

Abstract
Pseudomonas aeruginosa hemolytic phospholipase C (PlcH) degrades phosphatidylcholine (PC), an abundant lipid in cell membranes and lung surfactant. A ΔplcHR mutant, known to be defective in virulence in animal models, was less able to colonize epithelial cell monolayers and was defective in biofilm formation on plastic when grown in lung surfactant. Microarray analyses found that strains defective in PlcH production had lower levels of Anr-regulated transcripts than the wild type. PC degradation stimulated the Anr regulon in an Anr-dependent manner under conditions where Anr activity was submaximal because of the presence of oxygen. Two PC catabolites, choline and glycine betaine (GB), were sufficient to stimulate Anr activity, and their catabolism was required for Anr activation. The addition of choline or GB to glucose-containing medium did not alter Anr protein levels, growth rates, or respiratory activity, and Anr activation could not be attributed to the osmoprotectant functions of GB. The Δanr mutant was defective in virulence in a mouse pneumonia model. Several lines of evidence indicate that Anr is important for the colonization of biotic and abiotic surfaces in both P. aeruginosa PAO1 and PA14 and that increases in Anr activity resulted in enhanced biofilm formation. Our data suggest that PlcH activity promotes Anr activity in oxic environments and that Anr activity contributes to virulence, even in the acute infection phase, where low oxygen tensions are not expected. This finding highlights the relationships among in vivo bacterial metabolism, the activity of the oxygen-sensitive regulator Anr, and virulence.
INTRODUCTION
Pseudomonas aeruginosa is a virulent opportunistic pathogen that is frequently cultured from burn wound infections, implanted medical devices, and ocular infections and is estimated to cause 8 to 16% of the nosocomial infections that occur worldwide (1). P. aeruginosa infections occur in 80% of adults with the heritable disease cystic fibrosis (CF), who are particularly susceptible to bacterial and fungal infections, in part because of decreased mucociliary clearance (2). P. aeruginosa is the etiological agent of morbidity and mortality in CF patients because of its contribution to decreased lung function (3). Its role as the etiological agent of these distinct disease states can be attributed to its metabolic versatility, expansive array of virulence factors, and biofilm-mediated persistence within the host.
P. aeruginosa produces a number of well-characterized virulence factors that facilitate the establishment of infections. Hemolytic phospholipase C (PlcH) is a secreted hydrolase that degrades host-associated phosphatidylcholine (PC) and sphingomyelin (4–6). These choline-containing phospholipids are abundant macromolecules in eukaryotic membranes and host lung surfactant. PlcH adversely affects the integrity of the lung and contributes to decreased lung function (7, 8). PlcH antibodies have been isolated from the sputum of CF patients shortly after colonization with P. aeruginosa (9), and plcH transcripts can be detected in the serum of CF patients chronically colonized with P. aeruginosa (8), indicating connections between PlcH activity and P. aeruginosa biology in mammalian hosts. The sequential activities of PlcH and choline phosphatase (PchP) release choline from choline-containing lipids, and choline is imported by multiple P. aeruginosa transporters (10) before sequential oxidation to glycine betaine (GB), dimethylglycine, sarcosine, and glycine (11, 12). GbdR positively regulates plcH and pchP transcription, as well as that of the genes involved in the catabolism of choline (8, 12).
In the nutrient-rich but hostile inflammatory environment of the lung, P. aeruginosa is thought to enhance its survival by adopting a protective biofilm mode of growth (13). Biofilms promote chronic infection by decreasing susceptibility to antibiotics and host clearance (14, 15). Biofilm formation and anaerobic metabolism have been linked in late-stage biofilms. For example, genes involved in anaerobic respiration (16–18), which are regulated by the transcriptional regulator Anr, are abundant in P. aeruginosa RNA extracted from the sputum of individuals with CF-associated lung infections (19). Anr is active when the oxygen tension is low (17, 20), likely because of its dependence upon the formation of an oxygen-labile [4Fe-4S]2+ cofactor that must be assembled prior to Anr dimerization, DNA binding, and regulation of target genes, according to work on Fnr, the Escherichia coli Anr homolog (21). Oxygen can destabilize the labile Fe-S cluster, resulting in loss of the active dimer.
Within mature 48-h biofilms, P. aeruginosa fermentation pathways, which are regulated in part by Anr, are upregulated (22). The decreased oxygen tension in CF mucus is hypothesized to lead to increased Anr activity and the induction of these pathways in chronic infections (23). Anr homologs have been linked to pathogenesis in other species, including Shigella flexneri (24), Bordetella pertussis (25), and Actinobacillus pleuropneumoniae (26). In Neisseria meningitidis, deletion of two operons directly controlled by the Anr homolog Fnr resulted in attenuated virulence of the pathogen in an infant mouse model of infection (27). The contribution of Anr to P. aeruginosa virulence in vivo has not been examined, particularly in the context of the acute, rather than the late, stage of chronic infection.
In this report, we present the links among PlcH activity, choline catabolism, and expression of the Anr regulon in aerated cultures through microarray analysis. We show that Anr plays an important role in the colonization of plastic surfaces and airway epithelial cells and that an Δanr mutant was severely defective in virulence in a murine model of acute-phase pneumonia. These data present a previously uncharacterized connection between PlcH activity and Anr activity and reveal roles for Anr in biofilm formation, host colonization, and virulence in environments with where oxygen is present.
MATERIALS AND METHODS
Growth conditions.
All of the strains used in these studies are listed in Table 1. Strains were maintained on LB, and media were supplemented with gentamicin (60 μg/ml for P. aeruginosa and 10 μg/ml for E. coli) as required. For lacZ fusion studies, the medium was supplemented with carbenicillin (300 μg/ml, nirS-lacZ) or tetracycline (75 μg/ml, cgrA-lacZ). Unless otherwise noted, cultures were grown in morpholinepropanesulfonic acid (MOPS)-glucose medium (28), which contained 10 mM glucose and 8 μM ferric chloride. When specified, choline chloride and/or sodium nitrate were added to a final concentration of 5 mM. MOPS-glucose-surfactant medium consisted of MOPS, 10 mM glucose, 8 μM FeCl3, and 3% (vol/vol) Survanta (Abbott Nutrition), which contains 11.0 to 15.5 mg/ml disaturated PC. Cultures were grown in tubes with 5 ml of medium on a roller drum at 37°C or in 250-ml flasks with 20 ml of medium (microarray experiments).
Table 1.
Strains and plasmids used in this study
| Strain reference no. or plasmid | Description | Source and/or reference |
|---|---|---|
| Strain no. | ||
| P. aeruginosa | ||
| DH1856 | PAO1 WT | S. Dove; 37 |
| DH1297 | PAO1 WT | M. Schobert; 70 |
| DH1722 | PA14 WT | 48 |
| DH860 | PAO1 ΔplcHR; in-frame plcHR deletion | 29 |
| DH1219 | PAO1 ΔplcHR-RE; reconstruction of plcHR | 42 |
| DH2085 | PAO1 ΔplcHR + mini-Tn7-Gm; empty vector at Tn7 site | This study |
| DH2086 | PAO1 ΔplcHR + anr-D149A; expression of anr-D149A at Tn7 site | This study |
| DH543 | PAO1 ΔgbdR; in-frame gbdR deletion | 12 |
| DH1857 | PAO1 Δanr; in-frame anr deletion in DH1856 | 37 |
| DH1298 | PAO1 Δanr | 17 |
| DH1977 | PA14 Δanr; in-frame deletion of anr in DH1722 | L. Dietrich |
| DH1913 | PAO1 Δanr + anr; complementation of Δanr at attTn7 site in DH1856 | This study |
| DH1837 | PAO1 Δanr + anr-D149A; complementation of anr deletion with anr-D149A attTn7 in DH1297 | This study |
| DH2035 | PAO1 ΔgbcA; in-frame deletion of gbcA in PAO1 (DH1856) | This study |
| DH1829 | PAO1 Anr-TAP | This study |
| DH1472 | PAO1 cgrA-lacZ; cgrA-lacZ reporter integrated at ϕCTX attachment site | 37 |
| DH1791 | PAO1 ΔcgrC; in-frame deletion of cgrC in PAO1 | |
| DH1792 | PAO1 ΔcupA2; in-frame deletion of cupA2 in PAO1 | |
| E. coli DH522 | S17λpir | |
| Plasmids | ||
| pMQ30 | Suicide vector, Gmr | 30 |
| pHA531 | nirS promoter upstream of lacZYA in pQF50, Carb/Ampr | 36 |
| pGbcA-KO | pMQ30 with gbcA SOE KOa product | This study |
| puc18T-mini-Tn7T-Gm | attTn7 site insertion vector | 31 |
SOE KO, splicing by overlap extension knockout.
Construction of in-frame deletion mutants and plasmids.
