

Abstract
The large-conductance Ca2+- and voltage-activated K+ channel (BKCa, MaxiK), which is encoded by the Kcnma1 gene, is generally expressed at the plasma membrane of excitable and nonexcitable cells. However, in adult cardiomyocytes, a BKCa-like channel activity has been reported in the mitochondria but not at the plasma membrane. The putative opening of this channel with the BKCa agonist, NS1619, protects the heart from ischemic insult. However, the molecular origin of mitochondrial BKCa (mitoBKCa) is unknown because its linkage to Kcnma1 has been questioned on biochemical and molecular grounds. Here, we unequivocally demonstrate that the molecular correlate of mitoBKCa is the Kcnma1 gene, which produces a protein that migrates at ∼140 kDa and arranges in clusters of ∼50 nm in purified mitochondria. Physiological experiments further support the origin of mitoBKCa as a Kcnma1 product because NS1619-mediated cardioprotection was absent in Kcnma1 knockout mice. Finally, BKCa transcript analysis and expression in adult cardiomyocytes led to the discovery of a 50-aa C-terminal splice insert as essential for the mitochondrial targeting of mitoBKCa.
Keywords: BK channel, KCa1.1, splice variation
The large-conductance Ca2+- and voltage-activated K+ channel (BKCa) is ubiquitously expressed and especially abundant at the plasma membrane of smooth muscle and neuronal cells, where they control contractility, resting membrane potential, neurotransmitter release, and action potential repolarization. In the adult heart, BKCa expression at the plasma membrane of myocytes has not been reported; however, electrophysiological, biochemical, and pharmacological evidence support its presence in mitochondria (1, 2). The presumed activation of mitochondrial BKCa (mitoBKCa) with NS1619 protects the heart from ischemia and reperfusion (1), which highlights the significant role of this channel in cardiac function.
Cardiac mitoBKCa shares functional characteristics, such as large conductance and inhibition by paxilline and charybdotoxin, with its plasma membrane counterpart. This resemblance, together with the reported Ca2+/voltage-dependency of mitoBKCa from human glioma (3), indicates that the molecular structure of this protein is closely related to the plasmalemmal BKCa. The plasmalemmal BKCa is minimally assembled by four pore-forming α-subunits encoded by a single gene, Kcnma1, which can undergo significant splice variation. The minimal protein, which is encoded by 27 constitutive exons, has a molecular mass of ∼125–130 kDa, depending on the start site (4, 5). However, proteins that migrate at ∼55 kDa (1, 6)—but also at ∼125 kDa (2)—have been proposed to correspond to the α-subunit of cardiac BKCa. In addition, the cloning of cardiomyocyte BKCa cDNAs revealed the standard 27 constitutive exons of Kcnma1, but their expression in H9c2 cardiac cells produced a BKCa protein that was unable to reach the mitochondria (7). Taken together, these studies questioned whether mitoBKCa was related to Kcnma1.
Here, we investigated the molecular identity of mitoBKCa, tested whether splice variation of Kcnma1 gene underlies BKCa targeting to cardiac mitochondria, and addressed the role of Kcnma1 gene expression in NS1619-mediated cardioprotection.
Results
Fingerprinting mitoBKCa.
We first evaluated the size of cardiac mitoBKCa using immunoblotting and mass spectrometry. For this purpose, we used Percoll-purified mitochondria from the left ventricle (8) (Fig. 1A) or from cardiomyocytes and specific antibodies raised against BKCa C terminus that generated no signals in BKCa knockout (Kcnma1−/−) samples. Immunoblots using lysates of purified mitochondria from mouse ventricular myocytes showed two specific bands, one at ~140 kDa and a smaller one near 100 kDa; the latter is likely to be a proteolytic fragment. These bands were present in the WT but not in the knockout mitochondrial samples (Fig. 1B, arrows) (n = 3). For comparison, whole-brain lysates were also used; a band of ∼125 kDa was observed only in the WT samples (Fig. 1C, arrow). The differences in BKCa separation characteristics in ventriculocyte mitochondria and brain samples are consistent with the use of different start sites and/or the inclusion of splicing-derived sequences in the immunodetected BKCa proteins. As shown later in Fig. 4, analyses of ventriculocyte transcripts predict proteins containing large 50- and 58-aa inserts. Proteins starting from Met 1 or Met 3 (4) and containing both inserts would have a molecular mass of 145 kDa or 139 kDa, respectively, in close agreement with the larger-size band in mitochondria. The size of 125 kDa in the brain is similar to that found in smooth muscle (9) and matches the mass of a predominant protein starting at Met 3 without large inserts.
Fig. 1.

Identifying cardiac BKCa in purified mitochondria. (A) Electron micrograph of purified mitochondria. Dark spots correspond to mitochondrial granules. (B) Immunoblot for BKCa in mitochondrial lysates from WT and knockout (−/−) ventriculocytes. (C) Immunoblot of whole-brain lysate. Protein = 50 µg/lane. Arrows, specific bands. (D) Example of spectra identifying BKCa from mitoplasts. (E) Identified peptides (underlined) span the C terminus (gray area).
