

Abstract
Extended circulation of nanoparticles in blood is essential for most clinical applications. Nanoparticles are rapidly cleared by cells of the mononuclear phagocyte system (MPS). Approaches such as grafting polyethylene glycol onto particles (PEGylation) extend circulation times; however, these particles are still cleared, and the processes involved in this clearance remain poorly understood. Here, we present an intravital microscopy–based assay for the quantification of nanoparticle clearance, allowing us to determine the effect of mouse strain and immune system function on particle clearance. We demonstrate that mouse strains that are prone to Th1 immune responses clear nanoparticles at a slower rate than Th2-prone mice. Using depletion strategies, we show that both granulocytes and macrophages participate in the enhanced clearance observed in Th2-prone mice. Macrophages isolated from Th1 strains took up fewer particles in vitro than macrophages from Th2 strains. Treating macrophages from Th1 strains with cytokines to differentiate them into M2 macrophages increased the amount of particle uptake. Conversely, treating macrophages from Th2 strains with cytokines to differentiate them into M1 macrophages decreased their particle uptake. Moreover, these results were confirmed in human monocyte–derived macrophages, suggesting that global immune regulation has a significant impact on nanoparticle clearance in humans.
Introduction
The potential clinical applications of nanoparticles and nanoformulations have been investigated for more than 30 years. Nanoparticle approaches have the potential to revolutionize drug delivery by allowing for the encapsulation of drugs with poor solubility or stability in a stable carrier particle. In addition, targeting nanoparticles to specific pathological sites may allow an increased effective dose of drug at the needed site, while decreasing systemic drug exposure, and therefore side effects. However, to date, only 2 nanoformulations for cancer treatment have been approved for clinical use (liposomal doxorubicin [Doxil] and protein-bound paclitaxel [Abraxane]) (1–3). One major obstacle for the use of nanoparticles in vivo is rapid clearance by the cells of the reticuloendothelial system (RES)/mononuclear phagocyte system (MPS) (4–7). In addition to rapid clearance, variable activity of the MPS among patients leads to widely variable pharmacokinetics of nanoformulations in the clinic, reducing the efficacy of both approved and future experimental nanoformulations (8). The main strategy for extending the circulation time of nanoparticles is grafting of uncharged hydrophilic polymers onto the surface of particles for “stealthing,” with the most common polymer used being polyethylene glycol (PEG) (9). PEGylation of particles clearly extends their circulation time in vivo (4, 10); however, up to 25% of patients exhibit circulating anti-PEG antibodies prior to treatment or develop anti-PEG antibodies after the first administration of PEGylated particles (11, 12). These factors limit the utility of PEGylation of nanoparticles in the clinic and suggest that a better understanding of the biomolecular interactions of nanoparticles and the MPS is critical for the development of alternative methods to PEGylation that will extend nanoparticle circulation times in vivo.
The MPS is composed of macrophages and DCs located in the liver and spleen, as well as monocytes and other phagocytic cells in the blood and spleen. When nanoparticles are i.v. injected and begin to circulate in the blood, they make direct contact with these MPS cells. Once a particle is in contact with MPS cells, receptors on the cell surface either directly recognize the particle, or recognize opsonizing serum proteins that have become attached to the particle. This leads to internalization of the particle and sequestration in the MPS cells (13). Extensive work has been done to understand particle uptake at the cellular level, with various studies implicating scavenger receptors (14), complement (15), and Fc receptors (16). The majority of these studies have been conducted with immortalized macrophage cell lines in vitro and with various types and sizes of particles. However, the physiological relevance of particle uptake by immortalized cell lines in vitro for clearance in vivo is unclear.
Another relevant factor that has received little attention in the nanoparticle field is the effect that global immune status, such as the balance of Th1-Th2 cytokines and M1-M2 macrophages, has on the clearance process. During immune responses, helper T cells adopt distinct Th1 or Th2 identities, leading to the secretion of specific combinations of cytokines and chemokines that instruct a wide variety of immune cells, including macrophages (as reviewed in Murphy et. al., ref. 17). The presence of Th1 cytokines has a tendency to polarize macrophages toward a proinflammatory M1 phenotype (18). Conversely, Th2 immune responses can induce macrophage polarization toward an antiinflammatory M2 phenotype that promotes wound healing and the resolution of inflammation, yet is known to contribute to diseases such as allergies and asthma (18, 19). In addition, M1 and M2 macrophages express different repertoires of phagocytic receptors and may show differential efficiency of endocytosis and phagocytosis (18, 19). It appears that the effect of macrophage polarization on phagocytosis is target dependent: M1 macrophages show enhanced phagocytosis of S. aureus (20), while M2 macrophages are more phagocytic toward myelin (21), apoptotic cells, and latex beads (22). The development of T cell–based immune responses requires days to weeks and would not be immediately provoked by exposure to particles. However, it is possible that Th1/Th2 priming from previous immune responses or differences in genetic background could affect the clearance of nanoparticles via changes in the relative number of M1 versus M2 macrophages (17).
Current techniques for assaying quantitative or relative nanoparticle pharmacokinetics (PK) have significant drawbacks for screening multiple particle types and/or animal models due to issues with time, expense, sensitivity, and temporal resolution (see Discussion). Intravital microscopy (IVM) is an appealing alternative for assaying the relative PK of nanoparticles in vivo. IVM has been used to assay the accumulation of targeted particles in tumors (23), tissue (24), the circulation time of gold nanorods (25), the accumulation of particles in the liver (26), the effect of size and shape on tumor extravasation (27), and the circulation of hydrogel microparticles (28). In order to determine the roles of particle parameters and global immune status on nanoparticle PK, a calibration-quality nanoparticle that has low batch-to-batch variability, with very low polydispersity, is required. To this end, we used the particle replication in non-wetting templates (PRINT, Liquidia Technologies) technique to generate particles of varying sizes. PRINT provides superior control over particle geometry and physical properties by taking advantage of a novel soft lithographic technique for particle fabrication. This results in low batch-to-batch variability with very low polydispersity values (29, 30). Recent work has demonstrated that the PK of PRINT nanoparticles can be carefully and reproducibly calibrated by adjusting the density of PEG chains present on the surface (31).
In this study, we observed striking differences in nanoparticle clearance kinetics between commonly used WT mouse strains using IVM and flow cytometry. We have identified the cell types and anatomical locations of this differential clearance and identified the global (Th1 versus Th2) immune status as a critical factor in nanoparticle clearance in vivo.
Results
IVM allows for rapid screening of nanoparticle clearance.
The ability to easily and inexpensively screen different formulations of nanoparticles in animal models is an unmet need in the field of nanomedicine. To address this need, we have modified an IVM technique that we recently developed to measure the PK of red blood cell mimetic (RBCM) microparticles (28). This technique allows for screening the relative nanoparticle resident times in the blood using as few as 4 animals per condition. In order to reduce variation in nanoparticles for these experiments, we used the PRINT technique to generate monodisperse 300-nm cylindrical PEG hydrogel nanoparticles containing far-red fluorescent dyes for in vivo and in vitro imaging (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI66895DS1); these particles are similar in size and composition to those used in a recent study by our group (31). When i.v. injected, these particles produce bright fluorescence in the vasculature of a mouse and can be easily imaged in the ear (Figure 1, A–C, and Supplemental Video 1). A time versus fluorescence intensity profile can then be generated, and the AUC can be calculated (Figure 1D). By comparing the AUC from different mice, we can determine the relative exposure of nanoparticles to blood in vivo.