The PAO1 ΔplcHR and ΔgbdR mutants were constructed previously by Shortridge et al. and Wargo et al., respectively (12, 29). The gbcA knockout plasmid was constructed with pMQ30 as described in reference 30. To complement the Δanr mutant, anr was amplified from PAO1 with its native promoter (1680824 to 1681070) and ligated into puc18T-mini-Tn7T-Gm (31). The anr-D149A allele was synthesized by GenScript and then fused to the 300 bp upstream of the native anr gene by splice overlap extension PCR and ligated into puc18T-mini-Tn7T-Gm. The anr-D149A gene was placed at the attTn7 attachment site in the Δanr and ΔplcH mutant backgrounds.
Biofilm assays on plastic and airway epithelial cells.
Static biofilm assays were performed with 96-well microtiter plates in MOPS–10 mM glucose medium or M63 with 0.2% Casamino Acids with incubation for 24 h, and biofilms were measured as previously described (32). Purified PlcH or PlcH-T178A protein was added to MOPS-glucose-surfactant medium at to a final concentration of 5 μg/ml for the indicated experiments.
For culture on airway epithelial cells, 5 × 105 CFTRΔF508 homozygous human bronchial epithelial cells (CFBE41o−) (33) were grown in either six-well plates or glass bottom dishes (MatTek Corp., Ashland, MA) and then maintained in minimal essential medium (MEM) with serum for 9 to 10 days. Once a confluent monolayer formed, overnight cultures of P. aeruginosa strains grown in lysogeny broth were resuspended in MEM. Epithelial cells were washed once, inoculated with 1 ml containing 1 × 108 P. aeruginosa, and incubated for 1 h. Planktonic cells were removed, and cocultures were refed with serum-free MEM. The medium was exchanged every 1.5 h. Cocultures were imaged with a Zeiss Axiovert 200 microscope with a 63× differential interference contrast (DIC) objective. Phospholipase (PLC) activity was measured in cocultures that were not refed over the 5-h incubation period (Fig. 1B). To quantify bacterial cell attachment, cocultures were washed twice with Dulbecco's phosphate-buffered saline (DPBS) and then treated with 1 ml of 0.1% Triton X-100 to detach epithelial cells along with attached bacteria from the wells. The cultures were serially diluted and plated on Pseudomonas isolation agar, and CFU were counted after growth at 37°C.
Fig 1.

PlcH activity positively affects P. aeruginosa host colonization. (A) PAO1 WT, ΔplcHR, and ΔplcHR-RE strains were incubated with a confluent monolayer of CFBE airway cells for 6 h, and the associated bacteria were counted as CFU. (B) PLC activities in coculture supernatants were measured by NPPC hydrolysis. The nitrophenol product was measured by determining the OD410. (C) Biofilm formation by the WT, ΔplcHR, and ΔplcHR-RE strains in MOPS-glucose-surfactant medium. (D) Biofilm formation by the ΔplcHR mutant in MOPS-glucose-surfactant medium upon the addition of 5 μg/ml native PlcH or PlcH-T178A. (E) Cell density of WT and ΔplcHR strains grown planktonically in surfactant-containing medium. The data are the averages of three replicate cultures. *, P < 0.005 (difference from the WT in all panels).
NPPC assay.
To measure phospholipase C (PLC) activity, supernatants from P. aeruginosa wild-type (WT), ΔplcHR mutant, and ΔplcHR-RE mutant strains cocultured with airway epithelial cells as described above were incubated with the artificial substrate p-nitrophenylphosphorylcholine (NPPC) as described by Kurioka and Matsuda (34). The reaction buffer contained 100 mM Tris-HCl (pH 7.2), 25% glycerol, and 20 mM NPPC. NPPC hydrolysis was detected by measuring the absorbance at 410 nm and by comparison to a p-nitrophenol standard curve.
Microarray and quantitative PCR experiments.
Cultures were grown in MOPS–10 mM glucose–2 mM sodium pyruvate–8 μM FeCl3–3% Survanta. After 4 h, cells were harvested and RNA was isolated with an RNeasy kit (Qiagen). Contaminating DNA was removed through 1-h RQ1 DNase (Promega) treatments, and semiquantitative PCR with the RNA was performed to ensure the absence of contaminating DNA. The array data were generated from two independent experiments performed on two different days; in each experiment, the WT, ΔplcHR mutant, and ΔgbdR mutant strain cultures were grown in parallel. cDNA was synthesized with Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA) and NS5 primers instead of random hexamers.
The cDNAs were terminally labeled with biotin-ddUTP (Enzo BioArray terminal labeling kit; Affymetrix) and hybridized to Affymetrix Pseudomonas GeneChips according to the manufacturer's instructions with the GeneChip fluidics station 450 (Affymetrix), and GeneChips were scanned with GeneChip Scanner 3000 7G (Affymetrix) in the Dartmouth Genomics and Microarray Laboratory. We used the BioConductor Affy library to read in CEL file data, and data were normalized with RMA in BioConductor (35).
For quantitative real-time PCR experiments, cDNA was synthesized in a manner similar to that described above, according to the protocol accompanying the Invitrogen Taq polymerase. The cycling regimen was 94°C for 3 min; 30 cycles of 94°C for 30 s, 56°C for 30 s, and 72°C for 30 s; and extension at 72°C for 2 min; products were kept at 4°C. Quantitative reverse transcription (RT)-PCR was set up with Power SYBR in accordance with the manufacturer's instructions. The primers used were dnr-Forward (AGC CAG CTG TTC CGT TTC TC), dnr-Reverse (GTG GCG TTC TTC AGG GAA AG), nirS-Forward (CCA AGT ACA TCC AGC ACA CC), nirS-Reverse (GGT ATC GAT GAC CTT GAC GA), cgrA-Forward (CAT TAC CAG GAT GCC GAT GT), cgrA-Reverse (CCT CCA GGT CGA TGA ACA GT), ppiD-Forward (GGT GAG TCG TGG AAA GTG GT), and ppiD-Reverse (GTA CAT GGC CTT CTC GTC CT). Expression analysis was done via the Applied Biosystems 7500 Real-Time PCR system with an annealing temperature of 56°C and an extension temperature of 72°C. Experimental transcripts were normalized to the housekeeping gene ppiD.
β-Gal assays.
To monitor the induction of promoters under the control of Anr, we used a construct with the nirS or cgrA promoter region fused to lacZ (36, 37). Cells were grown overnight at 37°C in LB and then diluted into 5 ml of fresh MOPS medium supplemented as indicated in culture tubes at a starting optical density at 600 nm (OD600) of 0.05. Cultures were incubated on a roller drum at 37°C for 5 h. For studies of Anr activity under conditions with altered access to oxygen, cultures were grown in MOPS–10 mM glucose–5 mM sodium nitrate with or without 5 mM choline on a roller drum for 5 h at 37°C. We designated aerated cultures 5-ml cultures and those with low aeration 10-ml cultures. Cultures grown incubated quiescently in closed capped tubes were designated static. β-Galactosidase (β-Gal) activity was measured as described by Miller (38, 39). For osmoprotection studies, a ΔgbcA nirS-lacZ mutant strain was grown overnight at 37°C in MOPS–10 mM glucose and then diluted into 5 ml of 50 mM NaCl–MOPS or 0.3 M NaCl–MOPS–10 mM glucose with or without 5 mM choline. Cell growth was monitored over time, and β-Gal activity was measured once cells grown in 0.3 M NaCl–MOPS–10 mM glucose–5 mM choline were at a higher density than cells grown in 0.3 M NaCl–MOPS–10 mM glucose.
General statistics.
Experimental replicates were averaged for NPPC, β-Gal, and quantitative PCR experiments. The means were compared with a two-sample t test assuming unequal variance. P ≤ 0.05 was considered significant.
Cellular respiration assay.
WT cells were grown in MOPS–10 mM glucose–5 mM choline to mid-exponential phase (OD600 of 0.4). Cells were harvested, washed twice, and resuspended in MOPS medium with only 10 mM glucose, 10 mM glucose, and 5 mM choline or no substrate (resting). A 100-μl volume of the cell suspensions was added to 96-well black Costar plates in addition to 10 μl alamarBlue (Invitrogen) reagent. Fluorescence was monitored at 37°C for 2 h at a 560-nm excitation wavelength and a 590-nm emission wavelength.
Cytotoxicity assays.
Epithelial cell cytotoxicity was assayed by quantification of lactate dehydrogenase (LDH) release according to the manufacturer's instructions (CytoTox96 nonradioactive cytotoxicity assay kit; Promega).
Western blotting.
A PAO1 strain producing a C-terminally tandem affinity purification (TAP)-tagged Anr protein (Anr-TAP) was constructed by the methods described in reference 40. Anr-TAP-producing cells were grown as described for the transcript analysis assays. Whole-cell lysates were prepared by boiling for 10 min, and 20 μl of sample was subjected to SDS-PAGE for 45 min at 150 V. Proteins were transferred to a polyvinylidene fluoride membrane, washed, and then probed with a soluble peroxidase-antiperoxidase complex (Sigma-Aldrich) to detect the TAP tag. Proteins were visualized by SuperSignal West Pico chemiluminescent substrate (Pierce). The SDS-PAGE gel was stained with GelCode Blue Stain reagent (Pierce Biotechnology, Inc.) to visualize loaded-protein concentrations.
