

Abstract
Microbes play an essential role in ecosystem functions, including carrying out biogeochemical cycles, but are currently considered a black box in predictive models and all global biodiversity debates. This is due to (i) perceived temporal and spatial variations in microbial communities and (ii) lack of ecological theory explaining how microbes regulate ecosystem functions. Providing evidence of the microbial regulation of biogeochemical cycles is key for predicting ecosystem functions, including greenhouse gas fluxes, under current and future climate scenarios. Using functional measures, stable-isotope probing, and molecular methods, we show that microbial (community diversity and function) response to land use change is stable over time. We investigated the change in net methane flux and associated microbial communities due to afforestation of bog, grassland, and moorland. Afforestation resulted in the stable and consistent enhancement in sink of atmospheric methane at all sites. This change in function was linked to a niche-specific separation of microbial communities (methanotrophs). The results suggest that ecological theories developed for macroecology may explain the microbial regulation of the methane cycle. Our findings provide support for the explicit consideration of microbial data in ecosystem/climate models to improve predictions of biogeochemical cycles.
INTRODUCTION
Soil microbial communities are among the most diverse and complex natural communities and are responsible for many ecologically and economically important ecosystem processes (1). For example, soil microbes carry out key steps in global biogeochemical cycles, and their activities influence primary productivity, plant and animal diversity, and Earth's climate, such as greenhouse gas emissions (2). Despite their essential role in ecosystem function, microbial communities are considered a black box in predictive ecosystem and climate models. This neglect is mainly because (i) microbial communities are regarded as being omnipresent and functionally redundant, (ii) there is a lack of theoretical approaches to disentangle microbial regulation of ecosystem functions from other biotic and abiotic drivers, and (iii) temporal and spatial variation in environmental microbes is considered too large to be meaningful in the predictive models. However, detailed studies on the biodiversity-ecosystem function relationship for plant communities have demonstrated that both the magnitude and stability of ecosystem functions are sensitive to loss of diversity. On the other hand, more-diverse plant communities appear to be more productive (in terms of biomass) and more stable in the face of disturbance (3–5). Two hypotheses to explain the underlying basis of the relationship between ecosystem processes and diversity have been put forward. The “complementarity hypothesis” states that biotic interactions and niche differentiation collectively result in a positive biodiversity-ecosystem function relationship in plant communities, whereas the “selection hypothesis” suggests that the magnitude of the process may be the result of the presence of one to a few particularly productive (key) species (3, 5, 6). For plants, the evidence from a number of pioneering studies overwhelmingly supports the complementarity hypothesis. For the microbial community-ecosystem function relationship, distinguishing between these two competing hypotheses is key in order to determine whether all functional microbial communities, or only selected species, need to be conserved or restored to maintain soil functions. Additionally, if either of these hypotheses clearly demonstrates the microbial regulation of the biogeochemical cycle, such knowledge may be further used to develop parameterized microbial data for incorporation into predictive models, as has been done for plant communities (7–11).
However, understanding the role of soil microbial communities in biodiversity-ecosystem function is made more complex due to the diversity of functions mediated by microbes, whether they are rare or abundant and whether they mediate specific or general processes. For example, organic matter decomposition is carried out by a large number of microbial species while xenobiotic degradation capability is restricted to more-specialized species. Should the biodiversity-ecosystem function relationship also hold for the microbial community, diversity loss would have a greater effect on some functions than on others. Moreover, the global importance of microbes in soil function, combined with the lack of knowledge on how the variability in composition and functioning of these communities is affected, necessitates a detailed examination of consistent microbial response to disturbance over time and space. This is a fundamental requirement in order to understand and predict how microbial community and ecosystem functions will respond to global changes (12, 13). Here we used land use change (tree growth) as a treatment to provide evidence that both microbial community structure and the rate of biogeochemical cycles are stable over time and space. Our results provide correlative evidence that ecological theories developed for macroorganisms, such as plants and animals (macroecology), may explain the microbial regulation of ecosystem functions. We used methane (CH4) flux and associated microflora (methanogens and methanotrophs) in our study because this function is restricted to selected microbial taxa which are well characterized. This allowed us to test our hypothesis without manipulating species diversity or constructing artificial consortia, which has huge confounding effects due to the unculturability of the majority (>99%) of environmental microbes (14, 15).
