

Abstract
The draft genome sequence of a single orange Beggiatoa (“Candidatus Maribeggiatoa”) filament collected from a microbial mat at a hydrothermal site in Guaymas Basin (Gulf of California, Mexico) shows evidence of extensive genetic exchange with cyanobacteria, in particular for sensory and signal transduction genes. A putative homing endonuclease gene and group I intron within the 23S rRNA gene; several group II catalytic introns; GyrB and DnaE inteins, also encoding homing endonucleases; multiple copies of sequences similar to the fdxN excision elements XisH and XisI (required for heterocyst differentiation in some cyanobacteria); and multiple sequences related to an open reading frame (ORF) (00024_0693) of unknown function all have close non-Beggiatoaceae matches with cyanobacterial sequences. Sequences similar to the uncharacterized ORF and Xis elements are found in other Beggiatoaceae genomes, a variety of cyanobacteria, and a few phylogenetically dispersed pleiomorphic or filamentous bacteria. We speculate that elements shared among filamentous bacterial species may have been exchanged in microbial mats and that some of them may be involved in cell differentiation.
INTRODUCTION
Large vacuolate Beggiatoaceae are conspicuous members of microbial mats at sites where hydrothermal fluids reach the surface in Guaymas Basin, a sedimented midocean spreading center in the Gulf of California (1). They may belong to two or more candidate genus-level groups in a recently proposed reorganization of the group (2, 3). Beggiatoaceae are also found in a variety of freshwater and hypersaline environments; of particular relevance to this paper, they form multispecies hypersaline microbial mats in Guerrero Negro (Baja California, Mexico) together with Cyanobacteria, Chloroflexi, Bacteroidetes, and a diversity of other bacteria and archaea (2, 4). The near-complete genome sequence of a single orange Guaymas Basin vacuolate Beggiatoa (“Candidatus Maribeggiatoa”) filament, referred to here as the BOGUAY sequence, was recently obtained (B. J. MacGregor, J. F. Biddle, C. Harbort, A. G. Matthysse, A. Teske, submitted for publication) (5). A complete genome for the freshwater Beggiatoa alba B18LD (NCBI project ID 62137) and partial genomes for two filaments (BgP and BgS) from Baltic Sea harbor sediment (6) are available for comparison.
The BOGUAY genome annotation, like most others, identified a variety of possible mobile or formerly mobile elements (introns, inteins, homing endonucleases) and genes likely delivered by them (restriction-methylation and toxin-antitoxin systems). Their phylogenetic affiliations may provide clues to the evolution of the Beggiatoaceae and the mat communities they are found in. We present evidence here that the marine BOGUAY lineage, cyanobacteria, and diverse filamentous gliding bacteria have a history of genetic exchange, which appears to have been less extensive and/or less well preserved in the freshwater Beggiatoa alba. A first genome-wide comparison of BOGUAY-predicted proteins with the public databases suggests that signal transduction genes and some cell wall biogenesis genes have been especially successful in transfers.
MATERIALS AND METHODS
Retrieval of orange filaments for sequencing.
Core 4489-10 from RV Atlantis/HOV Alvin cruise AT15-40 (13 December 2008; latitude, 27°0.450300′N; longitude, 111°24.532320′W; depth, 2,001 m) at Guaymas Basin, Mexico, contained orange mat material, which was removed to a 50-ml Falcon tube with seawater and refrigerated overnight (also described in reference 5). The orange tuft was nicely reestablished the next day. A large portion was placed in sterile seawater-0.1% agar and incubated at room temperature with the motion of the ship, and then a portion of this was transferred to sterile seawater in a petri plate. Single filaments were selected using a 10-μl pipette tip. Three microliters of liquid was drawn up as the pipette tip was centered on a filament head under ×40 magnification in a dissecting microscope. Filaments were immediately expelled into sterile 500-μl tubes and frozen at −20°C. The sequenced filament had a diameter of 35 to 40 μm.
Amplification of single-filament DNA.
Filament DNA was initially amplified with the RepliG minikit (Qiagen, Germantown, MD) with the following modifications. Solution DLB was prepared by adding 500 μl of DNase/RNase-free water (Qiagen) to the DLB stock in the kit. Solution D2 was prepared by adding 5 μl dithiothreitol (DTT) from the kit to 55 μl resuspended DLB solution. PicoGreen dye stock (Invitrogen, Carlsbad, CA) was diluted 1:10 in PCR-grade water. Frozen filaments were thawed and centrifuged briefly. All liquid was removed from the tube (∼3 μl), and the filament was visible at the bottom. A total of 1 μl of 1× phosphate-buffered saline (PBS) and 1.5 μl solution D2 were added. Tubes were vortexed, centrifuged briefly, and incubated 10 min on ice, and then 1.5 μl of stop solution was added. The entire volume was transferred to a QPCR plate (Stratagene, La Jolla, CA), and 15 μl reaction buffer, 1 μl RepliG phi29 polymerase, and 0.25 μl diluted PicoGreen dye were added. The reaction was placed in an MX3500P QPCR machine (Stratagene) and observed every 10 min for 1.5 h at 30°C. Negative controls using water or the seawater sorting solution in place of the filament mixture showed no increase in PicoGreen fluorescence during this time. Reactions were stopped by incubation at 65°C for 3 min.
To dilute the phi29 polymerase founder effect, this initial amplification was split in 6 aliquots and amplified further using the RepliG Midi kit (Qiagen) according to the manufacturer's instructions with minor modifications, including the addition of 1 μl of diluted PicoGreen dye into the reaction mixture and incubation of the reaction at 30°C for 4 h with observation of PicoGreen fluorescence every 10 min by the QPCR machine. Negative controls of water or the seawater sorting solution again showed no increase in PicoGreen fluorescence during this time.
Confirmation of single-filament amplification.
Products from the RepliG reactions were diluted 1:10, and 2 μl of this was used as the template in a PCR using primers B4 and B7 (7), which amplify the intergenic spacer between the 16S and 23S rRNA genes. A single PCR product was seen for the BOGUAY filament sample in all amplifications, suggesting that a single species was present. No amplifications were seen for the negative controls. To confirm this, PCR was repeated using primers B8F and B1492R to amplify the full 16S rRNA gene of the amplified filament. PCR products were cloned in TOPO TA cloning vectors (Invitrogen) and transformed into Escherichia coli. Cloned products were sequenced via colony amplification by Genewiz (Plainsfield, NJ) using M13 priming sites. Over 30 clones were examined, all of which were identical products, confirming that most likely one species was present.
Preparation of DNA for genome sequencing.
To remove potential chimeric structures from the amplified DNA, individual RepliG Midi reactions were pooled and treated with S1 nuclease at 37°C for 1 h. DNAs were then precipitated and concentrated. Aliquots were run on a 0.8% Tris-acetate-EDTA (TAE) agarose gel and showed that most of the product was high-molecular-weight (HMW) DNA, and a total of 48 μg of DNA had been amplified from a single orange filament. Samples were then sent to the JCVI for further sequencing.
Sequencing at JCVI and open reading frame (ORF)-naming convention.
The sample was subjected to quality control by 16S rRNA gene PCR amplification and sequencing of 384 clones, all of which had identical sequences matching the one previously obtained at UNC. A 454 paired-end library was prepared and sequenced on a GSFLX titanium sequencer (454 Life Sciences). Assembly was performed using the Celera assembler, and annotation was done via the Newbler pipeline. A total of 99.3% of the sequence was assembled into 822 contigs, suggesting that good coverage was achieved. A total of 4.7 Mb of sequence was recovered, with 80% of the sequence forming large (≥15-kb) contigs. Annotated sequences are referred to by 5-digit contig and 4-digit ORF numbers, e.g., 00024_0691.
Annotation at UNC.
