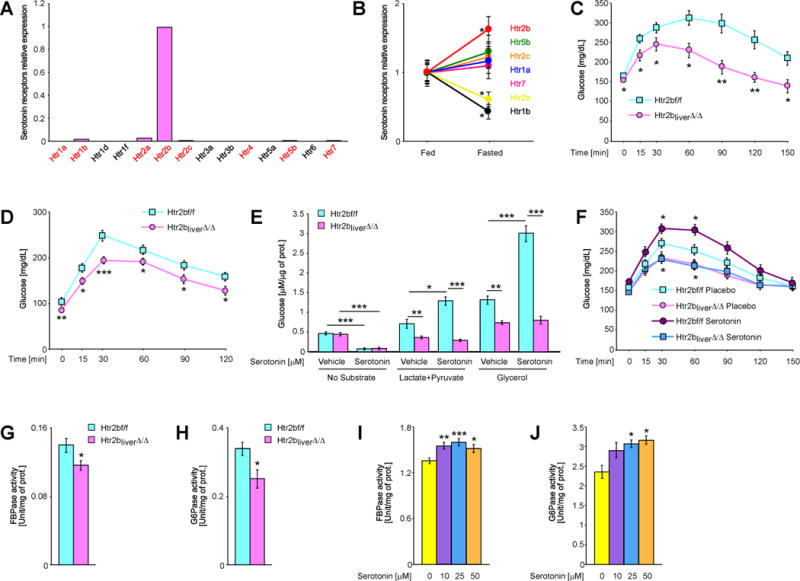
(A) Relative expression of indicated serotonin receptors in primary mouse hepatocytes. Red color indicates receptors for which the transcript was detected. (B) Relative expression of indicated serotonin receptors in liver of fed and fasted mice. (C) Glycerol tolerance test in Htr2bf/f and Htr2bliver∆/∆ mice (n ≥ 7). (D) Pyruvate tolerance test in Htr2bf/f and Htr2bliver∆/∆ mice (n ≥ 7). (E) Production of glucose from indicted substrates by hepatocytes isolated from Htr2bf/f and Htr2bliver∆/∆ mice and stimulated by 50μM serotonin (n = 5). (F) Glycerol tolerance test in Htr2bf/f and Htr2bliver∆/∆ mice implanted with placebo or serotonin-releasing pellets (n ≥ 6). Normalized (G) FBPase and (H) G6Pase activity in livers of fasted Htr2bf/f and Htr2bliver∆/∆ mice (n ≥ 6). Normalized (I) FBPase and (J) G6Pase activity in isolated hepatocytes stimulated with indicated doses of serotonin (n ≥ 3). Data represented as mean +/− SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
When fed ad libitum a normal diet Htr2bliver∆/∆ mice had normal body and organ weights, insulin, leptin, triglycerides and FFAs circulating levels (Figure S6B–G). However, glucose production measured following glycerol or pyruvate injections was attenuated in Htr2bliver∆/∆ compared to Htr2b fl/fl mice (Figure 4C and D). Albumin-Cre transgenic mice and littermate control animals responded similarly to glycerol and pyruvate challenge (Figure S3R and S). Two additional experiments support the notion that Htr2b expression in hepatocytes mediates GDS regulation of liver gluconeogenesis by GDS. First, serotonin could not induce glucose production from glycerol or pyruvate/lactate when added to Htr2b∆/∆ hepatocytes while it did when added to Htr2b fl/fl hepatocytes (Figure 4E). Second, implantation of serotonin-releasing pellets did not induce gluconeogenesis in Htr2bliver∆/∆ mice as it did in Htr2b fl/fl mice (Figure 4F). That the decrease of glucose production by serotonin in absence of substrate was not affected by Htr2b deletion, suggests that another receptor mediates this function of GDS in liver (Figure 4E).
In an effort to elucidate how GDS could promote liver gluconeogenesis we analyzed gene expression and protein levels of key determinants of glycerol-dependant gluconeogenesis such as FBPase, G6Pase, glycerol kinase and aquaporin 9 (Lin and Accili, 2011) but failed to observe any significant changes in Htr2bliver∆/∆ mice (Figure S6H and I). In contrast, when enzymatic activities were measured through classical bioassays (Alegre et al., 1988; Reyes et al., 1987) we observed that the activity of two rate limiting enzymes for gluconeogenesis, FBPase and G6Pase, was decreased 20 and 25%, respectively, in Htr2bliver∆/∆ mice (Figure 4K and L). Moreover, serotonin (10–50mM) treatment of isolated hepatocytes significantly increased activity of both G6Pase and FBPase (Figure 4I and J). These results suggest that one mechanism whereby serotonin favors gluconeogenesis is by enhancing activity of these two enzymes.
