

Background: Ligand binding to ionotropic glutamate receptors opens the channel gate to control synaptic activity.
Results: Placing glycine residues at pore-facing positions in the M3 domain of GluA2 confers a more readily activated channel.
Conclusion: Like potassium channels, GluA2 uses bending of a pore-lining helix to open its gate and conduct ions.
Significance: Flexibility of M3 domain in iGluRs is critical to their function.
Keywords: Gating; Glutamate; Glutamate Receptors Ionotropic (AMPA, NMDA); Ion Channels; Pharmacology; Desensitization; Kainate
Abstract
Ionotropic glutamate receptor (iGluR) channels control synaptic activity. The crystallographic structure of GluA2, the prototypical iGluR, reveals a clamshell-like ligand-binding domain (LBD) that closes in the presence of glutamate to open a gate on the pore lining α-helix. How LBD closure leads to gate opening remains unclear. Here, we show that bending the pore helix at a highly conserved alanine residue (Ala-621) below the gate is responsible for channel opening. Substituting Ala-621 with the smaller more flexible glycine resulted in a basally active, nondesensitizing channel with ∼39-fold increase in glutamate potency without affecting surface expression or binding. On GluA2(A621G), the partial agonist kainate showed efficacy similar to a full agonist, and competitive antagonists CNQX and DNQX acted as a partial agonists. Met-629 in GluA2 sits above the gate and is critical in transmitting LBD closure to the gate. Substituting Met-629 with the flexible glycine resulted in reduced channel activity and glutamate potency. The pore regions in potassium channels are structurally similar to iGluRs. Whereas potassium channels typically use glycines as a hinge for gating, iGluRs use the less flexible alanine as a hinge at a similar position to maintain low basal activity allowing for ligand-mediated gating.
Introduction
Ionotropic glutamate receptors (iGluRs),2 including the AMPA receptor family GluA1–4 (also called GluR1–4 or GluRA–D), play critical roles in controlling neuronal activity at the synapse (1). These receptors have extracellular N termini and intracellular C termini with four membrane domains (M1–4), one of which forms a reentrant loop. Crystal structures of GluA2 with intact membrane domains (2) and ligand binding domain structures (3) show tetrameric channels with 2-fold symmetry in the ligand-binding domain (LBD), which forms an open cleft that closes upon ligand binding. This Venus flytrap closure model accommodates binding of full agonists, partial agonists as well as antagonists with each causing a different degree of closure (4). Glutamate, for instance, closes the GluA2 LBD cleft by ∼20°, whereas the partial agonist kainate closes it by ∼12°. The competitive antagonist DNQX closes the cleft by ∼2.5°(4). It is speculated that the degree of domain closure translates to channel gating by specific ligands.
The transmembrane domains of iGluRs have 4-fold symmetry and resemble inverted potassium channels (1, 2, 5). Bending of a pore lining α-helical domain around a conserved glycine in potassium channels opens their gate (6, 7). We previously showed that in Kir3 channels mutation of this glycine to larger residues impaired channel gating whereas an alanine could substitute for the glycine and retain near full activity (7). Detailed requirements for the hinge were subsequently examined to show permissible substitutions at the hinge depend on the local environment of the channel pore and the interactions among neighboring residues (8, 9). Approximately 15% of potassium channels use small residues other than glycine, mostly alanine, serine, or asparagine, at the hinge position.
In iGluRs, the Ala to Thr, mutation at the lurcher site in the conserved SYTANLAAF region, results in a highly active channel (10–12). Cysteine substitutions in this motif can alter channel activity after modification with MTS reagents (13). Whereas a previous study suggested that iGluRs do not use a hinge for their gating (14), structural information for any full-length iGluR was not available for those studies, and glycines were the only residues considered as a potential hinge. Overall, these types of approaches have shed some light on channel pore behavior but have fallen short in elucidating the gating mechanism.
In this study, we set out to examine whether glutamate receptors use a pivoted bending mechanism for gating. Because there are no obvious glycines that can play the pivot role in the pore helix, we reasoned that a bulkier residue may serve as pivot; therefore, substitution with glycine at such a critical position would improve gating. In the conserved 615SYTANLAAF623 motif located below the channel gate in the M3 of GluA2, we found that glycine substitutions at several pore-facing residues result in highly active channels. Among these, the A621G mutation resulted in nondesensitizing channels with large agonist-independent basal activity and increased potency for glutamate. Glycine substitution at Met-629 above the channel gate impaired channel activity. Together, these results suggest a bending mechanism for channel opening that requires a hinge, most likely at amino acid position 621, on the pore lining face of the helix below the gate.
EXPERIMENTAL PROCEDURES
Expression of Recombinant Receptors in Xenopus Oocytes
All constructs for oocyte expression were subcloned into the pGEMHE plasmid vector (15). The flop splice variant of GluA2 was used for all experiments. Throughout the paper, the residue numbering of the mature protein without the 21-amino acid signal peptide is used. An HA epitope was inserted following glycine 382 in the N-terminal domain. As reported previously, addition of this epitope does not interfere with receptor function (16). Two mutations, L483Y and R586Q, were introduced to remove desensitization (17) and increase channel expression (18). The point mutants were generated using the QuikChange site-directed mutagenesis kit (Stratagene), and the HA epitope was inserted using splicing by overlap PCR as described (19). The resulting construct is hereafter referred to as GluA2**HA. All other point mutations were made in this background unless noted. The full sequences of all constructs were confirmed by automated DNA sequencing. All constructs were linearized with appropriate restriction enzymes, and cRNAs were transcribed in vitro using mMessage mMachine (Ambion). Oocytes were isolated and microinjected with 2 ng of cRNA. All oocytes were maintained at 18 °C in OR2 solution containing 82.5 mm NaCl, 2 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2, 5 mm HEPES (pH 7.5) supplemented with penicillin and streptomycin. Electrophysiological recordings were performed 1–3 days following injection.