Fig. 4.
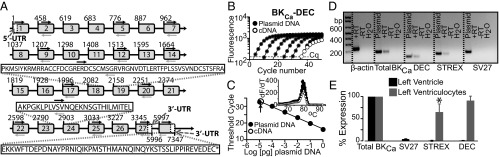
Molecular determination and quantification of cardiac BKCa. (A) Scheme of the detected 27 constitutive exons and translated alternatively-spliced exons (STREX, SV27, and DEC). Boxes, exons; line, introns. Numbers correspond to nucleotide numbers. White boxes, 5′UTR and alternative 3′UTRs. Black arrows, forward primers; gray arrows, reverse primers. Dashed gray arrows, 3′UTR primers used for cDNA synthesis (Table S2). (B) Example of real-time PCR fluorescence vs. cycle number plots for known amounts of DEC insert (closed circles) and in ventriculocytes’ cDNA (open circles). Dashed line, level of threshold cycles (Cq). (C) Corresponding dose–response curve together with experimental value (open circle). (Inset) Corresponding melting curves. (D) Agarose gel of PCR end-products for β-actin, total BKCa, and DEC, STREX, and SV27 exonic sequences. For explanation, see text. (E) Percentage expression of BKCa-SV27, BKCa-STREX, and BKCa-DEC normalized to total BKCa in left ventricle and in left ventriculocytes. *P < 0.05 vs. total BKCa.
For mass spectrometry analysis, Percoll-purified mitochondria from rat left ventricle were used to prepare inner mitochondrial membranes (mitoplasts). mitoBKCa was immunoprecipitated from mitoplast lysates using anti-BKCa antibodies that generated no signals in purified mitochondria of Kcnma1−/− (Fig. 2M). Immunopurified samples from 10 rat hearts were combined for each experiment (n = 5). LC/MS/MS spectra, such as the one shown in Fig. 1D, confirmed the presence of BKCa encoded by the Kcnma1 gene in the mitochondrial bands excised at ∼100–150 kDa (three of five experiments). Notably, we did not obtain BKCa sequences in the gel plugs that corresponded to molecular masses of <100 kDa or >150 kDa. In contrast, in gel plugs at ∼100–150 kDa, we identified 11 unique peptides throughout the C terminus of BKCa (Fig. 1E and Table S1). The coding sequences of the identified peptides were localized in constitutive exons 9, 12, 14, 17, 19, 21, 25, and 27, and spanned exons 21 and 22. In addition, we found a peptide of a 27-aa splice insert, which we refer to as SV27. The presence of peptides covering 778 residues, or 95% of the constitutive BKCa C terminus, ruled out the possibility that the cardiac mitoBKCa α-subunit consists of an ∼55-kDa protein (1). Supporting this view, LC/MS/MS analysis of mitoBKCa immunoprecipitated from lysates of mouse brain mitoplasts (from Percoll-purified mitochondria) yielded 21 unique peptides covering 1,113 residues, or 92% of the entire BKCa sequence. The peptides included one within the first intracellular loop (between S0 and S1) and two overlapping parts of the S1 and S4 transmembrane domains (Fig. S1 and Table S1). Taken together, these results support the presence of classic BKCa in mitochondria and revealed mitoBKCa as a protein of ∼140 kDa encoded by the Kcnma1 gene.
Fig. 2.

Localization of native BKCa in mitochondria. (A and B) Ventriculocytes loaded with MitoTracker, fixed, permeabilized and labeled with anti-BKCa Ab. (C) Overlay of A and B. (D and E) Ventriculocytes labeled with WGA under nonpermeabilized conditions, and subsequently permeabilized and labeled with the anti-BKCa Ab. (F) Overlay of D and E. (G–I) Purified mitochondria from left ventricle triple-labeled with anti-BKCa pAb (G), MitoTracker (Mito) (H), and anti-VDAC1 Ab (I). (J–L) Overlays of G and H, G and I, and H and I, respectively. (M–O) Kcnma1−/− mitochondria triple-labeled for BKCa (M), MitoTracker (N), and VDAC1 (O).
Direct Visualization of mitoBKCa.
We then wondered whether BKCa could be visualized within the mitochondria of adult ventriculocytes. Notably, BKCa immunolabeling indicated a longitudinal distribution pattern (Fig. 2 A and D) that largely coincided with MitoTracker labeling (Fig. 2 B and C) but not with surface membrane labeling by wheat germ agglutinin (WGA) (Fig. 2 D–F). Colocalization analysis using the protein proximity index (PPI) algorithm (10) indicated that the WGA to BKCa PPI was low (0.15 ± 0.01; n = 32 cells from five independent experiments). In contrast, the MitoTracker to BKCa PPI was considerable (0.52 ± 0.02; n = 56 cells from 10 independent experiments), which is consistent with the view that BKCa resides in mitochondria.