MATERIALS AND METHODS
A full description of the field sites and methodologies is provided in the supplemental material.
Field site description.
Four sites were investigated in Scotland (see Table S1 in the supplemental material). As a result, three types of land use change were examined: afforestation of bog with pine trees (Bad à Cheo), conversion of grassland to pine forest (Glensaugh), and natural colonization of moorland by birch stands (Craggan and Tulchan). For ease of analysis, afforestation of bog and grassland (at Bad à Cheo and Glensaugh) and tree invasion (at Craggan and Tulchan) were referred to as “tree growth” in this paper to describe the general change in land use. Craggan and Tulchan were sites with similar habitats and were ∼7 km apart, whereas the Bad à Cheo and Glensaugh sites both had pine woodland and were ∼178 km apart.
Soil sampling and soil analyses.
Briefly, for each habitat from each site and at each season, 12 intact soil cores were collected in stainless steel rings (10-cm diameter, 0- to 10-cm depth). They were ∼2 m apart from each other over 3 transects about 10 m apart and were grouped in triplicate for measurement of CH4 fluxes (n = 4 replicates per season). Following gas measurements (see below), the soils were processed for physicochemical analysis as well as for biochemistry and molecular biology work. Chemical properties (pH, total C and N, mineral N [NH4+-N and NO3−-N], and moisture) were measured periodically (autumn and summer). Physical properties (particle size, bulk density, and water-filled pore space [WFPS]) were measured once (summer). Soil property data from the different sites and habitats are presented in Table S2 in the supplemental material.
Function measurements and upscaling.
Atmospheric CH4 concentrations inside the headspace of the closed polyvinyl chloride (PVC) chambers were measured on a gas chromatograph (Trace GC; Thermo-Finnigan, Italy) using an analytical capillary column, PLOT Al2O3/KCl FS (length = 50 m; inner diameter [ID] = 0.53 mm; outer diameter [OD] = 0.70 mm; 10-μm-thick film) (Varian, United Kingdom), and a flame ionization detector (FID) for the detection of CH4. Four headspace measurements were made at 30-min intervals over a 90-min period (t0, t30, t60, and t90). A linear model (16, 17) was then applied in order to estimate the CH4 flux inside the chamber headspace.
Analysis of the functional microbial community using molecular ecology approaches. (i) DNA isolation.
Soil (∼250 mg) from each sample was used for DNA extraction using a PowerSoil-htp 96-well soil DNA isolation kit (Mobio). DNA concentrations were measured using a spectrophotometer (NanoDrop ND-1000; NanoDrop Technologies). We characterized the methanotrophic communities using the molecular marker from the functional gene pmoA. It encodes the putative active site of the particulate methane monooxygenase (pMMO) enzyme (18), which is specific to all known methanotrophs except Methylocella and Methyloferula spp. (19–21). We used primers targeting 16S rRNA genes for type II methanotrophs (22) to identify the relative abundance of methanotrophs which do not possess pmoA genes. Terminal restriction fragment length polymorphism (T-RFLP) analyses did not produce any T-RF assigned to Methylocella spp. The pmoA primers used in this study are described elsewhere (23). Methanogens were characterized by targeting methyl coenzyme M reductase (mcrA) genes using the primers and PCR conditions described previously (24).
(ii) T-RFLP analysis.
