

Abstract
Increased expression of chromosomal genes for resistance-nodulation-cell division (RND)-type efflux systems plays a major role in the multidrug resistance (MDR) of Acinetobacter baumannii. However, the relative contributions of the three most prevalent pumps, AdeABC, AdeFGH, and AdeIJK, have not been evaluated in clinical settings. We have screened 14 MDR clinical isolates shown to be distinct on the basis of multilocus sequence typing (MLST) and pulsed-field gel electrophoresis (PFGE) for the presence and overexpression of the three Ade efflux systems and analyzed the sequences of the regulators AdeRS, a two-component system, for AdeABC and AdeL, a LysR-type regulator, for AdeFGH. Gene adeB was detected in 13 of 14 isolates, and adeG and the intrinsic adeJ gene were detected in all strains. Significant overexpression of adeB was observed in 10 strains, whereas only 7 had moderately increased levels of expression of AdeFGH, and none overexpressed AdeIJK. Thirteen strains had reduced susceptibility to tigecycline, but there was no correlation between tigecycline MICs and the levels of AdeABC expression, suggesting the presence of other mechanisms for tigecycline resistance. No mutations were found in the highly conserved LysR regulator of the nine strains expressing AdeFGH. In contrast, functional mutations were found in conserved domains of AdeRS in all the strains that overexpressed AdeABC with two mutational hot spots, one in AdeS near histidine 149 suggesting convergent evolution and the other in the DNA binding domain of AdeR compatible with horizontal gene transfer. This report outlines the high incidence of AdeABC efflux pump overexpression in MDR A. baumannii as a result of a variety of single mutations in the corresponding two-component regulatory system.
INTRODUCTION
Acinetobacter baumannii is a ubiquitous nonfermentative Gram-negative bacterial species responsible for epidemics of nosocomial infections (1). This opportunistic pathogen is often resistant to a wide range of antimicrobials, which renders Acinetobacter infections difficult to eradicate (2). Resistance can be intrinsic, because of production of a chromosomally encoded cephalosporinase and of low membrane permeability, or acquired, following transfer of foreign genetic information or mutation in endogenous structural or regulatory genes. Of particular clinical importance are mutational events in the regulators of efflux systems which, in a single step, can confer to the host multidrug resistance (MDR) by overexpression of the pump.
The pumps of the resistance-nodulation-cell division (RND) superfamily are ubiquitous in Gram-negative bacteria and have the broadest substrate ranges (3). To date, overexpression of three RND systems, AdeABC (4), AdeFGH (5), and AdeIJK (6), has been associated with MDR in A. baumannii (7). The first pump described, AdeABC, confers resistance to aminoglycosides, tetracyclines, fluoroquinolones, chloramphenicol, and trimethoprim (4) and reduced susceptibility to tigecycline (8, 9, 10). The AdeFGH system has chloramphenicol, clindamycin, fluoroquinolones, trimethoprim, tetracyclines, tigecycline, and sulfonamide as substrates (5). AdeIJK effluxes β-lactams, chloramphenicol, tetracyclines, erythromycin, fluoroquinolones, fusidic acid, novobiocin, and trimethoprim (6).
AdeABC, primarily, and AdeFGH play a major role in acquired resistance (7), whereas AdeIJK is responsible for intrinsic resistance (6). Expression of each pump is tightly regulated but by different mechanisms. Production of AdeABC is controlled by a two-component regulatory system, AdeRS (11), that of AdeFGH by the LysR-type transcriptional regulator AdeL (5), and that of AdeIJK by the TetR transcriptional regulator AdeN (12).
Mutations in AdeRS selected in vitro (11) or in vivo (8, 13, 14, 15) lead to constitutive expression of the AdeABC efflux system, and in vitro mutations in AdeL (5) and AdeN (12) result in overexpression of AdeFGH and AdeIJK, respectively. However, the occurrence of these mutations in clinical settings is not well documented. Most studies have dealt with AdeRS, since AdeABC overexpression has been shown to be involved in tigecycline efflux leading to reduced susceptibility to the drug but with contradictory results (8). In that report, sequencing of AdeRS of a susceptible isolate and of two tigecycline-resistant clinical isolates revealed several point mutations in AdeR or AdeS but none that could be specifically correlated to overexpression of the pump. In addition, after culture in vitro in the presence of tigecycline, the susceptible strain became resistant and overexpressed AdeABC but no mutations were found in AdeRS.
In contrast, in a series of A. baumannii isolates longitudinally collected from a single patient, the first tigecycline-resistant isolate with a >7-fold increase in adeB expression had a D20N substitution in AdeR (13). An A94V substitution in AdeS was found in an A. baumannii isolate from a patient receiving tigecycline therapy that resulted in 6-fold adeB overexpression and an increase in the tigecycline MIC from 0.5 to 16 μg/ml compared to the susceptible strain isolated previously from the same patient (14); however, because of the presence of additional single nucleotide polymorphisms (SNPs) between the two strains, the possibility of nonisogenic clinical isolates was raised (15). Lately, no AdeRS mutations were found in 13 unrelated MDR A. baumannii isolates with increased AdeABC expression and decreased tigecycline susceptibility (16).