Figure 1. IVM-based assay for screening nanoparticle clearance rates in live animals.

(A) A blood vessel is positioned in the center of the scan area. (B) Cartoon showing the orientation of the mouse, the positioning of the objective, and the region of the mouse ear imaged. (C) Movies were analyzed with ImageJ software by selecting a straight-line region of interest (ROI) in the large vein, and the average fluorescence intensity for each time point was calculated. Scale bar: 50 μm. (D) A representative fluorescence versus time plot of nanoparticle clearance. Marked region represents the AUC, which is calculated to determine the relative exposure of the particles to blood.
Since the clearance of nanoparticles is mediated by the phagocytic cells of the MPS, we depleted these cells from female BALB/c mice using liposomal clodronate (Clod) (32) and continuously measured the blood fluorescence over a 2-hour period in treated and untreated mice (Figure 2A and compare Supplemental Videos 1 and 2). As expected, plots of average fluorescence intensity over time show a greater peak fluorescence intensity and longer nanoparticle residency in blood for Clod-treated mice compared with untreated controls (Figure 2B). To compare the relative exposure of nanoparticles with blood between the two conditions, the AUC was calculated (Figure 2C) and showed a greater than 6-fold increase in particle exposure to blood in the Clod-treated mice. This experiment validated the idea that IVM can be used to screen relative exposure of nanoparticles to blood.
Figure 2. Treatment of BALB/c mice with Clod increases the circulation of nanoparticles in vivo.

(A) Still images from a BALB/c mouse and a BALB/c mouse pretreated with Clod show significant differences in blood fluorescence from nanoparticles. Scale bar: 50 μm. (B) A plot of mean fluorescence intensity versus time shows the difference in particle clearance rates between the 2 conditions (n = 4). (C) Plotting AUC shows a significant (P < 0.003, unpaired 2-tailed t test) increase in fluorescence intensity in blood with Clod pretreatment (n = 4).
Interestingly, we observed that in all BALB/c mice, after a significant fraction of the particle dose had been cleared from the blood, areas containing concentrated particles became visible (Supplemental Video 1). Upon careful observation, it became clear that these visible areas of fluorescence were cells and not extracellular aggregates of particles, as some of them moved in a random manner, including some periods of persistent migration against the blood flow (Figure 3 and Supplemental Video 3). Based on observations from others, it is likely that these cells are a combination of neutrophils and peripheral monocytes (33). This uptake of particles in the peripheral vasculature could explain the common finding across many nanoparticle studies that a significant fraction of the injected particle dose is not recovered from the exsanguinated blood and major organs (34). Since a substantial proportion of blood vessel surface is left behind in the carcass and skin, particle clearance in the peripheral vasculature may account for some of the missing dose.
Figure 3. Peripheral immune cells clear nanoparticles in the vasculature.
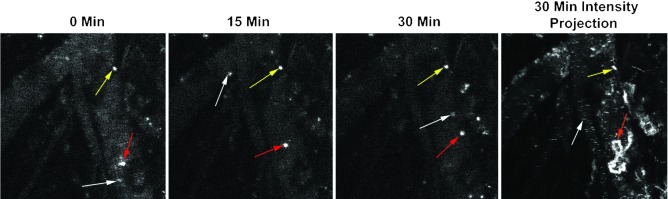
Still images from a representative movie (Supplemental Video 3) show the presence of 3 different types of cells made fluorescent by the uptake of labeled nanoparticles. White arrows indicate a cell that is free flowing in the blood and present for only a single frame of the movie, the red arrows show a single, randomly migrating cell, and the yellow arrows show a stationary cell. The 30-minute maximum intensity projection shows that the migrating cell (red arrows) moved in a circuitous route, including migrating against the blood flow.
Different mouse strains clear nanoparticles with different kinetics.
The removal of nanoparticles from blood is mediated by the MPS system and has been well studied on the cellular level by examining the role of various phagocytic receptors and endocytic/phagocytic mechanisms for particle uptake (14–16). These properties of MPS cells are regulated at the level of the whole animal by higher-order immune regulation mechanisms such as Th status (35). However, the role of these global immune properties on nanoparticle clearance kinetics has been poorly studied. We examined the effect of Th1/Th2 immune priming in mice by measuring the blood exposure of nanoparticles in 2 Th1-biased mouse strains (C57BL/6 and B10D2) (18), 2 Th2-biased mouse strains (BALB/c and DBA/2) (18), as well as BALB/c and C57BL/6 mice treated with Clod (Figure 4A). We observed that nanoparticle blood exposure was significantly higher in Th1-biased strains than in Th2-biased strains (Figure 4, B and C), suggesting that clearance mechanisms differ according to mouse strain. Clod treatment increased blood exposure in the BALB/c strain, but not in the C57BL/6 strain (Figure 4, B and C), indicating that little MPS-mediated clearance occurs in the first 2 hours for Th1-biased strains. In addition, we consistently observed that the initial peak fluorescence for BALB/c mice was significantly lower than in all other strains. Clod treatment resulted in BALB/c peak fluorescence levels similar to those of other strains, indicating that this lower peak fluorescence was due to the MPS. As peripheral immune cells containing particles were consistently observed to a greater extent in BALB/c mice compared with other strains tested, it is possible that peripheral clearance is stronger in BALB/c mice, resulting in a substantial amount of particles being cleared in the first pass through the circulatory system.
Figure 4. Th1 mouse strains clear nanoparticles slower than Th2 strains.

(A) Still images from Supplemental Videos 1–6 show the differences in particle circulation among the 4 mouse strains as well as the Clod-pretreated BALB/c and C57BL/6 mice. (B) Plots of fluorescence intensity versus time for all 4 mouse strains and Clod-treated mice (n = 4 mice per condition). (C) Plots of AUC for all conditions show a significant (P < 0.0001, 1-way ANOVA with Dunnett’s post test) increase in blood fluorescence in BALB/c and C57BL/6 and B10D2 mice, but not in DBA2 mice. Clod treatment significantly increases (P < 0.003, unpaired 2-tailed t test) blood fluorescence in BALB/c mice, but not in C57BL/6 mice.
To further address differences in clearance, we performed a biodistribution study by injecting BALB/c and C57BL/6 mice with particles and then sacrificing the mice at 5 minutes, 30 minutes, 2 hours, and 24 hours after injection, followed by measuring fluorescence in homogenates of tissues using a plate reader. Relative particle distribution was measured in the nonlymphoid organs (heart, lung, and kidneys; Supplemental Figure 2A), lymphoid organs (spleen and liver; Supplemental Figure 2B), whole blood (Supplemental Figure 2C), plasma (Supplemental Figure 2D), and blood cell fraction (red cells and leukocytes of whole blood) (Supplemental Figure 2E). Nonlymphoid organs showed low levels of particles at all time points, but C57BL/6 mice showed substantially higher levels of particles at 30 minutes and 2 hours in heart and kidney, likely due to more particles still being present in the blood contained in those organs. Spleen and liver showed a high level of particle uptake at all time points, with BALB/c mice having significantly more particles present at 5 minutes, 30 minutes, and 2 hours. By 24 hours, no significant differences were observed in any organ or compartment. Whole blood and plasma showed significantly more particles in C57BL/6 mice at 5 minutes, 30 minutes, and 2 hours, but no significant difference was observed by 24 hours. Blood cells showed significantly more particles in BALB/c mice at 5 and 30 minutes, consistent with the IVM results.
Some of the differential clearance of particles is due to uptake by granulocytes in Th2-prone strains.
