

Abstract
Inhibitors of peptide deformylase (PDF) represent a new class of antibacterial agents with a novel mechanism of action. Mutations that inactivate formyl methionyl transferase (FMT), the enzyme that formylates initiator methionyl-tRNA, lead to an alternative initiation of protein synthesis that does not require deformylation and are the predominant cause of resistance to PDF inhibitors in Staphylococcus aureus. Here, we report that loss-of-function mutations in FMT impart pleiotropic effects that include a reduced growth rate, a nonhemolytic phenotype, and a drastic reduction in production of multiple extracellular proteins, including key virulence factors, such as α-hemolysin and Panton-Valentine leukocidin (PVL), that have been associated with S. aureus pathogenicity. Consequently, S. aureus FMT mutants are greatly attenuated in neutropenic and nonneutropenic murine pyelonephritis infection models and show very high survival rates compared with wild-type S. aureus. These newly discovered effects on extracellular virulence factor production demonstrate that FMT-null mutants have a more severe fitness cost than previously anticipated, leading to a substantial loss of pathogenicity and a restricted ability to produce an invasive infection.
INTRODUCTION
In bacteria, protein synthesis initiates with formyl-methionyl-tRNAi, and therefore, all newly synthesized polypeptides contain an N-formyl-methionine terminal end that, in most cases, is not retained in mature proteins. As the polypeptides emerge from the ribosome, the N-formyl group is hydrolyzed by peptide deformylase (PDF), and subsequently, methionine aminopeptidase removes the N-terminal methionine. Deformylation plays a crucial role in protein maturation and has been shown to be essential for bacterial growth (1–4), as methionine aminopeptidase cannot hydrolyze N-blocked peptides (5). The ubiquitous nature of PDF, together with the fact that this function is not required for cytoplasmic protein synthesis in eukaryotes, has made this an attractive target for the development of new antibacterial agents. The successful purification and characterization of the native form of Escherichia coli PDF (6), as well as the discovery that the natural product actinonin is an inhibitor of PDF with weak antibacterial activity (7–9), triggered the search for additional PDF inhibitors. Since then, a large number of structurally diverse PDF inhibitors have been identified, including several compounds with demonstrated in vivo efficacy and good safety profiles (10). Three PDF inhibitors have progressed to clinical trials (11, 12), and one of them, GSK1322322, is in phase II clinical development for the treatment of respiratory tract and skin infections.
While mutations in FolD and GlyA, two enzymes involved in the synthesis of 10-formyl-tetrahydrofolate, have been described (13, 14), loss-of-function mutations in the gene encoding formyl-methionyl transferase (FMT), the enzyme that catalyzes the formylation of the initiator methionyl-tRNA, are the most common cause of resistance to PDF inhibitors in bacteria where FMT is not essential for viability, such as Staphylococcus aureus (3, 15), Bacillus subtilis (13), Pseudomonas aeruginosa (16), Salmonella enterica (14), and E. coli (15, 17, 18). In those organisms, protein synthesis can still initiate with unformylated methionyl-tRNA, bypassing the need for PDF function. However, FMT mutants show compromised growth in vitro (50% to 90% reduction in the growth rate compared to the wild type) (3, 14–18) and in vivo (3, 14). Whether these effects are simply due to slow translation initiation or additional factors was heretofore unknown.
In order to further understand the liability associated with mutations in FMT, a thorough comparative in vitro characterization of S. aureus FMT mutants versus the wild-type strain was undertaken, and their abilities to produce an infection in immunocompetent and immunocompromised animal models were investigated. These studies demonstrate that S. aureus FMT mutants show a substantial reduction in production of extracellular virulence factors and are significantly less pathogenic than their wild-type counterparts in animal models of infection.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in these studies include S. aureus WCUH29 (hospital-acquired methicillin-resistant S. aureus [HA-MRSA]) (19), S. aureus PVL-2 (USA-300; Panton-Valentine leukocidin [PVL] positive; community-acquired MRSA [CA-MRSA]) (20), and their characterized derivatives (Table 1), as well as S. aureus MW2, 90265/97, and PK1. Bacteria were grown in tryptic soy broth (TSB) or on tryptic soy agar (TSA) plates, in Mueller-Hinton (MH) broth, on CCY medium (21), or on agar-based MH plates, as indicated. To detect hemolysis, TSA plates supplemented with 5% sheep or rabbit blood were incubated at 37°C for 24 to 48 h and at 4°C for an additional 12 h when sheep blood was used. S. aureus WCUH29 strains used in the experimental animal models were grown on brain heart infusion (BHI) broth.
Table 1.
Characterized S. aureus strains used in these studies
| Strain | Genotype | Reference |
|---|---|---|
| WCUH29 | WT; HA-MRSA | 19 |
| Δfmt | Δfmt::tetM | This study |
| FMT1 | fmt +T550; frameshift | This study |
| FMT7 | fmt C577T Q193stop | This study |
| FMT8 | fmt −G434; frameshift | This study |
| FMT10 | fmt C319T H107Y | This study |
| PDF18 | pdf T176A V59D | This study |
| pYH4 | Plasmid pYH4 | 25, 26 |
| Δfmt (pYH4) | Δfmt::tetM; plasmid pYH4 | This study |
| Δfmt (pYH4-fmt) | Δfmt::tetM; fmt cloned in pYH4 | This study |
| PVL-2 | WT; CA-MRSA; USA-300; PVL+ | 20 |
| Δfmt | Δfmt::tetM | This study |
Antibacterial susceptibility testing.
MICs of antibiotics were determined using the broth microdilution methodology recommended by the Clinical and Laboratory Standards Institute (22). The MIC was the lowest concentration of antibiotic that suppressed visible growth.
Construction of fmt deletion/insertion mutants.
S. aureus WCUH29 fmt deletion mutants were constructed as described previously (23). A Δfmt::tetA(M) cassette was first integrated into the S. aureus RN4220 chromosome and then moved into S. aureus WCUH29 by transduction with ϕ11 phage. To construct CA-MRSA–PVL-2 Δfmt::tetA(M), a Δfmt::tetA(M) cassette was cloned into pMAD and used as described previously (24), except that, after overnight growth at 42°C, the allelic-replacement mutants were directly selected in the presence of tetracycline and PDF inhibitor. All fmt allelic replacements were confirmed by PCR amplification. pYH4 and pYH4-fmt, from an open reading frame expression library of the S. aureus genome (25, 26), were used to transform S. aureus WCUH29 Δfmt.