Mouse experiments.
Male C57BL/6 mice (Jackson Laboratories) were anesthetized with isoflurane and then inoculated with 6 × 107 P. aeruginosa cells via oropharyngeal aspiration (8). Mouse external body temperature (°C) was logged at 0, 3, and 24 h postinfection with an infrared thermometer (IR-101; Infrascan, La Crosse, WI). After 24 h, the mice were euthanized and then their lungs were surgically removed and homogenized in 1 ml phosphate-buffered saline. The homogenate was serially diluted on Pseudomonas isolation agar for CFU enumeration after incubation at 37°C. The mouse experiments were performed in compliance with all institutional and federal policies.
Microarray data accession number.
Microarray data sets were deposited in the GEO database (GSE41926).
RESULTS
PlcH requirement for P. aeruginosa colonization of epithelial cells and biofilm formation in lung surfactant-containing medium.
Previous work has shown that PlcH degrades phospholipids in host membranes and in lung surfactant both in vivo and in vitro (8, 41). Furthermore, we recently reported that the recovery of WT P. aeruginosa was 40-fold greater than that of ΔplcHR mutant cells after oropharyngeal infections of the mouse lung with 106 CFU of P. aeruginosa (42). To understand the role of PlcH, if any, in colonization of and growth in the lung, we examined P. aeruginosa WT and ΔplcHR mutant cells in an airway epithelial cell colonization assay. P. aeruginosa strains were coincubated with a confluent monolayer of CFTRΔF508 (CFBE41o−) homozygous human airway epithelial cells under conditions that lead to the formation of P. aeruginosa biofilms on epithelial cells (43). While the initial attachment of WT and ΔplcHR mutant cells was not different after 1 h (data not shown), the biofilms formed by the ΔplcHR mutant on the epithelial cell monolayer contained significantly fewer cells than did the biofilms formed by the WT, and the defect was complemented by restoration of plcHR at the native locus (ΔplcHR-RE) (P < 0.005, Fig. 1A). To determine if PlcH was produced in this system, we measured PLC activity in supernatants from airway cell cocultures. WT coculture supernatants had nine times as much PLC activity as the ΔplcHR mutant coculture supernatants, and complementation with plcHR restored PLC activity to 62% of the WT level (Fig. 1B).
To examine the effects of PlcH on surface colonization in PC-rich environments, we assessed P. aeruginosa growth and biofilm formation on plastic in a glucose-based medium with 3% Survanta, a synthetic lung surfactant, which contains 11.0 to 15.5 mg/ml disaturated PC. PlcH is produced in this medium, and catabolism of the released phosphorylcholine head group has been shown to occur (8). In lung surfactant-containing medium, the ΔplcHR biofilm in the microtiter dish well assay was significantly reduced by 7-fold compared to that of the WT or ΔplcHR-RE mutant strain (Fig. 1C). Biofilm formation by the ΔplcHR mutant could also be complemented with purified native PlcH protein but not an equivalent amount of a catalytically inactive PlcH variant (PlcH-T178A) (44), indicating that its activity rather than its surface association domains is required for the stimulation of biofilm formation (Fig. 1D). We found that WT and ΔplcHR mutant cell densities were not significantly different in surfactant-containing medium, suggesting that the difference in biofilm formation was not due to decreased growth (Fig. 1E).
PC availability-induced transcript levels of genes regulated by Anr.
To determine how PlcH activity impacted P. aeruginosa surface colonization (Fig. 1A and C), we compared the global expression profiles of the ΔplcHR mutant and the ΔgbdR mutant, a strain lacking a transcriptional regulator required for PlcH production and PC catabolism (8), to that of the WT in surfactant-containing medium at 4 h. The P. aeruginosa WT, ΔplcHR, and ΔgbdR strains had completely overlapping growth curves over 10 h in MOPS-glucose-surfactant medium, as determined by OD (data not shown) and consistent with data in Fig. 1E. The mRNA expression profiles were determined with the Affymetrix Pseudomonas GeneChip. While most of the transcripts did not differ among the three strains, one strong pattern emerged, i.e., lower levels of transcripts regulated by the transcription factor Anr in the ΔplcHR and ΔgbdR mutant strains than in the WT. Trunk et al. reported genes with Anr boxes in their promoter regions that are differentially expressed in an Anr-dependent manner (45). We found that, with few exceptions, transcripts within this defined Anr regulon were lower in strains that could not produce PlcH (ΔplcHR and ΔgbdR mutant strains) than in the WT (Fig. 2A).
Fig 2.

PlcH activity correlates with the induction of the Anr regulon. (A) The WT and ΔplcHR mutant strains were grown for 4 h at 37°C in MOPS-glucose-surfactant medium. RNA was profiled with Affymetrix Pseudomonas GeneChips (n = 2). Shown is a heat map of Anr-regulated transcripts clustered according to differential expression in the WT, ΔplcHR, and ΔgbdR strains. (B) Volcano plot depicting the average n-fold difference in transcripts in surfactant-containing medium between the ΔplcHR mutant and WT strains. Anr-regulated transcripts are highlighted in red. The horizontal line marks P = 0.05, and the vertical lines represent a 2-fold change. (C) To confirm the microarray data, the nirS-lacZ promoter fusion was used in the WT, ΔplcHR, and plcHR-RE strains in MOPS-glucose medium with (closed bars) and without (open bars) 3% Survanta after growth at 37°C for 6 h. The data are the means of three replicate cultures. *, P < 0.0005 (different from the WT without PC).
To better compare the WT to the ΔplcHR mutant, the P values and n-fold changes in more than 5,000 P. aeruginosa transcripts were displayed on a volcano plot (Fig. 2B). This presentation method showed that the bulk of the transcripts did not change significantly in abundance and that the differences in Anr-regulated transcripts were among the strongest trends in the data comparing ΔplcHR (Fig. 2B) or ΔgbdR (not shown) to the WT. The n-fold differences between the WT and the ΔplcHR mutant for Anr-regulated genes ranged from 5- to 36-fold (see Table S1 in the supplemental material), and differentially regulated transcripts include those involved in energy generation under low-oxygen conditions, such as those that encode high-affinity cytochrome oxidases and fermentation and denitrification enzymes. The Anr-regulated operon cgrA-cgrC, which encodes transcriptional regulators of CupA fimbriae (37, 46), was also differentially expressed. Transcripts that were more highly expressed in the ΔplcHR mutant but not known to be controlled by Anr are shown in Table S2 in the supplemental material; the majority of these transcripts encode proteins that have not yet been characterized.
P. aeruginosa PC catabolism yields the catabolic intermediates GB and dimethylglycine, inducing plcH and pchP transcription through activation of GbdR (8), in addition to genes involved in the GB degradation pathway (12). We observed that many of the choline catabolism-related transcripts were differentially expressed with 20-fold lower levels of transcripts involved in GB catabolism (gbc, dgc, and soxA-D) in the ΔgbdR mutant than in the WT (8). The lower abundance of these transcripts indicated that GB was being generated in WT cultures from the release of the phosphorylcholine head group by PlcH. PlcH activity also releases diacylglycerol, which can be oxidized by P. aeruginosa as a source of energy in the lung (19). Interestingly, the psrA transcript, which encodes a repressor of genes involved in the oxidation of lipids, was 12-fold more abundant in the WT than in the ΔplcHR mutant (47). Fatty acid oxidation-associated transcripts (fad genes) were not among those identified as being different among the WT, ΔplcHR, and ΔgbdR strains. These data suggest that in these cultures, catabolism of the choline head group, but not the lipid portion of PC, was under way.
PlcH activity is associated with higher levels of Anr activity in vitro and on airway epithelial cells.
Because of the surprising differences in the levels of Anr-controlled transcripts (Fig. 2A) in aerated cultures of the P. aeruginosa WT, ΔplcHR, and ΔgbdR strains that were at the same cell densities, we examined links between PlcH activity and Anr induction. Following the transcription of an Anr-regulated gene, nirS, strains carrying a plasmid with the nirS promoter driving lacZ expression were grown with high levels of aeration in MOPS medium with and without surfactant. We observed significantly higher levels of β-Gal activity in WT cultures with surfactant, while the ΔplcHR strain did not show a significant difference in nirS-lacZ expression. Complementation with the plcHR gene at the native locus restored the increase in the expression of the Anr-controlled gene in medium with surfactant (P < 0.0005, Fig. 2C). The stimulation of nirS promoter activity upon the addition of surfactant was Anr dependent (Fig. 3A). Because Anr also regulates the cgrA-cgrC operon (46), we assayed activity at the cgrA promoter in strains carrying cgrA-lacZ fusions. We observed higher levels of β-Gal activity in medium with surfactant (P < 0.001) in an Anr-dependent manner (Fig. 3B).
Fig 3.