Two-electrode Voltage Clamp Recording and Analysis
Whole-oocyte currents were measured by conventional two-microelectrode voltage clamp with a GeneClamp 500 amplifier (Molecular Devices). Agarose-cushioned microelectrodes with resistances of 0.1–1.0 megohms were used (20). Oocytes were constantly superfused with normal frog Ringer's with barium containing 125 mm NaCl, 2.5 mm KCl, 0.5 mm MgCl2, 1.8 mm BaCl2, 10 mm HEPES (pH 7.4). In some experiments, N-methyl-d-glucamine (NMDG) replaced NaCl in normal frog Ringer's. To block or activate currents, the oocyte chamber was perfused with normal frog Ringer's with various drugs illustrated by bars above the tracings in the figures. Typically, oocytes were held at −60 mV and recorded at 1 KHz, filtered at 500 Hz. Data were collected using voltage ramps from −100 to +50 or in the gap-free mode at −60 mV. Dose responses were measured using single oocytes expressing each construct as noted in the figures. EC50 values for glutamate were determined from dose-response curves constructed using GraphPad Prism 5.0 (GraphPad Software).
Cell Surface Expression Assay
Live oocytes were incubated in OR2 with 1% BSA for 60 min at 4 °C. An HRP-conjugated anti-HA antibody was then added to the oocytes and incubated for 60 min at 4 °C. A 96-well plate was loaded with 50 μl of ECL reagent mixture per well, and background luminescence was read using a plate reader. After three washes with OR2 + 1% BSA and three washes with OR2, individual oocytes were transferred into the wells of the 96-well plate containing the ECL reagent, and the plate was read again. Background luminescence for each well was subtracted to determine total surface signal. Un-injected, H2O-injected, and oocytes expressing non-HA-tagged GluA2** served as controls.
Ligand Binding
For ligand binding experiments, GluA2**HA and mutants were subcloned into pcDNA3.1(−). HEK293T cells from ATCC were grown on 100-mm dishes and maintained in DMEM with 10% FBS. Each construct was transfected using FuGENE HD (Roche Applied Science), and cells were collected 48 h after transfection. Crude membranes were prepared using homogenization in hypotonic lysis buffer (10 mm Tris, 5 mm EDTA, and 250 mm sucrose (pH 7.4)) with protease inhibitors. Membranes were collected by centrifugation, and protein amounts were determined using a BCA protein assay (Thermo). To determine binding affinity for glutamate for each mutant a competition assay was used displacing [3H]AMPA. Briefly, equal amounts of membrane from each group were incubated with 12 nm [3H]AMPA in the presence of varying concentrations of glutamate. After incubating with [3H]AMPA and glutamate for 90 min on ice, membranes were washed and collected on nitrocellulose GF/C filters using a Brandell harvester. Nonspecific binding was determined using supersaturating concentration of glutamate. Total binding was determined in the absence of glutamate. Filters were counted in scintillation fluid, and GraphPad Prism 5.0 was used to determine IC50 for displacement of [3H]AMPA by glutamate for each mutant.
Patch Clamp Recordings
Desensitizing GluA2(R586Q)HA and GluA2(R586QA621G)HA mutants were subcloned into pcDNA3.1(−). HEK293T cells from ATCC were grown on fibronectin-treated 12-mm coverslips and maintained in DMEM with 10% FBS. Each construct was transfected using FuGENE HD, and cells were recorded 48–72 h after transfection. Coverslips with transfected cells were moved to a chamber mounted onto an inverted Nikon microscope. Outside-out patch clamp recordings were performed in successfully transfected HEK cells identified using fluorescence from co-transfected GFP. Electrodes (3–5 megohms) were filled with intracellular solution: 15 mm CsCl, 125 mm CsMeSO3, 0.5 mm CaCl2, 3 mm MgCl2, 2 mm MgATP, 5 mm Cs4BAPTA, and 10 mm HEPES (pH 7.4). The cells were superfused with bath solution: 140 mm NaCl, 4 mm KCl, 2 mm CaCl2, 0.1 mm MgCl2, 10 mm HEPES, and 10 mm glucose (pH 7.4). Cells were held at −60 mV and recorded using gap-free protocol using a Multiclamp700B amplifier, digitized with a Digidata 1322B, sampled at 10 KHz, low pass filtered at 4 kHz, and collected using pClamp9.2 (all from Molecular Devices). To achieve fast perfusion in outside-out recordings, a θ-glass with two streams, one control and one with 10 mm glutamate, was used. The patch was placed in the control stream, and a fast piezo switcher (Siskiyou Corp.) was used to control the movement of the θ-glass and apply glutamate to the patch. Solution exchange had a τ < 500 μs determined using changes in junction potential.
RESULTS
Glycine Substitution of Pore-facing Residues in M3 Helix below the Gate Results in Highly Active Channels
According to the published crystal structure of GluA2 (2), the highly conserved region 615SYTANLAAF623 occupies the region in GluA2 pore that corresponds to a part of the water-filled cavity in potassium channels where a glycine hinge resides (Fig. 1A) (6, 7). Using the GluA2**HA (see “Experimental Procedures”), we mutated each amino acid to glycine starting with Ile-611 one turn of helix before the conserved region, up to Met-629, approximately two turns of the helix above this region. Based on the crystal structure, residues 627–629 are part of the helix in two of the four subunits in GluA2 (2), and the helix bundle crossing at Met-629 at this position may form the putative channel gate. We expressed each mutant in oocytes and tested for activation with a saturating concentration of glutamate. Fig. 1B shows summary data for these recordings. Many of the mutants were inactive, whereas glycine substitutions at Ala-618, Ala-621, Thr-625, and Arg-628 showed activity that was ≥50% of GluA2**HA. All active glycine mutants reside in pore-facing positions in a short stretch below the helix bundle crossing. Quantitative cell surface labeling with an anti-HA antibody in live oocytes showed that all mutants, except for I612G, were expressed at detectible levels that were generally similar to or more than the GluA2**HA (Fig. 1C). Overall, altered expression cannot account for the pattern of activity observed in the mutants.