Isolation and characterization of spontaneous resistant mutants.
Overnight cultures of S. aureus strains were plated onto MH agar plates containing 4 times the MIC of the PDF inhibitor SB-734453, a member of the hydrazide family of PDF inhibitors [10; K. M. Aubart, A. B. Benowitz, S. B. Christensen, I. V. J. M. Karpinski, J. Lee, and D. J. Silva, 2003, preparation of N-hydroxy-N-(3-hydrazino-3-oxopropyl)formamide derivatives as peptide deformylase inhibitors with antibacterial activity, PCT international patent application WO 03 101442]. Resistant colonies were purified once on PDF inhibitor-containing plates and once on MH plates. pdf and fmt genes from mutants and their parent strains were amplified by PCR and sequenced to identify mutations.
Analysis of extracellular proteins by SDS-PAGE and Western immunoblotting.
Culture supernatants of S. aureus strains WCUH29 and PVL-2, equivalent to overnight cultures with optical densities at 600 nm (OD600) of 0.4, were concentrated by Speed-Vac (Savant) and analyzed by SDS-PAGE and Western immunoblotting, as described previously (28). S. aureus PVL-2 lukF-PV and lukS-PV genes were cloned onto a pET vector and overexpressed in E. coli BL21(DE3). His-tagged LukF-PV and LukS-PV were purified as previously described (29) and used to generate rabbit polyclonal antibodies. Alpha-toxin antibodies were provided by M. Burnham (30).
Measurement of cell clumping and coagulase activity.
Cell clumping was qualitatively observed by mixing 20 μl of a saline suspension of bacterial cells (2 × 109 CFU/ml), prepared from freshly grown colonies, with 50 μl of serial 2-fold dilutions of a 2% (wt/vol) solution of commercial human fibrinogen in phosphate-buffered saline (PBS). Occurrence of clumping was noted after 3 min at room temperature. Coagulase activity was determined qualitatively by adding 0.5 ml of serial 2-fold dilutions of S. aureus culture supernatants in PBS to 0.5 ml of rabbit plasma diluted 1:3 in PBS. The mixtures were incubated at 37°C under static conditions and observed over a 24-h period.
Mice.
Male CBA/J mice (19 to 24 g; Jackson Laboratory), used in the experimental models, were allowed food and water ad libitum. All procedures were performed in accordance with protocols approved by the GlaxoSmithKline Institutional Animal Care and Use Committee and met or exceeded the standards of the American Association for the Accreditation of Laboratory Animal Care, the United States Department of Health and Human Services, and all local and federal animal welfare laws.
Immunocompromised mice.
Mice were dosed intraperitoneally (i.p.) once daily with 150 mg cyclophosphamide/kg of body weight (0.2 ml/mouse) on day 0 and with 100 mg/kg on days 3, 6, 8, 11, 13, and 15 of the study. On days 0, 4, 5, 7, 12, 14, 18, and 21, the mice were bled by cardiac puncture, and 200 to 300 μl of blood was placed into EDTA-containing tubes and gently mixed. Samples were analyzed on an Advia 120 Hematology System (Siemens Diagnostics) to determine peripheral blood counts. Red blood cell counts remained relatively constant, with a slight decrease at days 18 and 21. White blood cell (WBC) and neutrophil counts decreased by day 4 to ≤1,000 WBC/μl and ≤100 neutrophils/μl, respectively, and remained low until at least day 18. By day 21, 6 days after the last cyclophosphamide dose, white blood cell counts returned to baseline numbers and neutrophil counts increased to approximately four times baseline numbers. The mice maintained neutropenia (≤100 neutrophils/μl) from day 4 to day 18 of the study.
Murine hematogenous pyelonephritis infection model.
S. aureus WCUH29 cultures, grown overnight in BHI broth, were centrifuged and washed three times in PBS. Bacterial suspensions (OD600, ∼0.2) were diluted and plated onto TSA plates with 5% sheep blood to determine viable CFU. Five mice per group were inoculated with 0.1 ml of this suspension (containing approximately 107 CFU/mouse) by tail vein injection. The mice were monitored twice daily for signs of illness, and any that appeared moribund were euthanized prior to the end of the experiment. All remaining animals were euthanized by carbon dioxide overdose at days 3, 7, 14, 21, and 28 postinfection. Both kidneys were removed using an aseptic technique, placed in bags, and homogenized in PBS for 2 min in a Stomacher 80 Biomaster (Seward). Tenfold serial dilutions in sterile saline were plated on TSA supplemented with 5% sheep blood for enumeration of viable bacteria.
Infection in immunocompromised animals.
Mice were treated i.p. with cyclophosphamide as described above—150 mg/kg on day −4 and 100 mg/kg on days −1, 2, 4, 7, 9 and 11—to create and maintain neutropenia for the study duration. Enumeration of the day for cyclophosphamide treatment has been adjusted to encompass the day infection started, day 0. The experimental model was performed as described above, except mice were inoculated with 103 CFU/mouse and the animals were euthanized at days 0, 2, 4, 7, 10, and 14 postinfection.
Survival studies.
Infection was induced as described above. Ten immunocompetent animals per group, inoculated with approximately 107 CFU/mouse, or 20 neutropenic animals/group, inoculated with 103 CFU/mouse, were used in these studies. Mouse survival was followed for 14 days, but bacterial enumeration of kidneys was not performed.
RESULTS
In vitro characterization of S. aureus FMT mutants.
Spontaneously occurring S. aureus WCUH29 mutants resistant to the PDF inhibitor SB-734453 (Aubart et al., PCT international patent application WO 03 101442) were isolated at a frequency of 5 × 10−7, similar to that previously reported for different PDF inhibitors with other S. aureus strains (3, 15). From over 200 mutants, 37 small and 3 large colonies were selected for genetic characterization of the fmt and pdf genes. The strains with large-colony morphology were found to carry mutations in the pdf gene. This is the first time that mutations conferring resistance to PDF inhibitors in S. aureus have been identified in the target gene, although they occur at a much lower frequency (5 × 10−8 to 5 × 10−9) and confer a lower level of resistance than fmt mutations. Two of the three mutants had a V59D substitution in the PDF protein, which maps to the inhibitor-binding pocket in the PDF crystal structure (10). All strains with small-colony morphology carried mutations in the fmt gene that should lead to defective FMT proteins, including premature stop codons or frameshifts, or amino acid substitutions shown to disrupt the catalytic mechanism of the enzyme (31). As previously described (3, 15), FMT mutants, regardless of the type of mutation, showed a decrease in the growth rate with respect to the parent strain or the PDF mutants.