To further understand the differences in clearance of nanoparticles by Th1- and Th2-prone mouse strains, we performed flow cytometric analysis of BALB/c and C57BL/6 PBLs, splenocytes, and lymph nodes at 2 hours after injection. Lymph nodes were also assayed at 24 hours after injection to check for trafficking of phagocytes with particles to lymph nodes. We used a multimarker flow cytometry protocol to determine particle uptake by lymphocytes (T and B cells), macrophages, monocytes, granulocytes, and DCs (Supplemental Figure 3). Lymph nodes showed no particles present at 2 or 24 hours after injection (data not shown), suggesting that the PRINT particles used do not gain access to the lymphatic system after i.v. injection. Uptake by T and B cells was minimal in both strains at 2 hours in blood and spleen, with no significant difference between strains (Figure 5A). Uptake by DCs was observed in the spleen in both strains at 2 hours, with C57BL/6 DCs showing a significantly lower percentage of positive cells (Figure 5B). Uptake by tissue macrophages (fully differentiated monocytes defined by the marker F480) showed no significant difference between strains in the spleen (Figure 5B). Surprisingly, uptake by monocytes in blood and spleen was significantly higher in C57BL/6 mice (Figure 5, A and B), but uptake by granulocytes was dramatically lower in both blood and spleen in C57BL/6 mice (Figure 5, A and B). These data suggest that some of the differential clearance observed between these mouse strains may be due to differential use of monocytes versus granulocytes for particle uptake in the blood.
Figure 5. Flow cytometric analysis of PBLs and splenocytes shows dramatically higher uptake of particles by granulocytes in BALB/c mice.

(A) Flow cytometric analysis of PBLs shows significantly higher uptake of particles by granulocytes in BALB/c mice (P < 0.002, unpaired 2-tailed t test), and significantly higher uptake by monocytes in C57BL/6 mice (n = 4) (P < 0.05, unpaired 2-tailed t test). (B) Flow cytometric analysis of splenocytes shows significantly higher uptake of particles by DCs (P < 0.05, unpaired 2-tailed t test) and granulocytes (P < 0.003, unpaired 2-tailed t test) in BALB/c mice, while monocytes take up significantly more particles in C57BL/6 mice (n = 4) (P < 0.05, unpaired 2-tailed t test).
To address this idea directly, we depleted granulocytes from both strains by administering anti–Ly6-G antibodies, which have previously been shown to selectively deplete granulocytes in vivo (36). Flow analysis confirmed that after antibody depletion, granulocyte numbers were reduced to below detection levels in both strains (Supplemental Figure 4). IVM was performed on both strains after granulocyte depletion to determine the effect on particle blood exposure. After granulocyte depletion, BALB/c mice showed an equivalent peak fluorescence intensity compared with C57BL/6 mice (Figure 6A) and a significant increase in particle blood exposure compared with untreated BALB/c mice (Figure 6B). Conversely, C57BL/6 mice after granulocyte depletion showed no significant increase in peak fluorescence or particle blood exposure when compared with untreated C57BL/6 mice (Figure 6B). These data indicate that some of the lower particle blood exposure (i.e., faster clearance) and lower peak particle concentration in BALB/c mice during the early passage through the circulation are due to particle uptake by granulocytes in the blood.
Figure 6. Depletion of granulocytes significantly increases particle exposure to blood in BALB/c mice but not in C57BL/6 mice.

(A) Plots of fluorescence intensity versus time for BALB/c and C57BL/6 mice with and without monocyte depletion (n = 4 mice per condition). (B) Plots of AUC show a significant increase in particle exposure to blood in BALB/c mice (P < 0.01, 1-way ANOVA with Dunnett’s post test) but not in C57BL/6 mice (n = 4 mice per condition). (C) Flow cytometric analysis of changes in PBL particle distribution in BALB/c and C57BL/6 mice following granulocyte depletion. BALB/c monocytes take up significantly more particles after granulocyte depletion (P < 0.05, unpaired 2-tailed t test) (n = 3 mice for granulocyte-depleted conditions, and n = 4 mice for controls). (D) Flow cytometric analysis of splenocytes after granulocyte depletion in BALB/c and C57BL/6 mice. Macrophages in spleen take up significantly more particles after granulocyte depletion in both strains (P < 0.05, unpaired 2-tailed t test) (n = 3 mice for granulocyte-depleted conditions, and n = 4 mice for controls).
To determine the fate of particles in the blood and spleen in the absence of granulocytes, we assessed particle uptake by immune populations in granulocyte-depleted mice using flow cytometry. Particle uptake by T and B lymphocytes of blood and spleen were unaffected (Figure 6, C and D); however, increased particle uptake by blood monocytes was observed in the granulocyte-depleted BALB/c mice, but not in the C57BL/6 mice (Figure 6C). In the spleen, granulocyte depletion did not affect monocyte uptake, but did increase macrophage uptake, with a greater increase observed in the BALB/c mice (Figure 6D). Together, these data indicate that granulocytes account for some of the increased clearance seen in the BALB/c mice, but that in the absence of these cells, BALB/c mice still clear nanoparticles faster than C57BL/6 mice.
Differential clearance of particles in Th1 versus Th2 strains is dependent on particle type.
Since a wide variety of nanoformulations are being investigated for potential clinical use and often demonstrate very different properties in vivo, we tested whether the differential clearance observed in various mouse strains is specific to the PRINT particles (300 nm, PEG hydrogel). We tested the blood exposure of quantum dots (Qdots) in BALB/c and C57BL/6 mice. The Qdots were polymer coated with a carboxylated polymer that prevents aggregation in aqueous environments. The Qdots tested are a different class of nanoparticle (inorganic nanocrystal versus hydrogel) with a different surface (proprietary carboxylated polymer versus PEG), a different shape (sphere versus cylinder), and an order of magnitude smaller (average Qdot size of 30 nm versus 300-nm cylindrical PRINT hydrogels (Supplemental Figure 1, A–C). Upon i.v. injection, the Qdots were more rapidly removed from the blood by both BALB/c and C57BL/6 mice than were the PRINT particles. However, the Th1- versus Th2-biased strain clearance difference was still present (Figure 7, A and B), including a lower peak fluorescence intensity with Qdots, suggesting that a large number of Qdots are cleared in the periphery of BALB/c mice. To test whether microparticles display the same effect in Th1 versus Th2 strains, we used low elastic modulus PRINT microparticles (6-μm disks made of the same PEG hydrogel materials as the 300-nm cylindrical PRINT hydrogels) that were previously demonstrated to have long circulation times and are not cleared by mechanical filtration in the lungs (28). Interestingly, the BALB/c and C57BL/6 mice demonstrated the same microparticle exposure to blood (Figure 7, C and D), suggesting that the rapid removal of nanoparticles in Th2-prone strains is not observed with larger particles requiring phagocytic clearance.
Figure 7. The enhanced clearance of nanoparticles by Th2 strains is not observed with micro-sized particles.

(A) Plots of fluorescence intensity versus time for Qdots in blood of BALB/c and C57BL/6 mice show a similar trend to what was observed with 300-nm PRINT hydrogel particles (n = 4). (B) AUC shows a significant (P < 0.05, unpaired 2-tailed t test) increase in blood fluorescence from Qdots in C57BL/6 mice versus BALB/c mice. (C) Plot of fluorescence intensity in blood versus time for BALB/c and C57BL/6 mice show similar clearance rates for microparticles. (D) AUC analysis of blood fluorescence intensity from both strains shows no significant difference.