GDS inhibits glucose uptake through Htr2b receptor in hepatocytes
To test if GDS blocks glucose uptake in hepatocytes by acting on the Htr2b receptor, we subjected Htr2bliver∆/∆ mice to glucose tolerance test. Glucose clearance in Htr2bliver∆/∆ mice was markedly increased compared to Htr2b fl/fl mice (Figure 5A) while Albumin-Cre transgenic mice and littermate control animals responded similarly to glucose challenge (Figure S3T). Implantation of serotonin-releasing pellets did not induce glucose intolerance in Htr2bliver∆/∆ mice as it did in Htr2b fl/fl mice (Figure 5B) and serotonin failed to block glucose uptake in Htr2b-deficient hepatocytes (Figure 5C). Taken together these data indicate that GDS blocks glucose uptake in hepatocytes by acting on Htr2b receptor.
Fig. 5. GDS inhibits glucose uptake in liver by signaling through the Htr2b receptor.
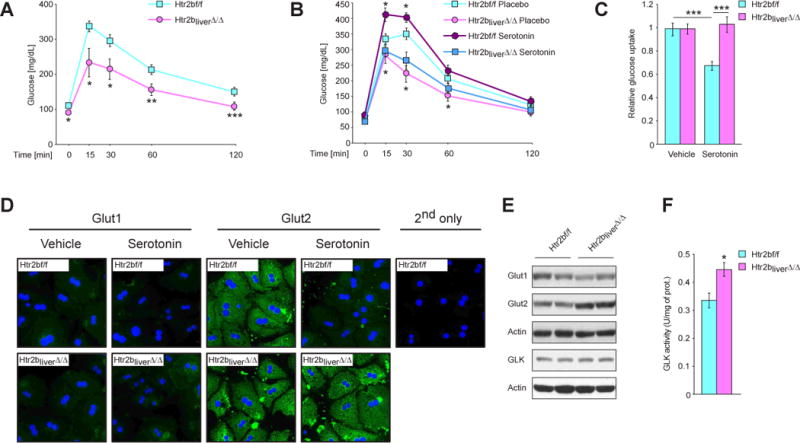
(A) Glucose tolerance test in Htr2bf/f and Htr2bliver∆/∆ mice (n ≥ 8). (B) Glucose tolerance test in Htr2bf/f and Htr2bliver∆/∆ mice implanted with placebo or serotonin-releasing pellets (n ≥ 6). (C) Relative glucose uptake in Htr2b-deficient hepatocytes stimulated with 50 μM serotonin. (D) Immunofluorescence with antibodies against Glut1 and Glut2 on primary hepatocytes of indicated genotypes stimulated with 50μM serotonin. (E) Western blot analysis of expression of indicated proteins in livers from fasted Htr2bf/f and Htr2bliver∆/∆ mice. (F) Normalized glucokinase activity in livers of fasted Htr2bf/f and Htr2bliver∆/∆ mice. Data represented as mean +/− SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
To unravel the molecular mechanisms underlying serotonin inhibition of glucose uptake we first examined expression and localization of two major glucose transporters (Gluts) in hepatocytes, Glut1 and Glut2 (Gonzalez-Rodriguez et al., 2008). Serotonin stimulation of hepatocytes isolated from Htr2b fl/fl or Htr2bliver∆/∆ mice did not alter subcellular localization of these two transporters (Figure 5D) but it decreased Glut2 abundance since the intensity of the Glut2 staining decreased markedly in serotonin-stimulated WT but not Htr2b-deficient hepatocytes. Moreover, Glut2 levels were up-regulated in Htr2bliver∆/∆ compared to Htr2b fl/fl livers during fasting while Glut1 levels were unaffected (Figure 5E). Glut1 and Glut2 expression was not changed in Htr2bliver∆/∆ (Figure S6I). Activity of glucokinase, the first rate limiting enzyme in hepatic glucose utilization (Matschinsky, 2009), was also significantly increased in fasted Htr2bliver∆/∆ mice (Figure 5F). These data suggest that GDS suppresses glucose uptake by hepatocytes in part by promoting Glut2 degradation and suppressing glucokinase activity.