Additional sequence analysis was carried out using a combination of the JCVI-supplied annotation, the IMG/ER (8) and RAST (9) platforms, and BLASTN, BLASTX, BLASTP, PSIBLAST, and DELTABLAST searches of the GenBank nr databases. Nucleic acid and amino acid sequence alignments were generally performed in MEGA5 (10) using MUSCLE (11); for several amino acid alignments, the NCBI COBALT aligner (12) gave a subjectively better result. Maximum-likelihood phylogenies were inferred in ARB (13) with RAxML rapid bootstrapping (14) and Bayesian phylogenies in MrBayes 3.2 (15).
RESULTS AND DISCUSSION
The various potential mobile elements identified to date in the BOGUAY genome exhibit two distinct types of phylogenetic history, as reconstructed from present-day sequences. Homing endonucleases, group I and group II introns, GyrB and DnaE inteins, fdxN excision-like elements, and multiple copies of an ORF of unknown function all have close cyanobacterial relatives and a limited range of other bacterial affiliations, while restriction-modification and toxin-antitoxin systems generally have (as expected) a much wider distribution. These patterns are discussed in the following sections. Putative transposons will not be discussed here; their identification and classification varied widely among automated annotations (JCVI, IMG/ER, UniProt), and many of them appear to be split between ORFs and thus may be relict elements.
A discussion of the evidence for the purity and completeness of the draft genome is presented first, as an essential basis for the rest of the discussion.
Genome identity: 16S rRNA gene sequence.
The orange Guaymas Beggiatoa genome sequenced (referred to here as the BOGUAY genome) includes a single set of rRNA genes, with the 16S rRNA gene sequence falling with the “Candidatus Maribeggiatoa” (3) group (see Fig. S1 in the supplemental material). Note that the orange Guaymas Beggiatoaceae are not monophyletic: the 16S rRNA gene from the genome-sequenced filament discussed here is highly similar to 16S rRNA gene sequences from filaments collected in 1996 and 2009 but not to a second orange filament collected in 2008. The four filaments came from different sites; whether mixed populations exist is not known. Near-complete (B. alba), partial (Beggiatoa PS, BgP) or very partial (Beggiatoa SS, BgS) genome sequences are available for three of the other species and strains shown. The BOGUAY 23S rRNA gene is interrupted by a homing endonuclease gene and an intron sequence, discussed below.
Genome completeness.
Because the BOGUAY genome is not completely assembled, the annotation of ribosomal protein, RNA polymerase, tRNA, and tRNA synthetase genes was examined as a test of genome coverage and purity.
Ribosomal proteins.
A complete set of ribosomal protein genes was identified (see Table S1 in the supplemental material), and six contigs have been provisionally connected (see Fig. S2 in the supplemental material) into an extended fragment with potential S10, spc, and alpha operons (16, 17). The remaining ribosomal protein genes are also generally found in conserved neighborhoods, except that L28 is downstream of the 5S rRNA gene rather than adjacent to L33, and S21 is separated from S9 and L13.
RNA polymerase.
A putative RNA polymerase α-subunit gene (rpoA) was found, as expected, in the alpha operon (16, 17). Single-copy RNA polymerase β and β′ subunit genes (rpoB and rpoC) are often found between an S12 gene downstream and additional ribosomal protein genes upstream, but in the BOGUAY sequence, rpoB and associated ribosomal protein genes are in the center of one contig, and copies of rpoC are annotated on two others (see Fig. S2 in the supplemental material). Two rpoC copies have also been noted in some cyanobacteria (18). The putative rpoC copies both have high-scoring BLAST hits to other gammaproteobacterial β′ subunit genes, with the top hit in both cases to the same BgP predicted protein (ZP_01998875.1; this is annotated as rpoA but is clearly mislabeled). The BOGUAY 00100_0018 predicted amino acid sequence has a gap of ∼75 amino acid (aa) residues beginning at approximately position 982 and another of ∼18 aa near the C terminus, relative to other β′ subunit sequences, so it may have an altered (or no) function. The other copy (BOGUAY 01343_3638) is near the end of a short contig and might be connected to the assemblage containing the α subunit gene, but there is no evidence for this in the existing sequence data. In light of this possible gene rearrangement, it is interesting that a transposase gene (BOGUAY 00680_3522) is annotated at the downstream end of the proposed alpha operon (see Fig. S2 in the supplemental material).
Candidate genes for several RNA polymerase sigma factors (σ70 [00397_1682], RpoN [00721_4589], RpoH [00906_2621, 00938_0742], RpoS [01092_1316], and RpoD [01192_0215]) have also been annotated but will not be further discussed here.
tRNAs.
Forty-six tRNA genes, covering all 20 standard amino acids, were identified by JCVI, RAST, and JGI using tRNAScan-SE-1.23 (19, 20), with three tRNAs (tRNA-Leu-GAG, tRNA-Tyr-GTA, tRNA-Val-TAC) represented twice (see Table S2 in the supplemental material). Initiator methionine (iMet, fMet), extension methionine (eMet), and lysylated isoleucine (kIle) tRNAs, which share the anticodon CAT, were identified via the TFAM Web server (21) and tRNA database (tRNdb) (22). Consistent with the identification of tRNA-kIle-CAT, the draft genome includes a predicted gene for tRNA(Ile)-lysidine synthetase (BOGUAY 00794_2073), which changes the specificity of one class of tRNA-CATs from methionine to isoleucine by modifying their anticodon C to lysidine (23). Methionyl-tRNA formyltransferase, for formylation of initiator methionine, was also identified (BOGUAY 00472_0536).
After application of the standard wobble-base rules (24), tRNA-Arg-TCT and tRNA-Leu-TAA remained missing. Directed BLASTN searches with the uninterrupted cognate genes from Beggiatoa alba B18LD revealed that these contain inserts, with the sequence coding for the first 38 tRNA bases (up to the end of the predicted anticodon loop) separated from the remainder of the genes by spacers of 272 and 316 nucleotides (nt), respectively (see footnote to Table S2 in the supplemental material). This is a common position for self-splicing group I introns in bacterial and chloroplast tRNAs (25). The BOGUAY inserts appear to have at least some of the conserved group I intron secondary structures (not shown), but proof that they can excise would require experimentation. The total of 48 tRNA genes is relatively few compared to other bacteria (26), a trait which has been correlated with slow growth and slow response to changes in nutrient concentrations (27, 28).
tRNA synthetase genes.
A complete set of tRNA synthetase genes was found (see Table S2 in the supplemental material). Like many species in all three domains of life (29), the BOGUAY genome apparently does not possess a dedicated asparaginyl-tRNA synthetase. Instead, tRNA-Asn may first be ligated with aspartate, which is then amidated by aspartyl-glutamyl-tRNA amidotransferase (GatCAB). A putative gatCA was located in the center of one relatively long contig (BOGUAY 00150; 40,422 bp) and gatB on a separate, smaller one (BOGUAY 00338; 3,292 bp). An apparent amplification or sequencing error splits gatA into two ORFS (00150_0829, 00150_0828), with both fragments having high-scoring matches to other putative gatA sequences (not shown).
In summary, although the single-filament BOGUAY genome is not closed, genes encoding complete sets of ribosomal protein, tRNA, and tRNA synthetase genes could be identified. While individual genes may certainly have been missed, it seems unlikely that entire multienzyme pathways were, but (as with any sequence-based predictions) experimental proof will be needed.
Homing endonuclease gene and a group I intron in the 23S rRNA gene.