Murine bone marrow–derived macrophages and primary human peripheral blood monocyte–derived macrophages take up more particles with M2 polarization.
We examined whether differential clearance by Th1- and Th2-prone strains can be reproduced in vitro by culturing primary bone marrow–derived macrophages (BMMs) from both strains and testing particle uptake by BMMs. We chose to use BMMs as opposed to granulocytes due to the fact that BMMs can be maintained in culture for longer periods of time, thus allowing treatment with Th1 and Th2 cytokine mixtures to polarize these macrophages toward M1 or M2 phenotypes, respectively. We imaged the accumulation of particles inside the macrophages and quantified the integrated fluorescence intensity of particles per cell. We also processed cells for analysis by flow cytometry. Consistent with our in vivo observations, untreated BALB/c macrophages take up significantly more particles than untreated C57BL/6 macrophages in vitro, as assayed by both confocal microscopy and flow cytometry (Figure 8, A and B). BMMs from both strains were also directed toward M1 or M2 polarization by specific mixtures of cytokines and immune stimulators (see Methods). M1 polarization of C57BL/6 macrophages had no effect on particle uptake, while differentiation into the M2 phenotype resulted in an increase in particle uptake (Figure 8, A and B). Conversely, M1 polarization of BALB/c macrophages reduced particle uptake to the same level as C57BL/6 macrophages, while M2 polarization showed no significant difference compared with BALB/c controls (Figure 8, A and B).
Figure 8. M2-polarized macrophages take up more particles than M1 macrophages in mice and humans.

(A) Average integrated fluorescence per cell showed a significant (P < 0.0001, 1-way ANOVA with Dunnett’s post test) increase in particle uptake by BALB/c untreated, BALB/c Th2-treated, and C57BL/6 Th2-treated versus C57BL/6 untreated cells. C57BL/6 M1-treated and BALB/c M1-treated cells showed no significant difference compared with C57BL/6 untreated cells (n = 4). (B) Flow cytometric analysis of uptake showed a significant (P < 0.05, 1-way ANOVA with Dunnett’s post test) increase in uptake by BALB/c untreated, BALB/c Th2-treated, and C57BL/6 Th2-treated versus C57BL/6 untreated cells. C57BL/6 M1-treated and BALB/c M1-treated cells showed no significant difference compared with C57BL/6 untreated cells (n = 4). (C) Flow cytometric analysis of uptake by human macrophages from volunteer A. M1 macrophages took up significantly fewer particles than control or M2 macrophages (n = 4) (P < 0.0001, 1-way ANOVA with Dunnett’s post test). (D) Flow cytometric analysis of uptake by human macrophages from volunteer B. M1 macrophages took up significantly fewer particles than the control macrophages (P < 0.0001, 1-way ANOVA with Dunnett’s post test). M2 macrophages took up significantly more particles than the control macrophages (n = 4) (P < 0.001, 1-way ANOVA with Dunnett’s post test). (E) Flow cytometric analysis of surface MMR expression on Balb/c and C57BL/6 mice. Balb/c mice showed significantly higher surface expression of MMR (n = 4) (P < 0.05, unpaired 2-tailed t test). (F) Microscopic analysis of uptake by BMMs after mannan blocking. The addition of mannan reduced uptake by Balb/c macrophages to the same levels as those of the C57BL/6 controls (n = 4) (P < 0.0001, 1-way ANOVA with Dunnett’s post test). (G) Representative flow cytometric histogram of particle uptake by BALB/c and C57BL/6 BMMs.
As significant differences are known to exist between mouse and human macrophages (37), we repeated particle uptake experiments with human monocyte–derived macrophages from 2 healthy human volunteers. Volunteer A’s macrophages showed high uptake of particles with no stimulation, a significant reduction in uptake with M1 polarization, and no difference with M2 polarization (Figure 8C). Volunteer B’s macrophages showed intermediate uptake of particles with no stimulation, a significant decrease in uptake with M1 polarization, and a significant increase in uptake with M2 polarization (Figure 8D). These results confirm that immune polarization fundamentally impacts cellular interactions with nanoparticles in human cells.
One important difference between M1 and M2 macrophages is that M2 macrophages are known to express higher levels of many different scavenger and lectin receptors than M1 macrophages. Flow cytometric analysis revealed increased macrophage manose receptor (MMR) expression on the surface of BALB/c macrophages BMMs compared with C57BL/6 macrophages (Figure 8E), which is consistent with previously published transcriptional profiling data (35, 38). To test whether the differential uptake of particles by BALB/c and C57BL/6 macrophages is MMR dependent, we treated the BMMs with the competitive MMR inhibitor mannan prior to the addition of particles. Mannan reduced particle uptake in the BMMs to the same level as the C57BL/6 macrophages, while showing no effect on C57BL/6 macrophages, suggesting that the increased level of MMR present on M2 macrophages in BALB/c mice may contribute to the differences in particle uptake in vitro (Figure 8F and Supplemental Figure 5). Figure 8G shows a representative flow cytometry histogram of particle uptake by BALB/c and C57BL/6 BBMs.
Discussion
In this study, we have identified a striking difference in nanoparticle clearance in different strains of mice that arises due to differences in global immune status. Th1-prone strains such as C57BL/6 clear particles more slowly and have a higher blood exposure compared with the Th2-prone BALB/c strain that demonstrated rapid particle clearance. Both monocytes, macrophages, and granulocytes in the peripheral vasculature and spleen are responsible for clearance differences between the strains. Interestingly, the differences in clearance were recapitulated in vitro using mouse and human macrophages treated with either Th1 or Th2 chemokine-cytokine mixtures. This effect may be due, in part, to differences in surface expression of scavenger receptors such as the MMR.
In most studies, nanoparticle PK is determined by blood draws at specific time points after injection, combined with appropriate assays for determining the particle concentration in the blood (39, 40). This method has the advantage of directly measuring the particle itself, rather than a reporter probe, and provides an absolute particle concentration in the blood. However, this approach requires large numbers of animals and particles to yield useful data. Most experiments of this type require 4 animals or more per time point, and at least 5 time points plus a control for a minimum of 24 animals needed for each condition tested. This makes blood-draw assays an unattractive option for screening multiple types of animals and/or particle types due to the large time and resource investments required. In addition, blood-draw PK tends to give very limited temporal resolution, usually 7 or fewer points per 24-hour experiment (41).
IVM offers a number of advantages for measuring nanoparticle clearance. With IVM, the relative amount of particles in the bloodstream is measured with a wide dynamic range, allowing for measurements spanning preinjection to peak particle concentration through clearance in a single animal. Fluorescence probes can be directly incorporated into hydrogel nanoparticles during synthesis, therefore eliminating the need for extensive postfabrication modifications. In addition, this technique is very fast, allowing for real-time or near–real-time measurement of relative particle concentrations. Finally, IVM can be multiplexed with different fluorescent dyes coupled with different particle types or formulations to measure differential clearance in the same animal. However, IVM is limited by the fact that only relative numbers are readily available from this type of experiment, the particle must be fluorescent, and it is difficult to run experiments longer than 3 hours due to the stress on the animal from extended anesthesia.