Inhibition of GDS synthesis protects against diet-induced type 2 diabetes
Elevation of FFAs levels can contribute to the development of insulin resistance, especially in conditions of hyperglycemia (Saltiel and Kahn, 2001; Samuel and Shulman, 2012). This notion, along with the positive effect of GDS on liver gluconeogenesis, led us to ask whether abrogating GDS synthesis or signaling in adipocytes will influence tolerance to insulin.
Insulin tolerance, measured by insulin tolerance test (ITT) or glucose infusion rates during hyperinsulinemic-euglycemic clamps, was significantly improved in Tph1gut∆/∆ and Htr2bfat∆/∆ mice compared to control animals (Figure 6A and B, Figure 3F). Likewise, Tph1gut∆/∆ and Htr2bliver∆/∆ mice were significantly more tolerant to a glucose challenge than Tph1 fl/fl or Htr2b fl/fl littermates (Figure 3F and 5A) while Cre expressing mouse lines tested (VillinCre, aP2Cre, AlbCre) responded to insulin and glucose challenge like WT mice (Figure S2F, G, M, N, T and U).
Fig. 6. Inhibition of GDS synthesis protects from type 2 diabetes.
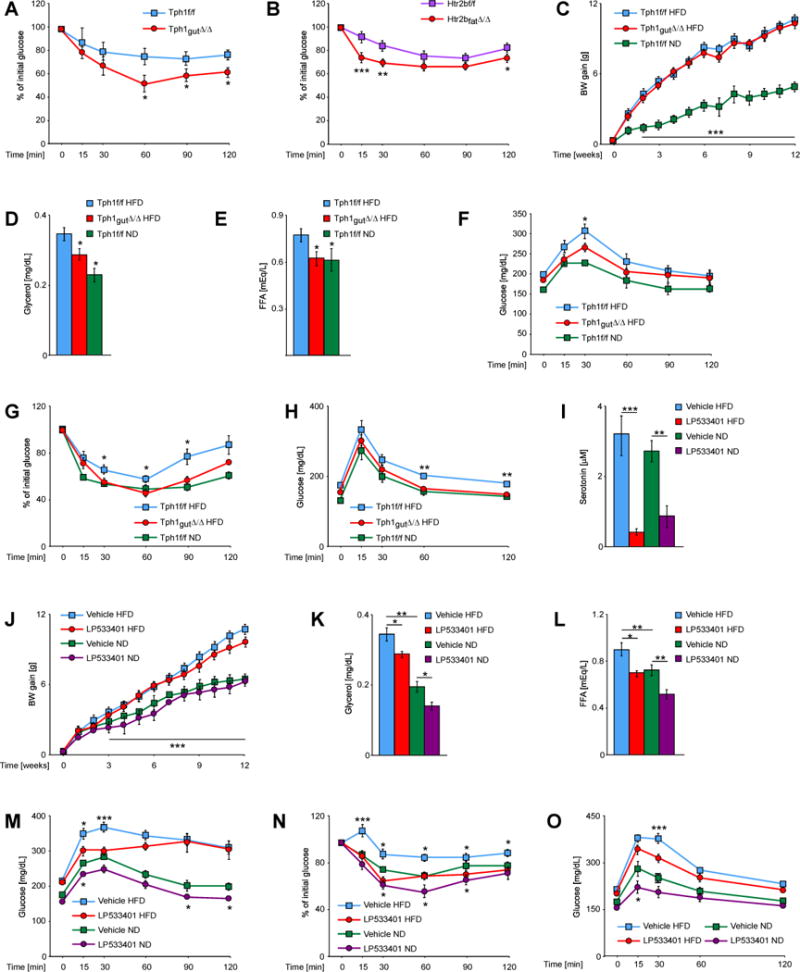
(A) Insulin tolerance test in Tph1f/f and Tph1gut∆/∆ mice (n ≥ 7). (B) Insulin tolerance test in Htr2bf/f and Htr2bfat∆/∆ mice (n ≥ 12). (C) Body weight gain of Tph1f/f and Tph1gut∆/∆ mice fed normal or high fat diet (n ≥ 5). Plasma levels of (D) glycerol and (E) FFAs in mice of indicated genotypes fed normal (ND) or high fat diet (HFD), fasted for 4h (n ≥ 5). (F) Pyruvate, (G) insulin and (H) glucose tolerance tests in Tph1f/f and Tph1gut∆/∆ mice on indicated diets (n ≥ 5). (I) Plasma serotonin concentrations in mice treated with vehicle or LP533401 inhibitor (n ≥ 8). (J) Body weight gain of mice treated with vehicle or LP533401 inhibitor, fed ND or HFD (n ≥ 8). Plasma levels of (K) glycerol and (L) FFAs in mice treated with vehicle or LP533401 inhibitor fed ND or HFD and fasted for 4h (n ≥ 7). (M) Pyruvate, (N) insulin and (O) glucose tolerance tests in mice treated with vehicle or LP533401 inhibitor fed ND or HFD (n > 7). Data represented as mean +/− SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
The results presented above implied that decreasing GDS synthesis might attenuate the severity of type 2 diabetes, a hypothesis tested genetically. Tph1gut∆/∆ and Tph1fl/fl mice were fed a high fat diet (HFD) for 12 weeks to induce obesity and glucose intolerance. This resulted in a twofold increase in body weight gain in Tph1gut∆/∆ and Tph1fl/fl mice compared to mice of the same genotype fed normal diet (Figure 6D). Both Tph1gut∆/∆ and Tph1fl/fl mice displayed markedly higher fasting glucose than control mice fed a normal diet (Figure 6G and I). As hypothesized, circulating glycerol levels were significantly reduced in Tph1gut∆/∆ mice fed HFD compared to control mice fed the same diet and FFAs levels in Tph1gut∆/∆ mice fed HFD were not different than levels observed in Tph1fl/fl mice fed a normal diet (Figure 6E and F). Gluconeogenesis, insulin and glucose tolerance were partially normalized in Tph1gut∆/∆ mice fed a HFD (Figure 6G–I).