Further characterization revealed that S. aureus WCUH29 FMT mutants (Table 1), when screened on sheep blood agar plates, showed a nonhemolytic phenotype, while wild-type (WT) S. aureus and PDF mutant strains were hemolytic (Fig. 1), indicating that FMT mutants had impaired hemolysin production (32). The hemolytic phenotype could be restored in a genetically engineered fmt deletion mutant, S. aureus WCUH29 Δfmt, by the addition of the fmt gene in the pYH4 plasmid (25, 26) (Fig. 1), implying that loss of FMT function resulted in lack of hemolysis. This was a characteristic of all S. aureus FMT mutants, irrespective of their parent strain, as lack of hemolysis on sheep blood agar plates was also observed with FMT mutants from 4 other S. aureus strains, PVL-2, MW2, 90265/97, and PK1, encompassing both HA-MRSA and CA-MRSA with different antibiotic resistance phenotypes, including multidrug-resistant S. aureus. Moreover, the hemolytic activity of S. aureus 90265/97 was enhanced when incubating the plates at 4°C while its corresponding FMT mutants were still nonhemolytic, suggesting β-hemolysin production was affected (32).
Fig 1.
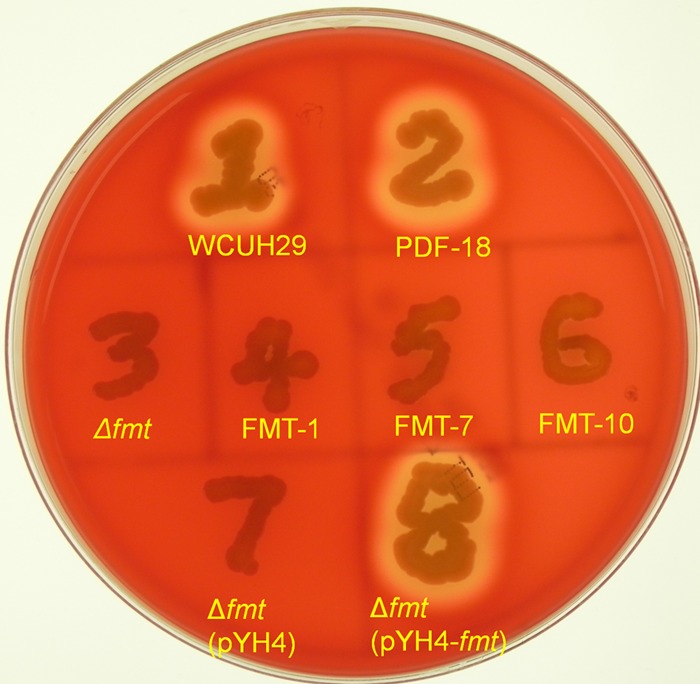
Nonhemolytic phenotypes of different S. aureus WCUH29 FMT mutants in sheep blood agar plates. S. aureus WCUH29 and its derivative strains (Table 1) were streaked onto a TSA plate containing 5% sheep blood agar and screened for hemolytic activity by incubation at 37°C for 24 to 48 h and were then left at 4°C for 12 h. FMT-null mutants isolated from S. aureus strains PVL-2, MW2, 90265/97, and PK1 showed identical nonhemolytic phenotypes on sheep blood agar plates screened under these conditions.
Identical results were obtained when hemolysis was evaluated on rabbit blood agar plates. While PDF mutants had a wild-type hemolytic phenotype, all S. aureus WCUH29 FMT mutants analyzed were nonhemolytic, implying that α-hemolysin production was compromised (32). Complementation of S. aureus WCUH29 Δfmt with the fmt gene in trans again restored hemolysis on rabbit blood agar plates. Western immunoblotting analysis, performed using culture supernatants, showed >95% reduction in production of α-hemolysin in the S. aureus FMT mutants, while the PDF mutant produced levels similar to those in the wild-type strain (Fig. 2A). Extracellular α-hemolysin production was restored to wild-type levels in S. aureus WCUH29 Δfmt(pYH4-fmt), where the fmt gene was supplied in trans. Production of α-hemolysin was similarly reduced in S. aureus FMT mutants from two additional strains tested, 90265/97 and PK1.
Fig 2.
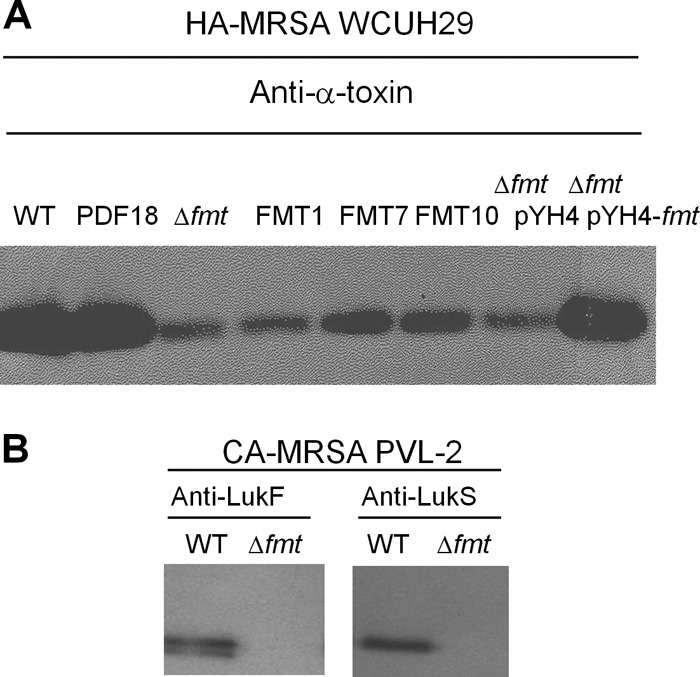
Reduction in alpha-toxin or PVL production in S. aureus FMT mutants. Culture supernatants from S. aureus WCUH29 strains grown in TSB or S. aureus PVL-2 strains grown in MH broth were fractionated by SDS-PAGE and subjected to Western immunoblotting with the indicated antisera. (A) Production of alpha-toxin in S. aureus WCUH29 strains. (B) Production of PVL subunits in S. aureus PVL-2 strains.