The nanoparticle research field operates under the implicit assumption that nanoparticles will circulate in similar ways in all strains of mice. We are unaware of any previous study that has directly compared the effect of mouse strain on nanoparticle circulation times. Our results indicate that mouse strain background is a critical factor for nanoparticle clearance and is likely important for the interpretation of all nanomedicine study results. In fact, the changes in circulation times observed between Th1- and Th2-prone mouse strains are equivalent to the changes that can be produced by heavily PEGylating particle surfaces (31). This demonstrates that changes in the immune status of patients and experimental animals may affect nanoparticle PK to at least the same degree as the material properties of particles. However, far less work has been done to study the effects of biology on nanoparticle PK than the effects of material properties on nanoparticle PK. It is perhaps not surprising that we see different results in different mouse strains for clearance, as mouse strain background has a significant impact on biological processes ranging from immune function (42), pain sensation (43), and cancer (44). Outbred WT mice show a large degree of heterogeneity in many measures of immune function, while inbred lab strains of mice show very little intrastrain variation (45). In the future, it would be beneficial for researchers in the nanoparticle field to begin explicitly considering mouse strain immunology when designing experiments and interpreting data.
Based on our results, global immune system regulation has a significant role in nanoparticle clearance. One main global immunological difference is the Th1 versus Th2 status. Numerous studies have established that Th1 and Th2 cytokines can induce macrophage differentiation into the corresponding M1 and M2 phenotypes (19). The fact that Th1-prone mice (e.g., C57BL/6) clear nanoparticles slower is somewhat surprising, as M1 macrophages are thought to be more inflammatory and involved in the destruction of pathogens, and have been shown to be more phagocytic toward S. aureus (20). The M2 macrophages prevalent in the fast-clearing Th2 strains are generally thought to be antiinflammatory, with involvement in wound healing and potentially less phagocytic activity (19). However, M2 macrophages are thought to have higher levels of endocytosis and may therefore take up small particles rapidly. In addition, M2 macrophages are known to express higher levels of scavenger and lectin receptors that could be responsible for the increased clearance (35). Our data on the elevated surface expression of MMR on M2 macrophages and the reduced nanoparticle uptake upon mannan treatment support this notion.
While the addition of mannan blocks the enhanced uptake of PEG hydrogel particles in M2 macrophages in vitro, a baseline uptake of particles still occurs with treatment. Indeed, clearance in vivo likely involves multiple receptors and mechanisms. Determining which receptors are responsible for nanoparticle clearance is an important next step. Unfortunately, the obvious genetic approach of using KO mice in candidate clearance receptors is severely complicated by the fact that most of these KO mice exist only in the C57BL/6 strain. Since treatment of the slow-clearing C57BL/6 mice with Clod (removing all MPS cells) did not show a statistically significant increase in nanoparticle circulation times in a 2-hour window, it is unlikely that the genetic loss of any single candidate clearance receptor would produce a measurable decrease in nanoparticle clearance. In the future, it will be essential to backcross these KO mice into the BALB/c background, where these experiments are likely to produce more informative results.
It is also worth considering the role of granulocytes (primarily neutrophils) in nanoparticle clearance. Although this cell type is rarely discussed as an MPS cell (46, 47), our data clearly indicate that granulocytes play a major role in the differential nanoparticle clearance observed between mouse strains. Previous reports indicate that BALB/c mice have greater numbers of circulating granulocytes than C57BL/6 mice (48). Our data indicate that mere differences in granulocyte numbers are unlikely to account for the clearance differences between strains, as particles are taken up by a smaller percentage of C57BL/6 granulocytes than of BALB/c granulocytes. Since neutrophils constitute a large fraction of circulating white blood cells in humans (49), and neutropenia is a common side effect of clinical treatments such as chemotherapy, it will be important to further explore the role of neutrophils in nanoparticle clearance in future studies.
Similar to our results with mice, the PK of nanoparticles is highly variable in human patients (41). Previous studies demonstrated that monocyte function, as assayed by phagocytic capacity and oxidative burst, can be used to predict the clearance of Doxil (41). Since both the phagocytic capacity and oxidative burst are partially controlled by the patient’s global immune status (18), it is likely that the Th1-Th2 balance is an important factor in human nanoparticle clearance as well. This is supported by our results with human macrophages that show a marked decrease in particle uptake from both volunteers’ macrophages following M1 polarization and an increase in uptake with M2 polarization in 1 volunteer’s macrophages. Future studies are needed to explore the role of previous immune-priming events such as infections or allergies on nanoparticle clearance. Our data suggest that a more comprehensive understanding of how global immune regulation affects nanoparticle clearance will be important both for nanoparticle studies in animals and eventual clinical use.
Methods
Particle materials and methods
Materials.
Poly(ethylene glycol) diacrylate (Mn 700) (PEG700DA), 2-aminoethyl methacrylate hydrochloride (AEM), diphenyl (2,4,6-trimethylbenzoyl)-phosphine oxide (TPO), and sucrose were purchased from Sigma-Aldrich. Thermo Scientific Dylight 650 Maleimide, PTFE syringe filters (13-mm membrane, 0.22-μm pore size), and methanol were obtained from Fisher Scientific. Conventional filters (2-μm) were purchased from Agilent Technologies, and polyvinyl alcohol (Mw 2000) (PVOH) was purchased from Acros Organics. PRINT molds (200 nm × 200 nm) were obtained from Liquidia Technologies. Tetraethylene glycol monoacrylate (HP4A) was synthesized in house.
PRINT nanoparticle fabrication.
The pre-particle solution was prepared by dissolving 5 weight percent (wt%) of the various reactive monomers in methanol. The reactive monomers included: a cure-site monomer (an oligomeric PEG with a nominal molar mass of 700 g/mol terminally functionalized on both end groups with an acryloxy functionality); a hydrophilic monomer used to make up the majority of the particle composition (HP4A); a polymerizable fluorescent tag (Dylight 650 Maleimide); and a photoinitiator. The pre-particle solution was composed of 87.5 wt% HP4A, 10 wt% PEG700DA, 1.5 wt% Dylight 650, and 1 wt% TPO. Using a number 3 Mayer rod (R.D. Specialties), a thin film of the pre-particle solution was drawn onto a roll of freshly corona-treated PET using a custom-made roll-to-roll lab line (Liquidia Technologies) running at 12 feet per minute. The solvent was evaporated from this delivery sheet by exposing the film to a hot air dam derived from heat guns. The delivery sheet was laminated (80 psi, 12 feet per minute) to the patterned side of the mold, followed by delamination at the nip. Particles were cured by passing the filled mold through a UV LED lamp (395 nm, 30 psi N2, 12 feet per minute; Phoseon). A plasdone harvesting sheet was heat laminated to the filled mold (140°C, 80 psi, 12 feet per minute). Upon cooling to room temperature, particles were removed from the mold by splitting the harvesting sheet from the mold. Particles were then harvested by dissolving the plasdone in a bead of water (1 ml of water per 5 feet of harvesting sheet). The particle suspension was passed through a 2-μm filter (Agilent Technologies) to remove large particulates. To remove the excess plasdone, particles were centrifuged at approximately 21,000 g for 15 minutes, the supernatant was removed and the particles were resuspended in sterile water. This purification process was repeated 4 times.
Nanoparticle characterization.