The inferred phylogeny of the BOGUAY 23S rRNA gene (see Fig. S3 in the supplemental material) is congruent with that of the 16S rRNA gene (see Fig. S1 in the supplemental material), at least so far as 23S rRNA sequences are available to date, but it contains two intervening elements (Fig. 1A) similar to ones found in the rRNA genes of diverse species (e.g., see references 30–32), including other giant sulfur bacteria (33). In orange Guaymas Beggiatoa, a putative group I catalytic intron is found at E. coli position 1917, in the loop of predicted helix 69, and a putative LAGLIDADG homing endonuclease at E. coli position 1931, in a predicted unstructured region between helices 69 and 71 (Fig. 1B). These positions and several nearby ones also harbor intervening sequences in a small but phylogenetically diverse group of other bacteria and archaea, hinting at a possibly complex history of exchange, decay, and preservation for these elements.
Fig 1.

Position of intervening sequences in the 23S rRNA gene. (A) The orange Guaymas Beggiatoa genome sequence includes a single set of rRNA genes. The figure combines JGI and RAST annotations; the 23S rRNA gene was not annotated by JGI and therefore has no number. It contains putative intron and homing endonuclease insertions, separated by 14 bp of apparent rRNA sequence. The intron sequence overlaps the upstream portion of the 23S gene by 4 bp. (B) The orange Guaymas Beggiatoa intervening sequences are in the predicted helix 69 loop and between helices 69 and 71 of the predicted rRNA structure, a region in which a variety of other bacteria and archaea also harbor putative introns and homing endonucleases, as outlined in the figure. These were identified primarily by searching the Silva (75) LSURef 111 database for 23S rRNA gene sequences longer than 3,000 bp and refining the alignment to place the intervening sequences precisely.
The phylogeny of the BOGUAY 23S homing endonuclease was inferred by both maximum-likelihood (Fig. 2) and Bayesian (see Fig. S4 in the supplemental material) methods, which yielded similar results. The BOGUAY 23S homing endonuclease gene is affiliated with those from Synechococcus sp. C9 and Synechococcus lividans C1 (34), with additional similar sequences in a few widely divergent bacteria (e.g., several Chlamydiales, some Thermotoga species, a few Coxiella burnetii strains) and eukaryotic organellar genomes (particularly in green algae). Close relatives of the intron sequence are also sporadically distributed (Fig. 3) but limited to bacteria, with the nearest relative so far in the giant sulfur bacterium Thiomargarita sp. NAM092. Given that sequences similar to both the intron and homing endonuclease genes are found together in BOGUAY, Synechococcus sp. C9, and several Coxiella burnetii strains, they may have been transmitted together, with the intron perhaps lost more easily over time. Synechococcus and Beggiatoaceae species can be found together in microbial mats (e.g., see reference 35), which may be favorable environments for gene exchange. Coxiella burnetii is a gammaproteobacterium only known to grow as an intracellular pathogen (36), but its DNA has been detected by PCR in a variety of terrestrial (37) and marine (38) environments, and one strain has been found infecting a Steller sea lion (39), making genetic exchange between Coxiella and Beggiatoaceae a possibility. As more giant sulfur bacterial chromosomal sequences become available, however, there may be evidence for vertical transmission of 23S rRNA introns within this group.
Fig 2.

Putative homing endonucleases encoded within 23S rRNA genes. Relatives of BOGUAY 0660_3608 were identified by BLASTX searches of the GenBank nr and JCVI IMG/ER databases (March 2013). Inferred amino acid sequences were aligned in MEGA5 (10) with MUSCLE (11). The first 100 hits were used for initial neighbor-joining trees, which were pruned to focus on the most closely BOGUAY-related sequences before further analysis. The maximum-likelihood phylogeny was inferred in ARB (13). All four combinations of the PROTGAMMA and PROTMIX rate substitution models and WAG and cpREV amino acid substitution models were tested with RAxML (14) new rapid hill climbing. The PROTGAMMA/cpREV combination gave the best likelihood score and was used for RAxML rapid bootstrap analysis with 1,000 bootstraps. Numbers represent bootstrap values for each node, and circles indicate uncertain branch points. Bayesian analysis also identified cpREV as the best-fitting amino acid substitution model for these data (see Fig. S4 in the supplemental material). Numbers at nodes represent bootstrap values for each node, and circles indicate uncertain branch points. The scale bar represents amino acid changes per position.
Fig 3.

Putative group I introns within 23S rRNA genes. Relatives of BOGUAY 0660_3609 were identified by BLASTN searches of the GenBank nr and JCVI IMG/ER databases; all are located within 23S rRNA genes. As shown, there were very few full-length or nearly full-length matches. Species which also have putative 23S homing endonucleases are indicated by black stars, and species whose close relatives do are indicated by open circles (compare to Fig. 2). Nucleic acid sequences were aligned in MEGA5 (10) using MUSCLE (11). Maximum-likelihood phylogenies were inferred in ARB (13) with RAxML rapid bootstrapping (14), using the GTRGAMMA rate distribution model and 1,000 bootstraps. Numbers at nodes represent bootstrap values for each node, and circles indicate uncertain branch points. The scale bar represents base changes per position.
Whether there is a selective advantage to producing rRNA with intervening elements is (so far as we are aware) an open question; perhaps it provides an additional control on the rate of ribosome production or protection against other nucleases recognizing the conserved target sites. It will be interesting to see if these elements are a general feature of deep-sea Beggiatoaceae; they are not found in B. alba L18D, and a BgP fragment (BGP_R0100; 329 bp) has similarity to BOGUAY 23S sequences to either side of the intron and homing endonuclease but not to the elements themselves. The BgS genome does not appear to include any contigs spanning the insert region.
Group I introns in tRNA genes.
The BOGUAY tRNA-Leu-TAA and tRNA-Arg-TCT genes contain inserts at position 38, the end of the anticodon loop, which may be group I self-splicing introns (see Table S2 in the supplemental material). The first self-splicing bacterial intron discovered was in the anticodon loop of a cyanobacterial tRNA-Leu-TAA (40, 41), and introns in this tRNA are widespread among cyanobacteria and chloroplasts (e.g., see references 42 and 43). At least one has been found in a gammaproteobacterium (44). Group I introns have also been identified in the tRNA-Arg-CCT of several alphaproteobacteria (45, 46), tRNA-Ile-CAT of a betaproteobacterium (45), and tRNA-fMet of some cyanobacteria (47). They are not easily found in databases because their primary sequences are variable, and automated annotation programs generally do not recognize the short-flanking host gene fragments. The closest relative of the BOGUAY tRNA-Leu-TAA was within the cognate gene of the cyanobacterium Leptolyngbya sp. PCC 6703 (AY768525). Directed searches with the BOGUAY tRNAs and their introns identified similar sequences in the BgP but not B. alba genomes.
Group II catalytic introns.
Five possible self-catalytic group II introns were found in the BOGUAY genome (Fig. 4; see also Fig. S5 in the supplemental material). Group II introns were originally discovered in eukaryotic organelles but have since been found in many bacteria and some archaea (reviewed in reference 48). They transpose as RNA: the intron self-catalytically splices from its host RNA, splices into a DNA target site, and is then reverse transcribed to become part of the genome at the new location. The enzymes required for reintegration are often encoded within the introns but may sometimes act in trans. The short (79- or 80-bp) lariat-encoding regions of the five BOGUAY group II intron candidates are highly similar to one another (two are identical) and have some close matches in the BgP and BgS, but not B. alba, genomes. The closest relatives of these sequences (only a few of which have been annotated as introns) are all from cyanobacteria, specifically Trichodesmium erythraeum IMS101 and several Arthrospira species. These do not seem to be embedded in similar genes (not shown), although identifying split genes is not always straightforward. The most parsimonious explanation would be a single transfer from a cyanobacterium into the lineage encompassing these three marine Beggiatoaceae (BOGUAY, BgP, BgS) after its divergence from the B. alba lineage, but additional genome sequences are needed to confirm this. It should also be pointed out that the several functions encoded by a single mobile element may be subject to different evolutionary pressures, so the true phylogeny may not be captured by simple sequence comparisons (49).