Lack of transformylase affected the production of additional virulence factors, with reductions in total extracellular proteins of at least 50% observed in supernatants of cultures from the different S. aureus WCUH29 FMT mutants. Separation of the proteins by SDS-PAGE showed a specific pattern, identical in all mutants, with substantial reductions seen in the production of at least six major exoproteins (Fig. 3A).
Fig 3.
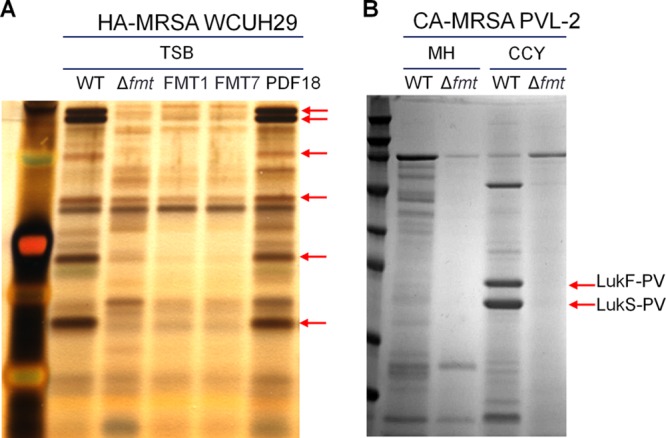
Reduction of extracellular protein production in S. aureus FMT-null mutants. Culture supernatants from S. aureus WCUH29 strains grown in TSB or S. aureus PVL-2 strains grown in MH broth or CCY medium were fractionated by SDS-PAGE, followed by silver staining (A) or SimplyBlue Safestain (B) for visualization.
As CA-MRSA infections in the United States are predominantly caused by the USA300 clone, which secretes PVL toxin, an fmt deletion mutant of a USA300 isolate was constructed. As expected, S. aureus PVL-2 Δfmt was nonhemolytic on sheep and rabbit blood agar plates, and its extracellular protein production was also severely impaired in different testing media (Fig. 3B). Specifically, production of the PVL subunits, LukS-PV and LukF-PV, could not be detected in S. aureus PVL-2 Δfmt when the strain was grown on CCY, a medium known to maximize extracellular production of PVL (33). This was further confirmed by Western blotting of PVL production in S. aureus PVL-2 Δfmt (Fig. 2B).
While diagnostic kits used clinically demonstrate that S. aureus WCUH29 FMT mutants are coagulase positive, like the parent strain, a qualitative evaluation of production of coagulase in culture supernatants and clumping factor in whole cells (the same strains indicated in Fig. 2A) showed that both activities were reduced in FMT mutants. Production could be restored to wild-type levels when the fmt gene was supplied in trans.
Finally, susceptibility testing of a representative set of S. aureus WCUH29 FMT mutants showed no cross-resistance with other marketed antibiotics. These mutants were 8- to 16-fold more susceptible to antibiotics that interact with 50S ribosomal subunits, such as macrolides, ketolides, lincosamides, streptogramins, and pleuromutilins, than the wild-type parent strain (Table 2). Interestingly, S. aureus FMT mutants could not be isolated when 1/4 MIC of erythromycin was added to a PDF inhibitor with an inoculum of 1.4 × 109 CFU of S. aureus WCUH29, indicating that subinhibitory concentrations of erythromycin can prevent spontaneous resistance development mediated by FMT mutation. This further suggests that all mutants with loss-of-function mutations in FMT are more susceptible to erythromycin regardless of their genotype.
Table 2.
S. aureus FMT-null mutations result in hypersensitivity to some 50S ribosomal inhibitors
| Antibiotic compound | Class | MICa (μg/ml) for S. aureus WCUH29 strain: |
|||
|---|---|---|---|---|---|
| Wild type | Δfmt | FMT-8 | FMT-10 | ||
| Azithromycin | Macrolide | 1 | 0.125 | 0.125 | 0.125 |
| Erythromycin | Macrolide | 0.25 | 0.016 | 0.016 | 0.016 |
| Tylosin | Macrolide | 1 | 0.063 | 0.063 | 0.063 |
| Telithromycin | Ketolide | 0.063 | 0.004 | 0.004 | 0.004 |
| Lincomycin | Lincosamide | 1 | 0.125 | 0.125 | 0.125 |
| Virginiamycin | Streptogramin | 2 | 0.125 | 0.25 | 0.25 |
| Tiamulin | Pleuromutilin | 0.25 | 0.03 | 0.03 | 0.03 |
No significant MIC changes (<2-fold variation) were observed against S. aureus FMT mutants with the following antibiotics: amoxicillin, bacitracin, ceftazidime, chloramphenicol, ciprofloxacin, gentamicin, imipenem, kanamycin, linezolid, mupirocin, novobiocin, oxacillin, phosphomycin, piperacillin, rifampin, streptomycin, trimethoprim, and vancomycin
Animal survival and culture studies comparing S. aureus wild-type and FMT mutant strains in a murine hematogenous pyelonephritis infection model.
Given the reduction in virulence factor production observed in S. aureus FMT mutants, it was anticipated that their ability to cause an invasive infection would be impaired. In order to evaluate if S. aureus FMT mutants could produce a lethal infection, mice were inoculated intravenously with wild-type S. aureus WCUH29 (4.5 × 107 CFU/mouse) or representative FMT mutants (Table 1): Δfmt (1.6 × 107 to 2.1 × 107 CFU/mouse), FMT8 (2.1 × 107 to 2.3 × 107 CFU/mouse), and FMT10 (1.5 × 107 CFU/mouse). The animals (10/group) were followed for 14 days in two separate experiments. All animals infected with wild-type organisms succumbed to infection by day 7, whereas those infected with S. aureus Δfmt, FMT8, or FMT10 showed 100%, 90%, and 95% survival rates, respectively (Fig. 4A).
Fig 4.