Stock particle concentrations were determined by thermogravimetric analysis (TGA) using a TA Instruments Q5000. TGA analysis was conducted by pipetting 20 μl of the stock particle solution into a tared aluminum sample pan. Samples suspended in water were heated at 30°C per minute to 130°C, followed by a 10-minute isotherm at 130°C. Samples were then cooled at 30°C per minute to 30°C, followed by a 2-minute isotherm at 30°C. TGA was also performed on a 20-μl aliquot of supernatant from a centrifuged sample of the stock nanoparticle suspension to account for the mass of any stabilizer remaining in each sample. The supernatant concentration was subtracted from the concentration of stock particle suspension to determine the actual particle concentration. Particles were visualized by scanning electron microscopy using a Hitachi S-4700 scanning electron microscope. Prior to imaging, scanning electron microscopic samples where coated with 1.5 nm of gold-palladium alloy using a Cressington 108auto sputter coater. Particle size and zeta potential were measured by dynamic light scattering (DLS) on a Zetasizer Nano ZS (Malvern Instruments).
Microparticle fabrication.
PRINT molds made of perfluoropolyether were obtained from Liquidia Technologies. The RBCMs — starting from a pre-polymer composition of 87% 2-hydroxyethyl acrylate (Sigma-Aldrich), 10% 2-carboxyethyl acrylate (Sigma-Aldrich), 1% poly(ethylene glycol) diacrylate (4,000 MW) (PolySciences), and 1% DyLight 650 Maleimide (Thermo Scientific) — were made using a mold that had discoid features with 2-μm diameters and 0.6-μm heights. Briefly, the pre-polymer mixture was spread onto a piece of the mold (0.5 × 0.5 feet) that was chilled to 2κC to 5°C on a custom-built laminator platform to avoid monomer evaporation prior to photocuring. A poly(ethylene terepthalate) (PET) sheet was laminated to the top of the mold and pre-polymer solution, wetting the total mold area. The sheet was peeled away at the nip point of the laminator, leaving the wells of the mold filled, while wicking away excess solution. The filled mold was immediately transferred into a chilled (2–5°C) nitrogen-purged UV oven and cured with UV light (λ = 365 nm, power = 20 mW/cm2) for 3 minutes. After curing, the filled mold was placed face down on a thin film of 0.1% 2,000 g/mol poly(vinyl alcohol) (PVOH; Acros Organics) in water on top of another PET sheet. This assembly was placed in a cooler filled with dry ice, and the water was allowed to freeze and adhere to the particles. After freezing, the mold was peeled away from the particles trapped in the ice layer, and the ice was allowed to melt. RBCMs and water were collected, washed, and concentrated via centrifugation at approximately 21,000 g (Eppendorf 5417R) to remove any soluble fraction from the solution. Particles were then suspended in 0.1% 2,000 g/mol PVOH in PBS, pH 7.4, for subsequent experiments.
IVM
IVM experiments were performed using an IV-100 laser scanning microscope (Olympus) on female mice weighing 19–24 g. BALB/c (Charles River), C57BL/6, DBA/2, and B10D2 (The Jackson Laboratory) mice were maintained in clean animal housing at the University of North Carolina. Hair was removed from the ear, and a tail vein catheter was applied. The mice were anesthetized with isofluorane and placed onto a heated stage (37°C), with their ear immobilized by 2-sided taping to an aluminum block. Blood vessels were located by illuminating the ear with a bright white light while imaging in the green fluorescence channel. Blood vessels appear dark on the field. A suspension of 300-nm PRINT particles, consisting of 15 mg of particles per kilogram of mouse weight via a 4 mg/ml solution of particles in isotonic 9% sucrose solution, was then injected and visualized using a 633-nm laser. For all imaging, blood vessels were selected from the same approximate region of the ear to reduce differences in intensity due to the depth of the vessel. For experiments with Clod, mice were both i.v. and i.p. injected with 100 μl of 5 mg/ml Clod 24 hours prior to the experiments. Clod was purchased from Encapsula. For microparticle injections, 15 mg/kg of particles in a 2-mg/ml PBS suspension was used. For Qdot experiments, a 4-nmol water-soluble carboxylated 665-nm emission suspension of Qdots was purchased from Ocean NanoTech. Qdots (5 μl) were suspended in 100 μl of PBS and then i.v. injected via the tail vein. Granulocyte depletion was performed by i.p. injection of 1 mg of rat anti-mouse Ly6-G (GR-1) monoclonal antibody 1a8 (Bio X Cell) 3 days and 1 day prior to the experiments.
The image files from each scan were exported to ImageJ software (ImageJ; NIH) for image analysis of the blood exposure of the particles. For ease of analysis, we followed the example of Merkel et al. (28) and stacked the images in groups of 5. We analyzed the region of interest containing vasculature for fluorescence signals prior to particle injection and in each scan after injection. The background fluorescence value prior to injection was then subtracted from all subsequent fluorescence values. The data were then exported to GraphPad Prism (GraphPad Software) to generate graphs and perform statistical analysis.
Biodistribution
BALB/c and C57BL6 mice were injected with 300 nm of cylindrical PRINT particles as described above. At 5 minutes, 30 minutes, 2 hours, and 24 hours after injection, 4 mice of each strain were sacrificed by CO2 asphyxiation followed by cardiac puncture to collect blood. The lungs, kidneys, heart, liver and spleen were collected and kept in PBS on ice. Solid organs were weighed and then placed in 2× PBS (weight/volume). The organs were then homogenized, and PBS was added to each tube to bring it to a known volume. Organ homogenate (50 μl) was pipetted into a 96-well plate. Whole blood (50 μl) was plated followed by centrifuging the remaining blood for 5 minutes at 210 g to separate the plasma and blood cells. Plasma (50 μl) and cell fraction were then plated. The plates were read using a fluorescent plate reader (BD), and the fluorescence values per gram of tissue were calculated.
Flow cytometric analysis of animal samples
Blood was collected by cardiac puncture and added to EDTA tubes. Spleen and inguinal lymph nodes were dissected and placed in FACS buffer (1× PBS plus 5% FCS) on ice. All samples were kept on ice or at 4°C for the duration of the experiment. Blood (300 μl) was taken from each sample; red cells were lysed by the addition of 2 ml of 0.1× PBS for 10 seconds, followed by 2 ml of 2× PBS and 10 ml of FACS buffer. Spleen and lymph nodes were gently dissociated between frosted slides to free cells and were filtered through 70-μm cell filters. Red cells in spleen samples were lysed by the addition of 1 ml of ACK lysis buffer (Gibco) for 1 minute followed by 14 ml of FACS buffer. Cells were blocked with anti-CD16/32 (Fc block; BioLegend) at 2 μl per sample in 10 μl for 10 minutes. Mouse cells were then stained with the following antibodies: anti–CD3-FITC (145-211C), CD19-FITC (eBio1D3), CD19-efluor450 (eBio1D3), and anti–TCRβ-APC-Cy7 (H57-591) from eBioscience; anti–CD11c-PacBlue (N418), anti–F4/80-PE (CI:A3-1), CD11b-PE-Cy7 (M1/70), and anti–GR-1-APC-Cy7 (1A8) from BioLegend; and anti–CD45-PacOrange (MCD4530) from Invitrogen. Cells were stained using antibodies at 1:200 for 30 minutes on ice. Cells were fixed using 2% PFA in PBS. Particle fluorescence in BMM was detected in the far red channel with a 633-nm laser. The average fluorescence intensity of live single cells was calculated and reported. All data were collected using LSRII (BD Biosciences) or CyAn (Beckman Coulter) flow cytometers. Data analysis was performed using FlowJo software (Tree Star).