We also asked whether a small molecule inhibitor of Tph1 activity and thereby of GDS synthesis, which does not cross the blood brain barrier, LP533401 (Liu et al., 2008), could improve glucose intolerance in WT mice fed a HFD. At the dose used for this study (100mg/kg/day) LP533401 decreased serotonin circulating levels 80% (Figure 6J). This resulted in a decrease in glycerol circulating levels and a normalization of circulating FFAs levels in WT mice fed HFD (Figure 6L and M). Insulin tolerance was also normalized while gluconeogenesis and glucose tolerance were significantly improved in LP533401-treated WT mice fed HFD (Figure 6N–P). Moreover, when given to WT mice fed normal diet LP533401 decreased glycerol and FFAs levels, decreased gluconeogenesis rate and improved glucose and insulin tolerance as in Tph1gut∆/∆ mice (Figure 6J–O). Although we cannot, at the present time, rule out that LP533401, used here as a proof of principle molecule, may have deleterious, effects we note that this small molecule did not affect body weight gain (Figure 6K).
Discussion
This study revealed that, in the mouse, GDS is multifunctional adaptor to food deprivation. Indeed, it favors lipolysis, liver gluconeogenesis and inhibits glucose uptake in liver by signaling through the same receptor, Htr2b, in adipocytes and hepatocytes.
Regulation of lipolysis by GDS
Lipolysis is an adaptation mechanism used by vertebrates to survive when food is scarce. Several convergent extracellular cues concur to increase lipolysis during fasting. One of them is the decline in the circulating levels of insulin, a hormone that inhibits lipolysis in the fed state (Saltiel and Kahn, 2001) while others are positive regulators of this survival function (Inagaki et al., 2007; Zechner et al., 2012).
We present here evidence that GDS synthesis is stimulated by fasting and that it favors lipolysis by signaling in adipocytes. Conversely, increasing GDS circulating levels in WT mice increases circulating glycerol and FFA levels. Our results identify Htr2b as being a receptor used by GDS in adipocytes to favor lipolysis. In that regard, we note that a recent high throughput screen identified Htr2b as a protein modulating lipid accumulation in adipocytes (Sohle et al., 2012). These observations do not exclude the possibility that other serotonin receptors may be implicated in this process, however, the fact that GDS fails to promote lipolysis in Htr2b-deficient adipocytes indicates that expression of this receptor is a critical role to mediate this function of GDS.
Our investigation indicates that GDS favors lipolysis by increasing the phosphorylation of hormone sensitive-lipase (HSL) on serine residues 563 and 660, two events that increase its activity (Anthonsen et al., 1998; Greenberg et al., 2001). This post-transcriptional effect of GDS on HSL distinguishes it from glucocorticoids that instead increase transcription of its gene (Xu et al., 2009). Although we have not identified any yet we do not exclude the possibility that other mechanisms may also be at work to account for GDS regulation of lipolysis.