S. aureus WCUH29 FMT mutants are attenuated in a murine pyelonephritis infection model. S. aureus WCUH29 wild type and FMT mutants, Δfmt, FMT8, and FMT10, were used in these studies. (A) Survival study combining two separate experiments, both with 10 animals/group. (B) Bacterial counts from kidneys of infected mice (5 animals/group) at different days postinfection. Kidneys from animals infected with WT organisms could not be cultured after day 3 due to death. The means ± standard deviations are indicated by the black lines.
In order to assess if the high survival rate observed with S. aureus FMT mutants was related to their inability to produce virulence factors or to impaired growth in the animals, the bacterial load present in the kidneys of mice infected with S. aureus WT or FMT mutant strains was enumerated at days 3, 7, 14, 21, and 28 postinfection (Fig. 4B). In this study, all mice infected with wild-type S. aureus died by day 7, while in the S. aureus FMT groups, only one animal, infected with S. aureus FMT10, died 10 days after infection. While similar bacterial counts were recovered from the kidneys of all animals infected with wild-type organisms, there were large variations in the bacterial loads recovered from the kidneys of animals infected with FMT mutant strains, with counts from animals in the same group differing by more than 103 CFU/kidney in many cases. Despite the variability, a very clear trend was observed in all groups, with mean kidney bacterial counts peaking between day 3 and day 7 and then decreasing over time. In fact, bacteria could not be detected in the kidneys of several animals infected with S. aureus Δfmt (2 animals each at days 21 and 28), FMT8 (3 animals at day 28), or FMT10 (1 animal at day 14).
Similar results were obtained in an additional experiment in which enumeration of bacteria from animals infected with these strains took place from day 28 to day 49 postinfection. In that study, all mice infected with wild-type S. aureus had succumbed to infection by day 8, while only one animal from the groups infected with S. aureus FMT mutants died by day 5, with all remaining animals surviving to day 49. In addition, kidney bacterial loads continued to decrease over time, and 60% of the mice had counts below the limit of detection at day 49.
While S. aureus FMT mutants were clearly capable of growing in the animals, albeit at a lower rate than wild-type organisms, they were substantially less pathogenic than the parent strain. In order to determine if the FMT isolates became more virulent after animal passage, an S. aureus Δfmt colony isolated from the 21-day-postinfection kidney homogenate was used to infect 10 animals, and survival was followed for 14 days. The survival rate was 100%.
Animal survival and culture studies comparing S. aureus wild-type and FMT mutant strains in a neutropenic murine hematogenous pyelonephritis infection model.
As immunocompromised animals are more vulnerable to infection, a neutropenic murine hematogenous pyelonephritis infection model that mimicked the conditions of the immunocompetent model previously used was developed. In this model, neutropenia was maintained for the duration of the study. The inoculum of the wild-type strain was adjusted to produce a survival curve similar to that obtained in the nonneutropenic model, and therefore, studies were performed with an inoculum of 103 CFU/mouse.
An initial study involving 20 mice/group showed 0% survival of those animals infected with wild-type S. aureus WCUH29 by day 8 postinfection (Fig. 5A). However, mice infected with S. aureus Δfmt or FMT1 displayed 100% and 90% survival on day 14, respectively (Fig. 5A). These results are almost identical to those obtained in the nonneutropenic model.
Fig 5.
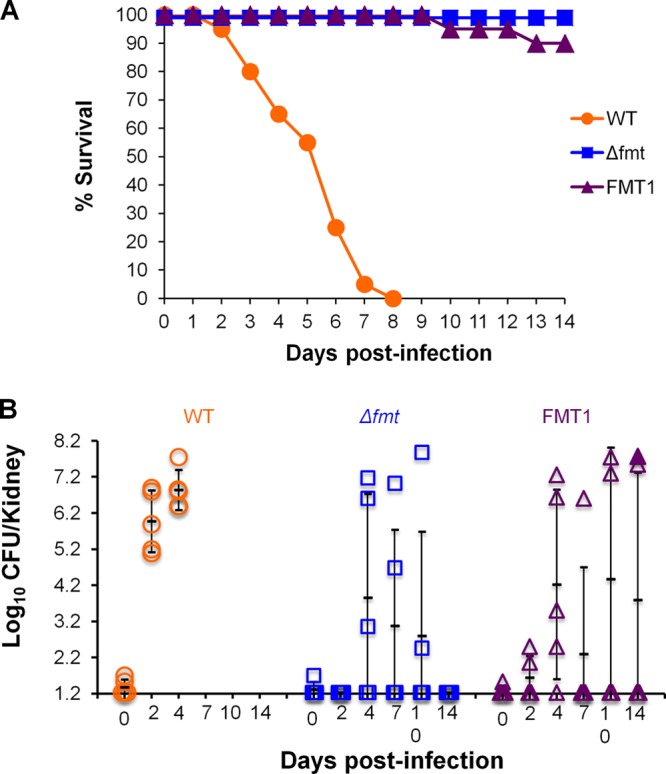
S. aureus WCUH29 FMT mutants are attenuated in a neutropenic murine pyelonephritis infection model. S. aureus WCUH29 wild type and FMT mutants, Δfmt and FMT1, were used in these studies. (A) Survival study, with 20 animals/group. (B) Bacterial counts from kidneys of infected mice (5 animals/group) at different days postinfection. Bacterial counts from the kidneys of one dead animal (FMT1; day 14) are marked with a filled triangle. Kidneys from animals infected with WT organisms could not be cultured after day 4 due to death. The means ± standard deviations are indicated by the black lines.
The bacterial load present in the kidneys of these animals was determined in an additional study (5 mice/group) that included bacterial enumeration at days 0, 2, 4, 7, 10, and 14 postinfection. Similar to the survival study, infection with the wild-type strain caused death of all animals by day 7, whereas no deaths occurred in the S. aureus Δfmt group and 2 animals died (days 6 and 14) in the S. aureus FMT1 group. Extremely large variations were observed in kidney bacterial counts of animals inoculated with FMT mutant strains (Fig. 5B). No bacteria were recovered from mice infected with S. aureus Δfmt at day 2, a maximum bacterial load was obtained at day 4, and counts decreased at days 7 and 10, reaching undetectable levels at day 14. There were differences of approximately 105 CFU/kidney from mice within the same group, and in all cases, there were animals with bacterial levels below the limit of detection (2/5 mice at day 4 and 3/5 mice at days 7 and 10). A similar trend was observed in the animals infected with S. aureus FMT1. At days 7, 10, and 14, bacteria were undetectable in ≥50% of the mice (4/5, 2/4, and 3/4 animals, respectively). As observed in the nonneutropenic-animal model, mice infected with wild-type S. aureus WCUH29 had similar bacterial counts in all groups enumerated.