Cell culture reagents
BMMs were generated by extracting bone marrow as described previously (50) from 4 female BALB/c or C57BL/6 mice and differentiated into macrophages using L media as previously reported (51). After 1 week of differentiation, cells were placed in serum-free DMEM media (Sigma-Aldrich) for 24 hours. Cells were then washed once with PBS, placed in ice-cold PBS, 5% FBS and 0.5 mM EDTA, and left on ice for 10 minutes. Cells were then lifted by gentle scraping and pelleted by centrifugation. Macrophages (350,000) were then plated on glass coverslips in DMEM supplemented with 10% FBS (HyClone), L-glutamine, and penicillin-streptomycin for microscopy, or plated under the same media conditions in a 24-well plate for flow cytometry experiments. For cytokine stimulation, we followed a previously published protocol for in vitro differentiation of macrophages (35). Briefly, cells were exposed for 24 hours to 1 μg/ml of LPS (InvivoGen) and 25 ng/ml interferon-γ (eBioscience) for M1 stimulation. Cells were cultured with 25 ng/ml IL-4 (eBioscience) for M2 stimulation. Human macrophages were generated using a previously published protocol (38). Briefly, peripheral blood monocytes were collected from 2 healthy male volunteers, then cells were plated on 15-cm dishes in DMEM supplemented with 10% FBS, L-glutamine, penicillin-streptomycin, and 100 ng/ml human M-CSF (eBioscience) for 7 days. After culture, cells were lifted from the 15-cm plates as described for BMMs and plated in 24-well plates for 48 hours. Macrophages were then left untreated (control) or supplemented with 1 μg/ml LPS and 25 ng/ml human interferon-γ (eBioscience) or human IL-4 25 ng/ml (eBioscience) for 24 hours to generate M1 or M2 macrophages, respectively. For MMR blocking, 5 mg/ml mannan from Saccharomyces cerevisiae (Invitrogen) was added to cells for 5 minutes prior to the addition of particles.
Particle uptake
For confocal microscopic particle uptake experiments, stock medium was made with 75 μg/ml of 300-nm PRINT particles. Medium on the cells was then exchanged for medium plus FBS and particles. Cells were returned to the incubator for 20 minutes. Medium with particles was removed and the cells were washed 3 times with warm PBS and 5% FBS to remove particles. Cells were then fixed with ice-cold 4% paraformaldehyde in Krebs buffer and permeabilized with 0.1% Triton in PBS for 5 minutes. F-actin was stained with Alexa 488 phalloidin (Invitrogen). Coverslips were mounted on glass slides. For flow cytometric analysis, cells were exposed to particles and washed as described for microscopy. After washing, cells were placed in ice-cold PBS supplemented with 5% FBS and 0.5 mM ETDA on ice for 10 minutes followed by gentle scraping to remove them from the plastic.
Fixed-cell imaging and analysis
Cells were imaged with an Olympus FV1000 laser scanning confocal microscope. Using a ×10 objective, Z stacks were taken in 3 random fields of the coverslip in the Alexa 488 channel to image the cell body and in the Alexa 633 channel to image the particles. Each Z stack was then exported to ImageJ software and turned into 2 maximum-intensity projections of the cell body (phalloidin) and the particles. Single cells were then outlined by automated thresholding in the phalloidin channel, and particle fluorescence intensity was acquired in the 633-nm channel. A total of 50 random cells were analyzed per coverslip. The results from cells from 4 mice were then averaged to determine the relative particle uptake.
Flow cytometry of cultured macrophages
Cells were analyzed using a Dako cyan ADP flow cytometer (Beckman Coulter). Live single cells were selected using forward scatter and side scatter. Particle fluorescence was detected in the far red channel with a 633-nm laser. The average fluorescence intensity of live single cells was calculated and reported. All analysis was performed using FlowJo software (Tree Star).
Statistics
All statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software). P values less than 0.05 were considered significant. An unpaired 2-tailed t test was used for t tests, and 1-way ANOVA with Dunnett’s post test was used for ANOVA tests. All error bars represent ± SEM.
Study approval
Studies with human samples were performed under IRB number 12-1858, with approval from the Office of Human Research Ethics at the University of North Carolina Chapel Hill. All animal experiments were performed with approval of the IACUC of the University of North Carolina.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from Liquidia Technologies, HHMI, NIH, the Carolina Center for Cancer Nanotechnology Excellence (U54CA151652), the University Cancer Research Fund, and the UNC-Olympus Imaging Research Center for funding and resources. We thank Barbra Villen from the University of North Carolina Department of Microbiology and Immunology for important advice and Bill Zamboni from the University of North Carolina School of Pharmacy for advice and help with data interpretation. We also thank members of the Bear and DeSimone laboratories for critical discussions.
Footnotes
Conflict of interest: Joseph DeSimone is a founder and member of the board of directors of Liquidia Technologies and maintains a financial interest in the company. Liquidia was founded in 2004 to commercialize PRINT technology and other discoveries of Professor Joseph DeSimone and colleagues at the University of North Carolina, Chapel Hill.
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Citation for this article:J Clin Invest. 2013;123(7):3061–3073. doi:10.1172/JCI66895.
References
- 1.Krown SE, Northfelt DW, Osoba D, Stewart JS. Use of liposomal anthracyclines in Kaposi’s sarcoma. Semin Oncol. 2004;31(6 suppl 13):36–52. doi: 10.1053/j.seminoncol.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Coleman RE, et al. A randomised phase II study of two different schedules of pegylated liposomal doxorubicin in metastatic breast cancer (EORTC-10993). Eur J Cancer. 2006;42(7):882–887. doi: 10.1016/j.ejca.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother. 2006;7(8):1041–1053. doi: 10.1517/14656566.7.8.1041. [DOI] [PubMed] [Google Scholar]
- 4.Zamboni WC, et al. Best Practices in Cancer Nanotechnology: Perspective from NCI Nanotechnology Alliance. Clin Cancer Res. 2012;18(12):3229–3241. doi: 10.1158/1078-0432.CCR-11-2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leuschner F, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29(11):1005–1010. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nel AE, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8(7):543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 7.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zamboni WC, et al. Pharmacokinetic study of pegylated liposomal CKD-602 (S-CKD602) in patients with advanced malignancies. Clin Pharmacol Ther. 2009;86(5):519–526. doi: 10.1038/clpt.2009.141. [DOI] [PubMed] [Google Scholar]
- 9.Owens DE. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 10.Papahadjopoulos D, et al. Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci U S A. 1991;88(24):11460–11464. doi: 10.1073/pnas.88.24.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garay RP, El-Gewely R, Armstrong JK, Garratty G, Richette P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin Drug Deliv. 2012;9(11):1319–1323. doi: 10.1517/17425247.2012.720969. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu T, Ichihara M, Yoshioka Y, Ishida T, Nakagawa S, Kiwada H. Intravenous administration of polyethylene glycol-coated (PEGylated) proteins and PEGylated adenovirus elicits an anti-PEG immunoglobulin M response. Biol Pharm Bull. 2012;35(8):1336–1342. doi: 10.1248/bpb.b12-00276. [DOI] [PubMed] [Google Scholar]
- 13.Yoo JW, Chambers E, Mitragotri S. Factors that control the circulation time of nanoparticles in blood: challenges, solutions and future prospects. Curr Pharm Des. 2010;16(21):2298–2307. doi: 10.2174/138161210791920496. [DOI] [PubMed] [Google Scholar]