Fig 4.

Inferred phylogeny of candidate BOGUAY group II catalytic introns and related sequences. Four sequences annotated as BOGUAY group II introns were used for BLASTN searches of the IMG/ER database (cutoff E value of 9e−06, cutoff score of 58 bits). For sequences not previously annotated (the majority), genome positions are indicated. Three Crocosphaera watsonii sequences were used to root the tree. Nucleic acid sequences were aligned in MEGA5 (10) using MUSCLE (11). Phylogenetic analysis was carried out in MrBayes 3.2 (15) (2 runs of 4 chains each, 1.5 million generations) using the GTR base substitution and invgamma rate variation models. With the first 25% of generations discarded, the final statistics indicated that convergence was reached between the two runs (data not shown). The scale bar represents base changes per position. See Fig. S5 in the supplemental material for the phylogeny inferred by neighbor joining.
Four of the five putative BOGUAY group II introns are adjacent to possible reverse transcriptase and/or transposase genes (see Table S3 in the supplemental material), but because three of the sequences are close to the ends of their contigs, it is not clear what the full complement of associated genes might be. The fourth (00500_2973) is located in the immediate neighborhood of three possible transposases, a possible DNA-binding protein, a possible endonuclease, and a possible retron-type reverse transcriptase. The fifth (00593; positions 280 to 358) is located on a short contig (1,600 bp) with a possible toxin-antitoxin gene pair as the only other identifiable ORFs. Whether these elements are still mobile is not known.
GyrB and DnaE inteins.
Inteins are amino acid sequences that excise self-catalytically from their host proteins posttranslationally, leaving a functional host protein (“extein”; reviewed in reference 50). They may encode homing endonucleases, which make double-stranded cuts in target genes into which the intein genes are spliced from an existing integrated copy. The BOGUAY genome encodes at least two possible inteins, one in the putative DNA gyrase B subunit gene gyrB (Fig. 5A; see also Fig. S6A in the supplemental material), and one in the putative DNA polymerase III alpha subunit gene dnaE (Fig. 5B; see also Fig. S6B in the supplemental material), which belong to an intein family also found in proteins, including replicative DNA helicase, GyrA, and ribonucleoside diphosphate reductases in a wide variety of bacterial and archaeal genomes. Both sequences encode possible homing endonucleases of the LAGLIDADG family (reviewed in reference 51), and both cluster with cyanobacterial sequences, although the identity of the closest relative varies with the phylogenetic inference method used (maximum likelihood or Bayesian analysis). The noncyanobacterial matches are more phylogenetically diverse than for some of the other BOGUAY elements discussed here. For the GyrB intein, these include halophilic archaea (notably Haloquadratum walsbyi, “Walsby's square bacterium” [52]) and two species of sulfur-oxidizing gammaproteobacteria (Fig. 5A), while the BOGUAY DnaE intein is related to a planctomycete and a Bacteroidetes sequence (Fig. 5B). GyrB inteins have not yet been found in other Beggiatoaceae species, but Beggiatoa alba B18LD encodes a possible DnaE intein (more distantly related than the sequences shown here), BgP encodes a DnaB one, and the partial BgS genome includes a short contig encoding a fragment of a replicative DNA helicase (BGS_0184) and a possible N-terminal intein fragment (BGS_0185). The inteins in the different Beggiatoaceae may have been introduced independently, but inferring the evolutionary history of inteins is complicated by the fact that the endonuclease function can be supplied in trans and could therefore decay rapidly in species that happen to bear or obtain a second, compatible intein.
Fig 5.
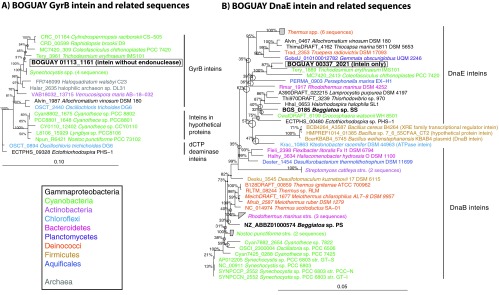
Inferred maximum-likelihood phylogeny of the putative BOGUAY GyrB and DnaE inteins. Relatives of the BOGUAY sequences (highlighted by boxes) were identified by BLASTX searches of the GenBank nr and JCVI IMG/ER databases. The first 100 hits were used for initial neighbor-joining trees, which were pruned to focus on the most closely Beggiatoa-related sequences before further analysis. Inferred amino acid sequences were aligned in MEGA5 (10) using MUSCLE (11). Maximum-likelihood phylogenies were inferred in ARB (13) with RAxML rapid bootstrapping (14), using the PROTMIX rate distribution model and WAG amino acid substitution model. Five hundred bootstraps were used for panel A, and 1,000 were used for panel B. Numbers at nodes represent bootstrap values for each node, and circles indicate uncertain branch points. The scale bar represents amino acid changes per position. Bayesian trees are presented for comparison in Fig. S6 in the supplemental material.
As expected, the exteins of the BOGUAY GyrB and DnaE inteins appear to be gammaproteobacterial and closely related to sequences from other Beggiatoaceae (see Fig. S7 in the supplemental material), and their positions are similar to that predicted from the 16S rRNA phylogeny (MacGregor et al., submitted) (53).
Additional N- and C-terminal intein fragments have been annotated in the BOGUAY genome (00251_2940 and 00127_3146, respectively), which are positioned at the end of their contigs in such an orientation that they could be joined end to end. The concatenated ORFs would encode the uncharacterized conserved protein RtcB (COG1690) containing an endonuclease-bearing intein, with the intein most similar to possible cyanobacterial inteins and the extein most similar to predicted proteins from BgP, B. alba, and many other gammaproteobacteria (not shown).
Sequences related to fdxN excision elements.
The BOGUAY genome includes multiple copies of ORFs similar to cyanobacterial XisH and XisI genes, most found in pairs. They have also been noted in the BgP and BgS genomes (6), and a single xisI-like ORF is found in B. alba L18D (BegalDRAFT_0886). These elements have been best studied in the diazotrophic cyanobacterium Anabaena (Nostoc) sp. strain PCC 7120, in which terminal differentiation of cells within filaments to nitrogen-fixing heterocysts requires excision of DNA fragments interrupting three nitrogen fixation genes, each of which is associated with a site-specific recombinase (nifD/XisA, hupL/XisC, fdxN/XisF) (54, 55). Excision of the fdxN element also requires XisH and XisI, which are encoded within the excised fragment along with XisF. Their mode of action is not known.
There are at least 12 sets of genes encoding putative XisHI homologs in the BOGUAY genome, plus a lone XisI gene at the beginning of a contig (see Table S4 in the supplemental material). The phylogenetic trees (Fig. 6) suggest that xisI and xisH were acquired as a pair on one or more occasions and have diverged as pairs, with xisH being lost or decaying more rapidly (for example, compare XisH 1 and 6 with XisI 1, 6, and 11). No associated recombinase could be identified, although one XisHI copy (on contig 00833) is flanked by putative genes for the RecB family and CRISPR-associated endonucleases. Sequences similar to xisHI have so far been found primarily in cyanobacteria and in only a few phylogenetically dispersed noncyanobacterial genomes, representing either filamentous or pleiomorphic species (see Table S5 in the supplemental material). Of genomes studied to date, the cyanobacterium Coleofasciculus (Microcoleus) chthonoplastes PCC 7420 (filamentous, nondiazotrophic) (56) and the sphingobacterium Haliscomenobacter hydrossis (filamentous) (57) have been annotated with a number of these elements comparable to that in the BOGUAY genome.
Fig 6.