FIGURE 1.

Glycine mutations in pore lining residues of M3 α-helix maintain channel activity. A, structures from the M1–M3 domain of GluA2 (2) and a closed potassium channels (30). Two opposing subunits are depicted for each protein. In GluA2, the M1 region is shown in green, the reentrant M2 region in purple, and M3 in gold. The GluA2 pore region clearly resembles an upside-down potassium channel. A glycine located in the middle of the pore lining α-helix for most potassium channels acts as a hinge to pivot and open the channel gate at the helix bundle crossing. On the right, the structure of the 3rd transmembrane domain (M3) of GluA2 is viewed from the side. Two opposing subunits (A and C) of the tetramer are depicted. Pore-lining residues in and above the conserved 615SYTANLAAF623 region are shown in red; Met-629, which sits just above the channel gate, is shown in blue. Side chains are depicted for some of the residues with molecular distance for α-carbons and side chains for the same residues in opposing subunits in purple and orange, respectively. B, glutamate (100 μm) activation of the glycine-substituted M3 residues in the GluA2**HA backbone. Activity of each mutant is expressed as percentage of the activity of GluA2**HA recorded in the same batch of the oocytes on the same day. C, surface expression for each M3 glycine mutant expressed as percentage of the GluA2**HA surface expression determined in the same batch of oocytes. RLU, relative luminescence units.
A621G Increases Ligand Sensitivity Not Affinity
Dose-response experiments showed that glycine substitutions at positions 618, 621, and 625 reduced the glutamate EC50 compared with GluA2**HA (sample traces in Fig. 2, A and B; summary in Fig. 2C). Most notably, the glutamate EC50 for the A621G mutant was shifted 39-fold. Met-629 resides just outside the membrane-spanning region (2) and the helix bundle crossing (Fig. 1A). This residue is critical in the transition from the 2-fold symmetry of the LBD to the 4-fold symmetry in the transmembrane domains. In contrast to the pore-facing residues below the gate, glycine mutation at Met-629 reduced channel activity and glutamate sensitivity (Figs. 1B and 2C).
FIGURE 2.
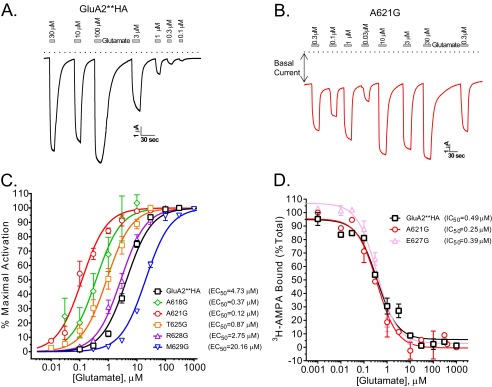
Effects of glycine substitutions on glutamate potency are independent of affinity. A and B, sample traces from oocytes expressing GluA2**HA (A) or A621G (B) recorded at −60 mV using varying concentrations of glutamate (Glu). Dotted line depicts zero current. Note large current in the absence of glutamate for A621G. C, glutamate dose-response curves for GluA2**HA, the active mutants, and M629G. EC50 values for each are shown. D, glutamate binding determined by displacing 12 nm [3H]AMPA in membranes from HEK293T cells expressing GluA2**HA, A621G, or E627G. There is no difference in IC50 values for ligand displacement in GluA2**HA and the highly active A621G or the inactive E627G (n = 4).
To determine whether the change in activity is due to corresponding changes in binding, we performed radioligand displacement experiments. We selected three constructs to test for binding. GluA2**HA, A621G, and E627G all had similar surface expression (Fig. 1C), whereas GluA2**HA and A621G were active, and E627G was completely inactive (Fig. 1B). We found no discernible difference in glutamate binding among the three (Fig. 2D). Furthermore, the ligand affinities were similar to those reported for the isolated LBD (21), indicating a lack of substantial effect of the channel pore on ligand binding affinity. The binding data show that improved gating is independent of ligand affinity.
Increased Activity by Glycine Substitutions Is Not Due to a Larger Pore
Due to the small size of glycine, substitutions in the pore may enlarge the channel cavity to increase ion flow. To test this, we substituted alanine 621 with residues of different side chains and determined the glutamate dose responses for each (Fig. 3A). The glutamate EC50 values for various substitutions at position 621 of GluA2**HA were as follows: Gly, 0.12 μm; Ser, 1.04 μm; Asn, 1.2 μm; Cys, 1.8 μm; Asp, 1.97 μm; Ala(WT), 4.73 μm; Thr, 8.1 μm; and Val, 13.2 μm. We also mutated Ala-621 to two larger amino acids, leucine and phenylalanine. Both mutants were expressed at levels similar to GluA2**HA, but they were less active than the wild-type receptors; A621F was more active than A621L despite its larger size (data not shown). Together, these data show that small substitutions at Ala-621 are permissible whereas amino acid size per se does not determine activity.
FIGURE 3.
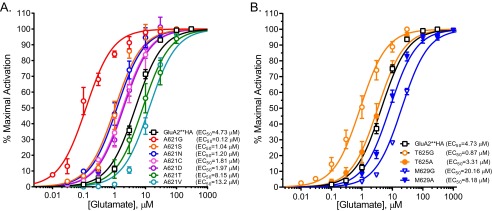
The size of the pore-facing residues does not dictate activity. A, the critical Ala-621 residue was mutated to amino acids of different size to test the effects of residue size at the pivot on channel activity. Glutamate dose-response curves for these mutants and corresponding EC50 values are shown. B, glutamate dose-response curves (EC50 values) for GluA2**HA, T625A, T625G, M629A, and M629G are shown.