The PCR products for the pmoA and mcrA genes for the T-RFLP analysis were fluorescently labeled with a forward primer (VIC-pmo189F and FAM-mcrA-F, respectively). A known amount of purified PCR amplicons (100 ng) was used with the restriction enzyme HhaI (25). T-RFLP analysis (n = 4 for each treatment and season) was carried out on an automated sequencer, an ABI Prism 3130xl genetic analyzer (Applied Biosystems, United Kingdom). Only peaks between 30 and 550 bp were considered to avoid terminal restriction fragments (T-RFs) caused by primer-dimers and obtain fragments within the linear range of the internal size standard (26).
(iii) Diagnostic pmoA microarray analysis.
In order to validate further our T-RFLP data, we performed diagnostic pmoA microarray analysis on all samples collected in summer (n = 4 for each treatment). Microarray analysis was carried out using a modified method of Stralis-Pavese et al. (27, 28). The different steps involved are briefly described here. The PCR products for the pmoA gene for the microarray assay were generated in the same way as detailed above except that no fluorescently labeled forward primer was used and the reverse primer (pmo650R) contained the T7 promoter site (5′-TAATACGACTCACTATAG-3′) at its 5′ end, which enabled T7 RNA polymerase-mediated in vitro transcription. Details of the in vitro transcription and hybridization procedures are in the supplemental material. Results were normalized to a positive-control probe, mtrof173. The hybridization signal for each probe was expressed as a percentage of the signal of the positive-control probe mtrof173 on the same array (29). The current version of the pmoA array contains 199 oligonucleotide probes targeting pmoA of methanotrophs, amoA (encoding a subunit of ammonia monooxygenase) of ammonia oxidizers, and other functionally related bacteria (28).
Identifying an active microbial community by PLFA-SIP.
Microcosm experiments were designed to enrich fresh soils (∼10 g) with 13CH4 (∼100 ppm) in 155-ml glass bottles, sealed with rubber septum, and crimped with an aluminum seal as described previously (26). Stable-isotope probing of phospholipid fatty acid (PLFA-SIP) was performed on the autumn and summer soils only (n = 4 for each treatment and each season). A subsample of all the 13C-enriched soils was freeze-dried, milled, and used for extracting PLFAs (26, 30). Standard nomenclature was used for PLFAs (31). Quantification of PLFA contents was based on the normalized peak area of each PLFA, all of which were compared to the peak area of the C19:0 PLFA internal standard, accounting for the weight of soil used for the PLFA extraction (32).
Statistical analysis. (i) T-RFLP data processing.
Raw data from GeneMapper were exported to be used with T-REX, an online software for the processing and analysis of T-RFLP data (http://trex.biohpc.org) (33). We used a standard approach for processing T-RFLP data (25, 33, 34). Briefly, quality control procedures included noise filtering and T-RF alignment (clustering threshold = 2 bp), and T-RFs were omitted if they occurred either in less than 2% of the total number of samples or with a relative abundance of less than 3% within a specific sample. This process led to 16 clean T-RFs, and all of them were included in the statistical analyses. Analysis of data matrices used the additive main effect and multiplicative interaction (AMMI) model based on analysis of variance (ANOVA), as discussed by Culman et al. (34). Interaction principal component (IPC) scores were obtained and used for further statistical analyses (see below). Only the T-RFs that were considered “true” by the T-REX analysis were used for subsequent analysis. The relative abundance of a detected T-RF within a given T-RFLP profile was calculated as the respective signal height of each peak divided by the total peak height of all the peaks of the T-RFLP profile.
(ii) Statistical tests.