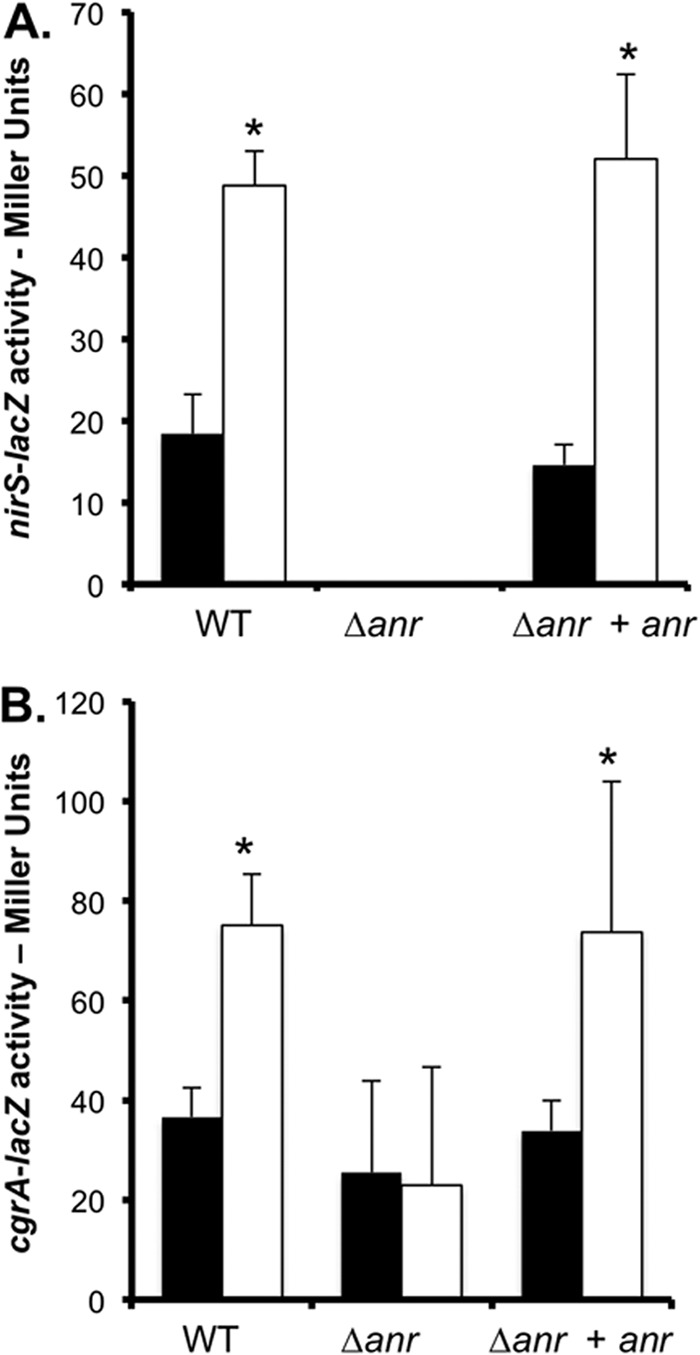
Anr is required for PC-mediated induction of nirS and cgrA. Shown are data for nirS-lacZ (A) and cgrA-lacZ (B) promoter fusions in the WT, Δanr mutant, and anr-complemented Δanr mutant strain backgrounds in MOPS-glucose without (closed bars) and with (open bars) 3% surfactant. The data are means of three replicate cultures with comparable results in independent experiments. *, P < 0.05 (difference from base medium).
To determine if PlcH activity impacted the activation of the Anr regulon when P. aeruginosa was cocultured with airway epithelial cells, we assayed nirS and cgrB levels in the WT, ΔplcHR, and ΔplcHR-RE strains. Both transcripts were lower in the ΔplcHR mutant than in the WT at 6 h (Fig. 4). The Δanr mutant had 40- and 30-fold lower levels of nirS activity than the WT and the complemented mutant, respectively (data not shown).
Fig 4.
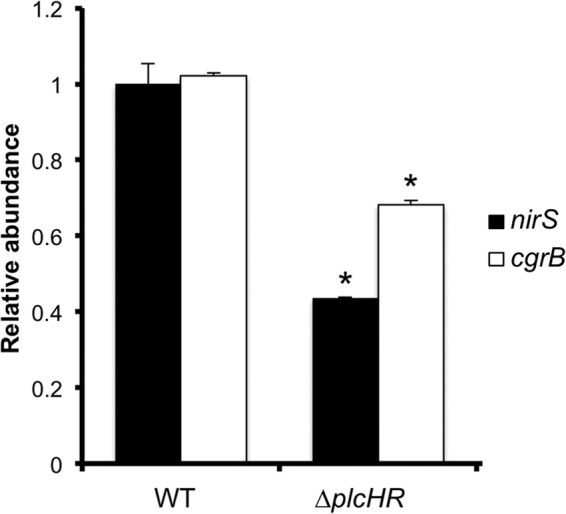
Anr is active early during P. aeruginosa colonization of epithelial cell monolayers. Shown are the results of a quantitative RT-PCR analysis of nirS and cgrB, normalized to ppiD, in the WT, the ΔplcHR mutant, and its complemented derivative. The data are means from three replicate cultures, and similar results were obtained in two independent experiments. *, P ≤ 0.001 (difference from the WT).
Choline catabolism positively regulates Anr activity in vitro.
In light of transcriptional evidence that choline derived from PlcH-mediated PC degradation (8) was being catabolized in WT cells when Anr-regulated transcripts were lower in ΔplcHR and ΔgbdR mutant cells than in the WT, we sought to determine if choline was sufficient to induce Anr activity in P. aeruginosa. We compared nirS promoter activity in a WT strain with that in the nirS-lacZ fusion grown with and without choline in MOPS-glucose medium. The inclusion of choline in the medium was sufficient to stimulate Anr activity in the WT by 3.3-fold (Fig. 5A). The levels of three Anr-regulated genes, dnr, cgrA, and nirS, assessed by quantitative RT-PCR in medium supplemented with the PC catabolite choline, were found to be significantly lower in the Δanr mutant strain than in the WT and the Δanr-complemented strain (see Fig. S1 in the supplemental material). It is important to note that the Δanr mutant was able to grow and catabolize choline; thus, the defect in PC-mediated induction was due solely to the absence of Anr activity at the given promoters. To determine if the catabolism of choline is required for the stimulation of the Anr-regulated transcript levels, we also monitored nirS-lacZ activity in the ΔgbcA mutant, which cannot catabolize choline or GB for energy or growth because of the inability to convert GB into dimethylglycine (12). In contrast to what was seen in WT cultures, activity at the nirS promoter was not higher in the ΔgbcA mutant upon the inclusion of choline in the growth medium (Fig. 5A). These data indicated that choline must be catabolized to induce Anr activity and suggested that the osmoprotectant properties of GB were not responsible for Anr induction. To exclude the possibility that the lack of response to choline in the ΔgbcA mutant was due to an inability to take up or accumulate GB, we grew the ΔgbcA mutant in MOPS-glucose medium with a concentration of salt that was partially inhibitory (0.3 M NaCl) in the absence of choline or another osmoprotectant. While 5 mM choline was able to rescue growth in 0.3 M NaCl to levels of control cultures (data not shown), choline did not influence the expression of nirS-lacZ in salt-containing medium (5.83 ± 2.5 Miller units without choline versus 8.40 ± 1.2 Miller units with choline).
Fig 5.

Induction of Anr activity is dependent on choline catabolism. (A) nirS promoter expression was assayed in the WT and ΔgbcA mutant backgrounds in MOPS-glucose (control) with or without 5 mM choline (Cho) and/or 5 mM nitrate (NO3−) after growth at 37°C for 5 h. (B) Respiratory activity of WT without a carbon source (Resting), with glucose, or with glucose and choline was measured with alamarBlue. Data are presented as percent reduction over time. (C) WT nirS-lacZ was grown in 20 mM glucose or choline alone at 37°C. β-Gal activity was measured at an OD600 of 0.2. (D) nirS-lacZ promoter fusion in WT cells grown in MOPS-glucose with 5 mM sodium nitrate with or without 5 mM choline in aerated, low-oxygen, and static cultures at 37°C. The data are the means of biological replicates. (E) Western blot analysis of an Anr-TAP-producing strain grown under high and low oxygen concentrations in MOPS-glucose medium with 5 mM nitrate with or without 5 mM choline. The data are the means of replicate cultures with comparable results between independent experiments. *, P < 0.05 (compared to control cultures).
One potential explanation for our finding that choline or surfactant catabolism stimulates Anr activity is that the addition of choline or the ability to degrade the head group from PC leads to increased respiratory activity or cell density. In our glucose-containing medium, the addition of choline did not result in a significant change in growth rates or final yields, suggesting that differences in OD did not explain the enhanced Anr activation seen (data not shown). Furthermore, when the respiratory activity of WT cells pregrown in choline-containing medium to induce the expression of choline transporters (10) was measured, it was found that there was similar respiratory activity in cells given glucose, even with the addition of choline as measured by alamarBlue (Invitrogen) reduction (Fig. 5B). Although P. aeruginosa grows more slowly on choline as a sole carbon source (12), WT cells grown solely on choline to an OD of 0.2 had higher levels of nirS-lacZ-derived β-Gal activity (6.75-fold) than did glucose-grown cells analyzed at the same cell density (Fig. 5C). Together, these data suggest a model in which choline-induced Anr activity was not dependent on cell density or changes in respiratory activity.