Sequences related to XisH and XisI. The trees include all sequences annotated as XisH, XisI, or fdxN excision-control proteins in IMG/ER (as of August 2012). A neighbor-joining tree was first constructed with all the inferred amino acid sequences, and the database was then divided into XisH- and XisI-like sequences. BOGUAY ORFs found adjacent to each other are indicated by boldface numbers; see Table S4 in the supplemental material for details. There is one additional putative partial XisI gene and 4 partial or split XisH genes in the BOGUAY genome. Phylogenetic analyses were conducted in MEGA5 (10), with sequences aligned by MUSCLE (11). Evolutionary histories were inferred by neighbor joining (76) with 500 bootstrap replicates (77). Evolutionary distances were computed by the Poisson correction method (78) and are in units of amino acid substitutions per site. All ambiguous positions were removed for each sequence pair. For XisH, the analysis involved 114 amino acid sequences, and there were 330 positions in the final data set. For XisI, 171 amino acid sequences and 260 positions were used. The scale bar represents amino acid changes per position. RAxML and Bayesian analyses gave similar groupings (not shown).
Whether any xisHI-like proteins are or were involved in gene inversions outside the cyanobacteria is not known, but gene rearrangements promoting cell differentiation (e.g., heterocyst formation) might be of particular benefit to filamentous species. Sacrificial dead cells (necrosomes) which serve as filament breakage points have been observed in several Beggiatoaceae strains (58), and as far as we are aware no mechanism governing their formation has been identified. The “White Point filament” strain can attach by holdfasts to solid substrates or form rosettes with other cells (59), suggesting specialization by terminal cells. Variable cell morphology has been reported for some close relatives, including several Thiothrix species (gliding gonidia, holdfasts, rosettes, spiral filaments) (60, 61). Any gene rearrangements induced by an Xis-like system might also be more easily tolerated in filamentous species, to the extent that they are able to share cellular contents between individuals, possibly making these elements more likely to persist and evolve new functions. Large bacteria such as the vacuolate Beggiatoaceae can also possess multiple nucleoids per cell, possibly allowing for coexistence of wild-type and mutant alleles.
Sequences similar to ORF 00024_0693.
ORF 00024_0693, found on the contig encoding an abundant orange cytochrome that helps give orange Guaymas Beggiatoaceae their color (5), may be derived from a mobile element. There are at least 28 more similar ORFs annotated in the BOGUAY genome, and they are found in the other sequenced Beggiatoaceae genomes as well (27 in BgP, 1 in BgS, and 1 in B. alba; Fig. 7). The BgP and especially BgS genomes are incomplete (6), so their lower copy numbers may be an artifact, but the B. alba genome is considered complete. A single copy has been annotated in one related sulfur oxidizer (Thiothrix nivea DSM 5205), but as previously noted in relation to some of the BgP copies (6), nearly all other annotated copies are found in cyanobacteria, often in multiple copies per genome. The cyanobacterial species represented include both unicellular (e.g., Cyanothece spp.) and filamentous (e.g., Nostoc punctiforme) types, but the noncyanobacterial species represented are all filamentous gliding bacteria (see Table S5 in the supplemental material). Only a few of these ORFs have been assigned a possible function. The BOGUAY ORF 01192_0187 has been annotated as a type I restriction enzyme R protein N terminus by IMG/ER, which would be consistent with a mobile-element origin(s) for these sequences. One of the two Cyanothece sp. PCC 7822 copies has been assigned to a plasmid (Fig. 7), and the “Nostoc azollae” ORF Aazo_2772 has an N-terminal 141-aa segment similar to the 0693 elements and a longer C-terminal segment which is a predicted IS4 family transposase. Database searches with this sequence yielded many related putative transposase-associated elements, largely in cyanobacteria (not shown). However, there is no obvious relationship between the 0693-like sequences and other mobile elements identified to date in the BOGUAY genome (transposases, XisHI genes, toxin-antitoxin or restriction-modification system genes). Where two 0693 elements are found on the same contig (01192_0187, 01192_0199; 00826_3884, 826_3885; 01185_3406, 01185_3412), their sequences are sufficiently different that they do not appear to be recent duplications.
Fig 7.

Sequences related to hypothetical protein BOGUAY 00024_0693 (arrow). The IMG/ER database was searched with the nucleic acid and inferred amino acid sequences of ORF 00024_0693 (BLASTX, cutoff score of 55 bits, E value of 9e−06; BLASTP, 56 bits, E value of 3e−06). Only BOGUAY 01192_0187 (star) has been annotated in IMG/ER with a possible function, containing a possible type I restriction enzyme R protein N-terminal domain (pfam04313). An ORF from Cyanothece sp. PCC 7822 (circle) has been assigned to a plasmid rather than a chromosome. Phylogenetic analyses were conducted as described for Fig. 6. The analysis involved 121 amino acid sequences. All ambiguous positions were removed for each sequence pair. There were a total of 328 positions in the final data set. The scale bar represents amino acid changes per position. RAxML and Bayesian analyses gave similar groupings (not shown).
The phylogenetic distribution of the xisHI and 00024_0693-like elements is therefore similar, including a wide range of cyanobacteria (motile and nonmotile, filamentous and unicellular, diazotrophic and nondiazotrophic, differentiating and nondifferentiating) but only a few other bacteria, whose shared characteristic appears to be filamentous or pleiomorphic cell morphology. Given the known habitats of the strains concerned (see Table S5 in the supplemental material), microbial mats or (very recently) activated sludge (57) seem a likely environment for gene exchange. The large surface area of filaments, particularly vacuolated ones, should increase the likelihood of encounters with mobile elements, which could mean higher uptake rates. There may also be elements (e.g., transducing phage) specializing in attachment to cell surface components shared among filamentous species, such as an element of the gliding motility apparatus.
Restriction-modification systems.
Restriction-modification systems generally combine methylation of specific DNA sequences with double-stranded DNA cutting by nucleases recognizing unmethylated copies of the same sequence, either within the recognition sequence or at a specific or undefined distance from it. The most common role of restriction-modification systems is thought to be defense against foreign DNA, such as phage: restriction sites on host DNA are protected by methylation, but incoming DNA is not, unless it derives from a source with a cognate system. There is extensive evidence for horizontal transfer of restriction-modification systems (e.g., see references 62 and 63), and there are often systems of multiple origins within a single genome (e.g., see reference 64). These may still be associated with evidence of mobile elements, including long inverted repeats and transposase genes (e.g., see reference 65). The pace of genetic exchange is such that even strains of the same species may have quite different restriction enzyme complements. There are also examples of restriction-modification system components that have apparently been adopted for other functions, such as methylation control of gene expression (e.g., see references 66 and 67).
For the BOGUAY genome, closest relatives of possible restriction-modification genes found by JCVI, IMG/ER, RAST, or BLASTX searches for one enzyme type in the neighborhood of the other were identified by BLASTX searches of REBASE, a curated database of DNA and RNA modification enzyme sequences (68), summarized in Table S6 (see Tables S7 to S9 for details) in the supplemental material. Unlike the mobile elements discussed above, these have a wide range of phylogenetic affiliations, both bacterial and archaeal, with no special concentration of cyanobacterium-like sequences. The likely role of restriction enzymes in defense against foreign DNA should favor maintenance of a varied arsenal.
Toxin-antitoxin systems.
Toxin-antitoxin (TA) systems typically consist of a cotranscribed long-lived bacteriostatic or bactericidal toxin and short-lived antitoxin (recently reviewed in references 69 and 70). Modes of toxin activity include translation inhibition, mRNA cleavage, and DNA gyrase inhibition. TA systems were originally discovered on plasmids, where they can serve as maintenance mechanisms; cells that lose the plasmid, or do not acquire it upon cell division, will be killed or inhibited by the toxin as the antitoxin decays. They have since been found on both mobile elements and chromosomes of many bacteria and archaea. A single chromosome may bear multiple copies of multiple classes of TA elements (see reference 71 for a compilation), which appears to be the case for the BOGUAY genome as well.