We also tested amino acid size at two other positions: Thr-625, below the gate, and Met-629, at the extracellular domain-channel interface. The dose-response relationship for glutamate activation T625A showed an EC50 value similar to the GluA2**HA (Fig. 3B). The glycine mutation at the same site reduced the EC50 ∼5-fold. Mutating Met-629 to alanine (M629A) actually improved glutamate sensitivity compared with M629G (Fig. 3B), suggesting that a less flexible residue between the LBD and the gate accommodates the force transfer similar to that described for potassium channels (7).
GluA2(A621G) Mutant Is Basally Active
Oocytes expressing the A621G mutants had a leak basal current (see Fig. 2B). We therefore reasoned that A621G might be basally open in the absence of exogenous glutamate. To test directly for agonist-independent activity, we used the open channel blocker IEM1460 (22) in the absence of exogenous glutamate to block basal currents. In oocytes expressing the A618G, A621G, and T625G mutants, IEM1460 blocked a significantly larger current compared with oocytes expressing GluA2**HA (Fig. 4, A and B). We did not observe an effect in uninjected oocytes (data not shown). In the presence of glutamate, IEM1460 blocked glutamate-induced current in both wild-type and mutant channels; however, the GluA2**HA receptor block was significantly less than the block of A618G, A621G, T625G, and R628G mutants (Fig. 4C). To examine further the basal activity in GluA2 pore mutant, we used a recording solution containing the impermeant NMDG in place of sodium and measured channel activity. In oocytes expressing the GluA2**HA, there was a negligible channel block when switching from sodium solution to NMDG (Fig. 4D); a similar effect was observed in uninjected oocytes (data not shown). In oocytes expressing A621G, the large basal current was blocked by NMDG (Fig. 4E). Summary data for NMDG-sensitive basal current expressed as a percentage of the glutamate-induced current for each construct are shown in Fig. 4F. Because the EC50 for glutamate activation of some of the glycine substitutions are significantly reduced, the basal activity that we observed may reflect channel opening by ambient glutamate levels (11, 12). Nevertheless, our data are consistent with a lower barrier for channel gating in these glycine mutants, most prominently in the A621G.
FIGURE 4.
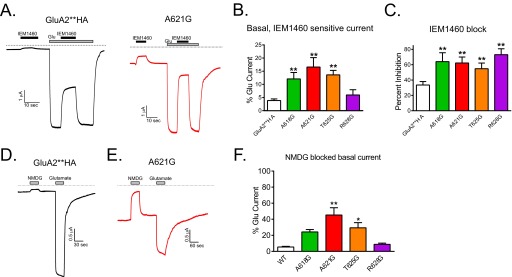
The A621G mutant has basal, agonist-independent activity. A, sample traces showing that the pore channel blocker IEM1460 (10 μm) has little effect on GluA2**HA in the absence of glutamate and blocks the channels after glutamate (100 μm)-induced activation. In contrast, the A621G channel has a large basal current that is blocked by IEM1460, and after glutamate activation, the channel is robustly blocked by IEM1460. B, summary data for IEM1460 effect on basal current in pore-lining glycine mutants (n = 7–26; **, p < 0.01 compared with GluA2**HA, one-way ANOVA with Dunnett's post hoc test). C, summary data for IEM1460 effect on glutamate-induced currents in pore-lining glycine mutants (n = 7–26; **, p < 0.01 compared with GluA2**HA, one-way ANOVA with Dunnett's post hoc test). D, sample trace showing that replacing sodium with NMDG does not significantly affect activity in the absence of agonist in GluA2**HA. E, replacing sodium in the recording buffer with the impermeant NMDG blocking the current in the absence of glutamate for oocytes expressing A621G. F, summary data showing the extent of NMDG-blocked basal current expressed as percentage of the glutamate-induced current in the same oocyte for GluA2**HA and the pore glycine mutants (n = 5–7; *, p < 0.05 compared with GluA2**HA, one-way ANOVA with Dunnett's post hoc test).
A621G Mutant Eliminates Receptor Desensitization
GluA2 receptors desensitize rapidly in the presence of glutamate, and cyclothiazide eliminates this desensitization (1). We had been using GluA2**HA which contains the L483Y mutant to eliminate this desensitization. To test the effect of the A621G mutation on desensitization, we used a GluA2 construct (carrying R586Q, without the L483Y (GluA2(R586Q)HA)) which readily desensitizes (Fig. 5A). Whereas the fast peak current cannot be detected in oocytes due to slow bath exchange kinetics, the steady-state desensitized current was readily observed in GluA2(R586Q)HA-expressing oocytes (Fig. 5B). This current was increased ∼5-fold in the presence of cyclothiazide (Fig. 5, A and C). In contrast, in GluA2(R586Q,A621G)HA-expressing oocytes, glutamate activated a large steady-state current that was only slightly increased by cyclothiazide (Fig. 5, B and C). In outside-out recordings from HEK293T cells expressing GluA2(R586Q)HA, ultrafast perfusion of glutamate gives a brief peak followed by a very small steady-state current (Fig. 5D). In contrast, patches from cells expressing GluA2(R586Q,A621G)HA did not desensitize (Fig. 5E). Summary data for desensitization in outside-patches are shown in Fig. 5F.
FIGURE 5.