The addition of nitrate to anoxic medium promotes nirS transcription via coordinate regulation by Anr and the secondary transcription factor Dnr (18, 45). While the addition of nitrate to aerated cultures in MOPS-glucose medium did not induce nirS promoter activity, choline with nitrate induced higher levels of nirS-lacZ transcription than choline alone (Fig. 5A), suggesting that nirS transcription could be promoted by the activation of coregulators once Anr was activated. As was observed in the absence of nitrate, there was no induction of nirS-lacZ in the ΔgbcA mutant.
A comparison of β-Gal activities in the WT nirS-lacZ strain in base medium without choline showed the expected increases as oxygen became less available (Fig. 5D). Increased liquid volumes in culture tubes (low-oxygen conditions) were sufficient to induce a 3-fold greater increase in nirS promoter activity over levels in aerated cultures, and static conditions led to a 25-fold increase in β-Gal levels (Fig. 5D). Thus, Anr was largely inhibited under our aerated and low-oxygen culture conditions because of the presence of oxygen. While choline significantly stimulated Anr activity at the nirS promoter under both aerated (Fig. 5A, 3.3-fold, and D, 2-fold) and low-oxygen (Fig. 5D, 2-fold) conditions (P < 0.05), there were no observable effects of choline on Anr activity under static culture conditions (Fig. 5D). To determine if Anr protein levels were affected by the addition of choline in cultures exposed to oxygen, we used a strain expressing a functional Anr fusion protein with a C-terminal TAP tag. The addition of choline did not lead to higher levels of Anr protein than in cells in medium with glucose alone under either aerated or low-oxygen conditions (Fig. 5E).
Anr activity contributes to P. aeruginosa biofilm formation on plastic and epithelial cells.
Because our interest in Anr came from an examination of the effects of PlcH activity in mouse lung colonization (8) and biofilm formation in lung surfactant-containing medium (Fig. 2), we assayed the Δanr mutant biofilm phenotype. The Δanr mutant strain, like the ΔplcHR mutant strain, was defective in biofilm formation compared to the WT in surfactant-containing medium, and biofilm formation was restored upon complementation with anr (Fig. 6A). Unlike the ΔplcHR mutant strain, which formed biofilms indistinguishable from those of the WT in medium without surfactant (data not shown), the Δanr mutant biofilm defect was not limited to surfactant-containing medium; its biofilm formation in the absence of surfactant was significantly lower than that of the WT in medium with glucose and amino acids as growth substrates (see Fig. S2A in the supplemental material). As PAO1 strains can vary between labs, the Δanr mutant biofilm defect was confirmed in two distinct PAO1 WT and Δanr mutant pairs (see Fig. S2) (17, 37) and was also observed in genetically distinct P. aeruginosa strain PA14 (see Fig. S3) (48), demonstrating that Anr was important for P. aeruginosa biofilm formation in different strain backgrounds. Because Anr controls CgrA-CgrC, regulators that control the expression of the genes that encode CupA fimbriae, appendages that promote cell-cell interactions and robust biofilms (37, 46, 49, 50), we assayed the contributions of cupA2 and cgrC to P. aeruginosa biofilms. In our assay, the absence of cupA2 or cgrC did not change the biofilm phenotype from that of the WT (see Fig. S4).
Fig 6.
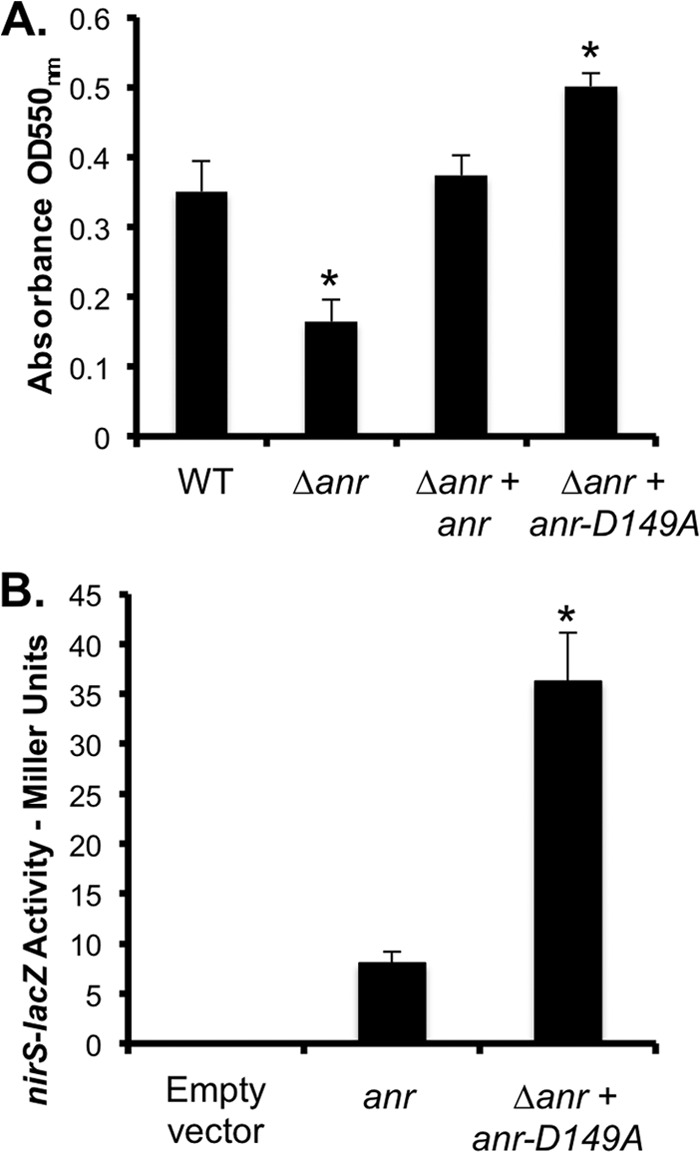
Anr activity promotes P. aeruginosa biofilm formation. (A) Biofilms formed by WT, Δanr mutant, and anr- and anr-D149A-complemented Δanr mutant strains after 24 h in MOPS-glucose-surfactant medium. (B) nirS-lacZ expression in the Δanr mutant strain carrying the empty vector, anr, or Δanr plus anr-D149A in MOPS-glucose medium. β-Gal activity was measured after growth at 37°C for 4 h. The data are the means of three replicate cultures. *, P ≤ 0.001 (difference from the WT).
To determine if induction of Anr activity affects P. aeruginosa biofilm formation, we constructed a strain expressing an Anr-D149A variant that was predicted to have constitutive activity on the basis of work on Fnr in E. coli (51). The Δanr mutant strain producing Anr-D149A formed a more robust biofilm than the WT and the complemented Δanr mutant in surfactant-containing medium (Fig. 6A), indicating that promotion of Anr activity increases biofilm formation. The activity of Anr-D149A was 4.5-fold higher than that of native Anr in MOPS-glucose medium lacking choline (Fig. 6B), which was fortuitously similar to the amount of stimulation of native Anr observed upon the addition of choline (Fig. 3A). Furthermore, as observed in surfactant-containing medium, enhanced biofilm formation upon the production of Anr-D149A was observed in MOPS-glucose medium (see Fig. S2 in the supplemental material).
To determine if the defects in biofilm formation in the ΔplcHR mutant in medium with surfactant was due in part to defects in Anr activation, the anr-D149A allele was transformed into the ΔplcHR strain. The ΔplcHR mutant strain expressing the hyperactive Anr-D149A variant had a 2.8-fold increase in biofilm formation compared to that of the ΔplcH mutant, whereas the appropriate vector control had an only 1.6-fold increase in biofilm formation. As a control, we confirmed that the anr-D149A allele stimulated biofilm formation in the WT background with native anr (not shown), as well as in the Δanr mutant (Fig. 6A). These findings are consistent with our model in which part of the ΔplcHR mutant's defect in biofilm formation in lung surfactant-containing medium is due to the inability to activate Anr under oxic conditions.
Anr is involved in the colonization of airway epithelial cells and the mouse lung.
To gain insight into how Anr may impact host interactions, we assessed P. aeruginosa colonization of CFBE cells. In this model, the Δanr mutant was observed to be defective in surface association (Fig. 7A). Compared to the abundant, robust microcolonies observed in WT cocultures, the Δanr mutant formed only small groups of bacterial cells on the epithelial cell monolayer after 6 h in a coculture (Fig. 7A). At this time point, the Δanr mutant showed a marked reduction in cytotoxicity relative to that of the WT, as measured by LDH release into the coculture supernatant (Fig. 7B). Both the airway cell colonization defect and the decrease in cytotoxicity in the Δanr mutant strain were rescued by complementation.
Fig 7.

Anr regulates microcolony formation on airway cells and cytotoxicity. (A) Images of WT, Δanr mutant, anr-complemented Δanr mutant biofilms on CFBE cells after 6 h. DIC images were taken at ×63 magnification. (B) Percentages of LDH release from CFBE cells following coculture with the WT, the Δanr mutant, and the anr-complement Δanr mutant for 6 h. The data are the means of three replicate cultures. *, P ≤ 0.05 (difference from the WT).