DISCUSSION
Resistance is expected to occur with any new antibacterial agent that is used clinically. The frequency and level of resistance are parameters that can be readily determined in vitro for any novel antibiotic. However, the rate at which resistance emerges once a compound is in the clinic will be determined by the controlled use of the antibiotic, the cost that the mutation inflicts on the fitness of the bacteria, and the ability of the bacteria to compensate for that cost while maintaining resistance.
As inactivation of FMT is the predominant cause of resistance to PDF inhibitors in certain pathogens, FMT mutants have been the focus of research by several groups assessing the validity of PDF as an antibacterial target (3, 4, 17). In our study, S. aureus FMT mutants were isolated at frequencies of 5 × 10−7 and showed, as previously described (3), a substantial reduction in in vitro growth rates, most likely due to the inefficient initiation of protein synthesis when unformylated methionyl-tRNAi is used (18). This slower initiation process may also account for the observation that these mutants are 8 to 16 times more susceptible than the wild-type strain to some protein synthesis inhibitors that bind to the 50S ribosomal subunit, such as macrolides, lincosamides, or pleuromutilins, while remaining equally susceptible to other antibacterial agents. Furthermore, we have discovered that loss-of-function mutations in S. aureus FMT have additional consequences, as FMT mutants are nonhemolytic and show a 50% reduction in total exoprotein production. Examination of some specific extracellular proteins revealed that key virulence factors associated with S. aureus pathogenicity were particularly impacted. In fact, Western immunoblotting analysis demonstrated a >95% reduction in alpha-toxin production in FMT mutants, as well as the elimination of PVL production in a CA-MRSA strain. These results are consistent with a slower translation process, as it has been demonstrated that protein synthesis inhibitors can reduce the synthesis of virulence factors even at concentrations that have no effect on bacterial growth (34, 35).
Although the molecular mechanisms that regulate the transition of S. aureus from a commensal bacterium to an invasive pathogen are not well understood, it is clear that production of a wide repertoire of virulence factors that allow S. aureus to attach to cells, degrade tissues, disseminate, and evade host defenses plays a critical role in its pathogenicity (36). In fact, alpha-toxin has been reported as a major cause of death in mice injected with S. aureus intraperitoneally (37) and in a murine model of S. aureus pneumonia (38, 39) and has also been described as a key virulence determinant in S. aureus brain abscess (40) and septic arthritis infections (41). In addition, deletion of agrA, a global regulatory gene responsible for the expression of most exoproteins, including all hemolysins and leukocidins (42), has an even more dramatic effect on the reduction of S. aureus pathogenicity (38, 43, 44). Due to the pronounced decrease in exotoxin production observed in S. aureus FMT mutants, we investigated their ability to establish an infection in a murine hematogenous pyelonephritis model. All mice inoculated with the S. aureus wild-type strain died by day 7 postinfection, whereas those infected with S. aureus FMT mutants showed a dramatic reduction in lethality, with 90 to 100% survival rates 14 days postinfection. This extreme difference in survival rates is consistent with the impaired exoprotein/virulence factor production associated with FMT mutations.
Analysis of the bacterial load in the kidneys of infected mice revealed that S. aureus FMT mutants could initially grow in vivo, although not at the same rate as wild-type organisms, and that kidney bacterial counts decreased over time. In fact, 20% of the animals infected with S. aureus FMT mutants did not have any detectable bacteria in their kidneys at either 14, 21, or 28 days postinfection. This could be due to the inability of the mutants to establish an invasive infection or to the ability of the immune system to eradicate them. As neutropenic animals are more susceptible to bacterial infections, we established a pyelonephritis animal model where neutropenia was maintained for the duration of the study (14 days). As seen in the immunocompetent animals, ≥90% of the mice infected with S. aureus FMT mutants survived while none of the animals infected with wild-type S. aureus lived beyond 8 days postinfection. In a culture study, S. aureus FMT mutants were capable of growing in some of the neutropenic animals but were clearly less pathogenic than the wild-type strain. Furthermore, bacteria could not be detected in the kidneys of two-thirds of the animals infected with FMT mutants 7 days after infection. The fact that similar results are observed in neutropenic and nonneutropenic animals suggests that the immune response is not the primary mechanism involved in the elimination of S. aureus FMT mutants.
The decreased pathogenicity demonstrated by S. aureus FMT mutants may be further accentuated in the presence of wild-type bacteria, since competition experiments performed in mice with S. enterica wild-type and FMT mutants showed that 2 days after injecting 105 CFU of both organisms, FMT mutants could be recovered at a frequency of only 5 × 10−5 to 4 × 10−6 (14). In addition, although mutations that partially compensated for the growth of S. enterica FMT mutants in vitro could be found after 50 to 150 serial passages (14, 45), these compensated mutants did not show improved growth rates in mice (14).
Our results agree with those of Margolis et al. (3), who reported the attenuation of S. aureus FMT mutants in a 4-day thigh abscess model of infection. In contrast, Zorzet et al. (46) and Gjertsson et al. (47) did not observe differences in the growth of S. aureus FMT mutants with respect to the wild-type strain in their animal models. These discrepancies are likely explained by the different experimental designs. The study duration used by Zorzet et al. (46) is too short, as we have demonstrated that periods longer than 3 days are necessary to observe differences between S. aureus WT and FMT mutant strains. Gjertsson et al. (47) used a wild-type S. aureus strain less virulent than WCUH29, which did not cause death after 16 days, making it difficult to compare the pathogenicities of S. aureus WT and FMT mutant strains.