We have screened 14 distinct MDR clinical isolates for overexpression of the three Ade efflux systems, determined the sequences of their respective regulators, and analyzed the contribution of mutations in the AdeRS two-component regulatory system to overexpression of AdeABC, the clinically most important pump (17).
MATERIALS AND METHODS
Bacterial strains.
A total of 14 MDR clinical isolates of A. baumannii collected in 2011 by the French National Reference Center for Antibiotic Resistance were studied. Genotyping by multilocus sequence typing (MLST) was performed as described previously (18). The sequences of amplified internal fragments of housekeeping genes cpn60, fusA, gltA, pyrG, recA, rplB, and rpoB were determined and compared with those in the A. baumannii MLST database of the Institut Pasteur (18). Cells were grown at 37°C in brain heart infusion broth and agar (Difco Laboratories, Detroit, MI). Antibiotic susceptibility was tested by disk diffusion on Mueller-Hinton agar (Bio-Rad, Marnes-la-Coquette, France). The MICs of tigecycline were determined by the Etest procedure (bioMérieux, Marcy l'Etoile, France) according to the manufacturer's recommendations and by microdilution using Mueller-Hinton II broth (Difco Laboratories).
Detection of the Ade RND systems.
Efflux pumps AdeABC, AdeFGH, and AdeIJK were screened for by PCR. A. baumannii genomic DNA extracted as described previously (19) was amplified in a GeneAmp PCR 9700 system (Perkin-Elmer Cetus, Norwalk, CT) with Taq DNA polymerase (MPbio, Illkirch, France) and previously designed primers specific for adeA, adeB, adeC, adeJ (20), adeF, adeG, and adeH (5).
RNA isolation and qRT-PCR.
A. baumannii total RNA was extracted from exponentially grown bacterial cells in antibiotic-free media (optical density at 600 nm of 0.8 to 0.9) using TRIzol reagent (Invitrogen, Carlsbad, CA). RNA samples were treated with a Turbo DNA-free kit (Applied Biosystems, Carlsbad, CA) to remove any genomic DNA carryover from RNA extraction. Expression of genes adeB, adeG, and adeJ was quantified by quantitative reverse transcriptase PCR (qRT-PCR) as described previously (12) using a LightCycler RNA amplification kit and SYBR green I (Roche Diagnostic GmbH, Mannheim, Germany) with the following cycle profile: 1 cycle at 95°C for 30 s followed by 45 cycles of 95°C for 5 s, 56°C for 10 s, and 72°C for 20 s. The expression level of the rpoB gene of A. baumannii BM4587 was used as the control with previously designed primers (20) for the genes, except those for adeG, which were as follows: forward, 5′-TTCATCTAGCCAAGCAGAAG-3′; reverse, 5′-CCTGCTAATGGTAGGGTTAAG-3′. Each experiment was performed in duplicate at least twice independently.
Nucleotide sequencing and data analysis.
Primer pair adeRS-Forward (5′-GGAGTAAGTGTGGAGAAATACGGA-3′) and adeRS-Reverse (5′-GAGAGTGAAGGATCACTTTAACTCTAAG-3′) and primer pair adeRS-m-Forward (5′-CATGATTGACCAACCCATAAAGTTTTTAC-3′) and adeRS-m-Reverse (5′-CAATAATTCCCTGTAAACGACCTTGTAA-3′) were designed to amplify the region upstream from the regulatory genes adeRS and to determine the sequence. For the adeL gene, the previously designed primer set L1 and L2 was used (5). Amplification was performed with Phusion High-Fidelity DNA polymerase (Finnzymes, Espoo, Finland) according to the manufacturer's recommendation, the PCR products were purified with a QIAQuick PCR purification kit (Qiagen, Inc., Chatsworth, CA), and sequencing was carried out with a CEQ 8000 DNA automated sequencer (Beckman Coulter, Fullerton, CA). The sequence was analyzed with the Genetic Computer Group sequence analysis software package (version 10.1; Genetics Computer Group, Madison, WI). BLAST program searches were performed using the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov). Multiple-sequence alignment of the deduced peptide sequences was carried out using the program ClustalW at the European Bioinformatics Institute website (http://www.ebi.ac.uk).
RESULTS AND DISCUSSION
Genotypic diversity of A. baumannii clinical isolates.