A role for Anr activity in chronic, late-stage infections associated with CF could be inferred from several studies of oxygen tension in CF patient airway mucus (52), induction of genes involved in denitrification in P. aeruginosa in sputum environments (53), and fermentation pathways in 48-h flow cell biofilms formed by P. aeruginosa (22). On the basis of the observation that the Δanr mutant had an early defect in biofilm formation on airway epithelial cells, we also hypothesized that the absence of Anr would lead to a defect in colonization or virulence in an acute-phase infection model. To this end, we used a mouse pneumonia model to assay the contribution of Anr regulation to the establishment of the acute phase of infection. Twenty-four hours after mice were inoculated with 6 × 107 P. aeruginosa cells by oropharyngeal aspiration, the Δanr mutant was recovered from the lungs at 1,000-fold lower levels than the WT and the anr-complemented Δanr mutant train (Fig. 8; P < 0.001). Furthermore, the mice infected with the WT and anr-complemented strains had lower external body temperatures, a marker of imminent death (54), than mice infected with the Δanr mutant strain. Mice infected with the Δanr mutant strain had a body temperature near the baseline (27.9 ± 0.9°C), while mice infected with the WT had a significantly lower body temperature (22.8 ± 0.4°C) at 24 h. The differences between the two groups in body temperatures and the number of CFU recovered suggest that Anr plays a role in this acute-phase infection model.
Fig 8.
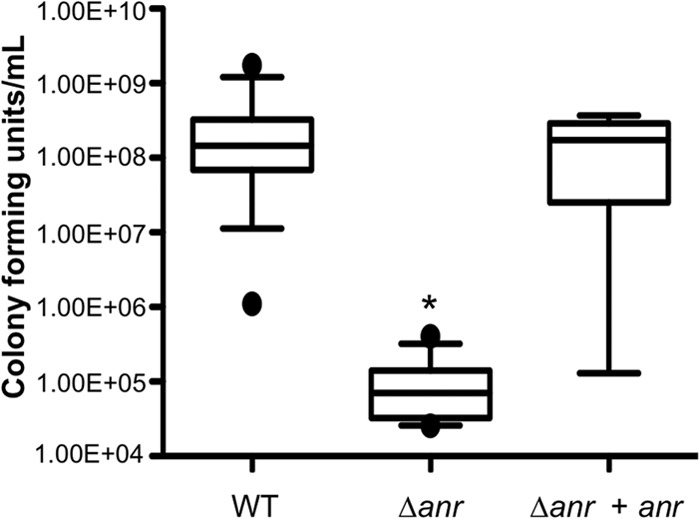
The Δanr mutant is severely defective in a murine pneumonia model. Male C57/BL6 mice received 6 × 107 WT and Δanr mutant cells by oropharyngeal inoculation. Mice were sacrificed at 24 h, and following growth at 37°C, CFU were enumerated. Data are the mean values of individual mice. Values from three separate experiments were pooled. *, P < 0.001 (difference from the WT and anr-complemented Δanr mutant strains).
DISCUSSION
Our analyses of P. aeruginosa strains grown in lung surfactant-containing medium showed significantly higher levels of Anr-regulated transcripts in the WT than in the ΔplcHR and ΔgbdR mutants, indicating that PlcH activity increased the Anr activation of its target promoters (Fig. 2). Evidence of increased Anr activity extended to several pathways involved in P. aeruginosa respiration, biofilm formation, and virulence (see Table S1 in the supplemental material). Choline was sufficient to increase activity at Anr-regulated promoters independent of increases in cell density or metabolic activity (Fig. 5). To our knowledge, this is the first report linking the catabolism of host-derived substrates and increased Anr activity under conditions where Anr is thought to be largely inactive because of the presence of oxygen (17, 55). A recent study has shown that P. aeruginosa betI and betB transcripts were particularly upregulated in CF patient lung infections, as well as burn wound samples, compared to those in planktonic controls (56). Because the betaine aldehyde dehydrogenase-encoding genes are responsible for the conversion of choline to GB (57), this implies choline uptake and utilization in vivo.
Biofilm formation was negatively impacted by the absence of Anr on plastic and on airway epithelial cells (Fig. 6 and 7) (58). While Anr is well known for its role in the regulation of denitrification (17), the defects in Anr biofilm formation extended to conditions without any sources of nitrate, nitrite, or nitric oxide, and thus, defects were not due to the inability to use these alternative electron acceptors (see Fig. S2 in the supplemental material). Recent studies have shown a role for pyruvate fermentation pathways in late stages of biofilm development in flow cells (22), and future studies will elucidate whether or not Anr is required for the control of these pathways and if these pathways play roles in early events in biofilm development on plastic and in host colonization. The Δanr mutant also had a severe defect in mouse lung colonization and appeared to be largely cleared from the lung within 24 h, in contrast to the WT (Fig. 8). The steep oxygen gradients that exist within the mucus plugs in the CF patient lung (52, 59), likely because of the combined respiratory and enzymatic activities of both bacterial and host cells (60), have already hinted at an important role for Anr in P. aeruginosa utilization of alternate electron acceptors during chronic CF lung infections.
Our data support a model in which Anr also plays an integral role in initial events governing P. aeruginosa colonization of the host (Fig. 7 and 8). The environments in which these events take place are not anoxic, and although Anr activity is repressed by oxygen, we have shown that the availability of choline, which cannot be synthesized by P. aeruginosa de novo, induces Anr activity only under aerated conditions (Fig. 5). Because choline catabolism is predicted to require oxygen at multiple steps (12), it is not surprising that choline does not stimulate Anr activity under anaerobic conditions. Induction of denitrification enzymes when oxygen is present may enable the detoxification of nitric oxide species produced in the highly inflammatory host environment (61). Small quantities of nitrate are available to be metabolized by P. aeruginosa in the CF lung (52, 62, 63), and nitric oxide, either provided exogenously or generated through denitrification, could stimulate the expression of the type III secretion system, which is associated with acute toxicity to epithelial cells and promotion of inflammation (64). The early induction of Anr may benefit P. aeruginosa in vivo through the induction of high-affinity terminal oxidases that can increase respiration at low oxygen tensions and may also give P. aeruginosa an advantage, as oxygen becomes limiting during biofilm formation or depletion of local oxygen by the host immune response (55). It is also interesting that a small RNA controlled by Anr impacts the expression of quorum sensing-controlled virulence factors such as phenazines (65). Anr also contributes directly to the regulation of genes involved in cyanide biosynthesis (66). Future studies will determine whether these or alternative Anr-regulated pathways clarify the nature of the Δanr defect in the mouse pneumonia model.
We put forward a model in which PlcH activity promotes Anr activity and thus promotes host colonization. Interestingly, PlcH shows little toxicity for epithelial cells (44); therefore, this potential role for PlcH in interactions with epithelial cells provides new insight into the mechanisms by which this virulence factor promotes disease. PlcH has been shown to contribute to virulence in many different hosts, including plants, invertebrates, and mammals. It is important to note that the plcH promoter has been reported to have an Anr consensus sequence, and thus, there is potential for there to be a positive feedback loop triggering this pathway (45). While our data suggest that PlcH activity stimulates Anr through the release of choline, it is challenging to determine whether abolition of choline catabolism is sufficient to prevent Anr activation in lung surfactant because of the regulation of PlcH production by choline catabolism intermediates (67). Our prior work has shown, however, that choline is readily catabolized in lung surfactant-containing medium (8), and even low concentrations of choline trigger the induction of multiple choline transporters (10). Because P. aeruginosa does not synthesize its own choline, we speculate that choline serves as an indicator of the presence of eukaryotic hosts. While the mechanism(s) by which choline stimulates Anr activity is unknown, our data show that Anr stimulation was not due to increased cell density or an increased respiratory rate (Fig. 1E and 5B). Oxygen consumption experiments did not show reproducible differences in oxygen uptake between cultures with choline and glucose and cultures with glucose alone (data not shown). Choline-derived GB is an osmoprotectant in P. aeruginosa and other species (10, 68), and while it is possible that it could stabilize the mature 4Fe-4S cluster-containing dimer in its soluble or DNA-bound state, its accumulation did not stimulate Anr activity (Fig. 5A). Because the activity of the E. coli Anr homolog, Fnr, is impacted by iron availability and nitric oxide (69), there is the potential for complex regulation of Anr activity in the context of mammalian hosts.
Together, these data suggest that Anr could be a viable target for the development of novel P. aeruginosa therapies. Since other pathogens require Anr homologs for virulence (24–27), such an approach may also be beneficial in the treatment of other diseases. Future work will determine if the inactivation of Anr during established infections can limit disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank Chinweike Okegbe, Lars Dietrich (Columbia University), and Elizabeth Sonnleitner (University of Vienna) for P. aeruginosa strains; Jenna Allard (University of Vermont) and Nicholas Jacobs (Geisel School of Medicine) for experimental advice; and Michael Vasil for providing purified PlcH. We acknowledge Jennifer Bomberger and Matthew Wargo for their efforts in initial experiments with the ΔplcHR mutant on airway cells.