Finally, over 50 in vivo efficacy studies have been performed in our laboratories, where S. aureus abscess groin infections (using an inoculum of 107 CFU/mouse) have been treated with efficacious doses of various PDF inhibitors twice daily for 4 days. At the end of every efficacy study, the antimicrobial susceptibilities of a number of isolates from randomly selected animals were tested, and FMT mutants were never detected. This indicates that the eradication of wild-type S. aureus by treatment with PDF inhibitors does not promote growth of S. aureus FMT mutants in animal models. This is in contrast to the in vitro situation, where breakthrough growth of FMT mutants is always observed after 24 h in cell-killing studies with PDF inhibitors using an inoculum of ∼106 CFU/ml. The different behaviors of these mutants in vitro versus in vivo further support the notion that lack of virulence, and not reduced growth, is primarily responsible for their compromised ability to cause productive infection in an animal.
In summary, this study shows that, although S. aureus FMT mutants can be isolated at high frequency in vitro, inactivation of FMT leads to a reduced growth rate both in vitro and in mice. More importantly, S. aureus FMT mutants show a significant decrease in production of virulence factors that translates into a substantial loss of pathogenicity in animal models. Therefore, it is anticipated that these mutants will have difficulty producing an invasive infection in humans.
ACKNOWLEDGMENTS
We thank Michelle Fuller and Janice Shih for their excellent technical assistance.
Footnotes
Published ahead of print 9 April 2013
REFERENCES
- 1. Mazel D, Pochet S, Marliere P. 1994. Genetic characterization of polypeptide deformylase, a distinctive enzyme of eubacterial translation. EMBO J. 13:914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meinnel T, Blanquet S. 1994. Characterization of the Thermus thermophilus locus encoding peptide deformylase and methionyl-tRNA(fMet) formyltransferase. J. Bacteriol. 176:7387–7390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Margolis PS, Hackbarth CJ, Young DC, Wang W, Chen D, Yuan Z, White R, Trias J. 2000. Peptide deformylase in Staphylococcus aureus: resistance to inhibition is mediated by mutations in the formyltransferase gene. Antimicrob. Agents Chemother. 44:1825–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Margolis P, Hackbarth C, Lopez S, Maniar M, Wang W, Yuan Z, White R, Trias J. 2001. Resistance of Streptococcus pneumoniae to deformylase inhibitors is due to mutations in defB. Antimicrob. Agents Chemother. 45:2432–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Solbiati J, Chapman-Smith A, Miller JL, Miller CG, Cronan JE., Jr 1999. Processing of the N termini of nascent polypeptide chains requires deformylation prior to methionine removal. J. Mol. Biol. 290:607–614 [DOI] [PubMed] [Google Scholar]
- 6. Giglione C, Pierre M, Meinnel T. 2000. Peptide deformylase as a target for new generation, broad spectrum antimicrobial agents. Mol. Microbiol. 36:1197–1205 [DOI] [PubMed] [Google Scholar]
- 7. Gordon JJ, Kelly BK, Miller GA. 1962. Actinonin: an antibiotic substance produced by an actinomycete. Nature 195:701–702 [DOI] [PubMed] [Google Scholar]
- 8. Chen DZ, Patel DV, Hackbarth CJ, Wang W, Dreyer G, Young DC, Margolis PS, Wu C, Ni ZJ, Trias J, White RJ, Yuan Z. 2000. Actinonin, a naturally occurring antibacterial agent, is a potent deformylase inhibitor. Biochemistry 39:1256–1262 [DOI] [PubMed] [Google Scholar]
- 9. Apfel C, Banner DW, Bur D, Dietz M, Hirata T, Hubschwerlen C, Locher H, Page MG, Pirson W, Rosse G, Specklin JL. 2000. Hydroxamic acid derivatives as potent peptide deformylase inhibitors and antibacterial agents. J. Med. Chem. 43:2324–2331 [DOI] [PubMed] [Google Scholar]
- 10. Aubart K, Zalacain M. 2006. Peptide deformylase inhibitors. Prog. Med. Chem. 44:109–143 [DOI] [PubMed] [Google Scholar]
- 11. Jain R, Chen D, White RJ, Patel DV, Yuan Z. 2005. Bacterial Peptide deformylase inhibitors: a new class of antibacterial agents. Curr. Med. Chem. 12:1607–1621 [DOI] [PubMed] [Google Scholar]
- 12. Ross JE, Scangarella-Oman NE, Miller LA, Sader HS, Jones RN. 2011. Determination of disk diffusion and MIC quality control ranges for GSK1322322, a novel peptide deformylase inhibitor. J. Clin. Microbiol. 49:3928–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duroc Y, Giglione C, Meinnel T. 2009. Mutations in three distinct loci cause resistance to peptide deformylase inhibitors in Bacillus subtilis. Antimicrob. Agents Chemother. 53:1673–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nilsson AI, Zorzet A, Kanth A, Dahlstrom S, Berg OG, Andersson DI. 2006. Reducing the fitness cost of antibiotic resistance by amplification of initiator tRNA genes. Proc. Natl. Acad. Sci. U. S. A. 103:6976–6981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clements JM, Beckett RP, Brown A, Catlin G, Lobell M, Palan S, Thomas W, Whittaker M, Wood S, Salama S, Baker PJ, Rodgers HF, Barynin V, Rice DW, Hunter MG. 2001. Antibiotic activity and characterization of BB-3497, a novel peptide deformylase inhibitor. Antimicrob. Agents Chemother. 45:563–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Newton DT, Creuzenet C, Mangroo D. 1999. Formylation is not essential for initiation of protein synthesis in all eubacteria. J. Biol. Chem. 274:22143–22146 [DOI] [PubMed] [Google Scholar]
- 17. Apfel CM, Locher H, Evers S, Takacs B, Hubschwerlen C, Pirson W, Page MG, Keck W. 2001. Peptide deformylase as an antibacterial drug target: target validation and resistance development. Antimicrob. Agents Chemother. 45:1058–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guillon JM, Mechulam Y, Schmitter JM, Blanquet S, Fayat G. 1992. Disruption of the gene for Met-tRNA(fMet) formyltransferase severely impairs growth of Escherichia coli. J. Bacteriol. 174:4294–4301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zalacain M, Biswas S, Ingraham KA, Ambrad J, Bryant A, Chalker AF, Iordanescu S, Fan J, Fan F, Lunsford RD, O'Dwyer K, Palmer LM, So C, Sylvester D, Volker C, Warren P, McDevitt D, Brown JR, Holmes DJ, Burnham MK. 2003. A global approach to identify novel broad-spectrum antibacterial targets among proteins of unknown function. J. Mol. Microbiol. Biotechnol. 6:109–126 [DOI] [PubMed] [Google Scholar]