Fourteen MDR A. baumannii isolates from our laboratory collection isolated in 2011 from different patients were selected on the basis of distinct geographical origins. They were all resistant to carbapenems by production of OXA-23, OXA-24, OXA-58, Ges-1, Ges-5, or PER β-lactamase (data not shown). Multilocus sequence typing (MLST) has a high potential to discriminate, besides the dominant sequence types (ST) of previously recognized international clones I to III, single-locus variants and novel clones. The ST of every isolate was determined by analysis of internal regions of seven housekeeping genes, and all the strains were found to belong to different STs (Table 1). The relationship among the various STs was disclosed using the minimum spanning tree (MStree) method of the Institut Pasteur (http://www.pasteur.fr/mlst). Four strains, BM4709 (ST1), BM4708 (ST7), BM4710 (ST20), and BM4711 (ST116), belonged to international clonal complex 1 (CC1), since ST7, ST20, and ST116 differ from ST1 by a single allelic mismatch. Strains BM4712 (ST2) and BM4713 (ST115) belonged to CC2; ST115 also differs from ST2 by a single mismatch. Regarding strains BM4701 (ST113), BM4702 (ST114), and BM4703 (ST25), ST114 and ST25 differ by one and two allelic changes from ST113, respectively, and therefore belong to related STs. ST112 (BM4705) differs by a single mutation from ST108 (BM4704). Remaining strains BM4706 (ST85), BM4707 (ST79), and BM4714 (ST107) belonged to STs which are remote from those of the other clones. All together, the 14 isolates were distinct from one another by MLST, a finding that was confirmed by pulsed field gel electrophoresis (data not shown). Although not extensively representative, this collection gathered clinical isolates belonging to the major epidemic clones reported recently (21).
Table 1.
Strains used in this study
| A. baumannii strain | Allelic profile | Sequence type | Clonal complex | Presence of Ade efflux pumpa |
Tigecycline MIC (μg/ml) |
|||
|---|---|---|---|---|---|---|---|---|
| AdeABC | AdeFGH | AdeIJK | Microdilution | Etest | ||||
| BM4701 | 3-3-3-4-7-4-4 | 113 | + | + | + | 0.5 | 0.75 | |
| BM4702 | 3-3-3-1-7-4-4 | 114 | + | + | + | 0.5 | 0.75 | |
| BM4703 | 3-3-2-4-7-2-4 | 25 | + | + | + | 0.5 | 0.19 | |
| BM4704 | 35-1-11-7-9-25-2 | 108 | + | + | + | 2 | 4 | |
| BM4705 | 35-36-11-7-9-25-2 | 112 | + | + | + | 0.25 | 0.19 | |
| BM4706 | 5-2-4-1-3-3-4 | 85 | + | + | + | 0.5 | 0.75 | |
| BM4707 | 26-2-2-2-29-4-5 | 79 | + | + | + | 0.5 | 2 | |
| BM4708 | 1-1-1-2-5-1-1 | 7 | CC1 | + | + | + | 2 | 12 |
| BM4709 | 1-1-1-1-5-1-1 | 1 | CC1 | + | + | + | 2 | 4 |
| BM4710 | 3-1-1-1-5-1-1 | 20 | CC1 | + | + | + | 1 | 4 |
| BM4711 | 1-1-1-4-5-1-1 | 116 | CC1 | + | + | + | 2 | 12 |
| BM4712 | 2-2-2-2-2-2-2 | 2 | CC2 | + | + | + | 1 | 2 |
| BM4713 | 2-2-2-1-2-2-2 | 115 | CC2 | + | + | + | 1 | 2 |
| BM4714 | 34-35-37-1-5-6-36 | 107 | − | + | + | 0.5 | 0.5 | |
The presence of the pumps was determined by PCR of the gene for the inner membrane protein.
Expression of the Ade efflux systems.
The presence of the AdeABC, AdeFGH, and AdeIJK RND systems was screened for by PCR. Since the inner membrane protein is an essential part of the tripartite efflux machinery, genes adeB, adeG, and adeJ were selected for amplification with specific primer pairs. Gene adeB was detected in 13 of 14 clinical isolates and adeG and intrinsic adeJ in all strains (Table 1). The absence of the adeABC operon in BM4714 was confirmed by lack of amplification of adeA or adeC.
(i) Expression levels of the Ade pumps.
The level of expression of structural genes adeB, adeG, and adeJ was measured by qRT-PCR. The mRNA of the constitutively expressed single-copy housekeeping gene rpoB was used as a control and the well-characterized susceptible clinical strain BM4587 as a reference (20).
Expression of the adeB gene was variable and depended on the strain. Ten isolates displayed levels of adeB expression that were ca. 20-fold higher than that of BM4587 (Fig. 1). Strain BM4702 showed the highest (ca. 80-fold-increased) expression. Four strains had elevated levels (between 40- and 50-fold increases), and six strains had moderate levels (around 20-fold increases), whereas three strains had low (from 2- to 5-fold) levels of overexpression.
Fig 1.