The significance of differences in soil characteristics, oxidation rates, and community structures (IPC scores) with different land use types and during different seasons was determined by nested ANOVA (followed by Tukey's test for multiple pairwise comparisons [α = 0.05]) and multivariate ANOVA (MANOVA). Regression analysis was used to explore the linear relationship (α = 0.05) between methanotroph diversity (community structure and operational taxonomic unit [OTU] richness) in each habitat and the corresponding net CH4 fluxes and ecosystem stability. The latter was calculated for each habitat as the mean net CH4 flux (between-season average) divided by the corresponding standard error (35). Data were arcsine (proportion of upland soil cluster alpha [USCα] members) and square root (methanotroph richness and ecosystem stability) transformed to obtain normal distributions. To examine the effect of soil abiotic properties on methanotrophs, we performed detrended correspondence analysis (DCA) followed by redundancy analysis (RDA) on T-RLP data to understand the influence of different environmental variables on individual T-RFs. First, DCA gave gradient lengths of 0.975 (Bad à Cheo), 1.241 (Glensaugh), 1.663 (Craggan), and 1.633 (Tulchan) on the first axis, which suggested that RDA was suitable for the analysis of relationships of T-RFs with environmental variables. Finally, following the square-root transformation of the PLFA data (percentage of total enriched PLFA content), a cluster analysis, based on a Bray-Curtis similarity matrix, was performed using the group average linking method to affiliate the active methanotroph population present in soils with published methanotrophs. The statistical analyses were carried out using GenStat 14th edition software (VSN International Limited, United Kingdom).
RESULTS
Land use change affects soil function.
We used four sites across Scotland, which included three land use categories per site (nonforested, young forest, and old forest) except for Glensaugh, which had no old forest, resulting in a total of five different habitats (bog, grassland, moorland, conifer forest, and birch woodland). We sampled each site four times over the period of a year, as a sampling campaign occurred once during each season: autumn (October/November 2008), winter (February 2009), spring (April 2009), and summer (July 2009). There was no significant seasonal effect on the CH4 fluxes measured at the four sites investigated (Fig. 1A to D) except at Tulchan in summer. Additionally, the age of the (pine/birch) forest did not influence the net CH4 flux (Fig. 1A, C, and D). The CH4 flux data from similar land uses (as well as from young/old forests) and from each seasonal sampling (total n = 176) were combined and upscaled to a yearly estimate. The seasonal estimates (total n = 44) were then averaged (final flux unit in kg CH4-C · ha−1 · year−1) as shown in Fig. 1E. Tree growth (afforestation of the bog and grassland with pine trees as well as heathland colonization by birch trees) significantly improved the CH4 sink in soils (P < 0.001). On average, tree growth induced 2.6-fold (conversion of heathland to birch woodland) and 4.5-fold (conversion of bog/grassland to pine forest) increases in CH4 sink.
Fig 1.

(A to D) Seasonal net CH4-C fluxes from soils at Bad à Cheo (A), Glensaugh (B), Craggan (C), and Tulchan (D); (E) yearly CH4 flux estimates from the nonforested and forested habitats. A positive value means that a production of CH4 occurs, whereas a negative flux denotes a sink of CH4. (A to D) The curves represent the flux means (error bars are standard errors of the means [SEM]; n = 4) of each season for each habitat from the closed-chamber experiment. Significance was analyzed through a nested ANOVA (land use by season). (E) Each histogram represents the yearly net CH4 flux average ± the SEM (n = 4 replicates) based on the upscaling of seasonal net CH4-C flux measurements. Data from the four sampling sites (Bad à Cheo, Glensaugh, Craggan, and Tulchan) which had similar land uses (bog, grassland, heathland, pine forest, or birch forest) were combined during upscaling. The method used for upscaling can be found within the text. For each site, statistical differences between seasons within each habitat are indicated by different Roman letters (a, b), while Greek letters (α, β, γ) indicate statistical differences between land uses according to multiple pairwise comparisons (α = 0.05).
Characterization of functional microbial communities.