Putative toxin and antitoxin genes were identified from the combined JGI and RAST annotations plus directed searches (BLASTX with flanking DNA, or BLASTP with unannotated nearby ORFs), mostly for antitoxins in the neighborhood of orphan toxins (summarized in Table S10 in the supplemental material). One antitoxin may neutralize more than one type of toxin, and toxins with different modes of action may have similar structures (reviewed in reference 72), so the groupings here should be considered provisional. There may be decayed, inactive loci (73) included. Toxins and their corresponding antitoxins are typically cotranscribed, at least in the well-studied cases. Many of the BOGUAY elements are in pairs or found at the ends of contigs, such that the paired element may simply not have been sequenced or connected (see Table S11 in the supplemental material). The pairs are not always the expected ones, however; the classification system and/or assignment of sequences to groups may need refinement.
Examples of the inferred phylogeny of a toxin and an antitoxin are shown in Fig. S8 in the supplemental material. They are similar to those of the restriction-modification systems (not shown) and in clear contrast to those of the other putative mobile elements discussed here. BOGUAY sequences may group with other Beggiatoaceae sequences, suggesting divergence within this lineage, but the next-closest neighbors for the examples shown include Gammaproteobacteria, Cyanobacteria, Deltaproteobacteria, and Alphaproteobacteria.
Genes possibly exchanged between cyanobacteria and the BOGUAY strain.
For a first overview of the genes that may have been transferred (in one direction or the other) with the various cyanobacterium-affiliated mobile elements, we have analyzed the results of a BLASTP search of the UniProt (74) database with the BOGUAY genome, which reported matches above a cutoff value (E value ≤ 1.00e−05), in order, to a maximum of five. Given the large number of sequenced gammaproteobacterial genomes, matches outside this group are suggestive (although not proof) of gene exchange. Once sequences putatively encoding elements such as restriction enzymes, transposases, XisH elements, and toxin-antitoxin systems were removed, some 228 ORFs with at least one high-scoring cyanobacterial match remained. Of these, 121 have so far been annotated only as a hypothetical protein or similar in BOGUAY (see Table S10 in the supplemental material). Of the remaining 107, some 33 can be classified as sensory and signal transduction proteins (see Table S11 in the supplemental material). Without knowing their specificity, it would be risky to speculate on possible evolutionary advantages, but species living in the same microbial mat might benefit from similar sensory systems. ORFs putatively encoding enzymes involved in cell wall and membrane biogenesis and possibly chromosome partitioning are also represented, as are possible proteins for Mn/Zn and ferrous iron uptake, Na+/Ca+ exchange, and multidrug efflux. The other major category of similar genes appears to be proteins possibly involved in secondary metabolite synthesis.
Perspectives.
One question arising from this consideration of possible mobile elements in the BOGUAY genome is their role over shorter and longer time scales. In the short term, perhaps there is cell differentiation along filaments by an Xis-like gene inversion or rearrangement mechanism. This might be investigated by single-cell sequencing or targeted gene amplification of dissected filaments. In the longer term, what mobile elements and gene rearrangements are seen in filaments of different colors or in Beggiatoaceae collected from different locations (for example, at hydrothermal sites compared to cold seeps)? If found, these might be clues to key physiological differences. In the very long term, how much of the evolution of microbial mat communities is recorded in the gene mosaics of their current inhabitants? Orange Guaymas Beggiatoa would seem to have spent some time in the company of cyanobacteria before colonizing the deep-ocean floor, not unexpected given the current geographical distribution of Beggiatoaceae and/or to have an active genetic connection with extant surface-dwelling species or strains. The inferred phylogeny of some carbon metabolism genes, on the other hand, has suggested interspecies gene transfers at hydrothermal sites (B. J. MacGregor, unpublished data). It may also be instructive to search for genes with phylogenies similar to that of the XisHI elements, which seem to be shared among filamentous species. Continued study of the Beggiatoaceae-hosting microbial mats of Baja California, Guaymas Basin, and elsewhere could help to answer some of these questions.
Supplementary Material
ACKNOWLEDGMENTS
Thanks to the captain and crews of the RV Atlantis and HOV Alvin and to the shipboard parties of legs AT15-40 and AT15-56.
Genome sequencing was performed by the J. Craig Venter Institute, with funding from The Gordon and Betty Moore Foundation Marine Microbial Genome Sequencing Project. The use of RAST was supported in part by National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services (NIAD), under contract HHSN266200400042C. The Guaymas Basin project was funded by NSF OCE 0647633.
Footnotes
Published ahead of print 19 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03821-12.
REFERENCES
- 1. Jannasch HW, Nelson DC, Wirsen CO. 1989. Massive natural occurrence of unusually large bacteria (Beggiatoa sp.) at a hydrothermal deep-sea vent site. Nature 342:834–836 [Google Scholar]
- 2. Hinck S, Mussmann M, Salman V, Neu TR, Lenk S, de Beer D, Jonkers HM. 2011. Vacuolated Beggiatoa-like filaments from different hypersaline environments form a novel genus. Environ. Microbiol. 13:3194–3205 [DOI] [PubMed] [Google Scholar]
- 3. Salman V, Amann R, Girnth AC, Polerecky L, Bailey JV, Hogslund S, Jessen G, Pantoja S, Schulz-Vogt HN. 2011. A single-cell sequencing approach to the classification of large, vacuolated sulfur bacteria. Syst. Appl. Microbiol. 34:243–259 [DOI] [PubMed] [Google Scholar]
- 4. Dillon JG, Miller S, Bebout B, Hullar M, Pinel N, Stahl DA. 2009. Spatial and temporal variability in a stratified hypersaline microbial mat community. FEMS Microbiol. Ecol. 68:46–58 [DOI] [PubMed] [Google Scholar]
- 5. MacGregor BJ, Biddle JF, Siebert JR, Staunton E, Hegg EL, Matthysse AG, Teske A. 2013. Why orange Guaymas Basin Beggiatoa (Maribeggiatoa) spp. are orange: single-filament genome-enabled identification of an abundant octaheme cytochrome with hydroxylamine oxidase, hydrazine oxidase, and nitrite reductase activities. Appl. Environ. Microbiol. 79:1183–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mussmann M, Hu FZ, Richter M, de Beer D, Preisler A, Jørgensen Huntemann BBM, Glöckner FO, Amann R, Koopman WJH, Lasken RS, Janto B, Hogg J, Stoodley P, Boissy R, Ehrlich GD. 2007. Insights into the genome of large sulfur bacteria revealed by analysis of single filaments. PLoS Biol. 5:1923–1937. 10.1371/journal.pbio.0050230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Biddle JF, House CH, Brenchley JE. 2005. Microbial stratification in deeply buried marine sediment reflects changes in sulfate/methane profiles. Geobiology 3:287–295 [Google Scholar]
- 8. Markowitz VM, Mavromatis K, Ivanova NN, Chen I-MA, Chu K, Kyrpides NC. 2009. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 25:2271–2278 [DOI] [PubMed] [Google Scholar]