To increase the sensitivity of our method, we directly visualized BKCa in purified mitochondria. Regular confocal and superresolution (stimulation emission depletion, STED) fluorescence microscopies were used. For regular confocal microscopy, mitochondria were triple-labeled for BKCa, with MitoTracker and for the voltage-dependent anion channel 1 (VDAC1) (Fig. 2 G–I). Supporting the presence of BKCa in mitochondria, BKCa signals frequently coincided with those of MitoTracker (Fig. 2J) and VDAC1 (Fig. 2K) in the same way as VDAC1 coincided with MitoTracker labeling (Fig. 2L). Furthermore, mitochondria from Kcnma1−/− mice produced no BKCa-specific signals (Fig. 2M) in MitoTracker-loaded mitochondria, in which MitoTracker and VDAC1 colabeling was preserved (Fig. 2 N and O) (n = 3). In fact, in the BKCa knockout samples, the PPI for mitoBKCa to MitoTracker and vice versa were identical and within background levels (0.14 ± 0.01, n = 12 experiments, three fields per experiment). In contrast, WT mitochondria displayed a PPI for mitoBKCa to MitoTracker of 0.84 ± 0.03 and for MitoTracker to mitoBKCa of 0.66 ± 0.04 (n = 8 experiments, three fields per experiment), which were comparable to the PPI values for VDAC1 to MitoTracker and for VDAC1 to mitoBKCa (0.63 ± 0.03 and 0.56 ± 0.03, n = 5 experiments, three fields per experiment). The results imply that the majority of mitoBKCa colocalizes with MitoTracker-loaded mitochondria but not all MitoTracker-labeled mitochondria express BKCa proteins.
Increasing resolution with STED microscopy allowed us to map mitoBKCa with unprecedented detail. STED transformed the indistinct regular confocal image (Fig. 3 A and D) into an image in which discrete mitoBKCa clusters were observed (Fig. 3 B and E). Overlaid images of the same field contrast the difference in resolution (Fig. 3 C and F). Random line scans (Fig. 3E) were used to construct line scan plots (Fig. 3G) to measure the full-width half-maximum of mitoBKCa clusters. A frequency histogram analyzing 500 clusters was fitted with a Gaussian function; the fit indicates that the majority of mitoBKCa arranges in clusters of 50 ± 1.4 nm (Fig. 3G, inset) (n = 3 independent preparations). Overall, the microscopy results established beyond doubt the presence of BKCa in cardiac mitochondria, and Kcnma1−/− experiments verified its Kcnma1 origin.
Fig. 3.

Nanovisualization of cardiac mitoBKCa. Confocal (A, green) and STED images (B, red) of same Percoll-purified mitochondria from mouse left ventricle labeled for BKCa and using Atto-647N secondary antibody conjugate. (C) Overlay of A and B. (D–F) Amplification of squared region in A–C, respectively. (G) STED intensity profile of the line scan in E (white line). (Inset) Cluster size histogram fitted with a Gaussian function (red line). Images were acquired at 3.5 nm/pixel and pseudocolored for presentation.
BKCa Exonic Composition in Ventriculocytes.
The molecular composition of mitoBKCa was addressed by first purifying mRNAs from isolated mouse ventriculocytes (avoiding contamination from other cardiac cell types) and performing a systematic exon scan of Kcnma1’s 41 predicted exons (National Center for Biotechnology Information Evidence Viewer, Gene ID 16531). Because the Kcnma1 gene has two predicted 3′UTRs (Fig. 4A), we used oligo-dT primers and primers from both 3′UTRs to reverse-transcribe mRNAs. All 41 predicted exons were scanned with specific primers designed to amplify exonic regions 200–400 bp in size (Table S2). This approach detected the standard 27 constitutive exons that encode the insertless BKCa beginning from the first possible start codon of Met 1 (NP_034740.2; n = 3) (4). Furthermore, we found three spliced exons in the C terminus encoding: a 57-aa insert corresponding to the stress-axis regulated exon (STREX) (11), SV27, and a 50-aa C-terminal sequence named after the three last amino acids, DEC (n = 3) (Fig. 4A and Fig. S2).