The T-RFLP analysis of the pmoA genes to characterize the methanotrophic community demonstrated that the T-RFs Hha-32, Hha-81, and Hha-129 were the most abundantly found at all sites (64 to 98% of all T-RFs detected) (Table 1). Based on our previous work involving cloning and sequencing (25, 36), these operational taxonomic units (OTUs) were related to type II methanotrophs of the Beijerinckiaceae and Methylocystacae families (Table 1). The identity of these OTUs was confirmed by pmoA microarray analysis (see next section). In particular, the T-RF Hha-32 was closely related to clones of the upland soil cluster alpha (USCα; first referred to as RA14 clade [37]) and the T-RF Hha-129 was associated with microorganisms of a clade called cluster 5 (38). Both USCα and cluster 5 are distant relatives of Methylocapsa sp., while the T-RF Hha-81 was related to members of the Methylocystaceae family (Table 1). At the Bad à Cheo and Glensaugh sites, the T-RFs Hha-32, Hha-81, and Hha-129 were equally present in nonforested (peatland, grass) and pine forest soils, although there was a trend toward an increase of the relative abundance of the T-RF Hha-32 in pine forests. Similarly, a dominant and significant trend was observed at both heathland sites, while the relative abundance of T-RF Hha-81 decreased in the birch woodland (Table 1). Birch invasion had a strong effect on specific OTUs. In particular, microorganisms distantly related to the Methylocystaceae family (T-RF Hha-81; P = 0.006 at Craggan and P < 0.001 at Tulchan) were found mostly in soils under moorland whereas bacteria related to Methylocapsa sp./USCα clones (T-RF Hha-32; P < 0.001) were dominant in soils under birch forest. Birch invasion also decreased the relative abundance of the T-RF Hha-129 (P = 0.038 at Craggan; P = 0.001 at Tulchan) (Table 1). Compared to cluster 5, USCα constituted the dominant microbial population in soils from forest except at Bad à Cheo (Table 1). No significant seasonal effect on the T-RF relative abundance was detected at any of the sites except a slight increase in the abundance of T-RF Hha-81 (P = 0.014) in spring in the old birch forest at Tulchan (see Table S4 in the supplemental material).
Table 1.
Relative annual abundance and phylogenetic affiliation of the most abundant T-RFs found in some Scottish soilsa
| Site and habitat | Relative annual abundance ± SEM (%) of indicated T-RF |
T-RF relative total (%) | ||
|---|---|---|---|---|
| Hha-32 | Hha-129 | Hha-81 | ||
| Bad à Cheo | ||||
| Bog | 17 ± 2 | 29 ± 2 | 31 ± 3 | 77 |
| Young pine | 21 ± 4 | 29 ± 3 | 38 ± 4 | 88 |
| Glensaugh | ||||
| Grassland | 61 ± 7 | 9.0 ± 2.3 | 3.8 ± 2.7 | 74 |
| Young pine | 71 ± 6 | 11 ± 1 | 0.8 ± 0.78 | 83 |
| Craggan | ||||
| Moorland | 18 ± 2α | 10 ± 3α | 36 ± 6αβ | 64 |
| Young birch | 45 ± 8β | 2.5 ± 1.2β | 46 ± 7α | 94 |
| Old birch | 58 ± 9β | 7.8 ± 2.7αβ | 17 ± 6β | 81 |
| Tulchan | ||||
| Moorland | 13 ± 3α | 14 ± 2α | 55 ± 6α | 82 |
| Young birch | 82 ± 8β | 2.7 ± 1.0β | 13 ± 7β | 98 |
| Old birch | 61 ± 9β | 10 ± 3α | 13 ± 8β | 84 |
| Associated organism | Distant relative of Methylocapsa sp./USCα | Distant relative of Methylocapsa sp./cluster 5 | Distant relative of Methylocystaceae | |
For each T-RF, Greek letters (α, β) indicate statistical differences between habitats within each site according to multiple pairwise comparisons (α = 0.05). n = 16 replicates for each habitat. Digestion of pmoA was performed with the restriction enzyme HhaI. Information for associated organisms was obtained from Nazaries et al. (25) and Singh et al. (36).