GDS is by no means the only extracellular cue favoring lipolysis (Inagaki et al., 2007; Zechner et al., 2012). This may explain why mice with a marked decrease in GDS synthesis can still increase, albeit less efficiently than control animals, FFAs and ketone bodies secretion during fasting. This illustrates the importance of lipolysis during fasting and therefore the need of multiple levels of regulation for this process.
Regulation of liver glucose metabolism by GDS
Given that glycerol may be a substrate for gluconeogenesis it was not surprising that by increasing lipolysis GDS would also increase gluconeogenesis. However, the use of pyruvate as a substrate revealed that GDS acts directly in hepatocytes to favor gluconeogenesis. In this cell type GDS mediates its signal also through Htr2b.
Molecular and genetic studies identified Htr2b as a receptor used by serotonin in hepatocytes to favor gluconeogenesis. Other serotonin receptors are expressed in hepatocytes, albeit at a lower level, and they may also be implicated in GDS-mediated liver gluconeogenesis. However, that the Htr2b deletion in hepatocytes disrupted liver gluconeogenesis as severely as in Tph1gut∆/∆ mice indicate that this receptor plays a prominent role in mediating this function of GDS. The only signaling event we could show to be affected by serotonin signaling in hepatocytes is an increase in the activity of two rate-limiting enzymes needed for gluconeogenesis, FBPase and G6Pase. Future investigations will aim at identifying signaling events elicited by GDS in hepatocytes and affecting the activity of these two enzymes. GDS also hampered glucose uptake by liver in part by stimulating degradation of Glut2, that is required for this function (Guillam et al., 1998). Since Glut2 does not appear to affect, at least in the mouse, glucose secretion from hepatocytes nor hepatic glucose production in vivo, its upregulation in the absence of GDS signaling should not interfere with hepatic glucose production (Guillam et al., 1998).
The spectrum of GDS functions described here in no way eclipse the paramount importance of glucagon and insulin as regulators of hepatic glucose output. We have in fact no evidence suggesting that GDS is a more important regulator of this aspect of glucose metabolism than these two hormones.
Another legitimate question raised by any metabolic study performed in the mouse is to know whether results of these works have relevance to other vertebrate species. For instance, it is possible, although it has not been tested yet in the same condition, that GDS may exert different influence on gluconeogenesis in mice and dogs (Moore et al., 2004). Alternatively the significant differences in experimental procedures and amounts of serotonin used may account, in part, for these differences. Of note, classical studies suggest that GDS favors lipolysis in humans, too (Carlson et al., 1967). The role that GDS may have in gluconeogenesis and glucose uptake by hepatocytes in human has not been thoroughly studied yet.
Tph1 functions in enterochromaffin cells
Our study revealed that ablation of Tph1 in the gut only has different consequences on glucose metabolism than its ablation in all cells. As such it illustrates the importance of cell-specific gene deletion to study complex physiological processes and to define all functions exerted by proteins that are expressed in several cell types. Indeed, Tph1 is expressed not only in enterochromaffin cells of the gut but also in β cells of the pancreas and cells of the pineal gland (Cote et al., 2007; Kim et al., 2010). In the pineal gland Tph1 initiates synthesis of melatonin, a hormone whose influence on glucose metabolism is not yet fully defined (Rios-Lugo et al., 2010).
Therapeutic implications of these functions of GDS
In type 2 diabetes there is often an increased lipolysis and liver gluconeogenesis that now occurs in the fed state. This aberrant activation of these two processes contributes to the progression of the disease since increasing liver gluconeogenesis leads to hyperglycemia while raising circulating FFAs levels contribute to peripheral insulin resistance (Saltiel and Kahn, 2001; Samuel and Shulman, 2012). Hence, the results presented here suggested that decreasing GDS synthesis could ameliorate type 2 diabetes.
That decreasing GDS circulating levels through genetic or pharmacological means improved glucose and insulin tolerance in mutant or WT mice rendered diabetic through a high fat diet supports this view although it does not mean that decreasing GDS synthesis could, alone, cure type 2 diabetes. The potential importance of this last aspect of our study stems, in part, from the fact that inhibition of GDS synthesis has already been shown to cure post-menopausal osteoporosis in rodents (Yadav et al., 2010). The steady increase in the incidence of osteoporosis and of type 2 diabetes implies that any therapeutic means improving the outcome of both diseases would have major advantages.
Experimental procedures
Animal experiments
Mice were housed under 12:12 light:dark cycle and chow and water were provided ad libitum. All animals were maintained according to NIH guidelines. The Htr2bflox/flox, Tph1flox/flox and Villin-cre mice have been described previously (Yadav et al., 2008). Alb-cre, aP2-cre and Adiponectin-Cre mice were obtained from the Jackon Labolatory.