To determine the relative contribution of the three spliced exons to cardiac BKCa mRNAs, we performed quantitative real-time PCR using cDNAs prepared from DNase pretreated total RNA of the left ventricle and isolated ventriculocytes. Controls included no reverse transcriptase (−RT), PCR primers spanning introns (Table S3), and H2O instead of cDNA. Standard curves using linearized BKCa plasmids were used to extrapolate the cDNA concentration in the cardiac samples. Fig. 4 B–D illustrates, as an example, the analysis for BKCa-DEC in ventriculocyte samples. Fig. 4B presents the fluorescence vs. PCR cycle curves for different concentrations of the BKCa-DEC standard (closed circles) together with the curve for a cardiac cDNA sample (open circles). Fig. 4C is the corresponding threshold cycle number (Cq, read at ∼100 relative fluorescence units) vs. the BKCa-DEC plasmid concentration curve (closed circles); also shown is the experimental point (open circle, arrow). The corresponding melting curves at the end of PCR cycles (Fig. 4C, Inset) featured a single peak, which is indicative of a single PCR product. The latter was confirmed by examining the PCR end-products using agarose gel electrophoresis as shown in Fig. 4D, which also shows results for β-actin, total BKCa, and the other two splice sequences (+RT refers to the PCR product in ventriculocyte cDNA samples, −RT and H2O are negative controls as described above, and plasmid refers to products from constructs BKCa, BKCa-DEC, BKCa-STREX, and BKCa-SV27). Absolute quantities of total BKCa were nearly four orders-of-magnitude greater in the left ventricle than in isolated ventriculocytes (1.5 ± 0.3, n = 4 vs. 5.4 × 10−4 ± 0.0002 pg cDNA/µg RNA, n = 6) pointing to the large abundance of BKCa transcripts in other cardiac cells. STREX and DEC were only 10- to 25-times more abundant in the left ventricle than in the ventriculocytes (STREX, 4.0 × 10−3 ± 0.001 vs. 3.8 × 10−4 ± 0.0001 pg cDNA/µg RNA, n = 4 each; DEC, 1.1 × 10−2 ± 0.003, n = 4 vs. 4.8 × 10−4 ± 0.0001, n = 6, pg cDNA/µg RNA). The abundance of SV27 was less than the detection limit in samples from isolated ventriculocytes (n = 6) but present in the left ventricle (4.7 × 10−2 ± 0.003 pg cDNA/µg RNA, n = 4). For comparison, Fig. 4E shows the percent expression with respect to total BKCa for all three species. It is clear that in the left ventricle, the STREX, SV27, or DEC transcript levels were minimal with respect to total BKCa (0.3 ± 0.03%, 3.8 ± 1.06% and 0.7 ± 0.1%, n = 4, respectively). In contrast, in ventriculocytes the DEC sequences were as abundant as total BKCa (91 ± 10%, n = 6), followed by STREX (65 ± 18%, n = 4) and SV27, which was negligible (n = 6). These results imply that ventriculocytes express BKCa transcripts containing primarily DEC splice sequences, whereas other cardiac cells (e.g., from vessels, fibroblasts, or neurons) express abundant insertless BKCa. In both cases, BKCa originates from the Kcnma1 gene.
DEC Splicing and mitoBKCa Targeting.
Because constitutive (insertless) BKCa fails to target BKCa to mitochondria in cardiac H9c2 cells (7), the finding of STREX, SV27, and DEC splice sequences in cardiomyocytes mRNAs prompted us to investigate whether a splicing mechanism could be responsible for targeting BKCa to mitochondria. We hypothesized that the most-abundant splice insert, DEC, should have a preponderant role in BKCa targeting; in accordance with this conjecture, the least-abundant insert, SV27, would play an insignificant role.
To test our hypothesis, adenovirus constructs containing insertless BKCa, and isoforms BKCa-SV27 and BKCa-DEC (with splice sequences in their native positions) were transduced into rat adult ventriculocytes for immunochemical analysis. All constructs contained a c-Myc epitope at the N terminus. Because the plasma membrane BKCa has an extracellular N terminus (12) (Fig. 5, schemes), the N-terminal c-Myc tag allowed us to examine nonpermeabilized and permeabilized cells to discern whether newly synthesized BKCa isoforms were targeted to the plasma membrane or found intracellularly, perhaps within mitochondria. To visualize mitochondria, cardiomyocytes were preloaded with MitoTracker. Using this strategy under nonpermeabilized conditions, we found that insertless BKCa was readily targeted to the plasma membrane (Fig. 5A) and that its signals were not linked to MitoTracker labeling (Fig. 5 B–D) (n = 3, 12 of 12 cells). In permeabilized cells, the peripheral expression pattern remained the same, and there was only a low intracellular signal that did not coincide with MitoTracker labeling (Fig. 5 E–H) (n = 9, 27 of 27 cells). Similarly, the BKCa-SV27 isoform displayed a pronounced plasmalemmal localization detected under nonpermeabilized conditions (Fig. 5 I–L) (n = 4, 10 of 10 cells) that was the same in permeabilized conditions and without overlap with MitoTracker labeling (Fig. 5 M–P) (n = 4, 15 of 15 cells).
Fig. 5.