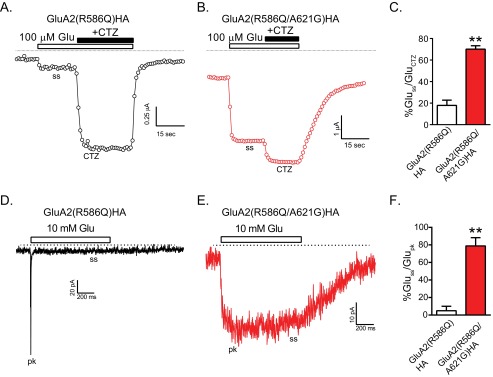
The A621G mutant does not desensitize. A, sample tracing shows glutamate-activated currents in an oocyte expressing the desensitizing receptor GluA2(R586Q). The current increased significantly once cyclothiazide was co-applied. Due to slow solution exchange, the peak current is not recorded. B, sample recording shows an oocyte expressing GluA2(R586Q,A621G). Currents increased slightly once cyclothiazide was co-applied. C, summary data show the ratio of the desensitized glutamate-induced steady-state currents (Gluss) to the total current recorded in the presence of cyclothiazide (Gluctz). The glutamate steady-state activation was significantly larger in receptors with the A621G mutation (**, p < 0.01, unpaired t test, n = 10–15). D, sample recording for glutamate-activated current in an outside-out patch from a HEK293 cell expressing GluA2(R586Q). A substantial peak current (pk) followed by a very small steady-state (ss) current is observed. E, glutamate-activated current in an outside-out patch from a HEK293 cell expressing GluA2(R586Q,A621G) did not desensitize. F, summary data expressing the desensitized glutamate-induced steady-state currents as percentage of peak current in the same patch (Glupk) are shown (**, p < 0.01, unpaired t test, n = 3).
Changes in Partial Agonist and Weak Antagonist Sensitivity of the Glycine Mutants
Partial agonists such as kainate do not fully close the LBD clamshell, which translates to partial channel activity. We found that kainate was more efficacious in activating the A621G mutant compared with GluA2**HA (Fig. 6). Other active pore-facing glycine mutants were also more kainate activated compared with GluA2**HA (Fig. 6B).
FIGURE 6.
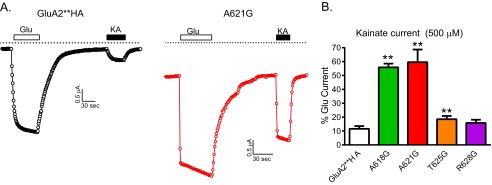
The partial agonist kainate acts as a nearly full agonist on A621G. A, sample recordings of 100 μm glutamate- and 500 μm kainate-induced current in oocytes expressing GluA2**HA or A621G. B, summary data for kainate activation of GluA2**HA and the active pore mutants expressed as percentage of glutamate-induced current from the same oocyte (n = 5–18; **, p < 0.01 compared with GluA2**HA, one-way ANOVA with Dunnett's post hoc test).
The competitive antagonists CNQX and DNQX bind to the same site as glutamate on GluAs but fail to induce adequate structural changes to open the channel (23). CNQX robustly inhibited the glutamate activated currents in GluA2**HA but was less effective on A621G or other pore-facing active glycine mutants (Fig. 7A). DNQX was also less effective in inhibiting the mutants compared with GluA2**HA (data not shown). To determine CNQX inhibition of each receptor, we stimulated with EC80 concentrations of glutamate (12 μm for GluA2**HA and 4 μm for A621G). The IC50 values for CNQX inhibition of GluA2**HA and A621G were 2.78 and 36.7 μm, respectively (Fig. 7B).
FIGURE 7.

Reduced potency of CNQX on glycine-substitute pore-facing residue. A, summary data for inhibition of 100 μm glutamate-induced current by 10 μm CNQX (n = 7–17; **, p < 0.01 compared with GluA2**HA, one-way ANOVA with Dunnett's post hoc test). B, dose response for CNQX inhibition of glutamate-activated current for GluA2**HA and A621G. For each construct varying doses of CNQX were applied in the presence of ∼EC80 concentration of glutamate. IC50 values for inhibition are shown on the figure.
Comparing the Active A621G with the A622T lurcher Site Mutant
Mutation of the 3rd alanine in the conserved SYTANLAAF region of GluD2 to a threonine in the lurcher mouse results in excess activity and ataxia (24, 25). Substituting the equivalent Ala to Thr in iGluRs has been show to results in channels with increased basal activity and left-shifted glutamate dose-response curves (10–12). We made the lurcher site mutant A622T in GluA2**HA and compared it with the active A621G mutant. Fig. 8A shows the glutamate dose-response curve for the two mutants compared with GluA2**HA. The EC50 values for glutamate activation of GluA2**HA, A621G, and A622T were 4.28, 0.11, and 2.2 μm, respectively. Fig. 8B shows sample tracings from oocytes expressing either A621G or A622T. In the absence of applied glutamate, A622T has a larger current compared with A621G determined by NMDG block. Application of CNQX and DNQX in the absence of glutamate activated small currents in the A621G-expressing oocytes, but no activation was observed in the A622T-expressing oocytes. Summary data for NMDG-sensitive current and CNQX and DNQX effects are shown in Fig. 8, C and D. Although there are functional similarities between A621G and the lurcher site A622T mutant, there are quantitative differences in basal activity, glutamate potency, and activity of competitive antagonist on the two mutants. CNQX induces a 6.4° closure in the binding cleft, and DNQX causes a 2.5° closure, giving both some partial agonist activity in GluA1 in the presence of stargazin (23). The relative activation of A621G by CNQX and DNQX compared with glutamate is similar to those reported for GluA1 co-expressed with stargazin, whereas NBQX acts as a true antagonist in both (23). Together, these data show that with the more flexible glycine residue in the M3 helix, a smaller closure of the LBD can open the channel.
FIGURE 8.