For glucose tolerance tests mice were fasted overnight before i.p. injection of 2g/kg BW d-glucose in PBS or 1g/kg BW when mice were fed high fat diet. For pyruvate tolerance tests mice were fasted overnight, injected i.p. with 2 g/kg BW sodium pyruvate in PBS. Glycerol tolerance test (3g/kg BW) was performed after 4h fasting. For insulin tolerance test mice were injected 0.75U/kg after 4h fasting. Glucose was measured by tail vein bleeds at the indicated intervals using an Accu-check glucometer.
All fasting experiments started at 9a.m. Measurement of glycerol, FFA and β–hydroxybutyrate were performed in plasma using triglycerides determination kit, (TR0100, Sigma), NEFA-HR(2) (WAKO Diagnostics) and beta Hydroxybutyrate assay kit (Abcam). Plasma concentration of serotonin and glucagon were determined by Glucagon ELISA, (Alpco, 48-GLUHU-E01) and Serotonin (Research) ELISA, (IBL-America, IB89540).
Hyperinsulinemic-euglycemic clamp was performed in the Mouse Phenotyping Center of University of Massachusetts Medical School. Prior to experiments mice were fasted for 4h; the insulin infusion rate was 1.25 mU/kg/min.
Enzymatic activity assays
FBPase, G6Pase and glucokinase enzymatic activities assays were performed on livers of fasted mice as described previously (Alegre et al., 1988; Reyes et al., 1987). All reagents were obtained from Sigma.
In vitro lipolysis of isolated fat pads
In vitro lipolysis assay was performed as described (Schweiger et al., 2006). Briefly, epididymal (or subcutaneous, where indicated) fat pads were isolated and washed several times in HBSS (Sigma). Tissue pieces (~ 25mg) were incubated in DMEM medium (Invitrogen) containing 2% fatty acids free bovine serum albumin (Sigma) at 37°C for 1h. Fat explants were then transferred into identical, fresh medium containing serotonin (Sigma) or 10μM isoproterenol as a positive control and incubated further for the indicated time-points at 37°C. Afterwards aliquots of medium were taken for measurements of FFAs and glycerol and tissue was lysed with RIPA buffer for subsequent Western blot analysis.
Statistical analysis
Data are shown as mean values ± s.e.m. Analyses of significant differences between means were performed using two-tailed Student’s t-tests in case of comparison of 2 groups or analysis of variance followed by Tukey’s post-hoc test, in case of multiple groups. n, number of independent cultures or animals used. In all cases *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary Material
Acknowledgments
We thank Drs P. Ducy, I. Goldberg and R. Ricci for experimental advice and critical reading of the manuscript, the Albert Einstein Diabetes Research and Training Center, Animal Physiology Core (NIH DK 020541) for performing MRI. This work was supported by grants from NIH and Institut MERIEUX (G.K.), and HFSP (G.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alegre M, Ciudad CJ, Fillat C, Guinovart JJ. Determination of glucose-6-phosphatase activity using the glucose dehydrogenase-coupled reaction. Anal Biochem. 1988;173:185–189. doi: 10.1016/0003-2697(88)90176-5. [DOI] [PubMed] [Google Scholar]
- Amireault P, Hatia S, Bayard E, Bernex F, Collet C, Callebert J, Launay JM, Hermine O, Schneider E, Mallet J, et al. Ineffective erythropoiesis with reduced red blood cell survival in serotonin-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13141–13146. doi: 10.1073/pnas.1103964108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthonsen MW, Ronnstrand L, Wernstedt C, Degerman E, Holm C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. The Journal of biological chemistry. 1998;273:215–221. doi: 10.1074/jbc.273.1.215. [DOI] [PubMed] [Google Scholar]
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annual review of medicine. 2009;60:355–366. doi: 10.1146/annurev.med.60.042307.110802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black A, Allison DB, Shapses SA, Tilmont EM, Handy AM, Ingram DK, Roth GS, Lane MA. Calorie restriction and skeletal mass in rhesus monkeys (Macaca mulatta): evidence for an effect mediated through changes in body size. J Gerontol A Biol Sci Med Sci. 2001;56:B98–107. doi: 10.1093/gerona/56.3.b98. [DOI] [PubMed] [Google Scholar]
- Carlson LA, Ekelund LG, Oro L. Metabolic and cardio-vascular effects of serotonin. Life sciences. 1967;6:261–271. doi: 10.1016/0024-3205(67)90155-5. [DOI] [PubMed] [Google Scholar]