- 9. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Papadopoulos JS, Agarwala R. 2007. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 23:1073–1079 [DOI] [PubMed] [Google Scholar]
- 13. Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, Buchner A, Lai T, Steppi S, Jobb G, Förster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, König A, Liss T, Lüssmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer K-H. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32:1363–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690 [DOI] [PubMed] [Google Scholar]
- 15. Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61:539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barloy-Hubler F, Lelaure V, Galibert F. 2001. Ribosomal protein gene cluster analysis in eubacterium genomics: homology between Sinorhizobium meliloti strain 1021 and Bacillus subtilis. Nucleic Acids Res. 29:2747–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coenye T, Vandamme P. 2005. Organisation of the S10, spc and alpha ribosomal protein gene clusters in prokaryotic genomes. FEMS Microbiol. Lett. 242:117–126 [DOI] [PubMed] [Google Scholar]
- 18. Lane WJ, Darst SA. 2010. Molecular evolution of multisubunit RNA polymerases: sequence analysis. J. Mol. Biol. 395:671–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mavromatis K, Ivanova NN, Chen I-MA, Szeto E, Markowitz VM, Kyrpides NC. 2009. The DOE-JGI standard operating procedure for the annotations of microbial genomes. Stand. Genome Sci. 1:63–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tåquist H, Cui Y, Ardell DH. 2007. TFAM 1.0: an online tRNA function classifier. Nucleic Acids Res. 35:W350–W353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jühling F, Mörl M, Hartmann RK, Sprinzl M, Stadler PF, Pütz J. 2009. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 37:D159–D162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Silva FJ, Belda E, Talens SE. 2006. Differential annotation of tRNA genes with anticodon CAT in bacterial genomes. Nucleic Acids Res. 34:6015–6022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grosjean H, de Crécy-Lagard V, Marck C. 2010. Deciphering synonymous codons in the three domains of life: co-evolution with specific tRNA modification enzymes. FEBS Lett. 584:252–264 [DOI] [PubMed] [Google Scholar]
- 25. Randau L, Söll D. 2008. Transfer RNA genes in pieces. EMBO Rep. 9:623–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee ZMP, Bussema C, Schmidt TM. 2009. rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res. 37:D489–D493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dethlefsen L, Schmidt TM. 2007. Performance of the translational apparatus varies with the ecological strategies of bacteria. J. Bacteriol. 189:3237–3245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rocha EPC. 2004. Codon usage bias from tRNA's point of view: redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 14:2279–2286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ibba M, Becker HD, Stathopoulos C, Tumbula DL, Söll D. 2000. The adaptor hypothesis revisited. Trends Biochem. Sci. 25:311–316 [DOI] [PubMed] [Google Scholar]
- 30. Marrs B, Kaplan S. 1970. 23S precursor ribosomal RNA of Rhodopseudomonas spheroides. J. Mol. Biol. 49:297–317 [DOI] [PubMed] [Google Scholar]
- 31. Haraszthy VI, Sunday GJ, Bobek LA, Motley TS, Preus H, Zambon JJ. 1992. Identification and analysis of the gap region in the 23S ribosomal RNA from Actinobacillus actinomycetemcomitans. J. Dent. Res. 71:1561–1568 [DOI] [PubMed] [Google Scholar]
- 32. Doolittle WF. 1973. Postmaturational cleavage of 23S ribosomal ribonucleic acid and its metabolic control in the blue-green alga Anacystis nidulans. J. Bacteriol. 113:1256–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Salman V, Amann R, Shub DA, Schulz-Vogt HN. 2012. Multiple self-splicing introns in the 16S rRNA genes of giant sulfur bacteria. Proc. Natl. Acad. Sci. U. S. A. 109:4203–4208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haugen P, Bhattacharya D, Palmer JD, Turner S, Lewis LA, Pryer KM. 2007. Cyanobacterial ribosomal RNA genes with multiple, endonuclease-encoding group I introns. BMC Evol. Biol. 7:159. 10.1186/1471-2148-7-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wood AM, Miller SR, Li WKW, Castenholz RW. 2002. Preliminary studies of cyanobacteria, picoplankton, and virioplankton in the Salton Sea with special attention to phylogenetic diversity among eight strains of filamentous cyanobacteria. Hydrobiologia 473:77–92 [Google Scholar]
- 36. Ghigo E, Pretat L, Desnues B, Capo C, Raoult D, Mege JL. 2009. Intracellular life of Coxiella burnetii in macrophages: an update. Ann. N. Y. Acad. Sci. 1166:55–66 [DOI] [PubMed] [Google Scholar]
- 37. Kersh GJ, Wolfe TM, Fitzpatrick KA, Candee AJ, Oliver LD, Patterson NE, Self JS, Priestley RA, Loftis AD, Massung RF. 2010. Presence of Coxiella burnetii DNA in the environment of the United States, 2006 to 2008. Appl. Environ. Microbiol. 76:4469–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pommier T, Canbäck B, Riemann L, Boström KH, Simu K, Lundberg P, Tunlid A, Hagström Å. 2007. Global patterns of diversity and community structure in marine bacterioplankton. Mol. Ecol. 16:867–880 [DOI] [PubMed] [Google Scholar]
- 39. Kersh GJ, Lambourn DM, Self JS, Akmajian AM, Stanton JB, Baszler TV, Raverty SA, Massung RF. 2010. Coxiella burnetii infection of a Steller sea lion (Eumetopias jubatus) found in Washington State. J. Clin. Microbiol. 48:3428–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuhsel MG, Strickland R, Palmer JD. 1990. An ancient group I intron shared by eubacteria and chloroplasts. Science 250:1570–1573 [DOI] [PubMed] [Google Scholar]
- 41. Xu M-Q, Kathe SD, Goodrich-Blair H, Nierzwicki-Bauer SA, Shub DA. 1990. Bacterial origin of a chloroplast intron: conserved self-splicing group I introns in cyanobacteria. Science 250:1566–1570 [DOI] [PubMed] [Google Scholar]
- 42. Olsson S, Kaasalainen U, Rikkinen J. 2012. Reconstruction of structural evolution in the trnL intron P6b loop of symbiotic Nostoc (Cyanobacteria). Curr. Genet. 58:49–58 [DOI] [PubMed] [Google Scholar]
- 43. Paquin B, Kathe SD, Nierzwicki-Bauer SA, Shub DA. 1997. Origin and evolution of group I introns in cyanobacterial tRNA genes. J. Bacteriol. 179:6798–6806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vepritskiy AA, Vitol IA, Nierzwicki-Bauer SA. 2002. Novel group I intron in the tRNALeu(UAA) gene of a γ-proteobacterium isolated from a deep subsurface environment. J. Bacteriol. 184:1481–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reinhold-Hurek B, Shub DA. 1992. Self-splicing introns in tRNA genes of widely divergent bacteria. Nature 357:173–176 [DOI] [PubMed] [Google Scholar]
- 46. Paquin B, Heinfling A, Shub DA. 1999. Sporadic distribution of introns among α-purple bacteria: evidence for horizontal transmission and transposition of a group I intron. J. Bacteriol. 181:1049–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bonocora RP, Shub DA. 2001. A novel group I intron-encoded endonuclease specific for the anticodon region of tRNAfmet genes. Mol. Microbiol. 39:1299–1306 [DOI] [PubMed] [Google Scholar]
- 48. Lambowitz AM, Zimmerly S. 2011. Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb. Perspect. Biol. 3:a003616. 10.1101/cshperspect.a003616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Simon DM, Kelchner SA, Zimmerly S. 2009. A broadscale phylogenetic analysis of group II intron RNAs and intron-encoded reverse transcriptases. Mol. Biol. Evol. 26:2795–2808 [DOI] [PubMed] [Google Scholar]