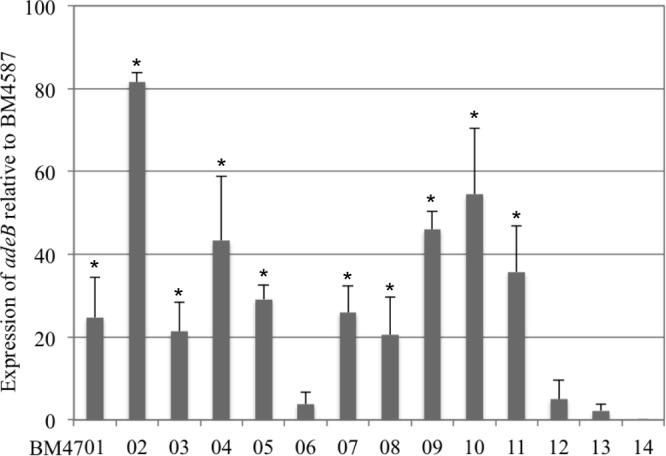
Expression of adeB. Gene expression relative to that of rpoB was determined by qRT-PCR. Results are presented relative to susceptible strain BM4587 taken as a reference. Each isolate was tested in duplicate or triplicate in two independent experiments. The bars represent the means and the error bars the standard deviations. The adeB gene was not detected in strain BM4714.
Eight of the 14 strains had 4- to 15-fold increases in adeG expression, and 6 had relative ranges of expression around 1 (data not shown). Recently, overexpression of this pump has been reported to be of clinical relevance in A. baumannii: among 11 isolates collected in Canada, all showed the presence of adeFGH, with increased expression of the pump in eight strains (22).
The relative range of adeJ expression was very narrow, ≤1-fold in six strains and between 1- and 2-fold in the remaining strains. These data (not shown) are consistent with the observation that overexpression of AdeIJK is lethal for the host (6).
(ii) Overexpression of Ade pumps and tigecycline resistance.
Overexpression of AdeABC has been shown to confer reduced susceptibility to tigecycline (2, 6, 10, 14, 15, 16). Thirteen of the 14 clinical isolates had a tigecycline MIC of ≥0.5 μg/ml (Table 1) and were considered resistant (23). However, in contrast to the report by Ruzin et al. (10), there was no obvious correlation between the MICs of tigecycline and the levels of expression of the adeABC operon (Fig. 1). Strains BM4709, BM4710, and BM4711 with adeB relative expression levels around 40 had tigecycline MICs of 1 or 2 μg/ml. In contrast, strains BM4712 and BM4713 with tigecycline MICs of 1 μg/ml displayed very low levels of adeB expression, and BM4702, with the higher level of adeB expression (80-fold), had a tigecycline MIC of 0.5 μg/ml similar to that of strain BM4714, which lacks the adeABC operon. In most previous reports, tigecycline MICs have been determined by Etest. However, a recent comparison of MIC determination methods indicated discrepancies between the techniques, with Etest providing higher values (24). We observed similar discrepancies with the 14 clinical strains studied (Table 1), but the differences did not alter the general observation of a lack of correlation between tigecycline MICs and AdeABC expression. Taken together, these observations support the hypothesis that other mechanisms can be responsible for tigecycline resistance.
Expression of efflux pumps can be induced by their substrates (3), but we did not observe any increase in tigecycline MIC or in AdeABC expression by qRT-PCR after growth of the cells in the presence of subinhibitory concentrations of the drug (data not shown).
Analysis of the regulators of the Ade systems.
Since we did not find any overexpression of the AdeIJK pump, only the adeL LysR-type regulator of adeFGH and the adeRS two-component system of adeABC were studied.
(i) Analysis of the AdeFGH regulator.
The LysR-type transcriptional regulator family is highly conserved and ubiquitous among bacteria (25). The AdeL sequences of the 14 clinical isolates were identical and were also identical to those of susceptible strains BM4587 (unpublished data), SDF (26), ATCC 17978 (27), AB307-0294 (27), and AB900 (27). In two strains, BM4708 and BM4709, a single Q256R polymorphism was found which was also present in MDR strain AYE, which does not overexpress adeG (data not shown). In addition, we did not find any mutations in the promoter region of the adeFGH operon of our collection of clinical isolates relative to prototype strain BM4587. Missense mutations AdeLQ326stop and AdeLV139G, which confer 300- and 200-fold increases in expression of adeG, respectively, have been reported (5). In the strains studied, we found less than 15.5-fold-increased expression of adeG and the expression rates in the majority of the strains were similar to that of the reference strain BM4587 (data not shown). These observations suggest the existence of an additional regulatory mechanism for overexpression of AdeFGH.
(ii) Involvement of AdeRS in overexpression of adeB.