Comparison of A621G and A622T for glutamate potency, basal activity, and conversion of antagonist to partial agonists. A, glutamate dose-response curves for GluA2**HA, A621G, and A622T that correspond to the lurcher mutation. EC50 values for each are shown. B, sample traces for effect of NMDG, CNQX, and DNQX on basal current for A621G and A622T. NMDG blocked a larger basal current (as percentage of glutamate-activated current) in A622T expressing oocytes compared with A621G. In the absence of agonist, CNQX and DNQX consistently activated small currents in oocytes expressing A621G but not A622T. C, summary of NMDG-sensitive basal current of GluA2**HA, A621G, and A622T expressed as percentage of the glutamate-induced current in the same oocyte (**, p < 0.01, one-way ANOVA with Tukey's post hoc test; n = 5–11). D, summary of CNQX activation of GluA2**HA, A621G, and A622T expressed as percentage of the glutamate-activated current in the same oocyte (**, p < 0.01, one-way ANOVA with Tukey's post hoc test; n = 3–12). E, summary of DNQX activation of GluA2**HA, A621G, and A622T expressed as percentage of the glutamate-activated current in the same oocyte (*, p < 0.05 compared with GluA2**HA, one-way ANOVA with Tukey's post hoc test; n = 3–7).
DISCUSSION
The 615SYTANLAAF623 motif in the α-helix that forms the M3 in GluA2 is highly conserved among all glutamate receptor channels (26). Our data are consistent with a critical role for this region in channel gating. Glycine substitutions in most residues within this region were inactive; however, most mutants were highly expressed and targeted to the cell surface. Four residues (Ala-618, Ala-621, Thr-625, and Arg-628), separated by ∼3.5 amino acids, tolerated glycine mutation. A618G, A621G, and T625G had lower EC50 values for activation by glutamate all without significant changes in expression or membrane targeting. The most active glycine mutant, A621G, had a 39-fold reduction in glutamate EC50 without any changes in glutamate binding affinity. These data are consistent with a critical role for this residue in translating ligand binding to efficient channel gating. We believe that alanine 621 most likely acts as the hinge for GluA2 gating, and its flexibility is critical to channel function. In contrast, placing a glycine residue at position Met-629 reduced channel activity. Because Met-629 resides at the interface between the LBD and the pore domain, we speculate that rigidity here may be critical in transmitting the mechanical force of LBD closure to opening the gate. In Kir3.4 channels, placing glycines between the gate and activation domains reduces channel activity (7), providing further evidence for a conserved gating mechanism between iGluRs and potassium channels. That glycine substitution in the GluA2 pore improved ligand-mediated activation suggests that a more flexible M3 has improved function, but may be physiologically unfavorable due to significant basal activity. A previous study showed that substitutions at a corresponding position on NMDA receptors can also affect their channel gating, but the hinging mechanism was not explicitly tested (13).
Three possible effects of glycine replacements in the M3 helix may explain their functional consequences. First, glycine residues can enhance close packing of adjacent α-helices (27, 28). Closer packing of the membrane domains may enlarge the pore. The side chain of the residues in the M3 that allowed glycine substitutions (Ala-618, Ala-621, Thr-625) all face the channel pore, therefore these glycine substitutions would not affect helix packing. A second possibility is replacing bulkier side chains in the pore with hydrogen, in glycine mutants, widens the pore cavity. If this were the case there would be a graded response based on the size of the side chain. We tested this explicitly by replacing Ala-621 with various residues. An asparagine substitution that is ∼29% larger than the native alanine has glutamate EC50 that is 3.6 times lower. A phenylalanine substitution that is 23 Å3 larger than a leucine resulted in much higher activity. Therefore, the size of the side chain per se was not the determining factor for activity in Ala-621 substituted mutants. Finally, glycine residues introduce flexibility in an α-helix (29). Whereas natively an alanine occupies the most likely hinge position at residue 621, substitutions with glycine result in channels where a smaller stimulus results in a larger activation, denoted by the 39-fold reduction for glutamate EC50. Substitutions with different sized amino acids resulted in activity that is consistent with the tolerance for small residues and the flexibility that they afford. Substitution with serine, cysteine, asparagine, and aspartate was highly active with low glutamate EC50 values. Substitution with valine resulted in a channel with low activity and elevated glutamate EC50. Substitution with a large amino acid such as phenylalanine resulted in channels that would open; however, they needed larger stimulus (more glutamate). This model is consistent with structural and functional data in potassium channels in that open and closed potassium channel structures clearly show a kink in the α-helical structure around a conserved glycine (6, 30). Closer analysis shows that in ∼81% of potassium channels the glycine at this position is conserved (8). Another ∼15% of the channels have other small residues such as alanine, serine, or asparagine. Interestingly, among the Ala-621 mutants, after A621G, A621S and A621N had the lowest glutamate EC50 values. Substituting the conserved glycine with alanine in potassium channels can be tolerated but may result in reduced function, whereas substitutions with larger residues result in channel that can only be opened with excessive stimulus such as co-expressed exogenous Gβγ (7). Whereas potassium channels are more easily gated due to their native glycine residues, glutamate receptors require external stimulus for gating. Coupled with a clamshell model for ligand binding, the semiflexible membrane gate fine-tunes response to stimuli.