Insertless BKCa and BKCa-SV27 do not colocalize with mitochondria in transduced adult ventriculocytes. (A) Insertless BKCa was observed at the plasma membrane of a nonpermeabilized cell. (B) Same cell showing clear MitoTracker signals. (C) Overlay of A and B. (D, H, L, and P) Amplified images (squares in C, G, K, and O, respectively). (E) In permeabilized cells insertless BKCa was predominantly observed at the cell periphery. (F) MitoTracker signals of same cell. (G) Overlay of E and F. (I) BKCa-SV27 labeling was also observed at the plasma membrane under nonpermeabilized conditions. (J) MitoTracker labeling of the same cardiomyocyte. (K) Overlay of I and J. (M) BKCa-SV27 at the cell periphery in a permeabilized cell. (N) MitoTracker signal of the cell in M. (O) Overlay of M and N. Cartoons show orientation of constructs and tag at the N terminus. PM, plasma membrane.
Finally, when cells were transduced with BKCa-DEC, nonpermeabilized cells displayed no BKCa-DEC signal at the surface membrane (Fig. 6A), which demonstrated that the DEC insert prevented expression of BKCa at the plasma membrane; as a positive control, MitoTracker labeling was clearly observed (Fig. 6 B–D) (n = 3, 12 of 12 cells). In contrast, permeabilized cardiomyocytes exhibited remarkable longitudinal labeling similar to mitochondria (Fig. 6 E and F). The coincidence in BKCa-DEC and MitoTracker is illustrated in the overlay (Fig. 6 G and H). Greater than 90% of the BKCa-DEC transduced cells examined exhibited this exceptional mitochondrial localization (n = 4, 14 of 15 cells), which resembled that observed in native cells (Fig. 2). In fact, image analysis demonstrated a PPI for BKCa-DEC to MitoTracker of 0.51 ± 0.04 (n = 4 experiments; 14 cells) which is close to the PPI of native BKCa to MitoTracker-loaded mitochondria. These results indicate that BKCa-DEC, but neither insertless BKCa nor BKCa-SV27, contains sequences recognized by ventriculocytes’ machinery for mitochondrial delivery.
Fig. 6.

The expression of DEC exon favors BKCa mitochondrial targeting in transduced adult ventriculocytes. (A) In nonpermeabilized cells, no significant BKCa-DEC signal was observed. (B) The same ventriculocyte readily loaded with MitoTracker. (C) Overlay of A and B. (D) Zoom-out of the square in C. (E) In permeabilized cells, a profuse intracellular BKCa-DEC labeling that resembled MitoTracker labeling (F) was observed. (G) Overlay with remarkable colocalization of BKCa-DEC and MitoTracker. (H) Region at higher magnification (square in G). The cartoon shows BKCa-DEC intracellular location.
Kcnma1 Is Essential for the Cardioprotective Action of NS1619.
mitoBKCa has been proposed to protect the heart from ischemic insult because a general BKCa opener, NS1619, increases mitochondrial K+ uptake and promotes heart protection against ischemia/reperfusion (1). Because our previous results indicate that mitoBKCa is a product of the Kcnma1 gene, we first investigated whether the hearts from Kcnma1−/− animals subjected to ischemia/reperfusion lost their characteristic functional improvement in response to NS1619. To test this point, isolated hearts from WT and Kcnma1−/− mice were preconditioned for 10 min without (−, vehicle alone) or with (+) NS1619 (10 µM); this period was followed by an 18-min period of ischemia and ∼1 h of reperfusion, as depicted in Fig. 7A.
Fig. 7.

BKCa protects the heart from ischemic injury. (A) Ischemia/reperfusion protocol. (B and C) Function traces of hearts preconditioned with vehicle (DMSO, control) or with NS1619 (10 µM) in WT and Kcnma1−/− mice. (D) NS1619 significantly improved mean RPP in WT but not in Kcnma1−/− mice. (E and G) WT hearts preconditioned with NS1619 exhibited less infarct size (white) compared with the control. (F and G) In Kcnma1−/−, infarct size was not reduced with NS1619. (H–J) Mitochondrial Ca2+ uptake. NS1619 preconditioning increased the amount of Ca2+ needed to induce a large Ca2+ release in WT but not in Kcnma1−/−samples. Black arrows, addition of mitochondria. Blue arrows, 40 nmol Ca2+ pulses. Arrowheads, massive release of Ca2+. *P < 0.05 vs. control (Ctrl); CRC, Ca2+ retention capacity.
Typical cardiac function traces for the left ventricular-developed pressure as a function of time demonstrate that preconditioning the heart from WT animals with NS1619 causes, as expected, an improvement in cardiac function during reperfusion compared with the decreased function observed in the control hearts that were preconditioned with vehicle alone (Fig. 7B). In contrast, Kcnma1−/− hearts lost their ability to be protected by NS1619 (Fig. 7C). Accordingly, the mean values for heart rate pressure product (RPP) (Fig. 7D) and the maximum rates of rise (dP/dt max) and fall (dP/dt min) (Fig. S3), which were measured as the mean of the last 10 min of reperfusion, indicated a significant improvement because of NS1619 in WT animals (n = 4), a property that was lost in Kcnma1−/− animals (n = 3–4).