In the two-component signaling systems, generally the membrane-associated sensor kinase autophosphorylates at an internal histidine in response to an environmental stimulus and the phosphate group is then transferred to an aspartate residue of the cytoplasmic response regulator (RR) which acts as a transcriptional activator. The phosphorylated regulator can also be dephosphorylated by the phosphatase activity of the sensor (28). The C-terminal portion of the sensor contains highly conserved sequences designated H, N, G1, F, and G2 boxes and three D boxes and one K box of the regulator (29) (Fig. 2 and Table 2). The AdeRS sequences of A. baumannii BM4587 (Fig. 2E), CIP70-10 (11), ATCC 17978 (27), AB307-0294 (27), AB900 (27), 36 (30), 38 (30), A24 (8), and A54 (8), which do not overexpress AdeABC, were used as references for sequence comparison and identification of SNPs that could have occurred in the clinical isolates.
Fig 2.

Mutations in AdeS and AdeR conferring AdeABC pump overexpression were mapped on structural models generated by homology modeling with the Swiss Model Server (34). (A and E) Conserved motifs (dark gray boxes) H, N, G1, F, and G2 in AdeS and D, D, D, and K in AdeR and the amino acid positions corresponding to the sequences presented in panel E are indicated. Mutations in clinical isolates (in black), previously described mutations (in green), and catalytic residues H149 in AdeS and D63 in AdeR (in red) are indicated. (B to D) The models are based on the following templates: solution structure of the histidine kinase, adenylyl cyclase, methyl-accepting protein, and phosphatase (HAMP) linker domain of the hypothetical transmembrane receptor Af1503 from Archaeoglobus fulgidus (35, 36) (Protein Data Bank identification no. [PDB ID] 2L7H) and the catalytic core (DHp and CA) of the histidine kinase HK853 from Thermotoga maritima (PDB ID 3DGE) (32) (B); structure of the complex between HK853 and its cognate response regulator RR468 (32) (C); and the complex between the Escherichia coli PhoB output domain and its target DNA sequence (PDB ID 1GXP) (37) (D). Mutated and catalytic residues, indicated with the same color code as described above, are illustrated in stick representations. TM, transmembrane domain; DHp, dimerization and histidine-containing phosphotransfer domain; CA, catalytic and ATP-binding domain; REC, receiver domain.
Table 2.
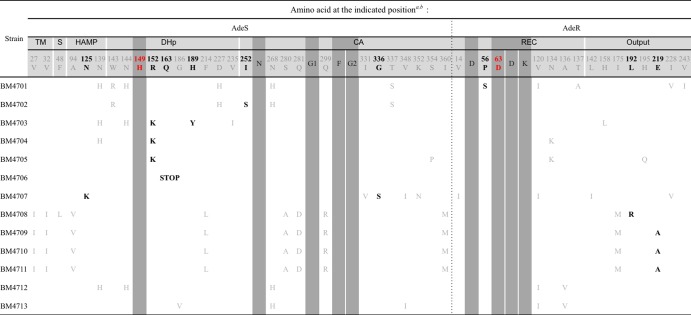
Amino acid substitutions are indicated using the one-letter code.
Conserved motifs are indicated by dark gray boxes, SNPs in gray lettering, and mutations putatively responsible for overexpression in bold characters.
To identify the putative amino acid substitutions affecting AdeABC expression, we first screened for the polymorphic sites in AdeRS. Since MLST can discriminate clonal types, we based our analysis of the AdeRS polymorphism on the STs of the strains. As mentioned above, BM4708 (ST7), BM4709 (ST1), BM4710 (ST20), and BM4711 (ST116) were found to belong to clonal complex CC1. Among the previously sequenced A. baumannii strains, susceptible strain AB307-0294 and MDR strain AYE also belong to CC1. We did not find any significant overexpression of the three Ade pumps in AYE (data not shown). Thus, the nine amino acid differences V27I, V32I, A94V, F214L, S280A, Q281D, Q299R, and I360M in AdeS and I175M in AdeR found in the CC1 reference strains and in BM4708, BM4709, BM4710, and BM4711 were considered SNPs characteristic of this clonal complex. Of note, among those, polymorphism A94V in the histidine kinase, adenylyl cyclase, methyl-accepting protein, and phosphatase (HAMP) linker domain has been erroneously reported as a mutation responsible for increased AdeABC expression (15).
We also found several polymorphisms characteristic of the other clonal complexes. Amino acid substitution N268H in AdeS of BM4701 (ST113) and BM4702 (ST114) and of two CC2 isolates, BM4712 and BM4713, was also present in ACICU, a CC2 MDR strain, and in susceptible strains BM4587, A24, and 38 of unknown STs and was therefore considered a polymorphism. Strains BM4701, BM4702, and BM4703 (ST25) belong to related STs and displayed the same W143R, D227H, and T337S polymorphisms in AdeS. In addition, strains BM4701 and BM4703 displayed AdeS polymorphisms N139H and N144H. These SNPs were also detected in BM4704 (ST108) and in BM4712 (ST2 of CC2), which does not overexpress AdeABC.