The additive main effect and multiplicative interaction (AMMI) model (34, 39) was used to identify an interaction between the pattern of the T-RFs (or OTUs) and the different habitats investigated (Fig. 2). This was confirmed by the multivariate analysis of variance (MANOVA) on the four interaction principal component (IPC) scores of the AMMI analysis, which gave an overall significant effect (P < 0.001) of land use change at all sites (see Table S5 in the supplemental material). Afforestation with pine trees had a consistent effect on the methanotrophic community structure at all sites, with a more dominant effect at Bad à Cheo than at Glensaugh (Fig. 2A; see also Table S5 in the supplemental material). Birch tree invasion and the age of the birch forest appeared to have had a significant effect on the community structure and resulted in the detection of a different methanotrophic community structure in each habitat (Fig. 2C and D). No significant seasonal effect on the community structure was detected at any of the sites except in summer in soils from grassland at Glensaugh (IPC = 2) (see Table S5 in the supplemental material) and in spring in soils from moorland at Craggan (IPC = 4) (see Table S5 in the supplemental material), which accounted for only 23.2% and 7.2% of variation, respectively, in community data. Redundancy analysis of T-RFs and environmental variables indicated that only a few environmental variables were found to significantly influence the T-RFs. The variables were porosity (at Craggan only) and soil moisture (at Tulchan only). At Craggan, soil porosity was found to significantly influence the T-RFs (P = 0.020) by having a positive impact on the T-RFs related to Methylocystaceae (T-RF Hha-81) and a negative impact on the T-RFs related to Methylocapsa sp. (T-RF Hha-32). At Tulchan, soil moisture and the age of the birch forest significantly influenced the T-RFs (P = 0.010). Soil moisture had a positive impact on the T-RFs related to Methylocystis/Methylosinus spp. (Hha-81), while it had a negative impact on the T-RFs related to Methylocapsa sp. (Hha-32). However, the C/N ratio and porosity showed weak influences at Bad à Cheo (P = 0.066) and Glensaugh (P = 0.060), respectively, on the T-RFs related to Methylocapsa sp. (T-RFs Hha-32 and Hha-129). Due to that general lack of pattern, we concluded that the abiotic properties investigated were not strong drivers of the changes observed.
Fig 2.

Relationship between CH4 flux and methanotrophic community structure at Bad à Cheo (A), Glensaugh (B), Craggan (C), and Tulchan (D). The empty shapes (IPCA versus IPCA) are data points representing the IPC scores after analysis of the pmoA-based T-RFLP profiles with the AMMI model. The filled shapes (IPCA versus CH4 flux) are data points corresponding to the strongest linear regression of the net seasonal CH4 fluxes (Fig. 1A to D) with one of the IPC scores. The data points within each habitat represent the averages over replicates (n = 4) of the IPC scores of each season.
We applied a high-throughput diagnostic pmoA microarray to further characterize the methanotrophic community. Type I methanotrophs were not detected at any sites (data not shown), a finding which confirms the pmoA-based T-RFLP findings (Table 1). Neither nitrifiers nor any Methylocapsa sp. was detected at either site (Fig. 3).
Fig 3.

Community analysis of type II methanotrophs and related organisms using the pmoA microarray (n = 44). Within each habitat, each row represents a replicate. The results were normalized first to the positive-control probe mtrof173 and then to the reference values determined individually for each probe (29). Color coding is as follows: red color indicates the maximum achievable signal for an individual probe, while blue color indicates that no detectable PCR product hybridized to that probe. The color gradient between blue and red reflects the proportion of hybridization.