Similarly, myocardial infarction measured at the end of the reperfusion period was less pronounced after preconditioning WT animals with NS1619. Fig. 7E illustrates typical heart cross-sections stained with 2,3,5-triphenyltetrazolium chloride, in which the pale areas that correspond to nonviable tissue are smaller with NS1619 preconditioning than in vehicle-treated hearts (control). In contrast, hearts from Kcnma1−/− were not protected by NS1619 and exhibited significant damage similar to that observed in the control hearts (Fig. 7F). Individual and mean infarct size values are given in Fig. 7G for WT (n = 4 each) and Kcnma1−/− (n = 3 each).
Next, we tested whether NS1619 preconditioning could also benefit mitochondria function and if this effect was retained in Kcnma1−/−. To this end, we measured the Ca2+ retention capacity of mitochondria isolated at 10 min of reperfusion. NS1619 (10 µM) treatment resulted in an increase in the number of Ca2+ pulses (Fig. 7H, blue arrows) needed to cause a massive Ca2+ release (Fig. 7H, arrowheads). This beneficial effect was abrogated in mitochondria from Kcnma1−/− animals (Fig. 7 I and J) (n = 4–5 each). Overall, these experiments clearly demonstrate that Kcnma1 gene expression is a prerequisite for NS1619-mediated cardioprotection from ischemia/reperfusion injury, and lead us to propose that mitoBKCa is a target of NS1619 in the heart.
Discussion
In this work, we uncovered the origin of mitoBKCa as the product of the Kcnma1 gene, the same gene that encodes the plasma membrane BKCa, and demonstrated that splicing of the DEC exon is a mechanism that allows the protein to be targeted to cardiac mitochondria.
The strategies used to reach these conclusions were: (i) use of Percoll-purified mitochondria from isolated ventriculocytes or the left ventricle to assess the molecular mass of mitoBKCa through immunoblotting with antibodies raised against the plasma membrane BKCa, for protein sequence analysis of BKCa immunoprecipitates, and for visualization of mitoBKCa clusters at superresolution; (ii) use of isolated cardiomyocytes for immunolabeling of native mitoBKCa, for isolation of mRNAs for exon scanning, for quantification of splice inserts, and for transduction of BKCa isoforms to monitor subcellular localization; and (iii) use of Kcnma1−/− mice for negative controls and to demonstrate loss of the protective action of a BKCa opener, NS1619, against ischemic injury.
Cardiac mitoBKCa and Kcnma1 Gene.
The finding that the mitoBKCa primary subunit has a size of ∼140 kDa is in contrast with an early study, reporting a smaller size protein of ∼55 kDa (1), and a more recent report of a larger protein with mass of ∼125 kDa (2); in both studies the protein was detected in mitochondrial lysates from isolated cardiomyocytes. In all instances, including the present study, mitochondrial lysates were examined with polyclonal antibodies (from Alomone Labs and Affinity Bioreagents) against the C-terminal end of the plasma membrane BKCa α-subunit encoded by the Kcnma1 gene. Because in some immunoblots, we have also observed a signal of ~50 kDa, it is possible that the observed smaller fragments, including the fragment near 100 kDa reported here, result from mitoBKCa proteolysis. This assumption is supported by the fact that BKCa is known to suffer C-terminal cleavage during purification, producing fragments of similar size (13). Conclusive evidence for the mitoBKCa α-subunit being a protein similar to its plasma membrane counterpart—encoded by the Kcnma1 gene—was given by sequence analysis of immunopurified BKCa from inner mitochondrial membranes (mitoplasts) purified from the left ventricle and from the brain. In both instances, peptides throughout the soluble C terminus were identified, whereas in brain mitoplast samples, the identified peptides spanned the whole BKCa protein from its N terminus to the end of its C terminus (Fig. 1 and Fig. S1).
Similar to our results (Fig. 4), a previous transcript analysis of ventricular myocytes reported expression of all 27 constitutive exons of the Kcnma1 gene (7), thereby supporting the view that mitoBKCa is indeed encoded by this gene. However, in those studies, expression of transcripts containing spliced exons, as we report here, was not detected, and thus expression of the insertless cDNA in H9c2 cells did not target BKCa to mitochondria.
Our conclusion that mitoBKCa is encoded by the Kcnma1 gene implies that all of the essential functional cassettes of the protein are conserved in both mitochondrial and plasma membrane versions. In fact, the previously reported biophysical and pharmacological properties of mitoBKCa from cardiomyocytes and other cell types resemble those of plasma membrane BKCa (3).
Mapping Cardiac BKCa to Mitochondria and Splice Variation as a Targeting Mechanism.