As already mentioned, AdeABC overexpression has been shown to be associated with specific point mutations in functional conserved domains of AdeRS. In AdeS, two alterations lead to increased expression of AdeABC: G103D in the HAMP linker between the sensor and the DHp domains (15) and T153M in the H box (11) (Fig. 2A). In AdeR, three substitutions resulting in overexpression of AdeABC have been reported, D20N in one of the three D boxes of the phosphorylation site (13), A91V located in the signal receiver domain (15), and P116L at the first residue of the helix α5 (11) required for the propagation of the phosphorylation-triggered signal (31). None of these point mutations were observed in the 13 clinical isolates studied.
We did not find any mutations in AdeRS of BM4712 and BM4713, and, as expected, the strains had only 5- and 2-fold-higher levels of adeB expression than the susceptible strain BM4587. In BM4706, with a 3.8-fold increase in adeB expression, the C487T nucleotide change led to a stop codon (Q163stop) near the autophosphorylation site in AdeS. Loss of AdeS kinase activity due to absence of the sensor has been reported to result in a decreased level of AdeABC expression (11). Surprisingly, constitutive overexpression of AdeABC observed in two clinical isolates was attributed to insertion of an ISABA-1 element in adeS in the absence of point mutations in genes adeR and adeS (10), but the site of insertion was not reported. We did not detect ISABA-1 in the vicinity of the adeABC operon in any of the 13 clinical isolates, but mutations which could be of functional importance have been found in AdeRS of the remaining 10 clinical isolates that overexpressed AdeABC.
Two-component signaling systems are relatively abundant in most eubacteria, usually playing important roles regulating housekeeping functions as well as pathogenic processes. Nevertheless, the structural data available on histidine kinases (HK), RR, and their complexes are relatively scarce. To get an insight into the functional relevance of the mutations observed, we mapped them on structures generated by homology modeling methods (Fig. 2B, C, and D). Surprisingly, the R152K substitution in AdeS was detected in the three overexpressing strains BM4703, BM4704, and BM4705, indicating a mutational hot spot compatible with convergent evolution of independent strains. Conserved arginine 152 is located in the helix α2 near phosphorylable histidine 149, one amino acid closer to the H box than the T153M substitution which also confers antibiotic resistance by overexpression of AdeABC (11). Its substitution by lysine could affect the interaction with either the catalytic domain or the receiver domain, which could account for overexpression. Of note, this mutation was also the only alteration in an entirely sequenced one-step in vitro mutant of BM4587 which overexpresses AdeABC (unpublished result).
Clinical isolate BM4707 harbored substitutions N125K and G336S in AdeS; substitution N125K in the HAMP linker domain might affect signal transmission.
BM4703 harbors mutation H189Y, a residue located at the C end of the DHp domain of the histidine kinase. Similar mutations in this domain lead to a phosphatase-defective phenotype without affecting RR phosphorylation or HK autokinase activity (32, 33). In BM4702, which had the highest AdeABC level of expression, mutation I252S in AdeS is adjacent to the N box in the catalytic domain. This substitution could affect ATP binding.
Three mutations occurred in AdeR. In BM4701, the P56S change was located near the phosphorylation site; in BM4708, the L192R substitution was in the effector domain of the regulator. Both mutations could alter protein stability. Finally, the substitution E219A in the DNA binding domain of AdeR in strains BM4709, BM4710, and BM4711 could be considered a second mutational hot spot. The observation that this functional substitution is associated, in the three distinct clinical isolates, with five SNPs in AdeS and one in AdeR (Table 2) strongly suggests horizontal transfer of the two-component regulatory system, possibly by transformation. Glutamate 219 is located at the positively charged surface facing the DNA helix, and its change to alanine is likely to interfere with DNA binding. Further structural and biochemical studies are needed to unravel the molecular mechanism of this two-component regulatory system.
Altogether, the study of a group of MDR A. baumannii isolates collected in clinical settings in 2011 indicated that AdeABC is the major efflux system implicated in resistance. Overexpression of the pump, known to lead, in one-step mutants, to increased resistance to several antibiotics of choice for the treatment of A. baumannii infections (i.e., aminoglycosides, fluoroquinolones, and carbapenems), results from a variety of mutations in the two-component system AdeRS. These in vivo-acquired regulatory mutations were mapped in a homology-generated model and should contribute to a better understanding of the mode of action of this two-component system.
ACKNOWLEDGMENTS
We thank K. Jeannot for selection of the strains, F. Depardieu and B. Perichon for helpful discussions, S. Brisse for reading of the manuscript, and D. Meziane-Cherif and A. Mechaly for help in the analysis of the mutations.
This work was supported in part by the European Union FP7-PAR grant that included a fellowship in support of E.-J.Y. and by an unrestricted grant from Pfizer.