- Cote F, Fligny C, Bayard E, Launay JM, Gershon MD, Mallet J, Vodjdani G. Maternal serotonin is crucial for murine embryonic development. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:329–334. doi: 10.1073/pnas.0606722104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dees C, Akhmetshina A, Zerr P, Reich N, Palumbo K, Horn A, Jungel A, Beyer C, Kronke G, Zwerina J, et al. Platelet-derived serotonin links vascular disease and tissue fibrosis. The Journal of experimental medicine. 2011;208:961–972. doi: 10.1084/jem.20101629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, et al. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. The EMBO journal. 2010;29:469–481. doi: 10.1038/emboj.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg C, et al. Increased bone formation in osteocalcin-deficient mice. Nature. 1996;382:448–452. doi: 10.1038/382448a0. [DOI] [PubMed] [Google Scholar]
- Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos-Flier E, Rosen ED. Transcriptional control of adipose lipid handling by IRF4. Cell metabolism. 2011;13:249–259. doi: 10.1016/j.cmet.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes & development. 2012;26:271–281. doi: 10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fligny C, Fromes Y, Bonnin P, Darmon M, Bayard E, Launay JM, Cote F, Mallet J, Vodjdani G. Maternal serotonin influences cardiac function in adult offspring. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2008;22:2340–2349. doi: 10.1096/fj.07-100743. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rodriguez A, Nevado C, Escriva F, Sesti G, Rondinone CM, Benito M, Valverde AM. PTP1B deficiency increases glucose uptake in neonatal hepatocytes: involvement of IRA/GLUT2 complexes. American journal of physiology Gastrointestinal and liver physiology. 2008;295:G338–347. doi: 10.1152/ajpgi.00514.2007. [DOI] [PubMed] [Google Scholar]
- Greenberg AS, Shen WJ, Muliro K, Patel S, Souza SC, Roth RA, Kraemer FB. Stimulation of lipolysis and hormone-sensitive lipase via the extracellular signal-regulated kinase pathway. The Journal of biological chemistry. 2001;276:45456–45461. doi: 10.1074/jbc.M104436200. [DOI] [PubMed] [Google Scholar]
- Guillam MT, Burcelin R, Thorens B. Normal hepatic glucose production in the absence of GLUT2 reveals an alternative pathway for glucose release from hepatocytes. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12317–12321. doi: 10.1073/pnas.95.21.12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell metabolism. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Iwata T, Taniguchi H, Kuwajima M, Taniguchi T, Okuda Y, Sukeno A, Ishimoto K, Mizusawa N, Yoshimoto K. The action of D-dopachrome tautomerase as an adipokine in adipocyte lipid metabolism. PloS one. 2012;7:e33402. doi: 10.1371/journal.pone.0033402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, Fujitani Y, Kawamori R, Miyatsuka T, Kosaka Y, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med. 2010;16:804–808. doi: 10.1038/nm.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–469. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesurtel M, Graf R, Aleil B, Walther DJ, Tian Y, Jochum W, Gachet C, Bader M, Clavien PA. Platelet-derived serotonin mediates liver regeneration. Science. 2006;312:104–107. doi: 10.1126/science.1123842. [DOI] [PubMed] [Google Scholar]
- Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell metabolism. 2011;14:9–19. doi: 10.1016/j.cmet.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Yang Q, Sun W, Vogel P, Heydorn W, Yu XQ, Hu Z, Yu W, Jonas B, Pineda R, et al. Discovery and characterization of novel tryptophan hydroxylase inhibitors that selectively inhibit serotonin synthesis in the gastrointestinal tract. J Pharmacol Exp Ther. 2008;325:47–55. doi: 10.1124/jpet.107.132670. [DOI] [PubMed] [Google Scholar]
- Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BH, Quast MJ, Gorenstein D, Chen KH, Chan L. Absence of perilipin results in leanness and reverses obesity in Lepr(db/db) mice. Nat Genet. 2000;26:474–479. doi: 10.1038/82630. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM. Assessing the potential of glucokinase activators in diabetes therapy. Nature reviews Drug discovery. 2009;8:399–416. doi: 10.1038/nrd2850. [DOI] [PubMed] [Google Scholar]
- Moore MC, Geho WB, Lautz M, Farmer B, Neal DW, Cherrington AD. Portal serotonin infusion and glucose disposal in conscious dogs. Diabetes. 2004;53:14–20. doi: 10.2337/diabetes.53.1.14. [DOI] [PubMed] [Google Scholar]