A previous study attempted to describe iGluR gating in terms of the glycine hinge requirement in potassium channels (14). No glycine residues on the pore region of GluRs mimic conserved glycines in potassium channels. This led to the conclusion that iGluR gating is different from potassium channel gating. However, we now know that a glycine is not an absolute requirement for gating in potassium channels, and any residue that maintains some flexibility and does not have deleterious interactions with adjacent residues can serve as a pivot (7, 9, 31). Alanine was initially shown as a good replacement for glycine in Kir3.4 (7). Others have shown that in Kv channels, a mutation of the pivot glycine can be overcome by placing other flexible residues close to the pivot (32). Overall structural and functional data point to a channel pore that needs some flexibility to open the gate. Moreover, although glycine seems to be the preferred residue at the hinge in voltage-gated channels or channels that receive cytoplasmic stimulus, in glutamate receptors, which receive extracellular stimulus, alanine may play a role in tempering activity. The naturally occurring threonine mutation found in the δ2 glutamate receptor in lurcher mice results in significant neuronal defects (24). This threonine replaces the third alanine in the SYTANLAAF region in the M3. Mutating the conserved Ala-622 in GluA2**HA resulted in a channel that showed characteristic changes similar but not equivalent to what we observed with the A621G mutation. The basal activity of A622T was more than A621G whereas the glutamate EC50 was higher, and the antagonists CNQX and DNQX acted as weak partial agonists on A621G and not on A622T. In GluA1, the lurcher site mutation, equivalent to A622T in GluA2, causes increase constitutive basal activity, increased agonist potency, changes in antagonist activity, changes in partial agonist activity, and desensitization (10–12). These changes are similar to our observations in the A622T mutant but may highlight the difference in the specific experimental parameters for each study. How placing a threonine in the lurcher position leads to structural alterations that cause these functional changes is not fully clear. A threonine can increase the helix bend angle and slightly open the helix at the turn preceding its position (33). Based on our model of pivoted bending around Ala-621 for GluA2 gating, change in helix bending angle by a threonine may explain the activity of the lurcher mutants. Interestingly a threonine at position 621 of GluA2 was not well tolerated.
The increased efficacy of the partial agonist kainate and partial agonist activity of the competitive antagonists CNQX and DNQX on the glycine pore mutants are consistent with a channel that opens more easily. Relative efficacy and antagonism in iGluRs have been very well described with the degree of clamshell closure in LBD (26). The LBD is directly connected to the M3 helix, whose rigidity may influence the state of domain closure. When there is sufficient closure due to binding of a full agonist, the energy associated with clamshell closure overcomes the pulling of the closed pore leading to channel gating. If the ligand-mediated clamshell closure is partial, the pulling force may not overcome the closed channel gate. Glycines make the pore domain more flexible, allowing a smaller closure of the clamshell to gate the channel.
GluA2 desensitization was described as a “rupture” in the extensive interactions below the LBD and above the M3 helix (34). A rigid M3 helix provides torsional pullback required for molecular rupture. We suggest that placing the more flexible glycine residue at the pivot point in the transmembrane domain renders a nondesensitizing channel by eliminating the somewhat rigid anchor needed for the interface rupture. This scenario may also happen when GluAs are co-expressed with TARPs such as stargazin. Interestingly, in the presence of stargazin, CNQX and DNQX act as weak partial agonists (23) similar to our findings with the A621G mutant. We speculate that TARPs change the flexibility and or packing of the membrane domains in iGluRs to increase agonist potency and reduce desensitization.
Although the mechanism for ligand interaction with GluRs has been well established, the exact mechanism for gating has remained elusive. Our data point to bending in the pore-facing domain of M3 as a mechanism for channel opening. This follows an evolutionary conserved model first described in potassium channels. Whereas in most potassium channels the flexibility is afforded by glycine residues lining the pore, an alanine occupies the most likely pivot point in glutamate receptors. A more rigidly hinged gate may be critical to glutamate receptor function to closely regulate activity upon proper stimulation and interaction with accessory proteins in neurons.
Acknowledgments
We thank Drs. D. Logothetis and J. Woodward for suggestions and comments on the manuscript, Drs. M. Hollmann and J. Boulter for the glutamate receptor clones, Dr. G. Breitwieser for use of the Brandell harvester, and A. Styer and M. Demszko for help in generating some of the GluA2 mutants.
This work was supported by funds from the Geisinger Clinic.
- iGluR
- ionotropic glutamate receptor
- ANOVA
- analysis of variance
- CNQX
- 6-cyano-7-nitroquinoxaline-2,3-dione
- DNQX
- 6,7-dinitroquinoxaline-2,3-dione
- LBD
- ligand-binding domain
- NBQX
- 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline
- NMDG
- N-methyl-d-glucamine
- TARP
- transmembrane AMPA receptor regulatory protein.