To our knowledge, BKCa activity has not been reported at the plasma membrane of adult ventriculocytes, which is consistent with our immunocytochemistry results. In contrast, we found profuse BKCa immunosignals coincident with MitoTracker-labeled mitochondria in these cells, a property that was confirmed beyond doubt using purified mitochondria and nanoimaging (Figs. 2 and 3). Compared with labeling cells, labeling-purified mitochondria had the advantage that we could rule out contamination and interference from signals originating from other intracellular organelles. The selective BKCa mitochondrial distribution vs. plasma membrane in adult ventriculocytes is not a property of neonatal cardiac cells (H9c2) in which BKCa can be visualized in both subcellular compartments (3). Dual targeting to the plasma membrane and mitochondria appears to be a common property of BKCa in other cell types.
In this work, we demonstrate that BKCa targeting to ventriculocytes mitochondria requires the inclusion of DEC exonic sequences at the C-terminal end of mitoBKCa. Thus, our work provides further evidence for a role of the C-terminal end in targeting proteins to mitochondria, unlike the canonical view of mitochondrial targeting by N-terminal sequences (14, 15). In agreement with the functional relevance of DEC sequences in mitochondria targeting, from the three splice inserts found in our ventriculocyte mRNA analysis, the DEC insert was the most abundant, with an abundance equal to the level of total BKCa. STREX transcripts were second in abundance to DEC transcripts, but the function of STREX is yet to be discovered. Nevertheless, our results demonstrate that the sole inclusion of DEC in BKCa is sufficient for mitochondrial delivery, which rules out an essential role for STREX in this function.
In contrast with our findings in cardiac myocytes, where BKCa-DEC is readily targeted to mitochondria, in COS cells a BKCa-DEC variant with additional splicing-derived sequences, NATRMTRMGQA, is found predominantly in the endoplasmic reticulum but also at the plasma membrane (16). These findings suggest that additional sequences may disrupt the targeting of BKCa-DEC to mitochondria or that cell-specific mechanisms are necessary to complement DEC in targeting BKCa to mitochondria. Whether the inclusion of STREX or SV27 modifies BKCa-DEC targeting to cardiomyocytes mitochondria is yet to be established. However, data using CHO cells indicate that SV27 does not have a major effect as a BKCa-DEC variant with additional splice inserts including SV27 can still be observed in mitochondria of CHO cells (17).
Cardiac BKCa and Cardioprotection.
Previous studies regarding the role of BKCa in protecting the heart from ischemic insult were based on pharmacology (1, 6, 18, 19). Because the most commonly used BKCa opener, NS1619, is able to inhibit Ca2+ currents when used at ∼30 µM (20), there was always the open question of whether NS1619 acts via BKCa or through some other mechanism (21). In our current study, we demonstrated that the protective effect of 10 µM NS1619 on the preischemic reperfused heart and on mitochondria isolated from these hearts was abolished in Kcnma1−/− mice (Fig. 7), thereby indicating that NS1619-mediated cardioprotection requires expression of BKCa. At present, we do not have an explanation of why the Kcnma1−/− mice exhibited reduced infarct size after ischemia/reperfusion under basal conditions. Nevertheless, the cardioprotective effects of NS1619 in the heart correlate with the activation of mitoBKCa (1) and thus, our studies using Kcnma1−/− mice support the view that activation of cardiomyocyte mitoBKCa is one mechanism that protects the heart against ischemia-reperfusion injury. However, additional mechanisms triggered by NS1619, including the opening of BKCa in nonmyocyte heart cells, cannot be ruled out.
Materials and Methods
Animals.
Three-month-old male C57BL/6NCrL mice and Sprague–Dawley (SD) rats were used. Kcnma1−/− mice were from Andrea Meredith (University of Maryland School of Medicine, Baltimore) and locally bred. All protocols received University of California, Los Angeles approval.
Antibodies and Procedures.
Antibodies and procedures are given in SI Materials and Methods.
Statistics.
The mean ± SEM for at least three individual experiments is given. A two-tailed Student t test value of P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank UCLA members Dr. Sirus Kohan for help with electron microscopy; Dr. Melissa Sondrej, Dr. Gregg Czerwieniec, and Dr. Arnab Chatopadhyay for their assistance and helpful discussion for mass spectrometry; and Dr. Vicente Ribas for helpful discussion in adenovirus construction. This work was supported by National Institutes of Health Grants HL107418 (to E.S. and L.T.) and HL088640 (to E.S.); American Heart Association Fellowship 09POST219008 (to J.C.B.); and National Scientist Development Award 11SDG7230059 (to H.S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1302028110/-/DCSupplemental.