- 50. Elleuche S, Poggeler S. 2010. Inteins, valuable genetic elements in molecular biology and biotechnology. Appl. Microbiol. Biotechnol. 87:479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stoddard BL. 2005. Homing endonuclease structure and function. Q. Rev. Biophys. 38:49–95 [DOI] [PubMed] [Google Scholar]
- 52. Bolhuis H, Palm P, Wende A, Falb M, Rampp M, Rodriguez-Valera F, Pfeiffer F, Oesterhelt D. 2006. The genome of the square archaeon Haloquadratum walsbyi: life at the limits of water activity. BMC Genomics 7:12. 10.1186/1471-2164-7-169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McKay LJ, MacGregor BJ, Biddle JF, Albert DB, Mendlovitz HP, Hoer DR, Lipp JS, Lloyd KG, Teske AP. 2012. Spatial heterogeneity and underlying geochemistry of phylogenetically diverse orange and white Beggiatoa mats in Guaymas Basin hydrothermal sediments. Deep Sea Res. 67:21–31 [Google Scholar]
- 54. Carrasco CD, Holliday SD, Hansel A, Lindblad P, Golden JW. 2005. Heterocyst-specific excision of the Anabaena sp. strain PCC 7120 hupL element requires xisC. J. Bacteriol. 187:6031–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Henson BJ, Pennington LE, Watson LE, Barnum SR. 2008. Excision of the nifD element in the heterocystous cyanobacteria. Arch. Microbiol. 189:357–366 [DOI] [PubMed] [Google Scholar]
- 56. Rippka R, Deruelles J, Waterbury JB, Herdman M, Stanier RY. 1979. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J. Gen. Microbiol. 111:1–61 [Google Scholar]
- 57. Nielsen PH, Kragelund C, Seviour RJ, Nielsen JL. 2009. Identity and ecophysiology of filamentous bacteria in activated sludge. FEMS Microbiol. Rev. 33:969–998 [DOI] [PubMed] [Google Scholar]
- 58. Kamp A, Røy H, Schulz-Vogt HN. 2008. Video-supported analysis of Beggiatoa filament growth, breakage, and movement. Microb. Ecol. 56:484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kalanetra KM, Huston SL, Nelson DC. 2004. Novel, attached, sulfur-oxidizing bacteria at shallow hydrothermal vents possess vacuoles not involved in respiratory nitrate accumulation. Appl. Environ. Microbiol. 70:7487–7496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Aruga S, Kamagata Y, Kohno T, Hanada S, Nakamura K, Kanagawa T. 2002. Characterization of filamentous Eikelboom type 021N bacteria and description of Thiothrix disciformis sp. nov. and Thiothrix flexilis sp. nov. Int. J. Syst. Evol. Microbiol. 52:1309–1316 [DOI] [PubMed] [Google Scholar]
- 61. Chernousova E, Gridneva E, Grabovich M, Dubinina G, Akimov V, Rossetti S, Kuever J. 2009. Thiothrix caldifontis sp. nov. and Thiothrix lacustris sp. nov., gammaproteobacteria isolated from sulfide springs. Int. J. Syst. Evol. Microbiol. 59:3128–3135 [DOI] [PubMed] [Google Scholar]
- 62. Jeltsch A, Pingoud A. 1996. Horizontal gene transfer contributes to the wide distribution and evolution of type II restriction-modification systems. J. Mol. Evol. 42:91–96 [DOI] [PubMed] [Google Scholar]
- 63. Naderer M, Brust JR, Knowle D, Blumenthal RM. 2002. Mobility of a restriction-modification system revealed by its genetic contexts in three hosts. J. Bacteriol. 184:2411–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nobusato A, Uchiyama I, Kobayashi I. 2000. Diversity of restriction-modification gene homologues in Helicobacter pylori. Gene 259:89–98 [DOI] [PubMed] [Google Scholar]
- 65. Furuta Y, Abe K, Kobayashi I. 2010. Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res. 38:2428–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fox KL, Dowideit SJ, Erwin AL, Srikhanta YN, Smith AL, Jennings MP. 2007. Haemophilus influenzae phasevarions have evolved from type III DNA restriction systems into epigenetic regulators of gene expression. Nucleic Acids Res. 35:5242–5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Srikhanta YN, Maguire TL, Stacey KJ, Grimmond SM, Jennings MP. 2005. The phasevarion: a genetic system controlling coordinated, random switching of expression of multiple genes. Proc. Natl. Acad. Sci. U. S. A. 102:5547–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Roberts RJ, Vincze T, Posfai J, Macelis D. 2010. REBASE—a database for DNA restriction and modification: enzymes, genes, and genomes. Nucleic Acids Res. 38:D234–D236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Blower TR, Salmond GPC, Luisi B. 2011. Balancing at survival's edge: the structure and adaptive benefits of prokaryotic toxin-antitoxin partners. Curr. Opin. Struct. Biol. 21:109–118 [DOI] [PubMed] [Google Scholar]
- 70. Van Melderen L. 2010. Toxin-antitoxin systems: why so many, what for? Curr. Opin. Microbiol. 13:781–785 [DOI] [PubMed] [Google Scholar]
- 71. Shao YC, Harrison EM, Bi DX, Tai C, He XY, Ou HY, Rajakumar K, Deng ZX. 2011. TADB: a Web-based resource for type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res. 39:D606–D611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Arbing MA, Handelman SK, Kuzin AP, Verdon G, Wang C, Su M, Rothenbacher FP, Abashidze M, Liu MH, Hurley JM, Xiao R, Acton T, Inouye M, Montelione GT, Woychik NA, Hunt JF. 2010. Crystal structures of Phd-Doc, HigA, and YeeU establish multiple evolutionary links between microbial growth-regulating toxin-antitoxin systems. Structure 18:996–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mine N, Guglielmini J, Wilbaux M, Van Melderen L. 2009. The decay of the chromosomally encoded ccdO157 toxin-antitoxin system in the Escherichia coli species. Genetics 181:1557–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Apweiler R, Martin MJ, O'Donovan C, Magrane M, Alam-Faruque Y, Antunes R, Barrell D, Bely B, Bingley M, Binns D, Bower L, Browne P, Chan WM, Dimmer E, Eberhardt R, Fazzini F, Fedotov A, Foulger R, Garavelli J, Castro LG, Huntley R, Jacobsen J, Kleen M, Laiho K, Legge D, Lin QA, Liu WD, Luo J, Orchard S, Patient S, Pichler K, Poggioli D, Pontikos N, Pruess M, Rosanoff S, Sawford T, Sehra H, Turner E, Corbett M, Donnelly M, van Rensburg P, Xenarios I, Bougueleret L, Auchincloss A, Argoud-Puy G, Axelsen K, Bairoch A, Baratin D, Blatter MC, Boeckmann B, Bolleman J, Bollondi L, Boutet E, Quintaje SB, Breuza L, Bridge A, deCastro E, Coudert E, Cusin I, Doche M, Dornevil D, Duvaud S, Estreicher A, Famiglietti L, Feuermann M, Gehant S, Ferro S, Gasteiger E, Gateau A, Gerritsen V, Gos A, Gruaz-Gumowski N, Hinz U, Hulo C, Hulo N, James J, Jimenez S, Jungo F, Kappler T, Keller G, Lara V, Lemereier P, Lieberherr D, Martin X, Masson P, Moinat M, Morgat A, Paesano S, Pedruzzi I, Pilbout S, Poux S, Pozzato M, Redaschi N, Rivoire C, Roechert B, Schneider M, Sigrist C, Sonesson K, Staehli S, Stanley E, Stutz A, Sundaram S, Tognolli M, Verbregue L, Veuthey AL, Wu CH, Arighi CN, Arminski L, Barker WC, Chen CM, Chen YX, Dubey P, Huang HZ, Mazumder R, McGarvey P, Natale DA, Natarajan TG, Nchoutmboube J, Roberts NV, Suzek BE, Ugochukwu U, Vinayaka CR, Wang QH, Wang YQ, Yeh LS, Zhang JA. 2011. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 39:D214–D219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and Web-based tools. Nucleic Acids Res. 41:D590–D596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406–425 [DOI] [PubMed] [Google Scholar]
- 77. Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791 [DOI] [PubMed] [Google Scholar]
- 78. Zuckerkandl E, Pauling L. 1965. Evolutionary convergence and divergence in proteins, p 97–166 In Bryson V, Vogel HJ. (ed), Evolving genes and proteins. Academic Press, New York, NY [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.