REFERENCES
- 1. Traynelis S. F., Wollmuth L. P., McBain C. J., Menniti F. S., Vance K. M., Ogden K. K., Hansen K. B., Yuan H., Myers S. J., Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sobolevsky A. I., Rosconi M. P., Gouaux E. (2009) X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462, 745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Armstrong N., Sun Y., Chen G. Q., Gouaux E. (1998) Structure of a glutamate receptor ligand-binding core in complex with kainate. Nature 395, 913–917 [DOI] [PubMed] [Google Scholar]
- 4. Armstrong N., Gouaux E. (2000) Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28, 165–181 [DOI] [PubMed] [Google Scholar]
- 5. Bennett J. A., Dingledine R. (1995) Topology profile for a glutamate receptor: three transmembrane domains and a channel-lining reentrant membrane loop. Neuron 14, 373–384 [DOI] [PubMed] [Google Scholar]
- 6. Jiang Y., Lee A., Chen J., Cadene M., Chait B. T., MacKinnon R. (2002) The open pore conformation of potassium channels. Nature 417, 523–526 [DOI] [PubMed] [Google Scholar]
- 7. Jin T., Peng L., Mirshahi T., Rohacs T., Chan K. W., Sanchez R., Logothetis D. E. (2002) The βγ subunits of G proteins gate a K+ channel by pivoted bending of a transmembrane segment. Mol. Cell 10, 469–481 [DOI] [PubMed] [Google Scholar]
- 8. Rosenhouse-Dantsker A., Logothetis D. E. (2006) New roles for a key glycine and its neighboring residue in potassium channel gating. Biophys. J. 91, 2860–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rosenhouse-Dantsker A., Logothetis D. E. (2007) Potassium channel gating in the absence of the highly conserved glycine of the inner transmembrane helix. Channels 1, 189–197 [DOI] [PubMed] [Google Scholar]
- 10. Kohda K., Wang Y., Yuzaki M. (2000) Mutation of a glutamate receptor motif reveals its role in gating and δ2 receptor channel properties. Nat. Neurosci. 3, 315–322 [DOI] [PubMed] [Google Scholar]
- 11. Taverna F., Xiong Z. G., Brandes L., Roder J. C., Salter M. W., MacDonald J. F. (2000) The lurcher mutation of an α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor subunit enhances potency of glutamate and converts an antagonist to an agonist. J. Biol. Chem. 275, 8475–8479 [DOI] [PubMed] [Google Scholar]
- 12. Klein R. M., Howe J. R. (2004) Effects of the lurcher mutation on GluR1 desensitization and activation kinetics. J. Neurosci. 24, 4941–4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang H. R., Kuo C. C. (2008) The activation gate and gating mechanism of the NMDA receptor. J. Neurosci. 28, 1546–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sobolevsky A. I., Yelshansky M. V., Wollmuth L. P. (2003) Different gating mechanisms in glutamate receptor and K+ channels. J. Neurosci. 23, 7559–7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liman E. R., Tytgat J., Hess P. (1992) Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 9, 861–871 [DOI] [PubMed] [Google Scholar]
- 16. Man H. Y., Lin J. W., Ju W. H., Ahmadian G., Liu L., Becker L. E., Sheng M., Wang Y. T. (2000) Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron 25, 649–662 [DOI] [PubMed] [Google Scholar]
- 17. Stern-Bach Y., Russo S., Neuman M., Rosenmund C. (1998) A point mutation in the glutamate binding site blocks desensitization of AMPA receptors. Neuron 21, 907–918 [DOI] [PubMed] [Google Scholar]
- 18. Greger I. H., Khatri L., Ziff E. B. (2002) RNA editing at Arg-607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 34, 759–772 [DOI] [PubMed] [Google Scholar]
- 19. Mirshahi T., Robillard L., Zhang H., Hébert T. E., Logothetis D. E. (2002) Gβ residues that do not interact with Gα underlie agonist-independent activity of K+ channels. J. Biol. Chem. 277, 7348–7355 [DOI] [PubMed] [Google Scholar]
- 20. Schreibmayer W., Lester H. A., Dascal N. (1994) Voltage clamping of Xenopus laevis oocytes utilizing agarose-cushion electrodes. Pflugers Arch. 426, 453–458 [DOI] [PubMed] [Google Scholar]
- 21. Chen G. Q., Sun Y., Jin R., Gouaux E. (1998) Probing the ligand binding domain of the GluR2 receptor by proteolysis and deletion mutagenesis defines domain boundaries and yields a crystallizable construct. Protein Sci. 7, 2623–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schlesinger F., Tammena D., Krampfl K., Bufler J. (2005) Two mechanisms of action of the adamantane derivative IEM-1460 at human AMPA-type glutamate receptors. Br. J. Pharmacol. 145, 656–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Menuz K., Stroud R. M., Nicoll R. A., Hays F. A. (2007) TARP auxiliary subunits switch AMPA receptor antagonists into partial agonists. Science 318, 815–817 [DOI] [PubMed] [Google Scholar]
- 24. Zuo J., De Jager P. L., Takahashi K. A., Jiang W., Linden D. J., Heintz N. (1997) Neurodegeneration in lurcher mice caused by mutation in δ2 glutamate receptor gene. Nature 388, 769–773 [DOI] [PubMed] [Google Scholar]
- 25. Wollmuth L. P., Kuner T., Jatzke C., Seeburg P. H., Heintz N., Zuo J. (2000) The lurcher mutation identifies δ2 as an AMPA/kainate receptor-like channel that is potentiated by Ca2+. J. Neurosci. 20, 5973–5980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mayer M. L. (2011) Emerging models of glutamate receptor ion channel structure and function. Structure 19, 1370–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Javadpour M. M., Eilers M., Groesbeek M., Smith S. O. (1999) Helix packing in polytopic membrane proteins: role of glycine in transmembrane helix association. Biophys. J. 77, 1609–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eilers M., Shekar S. C., Shieh T., Smith S. O., Fleming P. J. (2000) Internal packing of helical membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 5796–5801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dong H., Sharma M., Zhou H. X., Cross T. A. (2012) Glycines: role in α-helical membrane protein structures and a potential indicator of native conformation. Biochemistry 51, 4779–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Doyle D. A., Morais Cabral J., Pfuetzner R. A., Kuo A., Gulbis J. M., Cohen S. L., Chait B. T., MacKinnon R. (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity [see comments]. Science 280, 69–77 [DOI] [PubMed] [Google Scholar]
- 31. Hardman R. M., Stansfeld P. J., Dalibalta S., Sutcliffe M. J., Mitcheson J. S. (2007) Activation gating of hERG potassium channels: S6 glycines are not required as gating hinges. J. Biol. Chem. 282, 31972–31981 [DOI] [PubMed] [Google Scholar]
- 32. Ding S., Ingleby L., Ahern C. A., Horn R. (2005) Investigating the putative glycine hinge in Shaker potassium channel. J. Gen. Physiol. 126, 213–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deupi X., Olivella M., Sanz A., Dölker N., Campillo M., Pardo L. (2010) Influence of the G-conformation of Ser and Thr on the structure of transmembrane helices. J. Struct. Biol. 169, 116–123 [DOI] [PubMed] [Google Scholar]
- 34. Armstrong N., Jasti J., Beich-Frandsen M., Gouaux E. (2006) Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell 127, 85–97 [DOI] [PubMed] [Google Scholar]