Footnotes
Published ahead of print 15 April 2013
REFERENCES
- 1. Dijkshoorn L, Nemec A, Seifert H. 2007. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat. Rev. Microbiol. 5:939–951 [DOI] [PubMed] [Google Scholar]
- 2. Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. 2007. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 51:3471–3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li XZ, Nikaido H. 2009. Efflux-mediated drug resistance in bacteria: an update. Drugs 69:1555–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Magnet S, Courvalin P, Lambert T. 2001. Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob. Agents Chemother. 45:3375–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coyne S, Rosenfeld N, Lambert T, Courvalin P, Perichon B. 2010. Overexpression of resistance-nodulation-cell division pump AdeFGH confers multidrug resistance in Acinetobacter baumannii. Antimicrob. Agents Chemother. 54:4389–4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Damier-Piolle L, Magnet S, Bremont S, Lambert T, Courvalin P. 2008. AdeIJK, a resistance-nodulation-cell division pump effluxing multiple antibiotics in Acinetobacter baumannii. Antimicrob. Agents Chemother. 52:557–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coyne S, Courvalin P, Perichon B. 2011. Efflux-mediated antibiotic resistance in Acinetobacter spp. Antimicrob. Agents Chemother. 55:947–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peleg AY, Adams J, Paterson DL. 2007. Tigecycline efflux as a mechanism for nonsusceptibility in Acinetobacter baumannii. Antimicrob. Agents Chemother. 51:2065–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ruzin A, Immermann FW, Bradford PA. 2010. RT-PCR and statistical analyses of adeABC expression in clinical isolates of Acinetobacter calcoaceticus-Acinetobacter baumannii complex. Microb. Drug Resist. 16:87–89 [DOI] [PubMed] [Google Scholar]
- 10. Ruzin A, Keeney D, Bradford PA. 2007. AdeABC multidrug efflux pump is associated with decreased susceptibility to tigecycline in Acinetobacter calcoaceticus-Acinetobacter baumannii complex. J. Antimicrob. Chemother. 59:1001–1004 [DOI] [PubMed] [Google Scholar]
- 11. Marchand I, Damier-Piolle L, Courvalin P, Lambert T. 2004. Expression of the RND-type efflux pump AdeABC in Acinetobacter baumannii is regulated by the AdeRS two-component system. Antimicrob. Agents Chemother. 48:3298–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosenfeld N, Bouchier C, Courvalin P, Perichon B. 2012. Expression of the resistance-nodulation-cell division pump AdeIJK in Acinetobacter baumannii is regulated by AdeN, a TetR-type regulator. Antimicrob. Agents Chemother. 56:2504–2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Higgins PG, Schneiders T, Hamprecht A, Seifert H. 2010. In vivo selection of a missense mutation in adeR and conversion of the novel blaOXA-164 gene into blaOXA-58 in carbapenem-resistant Acinetobacter baumannii isolates from a hospitalized patient. Antimicrob. Agents Chemother. 54:5021–5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hornsey M, Ellington MJ, Doumith M, Thomas CP, Gordon NC, Wareham DW, Quinn J, Lolans K, Livermore DM, Woodford N. 2010. AdeABC-mediated efflux and tigecycline MICs for epidemic clones of Acinetobacter baumannii. J. Antimicrob. Chemother. 65:1589–1593 [DOI] [PubMed] [Google Scholar]
- 15. Hornsey M, Loman N, Wareham DW, Ellington MJ, Pallen MJ, Turton JF, Underwood A, Gaulton T, Thomas CP, Doumith M, Livermore DM, Woodford N. 2011. Whole-genome comparison of two Acinetobacter baumannii isolates from a single patient, where resistance developed during tigecycline therapy. J. Antimicrob. Chemother. 66:1499–1503 [DOI] [PubMed] [Google Scholar]
- 16. Sun JR, Chan MC, Chang TY, Wang WY, Chiueh TS. 2010. Overexpression of the adeB gene in clinical isolates of tigecycline-nonsusceptible Acinetobacter baumannii without insertion mutations in adeRS. Antimicrob. Agents Chemother. 54:4934–4938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nemec A, Maixnerova M, van der Reijden TJ, van den Broek PJ, Dijkshoorn L. 2007. Relationship between the AdeABC efflux system gene content, netilmicin susceptibility and multidrug resistance in a genotypically diverse collection of Acinetobacter baumannii strains. J. Antimicrob. Chemother. 60:483–489 [DOI] [PubMed] [Google Scholar]