- Osuga J, Ishibashi S, Oka T, Yagyu H, Tozawa R, Fujimoto A, Shionoiri F, Yahagi N, Kraemer FB, Tsutsumi O, et al. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:787–792. doi: 10.1073/pnas.97.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulmann N, Grohmann M, Voigt JP, Bert B, Vowinckel J, Bader M, Skelin M, Jevsek M, Fink H, Rupnik M, et al. Intracellular serotonin modulates insulin secretion from pancreatic beta-cells by protein serotonylation. PLoS biology. 2009;7:e1000229. doi: 10.1371/journal.pbio.1000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea A, Clemente F, Martinell J, Villanueva-Penacarrillo ML, Valverde I. Physiological effect of glucagon in human isolated adipocytes. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 1995;27:372–375. doi: 10.1055/s-2007-979981. [DOI] [PubMed] [Google Scholar]
- Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, Mohammadi M, Finck BN, Mangelsdorf DJ, Kliewer SA, et al. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:10853–10858. doi: 10.1073/pnas.0904187106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Burgos ME, Hubert E, Slebe JC. Selective thiol group modification renders fructose-1,6-bisphosphatase insensitive to fructose 2,6-bisphosphate inhibition. The Journal of biological chemistry. 1987;262:8451–8454. [PubMed] [Google Scholar]
- Rios-Lugo MJ, Cano P, Jimenez-Ortega V, Fernandez-Mateos MP, Scacchi PA, Cardinali DP, Esquifino AI. Melatonin effect on plasma adiponectin, leptin, insulin, glucose, triglycerides and cholesterol in normal and high fat-fed rats. Journal of pineal research. 2010;49:342–348. doi: 10.1111/j.1600-079X.2010.00798.x. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. Molecular regulation of adipogenesis. Annual review of cell and developmental biology. 2000;16:145–171. doi: 10.1146/annurev.cellbio.16.1.145. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger M, Schreiber R, Haemmerle G, Lass A, Fledelius C, Jacobsen P, Tornqvist H, Zechner R, Zimmermann R. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. The Journal of biological chemistry. 2006;281:40236–40241. doi: 10.1074/jbc.M608048200. [DOI] [PubMed] [Google Scholar]
- Sohle J, Machuy N, Smailbegovic E, Holtzmann U, Gronniger E, Wenck H, Stab F, Winnefeld M. Identification of new genes involved in human adipogenesis and fat storage. PloS one. 2012;7:e31193. doi: 10.1371/journal.pone.0031193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. The Journal of clinical investigation. 2012;122:4–12. doi: 10.1172/JCI60016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegiopoulos A, Herzig S. Glucocorticoids, metabolism and metabolic diseases. Molecular and cellular endocrinology. 2007;275:43–61. doi: 10.1016/j.mce.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Vestergaard ET, Gormsen LC, Jessen N, Lund S, Hansen TK, Moller N, Jorgensen JO. Ghrelin infusion in humans induces acute insulin resistance and lipolysis independent of growth hormone signaling. Diabetes. 2008;57:3205–3210. doi: 10.2337/db08-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahren J, Ekberg K. Splanchnic regulation of glucose production. Annual review of nutrition. 2007;27:329–345. doi: 10.1146/annurev.nutr.27.061406.093806. [DOI] [PubMed] [Google Scholar]
- Wei W, Dutchak PA, Wang X, Ding X, Wang X, Bookout AL, Goetz R, Mohammadi M, Gerard RD, Dechow PC, et al. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor gamma. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3143–3148. doi: 10.1073/pnas.1200797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, He J, Jiang H, Zu L, Zhai W, Pu S, Xu G. Direct effect of glucocorticoids on lipolysis in adipocytes. Molecular endocrinology. 2009;23:1161–1170. doi: 10.1210/me.2008-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav VK, Balaji S, Suresh PS, Liu XS, Lu X, Li Z, Guo XE, Mann JJ, Balapure AK, Gershon MD, et al. Pharmacological inhibition of gut-derived serotonin synthesis is a potential bone anabolic treatment for osteoporosis. Nat Med. 2010;16:308–312. doi: 10.1038/nm.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G, Glorieux FH, Chiang CY, Zajac JD, Insogna KL, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135:825–837. doi: 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F. FAT SIGNALS - Lipases and Lipolysis in Lipid Metabolism and Signaling. Cell metabolism. 2012;15:279–291. doi: 10.1016/j.cmet.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.