Research reported in this publication was supported by National Institutes of Health (NIH) grant RO1A109170 to D.A.H., the National Institute of General Medical Sciences (NIGMS) of the NIH as P20GM103413, and the Renal Function and Disease training grant from the NIGMS of the NIH under award 5P20-RR018787 to A.A.J. This work was also supported by grants from the Cystic Fibrosis Foundation Research Development Program (STANTO07R0 to D.A.H.) and NIH R01-HL074175 to B.A.S. and T.H.H.
The content is solely our responsibility and does not necessarily represent the official views of the NIH.
Footnotes
Published ahead of print 10 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02169-12.
REFERENCES
- 1. Van Delden C, Iglewski BH. 1999. Cell-to-cell signaling and Pseudomonas aeruginosa infections. Emerg. Infect. Dis. 4:551–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rajan S, Saiman L. 2002. Pulmonary infections in patients with cystic fibrosis. Semin. Respir. Infect. 17:47–56 [DOI] [PubMed] [Google Scholar]
- 3. Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, Burns JL, Kaul R, Olson MV. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. U. S. A. 103:8487–8492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bomberger J, MacEachran DP, Coutermarsh BA, Ye S, O'Toole GA, Stanton B. 2009. Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog. 5:e1000382. 10.1371/journal.ppat.1000382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berka RM, Vasil ML. 1982. Phospholipase C (heat-labile hemolysin) of Pseudomonas aeruginosa: purification and preliminary characterization. J. Bacteriol. 152:239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vasil M. 2006. Pseudomonas aeruginosa phospholipases and phospholipids, p 69–97 In Ramos J, Levesque R. (ed), Pseudomonas, vol 4 Springer, Houten, The Netherlands [Google Scholar]
- 7. Meyers DJ, Palmer KC, Bale LA, Kernacki K, Preston M, Brown T, Berk RS. 1992. In vivo and in vitro toxicity of phospholipase C from Pseudomonas aeruginosa. Toxicon 30:161–169 [DOI] [PubMed] [Google Scholar]
- 8. Wargo MJ, Ho TC, Gross MJ, Whittaker LA, Hogan DA. 2009. GbdR regulates Pseudomonas aeruginosa plcH and pchP transcription in response to choline catabolites. Infect. Immun. 77:1103–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Granström M, Ericsson A, Strandvik B, Wretlind B, Pavlovskis OR, Berka R, Vasil ML. 1984. Relation between antibody response to Pseudomonas aeruginosa exoproteins and colonization/infection in patients with cystic fibrosis. Acta Paediatr. Scand. 73:772–777 [DOI] [PubMed] [Google Scholar]
- 10. Malek A, Chen C, Wargo MJ, Beattie GA, Hogan DA. 2011. Roles of three transporters, CbcXWV, BetT1, and BetT3, in Pseudomonas aeruginosa choline uptake for catabolism. J. Bacteriol. 193:3033–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Serra A, Mariscotti JF, Barra JL, Lucchesi GI, Domenech CE, Lisa AT. 2002. Glycine betaine transmethylase mutant of Pseudomonas aeruginosa. J. Bacteriol. 184:4301–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wargo MJ, Szwergold BS, Hogan DA. 2008. Identification of two gene clusters and a transcriptional regulator required for Pseudomonas aeruginosa glycine betaine catabolism. J. Bacteriol. 190:2690–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Høiby N, Krogh Johansen H, Moser C, Song Z, Ciofu O, Kharazmi A. 2001. Pseudomonas aeruginosa and the in vitro and in vivo biofilm mode of growth. Microbes Infect. 3:23–35 [DOI] [PubMed] [Google Scholar]
- 14. Brooun A, Liu S, Lewis K. 2000. A dose-response study of antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 44:640–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Costerton JW. 2001. Cystic fibrosis pathogenesis and the role of biofilms in persistent infection. Trends Microbiol. 9:50–52 [DOI] [PubMed] [Google Scholar]
- 16. Sawers R. 1991. Identification and molecular characterization of a transcriptional regulator from Pseudomonas aeruginosa PAO1 exhibiting structural and functional similarity to the Fnr protein of Escherichia coli. Mol. Microbiol. 5:1469–1481 [DOI] [PubMed] [Google Scholar]
- 17. Ye RW, Haas D, Ka JO, Krishnapillai V, Zimmermann A, Baird C, Tiedje JM. 1995. Anaerobic activation of the entire denitrification pathway in Pseudomonas aeruginosa requires Anr, an analog of Fnr. J. Bacteriol. 177:3606–3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schreiber K, Krieger R, Benkert B, Eschbach M, Arai H, Schobert M, Jahn D. 2007. The anaerobic regulatory network required for Pseudomonas aeruginosa nitrate respiration. J. Bacteriol. 189:4310–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Son MS, Matthews WJ, Jr, Kang Y, Nguyen DT, Hoang TT. 2007. In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infect. Immun. 75:5313–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Galimand M, Zimmermann GMA, Haas D. 1991. Positive Fnr-like control of anaerobic arginine degradation and nitrate respiration in Pseudomonas aeruginosa. J. Bacteriol. 173:1598–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lazazzera BA, Beinert H, Khoroshilova N, Kennedy MC, Kiley PJ. 1996. DNA binding and dimerization of the Fe-S-containing FNR protein from Escherichia coli are regulated by oxygen. J. Biol. Chem. 271:2762–2768 [DOI] [PubMed] [Google Scholar]
- 22. Petrova O, Shurr JR, Shurr MJ, Sauer K. 2012. Microcolony formation by the opportunistic pathogen Pseudomonas aeruginosa requires pyruvate and pyruvate fermentation. Mol. Microbiol. 86:819–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hassett DJ, Sutton MD, Schurr MJ, Herr AB, Caldwell CC, Matu JO. 2009. Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol. 17:130–138 [DOI] [PubMed] [Google Scholar]
- 24. Marteyn B, West NP, Browning DF, Cole JA, Shaw JG, Palm F, Mounier J, Prevost MC, Sansonetti P, Tang CM. 2010. Modulation of Shigella virulence in response to available oxygen in vivo. Nature 465:355–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mattoo S, Ming YH, Huang LL, Miller JF. 2004. Regulation of type III secretion in Bordetella. Mol. Microbiol. 52:1201–1214 [DOI] [PubMed] [Google Scholar]
- 26. Baltes N, N′diaye M, Jacobsen ID, Maas A, Buettner FF, Gerlach GF. 2005. Deletion of the anaerobic regulator HlyX causes reduced colonization and persistence of Actinobacillus pleuropneumoniae in the porcine respiratory tract. Infect. Immun. 73:4614–4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bartolini E, Frigimelica E, Giovinazzi S, Galli G, Shaik Y, Genco C, Welsch JA, Granoff DM, Grandi G, Grifantini R. 2006. Role of FNR and FNR-regulated, sugar fermentation genes in Neisseria meningitidis infection. Mol. Microbiol. 60:963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neidhardt FC, Bloch PL, Smith DF. 1974. Culture medium for enterobacteria. J. Bacteriol. 119:736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shortridge VD, Lazdunski A, Vasil ML. 1992. Osmoprotectants and phosphate regulate expression of phospholipase C in Pseudomonas aeruginosa. Mol. Microbiol. 6:863–871 [DOI] [PubMed] [Google Scholar]
- 30. Shanks R, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl. Environ. Microbiol. 72:5027–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choi KH, Schweizer HP. 2006. Mini-Tn7 insertion with single attTn7 sites: example Pseudomonas aeruginosa. Nat. Protoc. 1:153–161 [DOI] [PubMed] [Google Scholar]
- 32. O'Toole GA, Kolter R. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449–461 [DOI] [PubMed] [Google Scholar]
- 33. Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. 1994. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 10:38–47 [DOI] [PubMed] [Google Scholar]
- 34. Kurioka S, Matsuda M. 1976. Phospholipase C assay using p-nitrophenylphosphoryl-choline together with sorbitol and its application to studying the metal and detergent requirement of the enzyme. Anal. Biochem. 75:281–289 [DOI] [PubMed] [Google Scholar]
- 35. Irizarry R, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264 [DOI] [PubMed] [Google Scholar]
- 36. Arai H, Igarashi Y, Kodama T. 1995. Expression of the nir and nor genes for denitrification of Pseudomonas aeruginosa requires a novel CRP/FNR related transcriptional regulator, DNR, in addition to ANR. FEBS Lett. 371:73–76 [DOI] [PubMed] [Google Scholar]