- 14.Patel PC, Giljohann DA, Daniel WL, Zheng D, Prigodich AE, Mirkin CA. Scavenger receptors mediate cellular uptake of polyvalent oligonucleotide-functionalized gold nanoparticles. Bioconjug Chem. 2010;21(12):2250–2256. doi: 10.1021/bc1002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang A, Liu W, Li Z, Jiang L, Xu H, Yang X. Influence of polyethyleneglycol modification on phagocytic uptake of polymeric nanoparticles mediated by immunoglobulin G and complement activation. J Nanosci Nanotechnol. 2010;10(1):622–628. doi: 10.1166/jnn.2010.1738. [DOI] [PubMed] [Google Scholar]
- 16.Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145(3):182–195. doi: 10.1016/j.jconrel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2(12):933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 18.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–6173. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 19.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Krysko O, et al. Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy. 2011;66(3):396–403. doi: 10.1111/j.1398-9995.2010.02498.x. [DOI] [PubMed] [Google Scholar]
- 21.Durafourt BA, et al. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012;60(5):717–727. doi: 10.1002/glia.22298. [DOI] [PubMed] [Google Scholar]
- 22.Chinetti-Gbaguidi G, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ Res. 2011;108(8):985–995. doi: 10.1161/CIRCRESAHA.110.233775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith BR, Cheng Z, De A, Koh AL, Sinclair R, Gambhir SS. Real-time intravital imaging of RGD-quantum dot binding to luminal endothelium in mouse tumor neovasculature. Nano Lett. 2008;8(9):2599–2606. doi: 10.1021/nl080141f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hak S, Reitan NK, Haraldseth O, de Lange Davies C. Intravital microscopy in window chambers: a unique tool to study tumor angiogenesis and delivery of nanoparticles. Angiogenesis. 2010;13(2):113–130. doi: 10.1007/s10456-010-9176-y. [DOI] [PubMed] [Google Scholar]
- 25.Tong L, He W, Zhang Y, Zheng W, Cheng JX. Visualizing systemic clearance and cellular level biodistribution of gold nanorods by intrinsic two-photon luminescence. Langmuir. 2009;25(21):12454–12459. doi: 10.1021/la902992w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng SH, et al. Visualizing dynamics of sub-hepatic distribution of nanoparticles using intravital multiphoton fluorescence microscopy. ACS Nano. 2012;6(5):4122–4131. doi: 10.1021/nn300558p. [DOI] [PubMed] [Google Scholar]
- 27.Smith BR, et al. Shape matters: intravital microscopy reveals surprising geometrical dependence for nanoparticles in tumor models of extravasation. Nano Lett. 2012;12(7):3369–3377. doi: 10.1021/nl204175t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merkel TJ, et al. Using mechanobiological mimicry of red blood cells to extend circulation times of hydrogel microparticles. Proc Natl Acad Sci U S A. 2011;108(2):586–591. doi: 10.1073/pnas.1010013108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rolland JP, Maynor BW, Euliss LE, Exner AE, Denison GM, DeSimone JM. Direct fabrication and harvesting of monodisperse, shape-specific nanobiomaterials. J Am Chem Soc. 2005;127(28):10096–10100. doi: 10.1021/ja051977c. [DOI] [PubMed] [Google Scholar]
- 30.Gratton SE, et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci U S A. 2008;105(33):11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perry JL, et al. PEGylated PRINT nanoparticles: the impact of PEG density on protein binding, macrophage association, biodistribution, and pharmacokinetics. Nano Lett. 2012;12(10):5304–5310. doi: 10.1021/nl302638g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camilleri JP, Williams AS, Amos N, Douglas-Jones AG, Love WG, Williams BD. The effect of free and liposome-encapsulated clodronate on the hepatic mononuclear phagocyte system in the rat. Clin Exp Immunol. 1995;99(2):269–275. doi: 10.1111/j.1365-2249.1995.tb05544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Auffray C, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317(5838):666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 34.Vasquez KO, Casavant C, Peterson JD. Quantitative whole body biodistribution of fluorescent-labeled agents by non-invasive tomographic imaging. PLoS One. 2011;6(6):e20594. doi: 10.1371/journal.pone.0020594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177(10):7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 36.Arnold L, Tyagi RK, Mejia P, Van Rooijen N, Perignon JL, Druilhe P. Analysis of innate defences against Plasmodium falciparum in immunodeficient mice. Malar J. 2010;9:197. doi: 10.1186/1475-2875-9-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 38.Beyer M, et al. High-resolution transcriptome of human macrophages. PLoS One. 2012;7(9):e45466. doi: 10.1371/journal.pone.0045466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kulkarni SA, Feng SS. Pharm Res. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery [published online ahead of print January 13, 2013]. doi: 10.1007/s11095-012-0958-3 . [DOI] [PubMed] [Google Scholar]
- 40.Chu KS, et al. Plasma, tumor and tissue pharmacokinetics of Docetaxel delivered via nanoparticles of different sizes and shapes in mice bearing SKOV-3 human ovarian carcinoma xenograft. Nanomedicine. 2012;pii:S1549-9634(12)00691-0. doi: 10.1016/j.nano.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caron WP, Song G, Kumar P, Rawal S, Zamboni WC. Interpatient pharmacokinetic and pharmacodynamic variability of carrier-mediated anticancer agents. Clin Pharmacol Ther. 2012;91(5):802–812. doi: 10.1038/clpt.2012.12. [DOI] [PubMed] [Google Scholar]
- 42.Kastrukoff LF, Lau AS, Thomas EE. The effect of mouse strain on herpes simplex virus type 1 (HSV-1) infection of the central nervous system (CNS). Herpesviridae. 2012;3:4. doi: 10.1186/2042-4280-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mogil JS, et al. Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. Pain. 1999;80(1–2):67–82. doi: 10.1016/S0304-3959(98)00197-3. [DOI] [PubMed] [Google Scholar]
- 44.Mesquita RC, et al. Tumor blood flow differs between mouse strains: consequences for vasoresponse to photodynamic therapy. PLoS One. 2012;7(5):e37322. doi: 10.1371/journal.pone.0037322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abolins SR, Pocock MJ, Hafalla JC, Riley EM, Viney ME. Measures of immune function of wild mice, Mus musculus. Mol Ecol. 2011;20(5):881–892. doi: 10.1111/j.1365-294X.2010.04910.x. [DOI] [PubMed] [Google Scholar]
- 46.Rabinovitch M. Professional and non-professional phagocytes: an introduction. Trends Cell Biol. 1995;5(3):85–87. doi: 10.1016/S0962-8924(00)88955-2. [DOI] [PubMed] [Google Scholar]
- 47.Silva MT, Correia-Neves M. Neutrophils and macrophages: the main partners of phagocyte cell systems. Front Immunol. 2012;3:174. doi: 10.3389/fimmu.2012.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petkova SB, Yuan R, Tsaih SW, Schott W, Roopenian DC, Paigen B. Genetic influence on immune phenotype revealed strain-specific variations in peripheral blood lineages. Physiol Genomics. 2008;34(3):304–314. doi: 10.1006/geno.1996.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bishton M, Chopra R. The role of granulocyte transfusions in neutropenic patients. Br J Haematol. 2004;127(5):501–508. doi: 10.1111/j.1365-2141.2004.05221.x. [DOI] [PubMed] [Google Scholar]
- 50.Bennett B. Isolation and cultivation in vitro of macrophages from various sources in the mouse. Am J Pathol. 1966;48(1):165–181. [PMC free article] [PubMed] [Google Scholar]
- 51.Englen MD, Valdez YE, Lehnert NM, Lehnert BE. Granulocyte/macrophage colony-stimulating factor is expressed and secreted in cultures of murine L929 cells. J Immunol Methods. 1995;184(2):281–283. doi: 10.1016/0022-1759(95)00136-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.