References
- 1.Xu W, et al. Cytoprotective role of Ca2+- activated K+ channels in the cardiac inner mitochondrial membrane. Science. 2002;298(5595):1029–1033. doi: 10.1126/science.1074360. [DOI] [PubMed] [Google Scholar]
- 2.Shi Y, et al. Mitochondrial big conductance KCa channel and cardioprotection in infant rabbit heart. J Cardiovasc Pharmacol. 2007;50(5):497–502. doi: 10.1097/FJC.0b013e318137991d. [DOI] [PubMed] [Google Scholar]
- 3.Singh H, Stefani E, Toro L. Intracellular BK(Ca) (iBK(Ca)) channels. J Physiol. 2012;590(Pt 23):5937–5947. doi: 10.1113/jphysiol.2011.215533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallner M, et al. Characterization of and modulation by a beta-subunit of a human maxi KCa channel cloned from myometrium. Receptors Channels. 1995;3(3):185–199. [PubMed] [Google Scholar]
- 5.Lu R, et al. MaxiK channel partners: Physiological impact. J Physiol. 2006;570(Pt 1):65–72. doi: 10.1113/jphysiol.2005.098913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Yin C, Xi L, Kukreja RC. Opening of Ca2+-activated K+ channels triggers early and delayed preconditioning against I/R injury independent of NOS in mice. Am J Physiol Heart Circ Physiol. 2004;287(5):H2070–H2077. doi: 10.1152/ajpheart.00431.2004. [DOI] [PubMed] [Google Scholar]
- 7.Ko JH, et al. Cloning of large-conductance Ca(2+)-activated K(+) channel alpha-subunits in mouse cardiomyocytes. Biochem Biophys Res Commun. 2009;389(1):74–79. doi: 10.1016/j.bbrc.2009.08.087. [DOI] [PubMed] [Google Scholar]
- 8.Singh H, et al. Visualization and quantification of cardiac mitochondrial protein clusters with STED microscopy. Mitochondrion. 2012;12(2):230–236. doi: 10.1016/j.mito.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M, et al. Thromboxane A2 receptor and MaxiK-channel intimate interaction supports channel trans-inhibition independent of G-protein activation. Proc Natl Acad Sci USA. 2010;107(44):19096–19101. doi: 10.1073/pnas.1002685107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Y, et al. Quantitative determination of spatial protein-protein correlations in fluorescence confocal microscopy. Biophys J. 2010;98(3):493–504. doi: 10.1016/j.bpj.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie J, McCobb DP. Control of alternative splicing of potassium channels by stress hormones. Science. 1998;280(5362):443–446. doi: 10.1126/science.280.5362.443. [DOI] [PubMed] [Google Scholar]
- 12.Meera P, Wallner M, Song M, Toro L. Large conductance voltage- and calcium-dependent K+ channel, a distinct member of voltage-dependent ion channels with seven N-terminal transmembrane segments (S0-S6), an extracellular N terminus, and an intracellular (S9-S10) C terminus. Proc Natl Acad Sci USA. 1997;94(25):14066–14071. doi: 10.1073/pnas.94.25.14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knaus HG, et al. Characterization of tissue-expressed alpha subunits of the high conductance Ca(2+)-activated K+ channel. J Biol Chem. 1995;270(38):22434–22439. doi: 10.1074/jbc.270.38.22434. [DOI] [PubMed] [Google Scholar]
- 14.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: Machineries and mechanisms. Cell. 2009;138(4):628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li M, et al. Identification and characterization of mitochondrial targeting sequence of human apurinic/apyrimidinic endonuclease 1. J Biol Chem. 2010;285(20):14871–14881. doi: 10.1074/jbc.M109.069591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma D, et al. Differential trafficking of carboxyl isoforms of Ca2+-gated (Slo1) potassium channels. FEBS Lett. 2007;581(5):1000–1008. doi: 10.1016/j.febslet.2007.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kathiresan T, Harvey M, Orchard S, Sakai Y, Sokolowski B. A protein interaction network for the large conductance Ca(2+)-activated K(+) channel in the mouse cochlea. Mol Cell Proteomics. 2009;8(8):1972–1987. doi: 10.1074/mcp.M800495-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stowe DF, et al. Cardiac mitochondrial preconditioning by Big Ca2+-sensitive K+ channel opening requires superoxide radical generation. Am J Physiol Heart Circ Physiol. 2006;290(1):H434–H440. doi: 10.1152/ajpheart.00763.2005. [DOI] [PubMed] [Google Scholar]
- 19.Bentzen BH, et al. Activation of big conductance Ca(2+)-activated K (+) channels (BK) protects the heart against ischemia-reperfusion injury. Pflugers Arch. 2009;457(5):979–988. doi: 10.1007/s00424-008-0583-5. [DOI] [PubMed] [Google Scholar]
- 20.Edwards G, Niederste-Hollenberg A, Schneider J, Noack T, Weston AH. Ion channel modulation by NS 1619, the putative BKCa channel opener, in vascular smooth muscle. Br J Pharmacol. 1994;113(4):1538–1547. doi: 10.1111/j.1476-5381.1994.tb17171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aldakkak M, Stowe DF, Cheng Q, Kwok WM, Camara AK. Mitochondrial matrix K+ flux independent of large-conductance Ca2+-activated K+ channel opening. Am J Physiol Cell Physiol. 2010;298(3):C530–C541. doi: 10.1152/ajpcell.00468.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.