- 37. Vallet-Gely I, Sharp JS, Dove SL. 2007. Local and global regulators linking anaerobiosis to cupA fimbrial gene expression in Pseudomonas aeruginosa. J. Bacteriol. 189:8667–8676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miller JH. 1992. A short course in bacterial genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 39. Zhang X, Bremer H. 1995. Control of the Escherichia coli rrnB P1 promoter strength by ppGpp. J. Biol. Chem. 270:11181–11189 [DOI] [PubMed] [Google Scholar]
- 40. Castang S, McManus HR, Turner KH, Dove SL. 2008. H-NS family members function coordinately in an opportunistic pathogen. Proc. Natl. Acad. Sci. U. S. A. 105:18947–18952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vasil M, Graham L, Ostroff RM, Shortridge VD, Vasil AI. 1991. Phospholipase C: molecular biology and contribution to the pathogenesis of Pseudomonas aeruginosa, p 34–47 In Homma J, Tanimoto H, Holder I, Høiby N, Doring G. (ed), Pseudomonas aeruginosa in human diseases, vol 44 Karger, Basel, Switzerland: [DOI] [PubMed] [Google Scholar]
- 42. Wargo MJ, Gross MJ, Rajamani S, Allard JL, Lundblad LK, Allen GB, Vasil ML, Leclair LW, Hogan DA. 2011. Hemolytic phospholipase C inhibition protects lung function during Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 184:345–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anderson GG, Moreau-Marquis S, Stanton BA, O'Toole GA. 2008. In vitro analysis of tobramycin-treated Pseudomonas aeruginosa biofilms on cystic fibrosis-derived airway epithelial cells. Infect. Immun. 76:1423–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vasil ML, Stonehouse MJ, Vasil AI, Wadsworth SJ, Goldfine H, Bolcome RE, Chan J. 2009. A complex extracellular sphingomyelinase of Pseudomonas aeruginosa inhibits angiogenesis by selective cytotoxicity to endothelial cells. PLoS Pathog. 5:e1000420. 10.1371/journal.ppat.1000420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trunk K, Benkert B, Quack N, Munch R, Scheer M, Garbe J, Jansch L, Trost M, Wehland J, Buer J, Jahn M, Schobert M, Jahn D. 2010. Anaerobic adaptation in Pseudomonas aeruginosa: definition of the Anr and Dnr regulons. Environ. Microbiol. 12:1719–1733 [DOI] [PubMed] [Google Scholar]
- 46. McManus H, Dove SL. 2011. The CgrA and CgrC proteins form a complex that positively regulates cupA fimbrial gene expression in Pseudomonas aeruginosa. J. Bacteriol. 193:6152–6161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang Y, Nguyen David T, Son Mike S, Hoang TT. 2008. The Pseudomonas aeruginosa PsrA responds to long-chain fatty acid signals to regulate the fadBA5 beta-oxidation operon. Microbiology 154(Pt 6):1584–1598 [DOI] [PubMed] [Google Scholar]
- 48. Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899–1902 [DOI] [PubMed] [Google Scholar]
- 49. Vallet-Gely I, Donovan KE, Fang R, Joung JK, Dove SL. 2005. Repression of phase-variable cup gene expression by H-NS-like proteins in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 102:11082–11087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Meissner A, Wild V, Simm R, Rohde M, Erck C, Bredenbruch F, Morr M, Romling U, Haussler S. 2007. Pseudomonas aeruginosa cupA-encoded fimbriae expression is regulated by a GGDEF and EAL domain-dependent modulation of the intracellular level of cyclic diguanylate. Environ. Microbiol. 9:2475–2485 [DOI] [PubMed] [Google Scholar]
- 51. Bates DM, Lazazzera BA, Kiley PJ. 1995. Characterization of Fnr* mutant proteins indicates two distinct mechanisms for altering oxygen regulation of the Escherichia coli transcription factor Fnr. J. Bacteriol. 177:3972–3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, Birrer P, Bellon G, Berger J, Weiss T, Botzenhart K, Yankaskas JR, Randell S, Boucher RC, Doring G. 2002. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Invest. 109:317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Platt MD, Schurr MJ, Sauer K, Vazquez G, Kukavica-Ibrulj I, Potvin E, Levesque RC, Fedynak A, Brinkman FSL, Schurr J, Hwang SH, Lau GW, Limbach PA, Rowe JJ, Lieberman MA, Barraud N, Webb J, Kjelleberg S, Hunt DF, Hassett DJ. 2008. Proteomic, microarray, and signature-tagged mutagenesis analyses of anaerobic Pseudomonas aeruginosa at pH 6.5, likely representing chronic, late-stage cystic fibrosis airway conditions. J. Bacteriol. 190:2739–2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ray MA, Johnston NA, Verhulst S, Trammell RA, Toth LA. 2010. Identification of markers for imminent death in mice used in longevity and aging research. J. Am. Assoc. Lab. Anim. Sci. 49:282–288 [PMC free article] [PubMed] [Google Scholar]
- 55. Kawakami T, Kuroki M, Ishii M, Igarashi Y, Arai H. 2010. Differential expression of multiple terminal oxidases for aerobic respiration in Pseudomonas aeruginosa. Environ. Microbiol. 12:1399–1412 [DOI] [PubMed] [Google Scholar]
- 56. Bielecki P, Komor U, Bielecka A, Musken M, Puchalka J, Pletz M, Ballmann M, Martins dos Santos VAP, Weiss S, Haussler S. 2013. Ex vivo transcriptional profiling reveals a common set of genes important for the adaptation of Pseudomonas aeruginosa to chronically infected host sites. Environ. Microbiol. 15:570–597 [DOI] [PubMed] [Google Scholar]
- 57. Velasco-García R, Villalobos MA, Ramirez-Romero MA, Mujica-Jimenez C, Iturriaga G, Munoz-Clares RA. 2006. Betaine aldehyde dehydrogenase from Pseudomonas aeruginosa: cloning, over-expression in Escherichia coli, and regulation by choline and salt. Arch. Microbiol. 185:14–22 [DOI] [PubMed] [Google Scholar]
- 58. O'Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30:295–304 [DOI] [PubMed] [Google Scholar]
- 59. Yoon SS, Hennigan RF, Hilliard GM, Ochsner UA, Parvatiyar K, Kamani MC, Allen HL, DeKievit TR, Gardner PR, Schwab U, Rowe JJ, Iglewski BH, McDermott TR, Mason RP, Wozniak DJ, Hancock RE, Parsek Noah TL, Boucher RC, Hassett DJ. 2002. Pseudomonas aeruginosa anaerobic respiration in biofilms: relationships to cystic fibrosis pathogenesis. Dev. Cell 3:593–603 [DOI] [PubMed] [Google Scholar]
- 60. Kolpen M, Hansen CR, Bjarnsholt T, Moser C, Christensen LD, van Gennip M, Ciofu O, Mandsberg L, Kharazmi A, Döring G, Givskov M, Høiby N, Jensen PØ. 2010. Polymorphonuclear leucocytes consume oxygen in sputum from chronic Pseudomonas aeruginosa in cystic fibrosis. Thorax 65:57–62 [DOI] [PubMed] [Google Scholar]
- 61. Aldallal N, McNaughton EE, Manzel LJ, Richards AM, Zabner J, Ferkol TW, Look DC. 2002. Inflammatory response in airway epithelial cells isolated from patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 166:1248–1256 [DOI] [PubMed] [Google Scholar]
- 62. Grasemann H. 1998. Nitric oxide metabolites in cystic fibrosis lung disease. Arch. Dis. Child. 78:49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Linnane SJ. 1998. Total sputum nitrate plus nitrite is raised during acute pulmonary infection in cystic fibrosis. Am. J. Respir. Crit. Care Med. 158:207–212 [DOI] [PubMed] [Google Scholar]
- 64. Van Alst NE, Wellington M, Clark VL, Haidaris CG, Iglewski BH. 2009. Nitrite reductase NirS is required for type III secretion system expression and virulence in the human monocyte cell line THP-1 by Pseudomonas aeruginosa. Infect. Immun. 77:4446–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sonnleitner EGN, Sorger-Domenigg T, Heeb S, Richter AS, Backofen R, Williams P, Hüttenhofer A, Haas D, Bläsi U. 2011. The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol. Microbiol. 80:868–885 [DOI] [PubMed] [Google Scholar]
- 66. Pessi G, Haas D. 2000. Transcriptional control of the hydrogen cyanide biosynthetic genes hcnABC by the anaerobic regulator ANR and the quorum-sensing regulators LasR and RhlR in Pseudomonas aeruginosa. J. Bacteriol. 182:6940–6949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fitzsimmons L, Hampel KJ, Wargo MJ. 2012. Cellular choline and glycine betaine pools impact osmoprotection and phospholipase C production in Pseudomonas aeruginosa. J. Bacteriol. 194:4718–4726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen C, Beattie GA. 2008. Pseudomonas syringae BetT is a low-affinity choline transporter that is responsible for superior osmoprotection by choline over glycine betaine. J. Bacteriol. 190:2717–2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fleischhacker A, Kiley PJ. 2011. Iron-containing transcription factors and their roles as sensors. Curr. Opin. Chem. Biol. 15:335–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Krieger R, Rompf A, Schobert M, Jahn D. 2002. The Pseudomonas aeruginosa hemA promoter is regulated by Anr, Dnr, NarL and integration host factor. Mol. Genet. Genomics 267:409–417 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.