- 18. Diancourt L, Passet V, Nemec A, Dijkshoorn L, Brisse S. 2010. The population structure of Acinetobacter baumannii: expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS One 5:e10034. 10.1371/journal.pone.0010034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sambrook J, Russell W. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 20. Coyne S, Guigon G, Courvalin P, Perichon B. 2010. Screening and quantification of the expression of antibiotic resistance genes in Acinetobacter baumannii with a microarray. Antimicrob. Agents Chemother. 54:333–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mezzatesta ML, D'Andrea MM, Migliavacca R, Giani T, Gona F, Nucleo E, Fugazza G, Pagani L, Rossolini GM, Stefani S. 2012. Epidemiological characterization and distribution of carbapenem-resistant Acinetobacter baumannii clinical isolates in Italy. Clin. Microbiol. Infect. 18:160–166 [DOI] [PubMed] [Google Scholar]
- 22. Cortez-Cordova J, Kumar A. 2011. Activity of the efflux pump inhibitor phenylalanine-arginine beta-naphthylamide against the AdeFGH pump of Acinetobacter baumannii. Int. J. Antimicrob. Agents 37:420–424 [DOI] [PubMed] [Google Scholar]
- 23. British Society for Antimicrobial Chemotherapy May 2011, posting date BSAC methods for antimicrobial susceptibility testing version 10.2. http://bsac.org.uk/wp-content/uploads/2012/02/Version-10.2-2011-final-May-20111.pdf
- 24. Casal M, Rodriguez F, Johnson B, Garduno E, Tubau F, de Lejarazu RO, Tenorio A, Gimenez MJ, Bartolome R, Garcia-Rey C, Aguilar L, Garcia-Escribano N. 2009. Influence of testing methodology on the tigecycline activity profile against presumably tigecycline-non-susceptible Acinetobacter spp. J. Antimicrob. Chemother. 64:69–72 [DOI] [PubMed] [Google Scholar]
- 25. Maddocks SE, Oyston PC. 2008. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154:3609–3623 [DOI] [PubMed] [Google Scholar]
- 26. Vallenet D, Nordmann P, Barbe V, Poirel L, Mangenot S, Bataille E, Dossat C, Gas S, Kreimeyer A, Lenoble P, Oztas S, Poulain J, Segurens B, Robert C, Abergel C, Claverie JM, Raoult D, Medigue C, Weissenbach J, Cruveiller S. 2008. Comparative analysis of Acinetobacters: three genomes for three lifestyles. PLoS One 3:e1805. 10.1371/journal.pone.0001805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adams MD, Goglin K, Molyneaux N, Hujer KM, Lavender H, Jamison JJ, MacDonald IJ, Martin KM, Russo T, Campagnari AA, Hujer AM, Bonomo RA, Gill SR. 2008. Comparative genome sequence analysis of multidrug-resistant Acinetobacter baumannii. J. Bacteriol. 190:8053–8064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Depardieu F, Podglajen I, Leclercq R, Collatz E, Courvalin P. 2007. Modes and modulations of antibiotic resistance gene expression. Clin. Microbiol. Rev. 20:79–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stock AM, Robinson VL, Goudreau PN. 2000. Two-component signal transduction. Annu. Rev. Biochem. 69:183–215 [DOI] [PubMed] [Google Scholar]
- 30. Bratu S, Landman D, Martin DA, Georgescu C, Quale J. 2008. Correlation of antimicrobial resistance with beta-lactamases, the OmpA-like porin, and efflux pumps in clinical isolates of Acinetobacter baumannii endemic to New York City. Antimicrob. Agents Chemother. 52:2999–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Allen MP, Zumbrennen KB, McCleary WR. 2001. Genetic evidence that the alpha5 helix of the receiver domain of PhoB is involved in interdomain interactions. J. Bacteriol. 183:2204–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Casino P, Rubio V, Marina A. 2009. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 139:325–336 [DOI] [PubMed] [Google Scholar]
- 33. Depardieu F, Courvalin P, Msadek T. 2003. A six amino acid deletion, partially overlapping the VanSB G2 ATP-binding motif, leads to constitutive glycopeptide resistance in VanB-type Enterococcus faecium. Mol. Microbiol. 50:1069–1083 [DOI] [PubMed] [Google Scholar]
- 34. Schwede T, Kopp J, Guex N, Peitsch MC. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ferris HU, Dunin-Horkawicz S, Mondejar LG, Hulko M, Hantke K, Martin J, Schultz JE, Zeth K, Lupas AN, Coles M. 2011. The mechanisms of HAMP-mediated signaling in transmembrane receptors. Structure 19:378–385 [DOI] [PubMed] [Google Scholar]
- 36. Hulko M, Berndt F, Gruber M, Linder JU, Truffault V, Schultz A, Martin J, Schultz JE, Lupas AN, Coles M. 2006. The HAMP domain structure implies helix rotation in transmembrane signaling. Cell 126:929–940 [DOI] [PubMed] [Google Scholar]
- 37. Blanco AG, Sola M, Gomis-Ruth FX, Coll M. 2002. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10:701–713 [DOI] [PubMed] [Google Scholar]