- 20. Tenover FC, McDougal LK, Goering RV, Killgore G, Projan SJ, Patel JB, Dunman PM. 2006. Characterization of a strain of community-associated methicillin-resistant Staphylococcus aureus widely disseminated in the United States. J. Clin. Microbiol. 44:108–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Labandeira-Rey M, Couzon F, Boisset S, Brown EL, Bes M, Benito Y, Barbu EM, Vazquez V, Hook M, Etienne J, Vandenesch F, Bowden MG. 2007. Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumonia. Science 315:1130–1133 [DOI] [PubMed] [Google Scholar]
- 22. Clinical and Laboratory Standards Institute 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 8th ed Approved standard M07-A8. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 23. Fan F, Lunsford RD, Sylvester D, Fan J, Celesnik H, Iordanescu S, Rosenberg M, McDevitt D. 2001. Regulated ectopic expression and allelic-replacement mutagenesis as a method for gene essentiality testing in Staphylococcus aureus. Plasmid 46:71–75 [DOI] [PubMed] [Google Scholar]
- 24. Arnaud M, Chastanet A, Debarbouille M. 2004. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70:6887–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang J, O'Toole PW, Shen W, Amrine-Madsen H, Jiang X, Lobo N, Palmer LM, Voelker L, Fan F, Gwynn MN, McDevitt D. 2004. Novel chromosomally encoded multidrug efflux transporter MdeA in Staphylococcus aureus. Antimicrob. Agents Chemother. 48:909–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ji Y, Yin D, Fox B, Holmes DJ, Payne D, Rosenberg M. 2004. Validation of antibacterial mechanism of action using regulated antisense RNA expression in Staphylococcus aureus. FEMS Microbiol. Lett. 231:177–184 [DOI] [PubMed] [Google Scholar]
- 27. Reference deleted.
- 28. Zhang L, Fan F, Palmer LM, Lonetto MA, Petit C, Voelker LL, St John A, Bankosky B, Rosenberg M, McDevitt D. 2000. Regulated gene expression in Staphylococcus aureus for identifying conditional lethal phenotypes and antibiotic mode of action. Gene 255:297–305 [DOI] [PubMed] [Google Scholar]
- 29. Du W, Brown JR, Sylvester DR, Huang J, Chalker AF, So CY, Holmes DJ, Payne DJ, Wallis NG. 2000. Two active forms of UDP-N-acetylglucosamine enolpyruvyl transferase in gram-positive bacteria. J. Bacteriol. 182:4146–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ji Y, Marra A, Rosenberg M, Woodnutt G. 1999. Regulated antisense RNA eliminates alpha-toxin virulence in Staphylococcus aureus infection. J. Bacteriol. 181:6585–6590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schmitt E, Blanquet S, Mechulam Y. 1996. Structure of crystalline Escherichia coli methionyl-tRNA (f) Met formyltransferase: comparison with glycinamide ribonucleotide formyltransferase. EMBO J. 15:4749–4758 [PMC free article] [PubMed] [Google Scholar]
- 32. Dinges MM, Orwin PM, Schlievert PM. 2000. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 13:16–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Woodin AM. 1959. Fractionation of a leucocidin from Staphylococcus aureus. Biochem. J. 73:225–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohlsen K, Ziebuhr W, Koller KP, Hell W, Wichelhaus TA, Hacker J. 1998. Effects of subinhibitory concentrations of antibiotics on alpha-toxin (hla) gene expression of methicillin-sensitive and methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 42:2817–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gemmell CG, Ford CW. 2002. Virulence factor expression by Gram-positive cocci exposed to subinhibitory concentrations of linezolid. J. Antimicrob. Chemother. 50:665–672 [DOI] [PubMed] [Google Scholar]
- 36. Archer GL. 1998. Staphylococcus aureus: a well-armed pathogen. Clin. Infect. Dis. 26:1179–1181 [DOI] [PubMed] [Google Scholar]
- 37. Patel AH, Nowlan P, Weavers ED, Foster T. 1987. Virulence of protein A-deficient and alpha-toxin-deficient mutants of Staphylococcus aureus isolated by allele replacement. Infect. Immun. 55:3103–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bubeck Wardenburg J, Patel RJ, Schneewind O. 2007. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 75:1040–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bartlett AH, Foster TJ, Hayashida A, Park PW. 2008. Alpha-toxin facilitates the generation of CXC chemokine gradients and stimulates neutrophil homing in Staphylococcus aureus pneumonia. J. Infect. Dis. 198:1529–1535 [DOI] [PubMed] [Google Scholar]
- 40. Kielian T, Cheung A, Hickey WF. 2001. Diminished virulence of an alpha-toxin mutant of Staphylococcus aureus in experimental brain abscesses. Infect. Immun. 69:6902–6911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nilsson IM, Hartford O, Foster T, Tarkowski A. 1999. Alpha-toxin and gamma-toxin jointly promote Staphylococcus aureus virulence in murine septic arthritis. Infect. Immun. 67:1045–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Novick RP. 2003. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 48:1429–1449 [DOI] [PubMed] [Google Scholar]
- 43. Abdelnour A, Arvidson S, Bremell T, Ryden C, Tarkowski A. 1993. The accessory gene regulator (agr) controls Staphylococcus aureus virulence in a murine arthritis model. Infect. Immun. 61:3879–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect. Immun. 63:3373–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zorzet A, Pavlov MY, Nilsson AI, Ehrenberg M, Andersson DI. 2010. Error-prone initiation factor 2 mutations reduce the fitness cost of antibiotic resistance. Mol. Microbiol. 75:1299–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zorzet A, Andersen JM, Nilsson AI, Moller NF, Andersson DI. 2012. Compensatory mutations in agrC partly restore fitness in vitro to peptide deformylase inhibitor-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 67:1835–1842 [DOI] [PubMed] [Google Scholar]
- 47. Gjertsson I, Jonsson IM, Peschel A, Tarkowski A, Lindholm C. 2012. Formylated peptides are important virulence factors in Staphylococcus aureus arthritis in mice. J. Infect. Dis. 205:305–311 [DOI] [PubMed] [Google Scholar]