Type II methanotrophs of different clades were detected in the nonforested and forested habitats. In soils from peatland and heathland (Bad à Cheo, Craggan, and Tulchan), the type II methanotroph community consisted of members of the Methylocystaceae family (probes McyM309, Mcy413, Mcy459, Mcy522, and Msi233), peat-related type II methanotrophs (probe Peat264), and watershed 1 clade (probe Wsh1-566) organisms (40) (Fig. 3; see also Table S6 in the supplemental material). Conversely, the type II methanotroph community consisted of organisms of the RA14/USCα (probes RA14-299, RA14-594, and RA14-591) and watershed 2 (probe Wsh2-491) clades (40) in soils from pine and birch forests (Bad à Cheo, Craggan, and Tulchan) (Fig. 3; see also Table S6 in the supplemental material). Overall, principal component analysis (PCA) and subsequent MANOVA on the first five principal components (PCs) indicated that tree growth (afforestation/tree invasion) changed the soil methanotrophic community at Bad à Cheo (P = 0.003), Craggan (P = 0.002), and Tulchan (P < 0.001) (see Table S7 in the supplemental material). The pmoA microarray data from the Bad à Cheo, Craggan, and Tulchan sites were congruent with the pmoA-based T-RFLP data presented in Fig. 2A, C, and D. Similarly, MANOVA did not detect a significant difference in the soil methanotrophic communities between habitats at Glensaugh (P = 0.299) (see Table S7 in the supplemental material), a finding which reflects the weak community shift observed with the pmoA-based T-RFLP analysis (Fig. 2B).
We also used T-RFLP analysis of the mcrA genes to characterize the methanogenic community. Most of the samples in afforested plots did not produce any PCR products for this gene despite several attempts to optimize DNA concentrations and PCR conditions. Only the bog, moorland, and young forest at Bad à Cheo and Tulchan produced PCR products and T-RFLP profiles. High aerobic conditions in the top 10 cm of soils in the afforested plots may explain this failure, as methanogens are anaerobes. Data analysis from available samples again suggested that season had no impact on the community composition, which was driven mainly by land use treatment (P < 0.001 [ANOVA] on PC axis 1, which explained 51% of total variation). Because the data on methanogens were not available for a significant number of land uses, sites, and times, these data were not used for regression analysis (to link community with function) or statistical treatment.
Linking community structure and biodiversity with soil function and evidence for ecological theory.
This was first investigated by a simple linear regression analysis (Fig. 2) between the pmoA IPC scores of each habitat and the corresponding net CH4 flux values. At all sites, tree growth and change in CH4 flux were significantly related to a shift in the community structure (P = 0.022 at Bad à Cheo, P = 0.027 at Glensaugh, P = 0.027 at Craggan, and P = 0.015 at Tulchan). However, the relationship was stronger at Bad à Cheo and Tulchan than at Glensaugh and was the lowest at Craggan (Fig. 2).
Stable-isotope probing of phospholipid fatty acid (PLFA-SIP) analysis was used to link methanotroph communities with their activity, i.e., oxidation of atmospheric CH4. Based on the enriched phospholipid fatty acid (PLFA) content (percentage of 13C incorporation) (see Fig. S1 in the supplemental material) extracted from the incubated soils, a cluster analysis was performed (see Fig. S2 in the supplemental material). It suggested that the active methanotrophs in the soils from the different habitats were all distant relatives (<90% similarity) of Methylosinus sporium and the Beijerinckiaceae family (containing Methylocapsa spp.) due to a high incorporation of 13C in the PLFA 18:1ω7 (41, 42) (see Fig. S1 in the supplemental material), which confirms the findings from the pmoA-based T-RFLP and pmoA microarray data (see previous section). However, it should be emphasized that PLFA-SIP analysis does not provide a high level of resolution (42) and was used to identify which organisms are responsible for the oxidation of CH4.
Because the above-mentioned results described an overall lack of temporal and spatial variation, the data from the different sites (and seasons) were grouped into the main habitats explored: heathland, peatland, grassland, pine forest, and (young/old) birch forest. Further linear regressions were performed on the proportions of members of the USCα and cluster 5 clades, methanotroph richness, and CH4 flux (Fig. 4). There was a strong relationship (R2 = 0.4755, P < 0.001) between the increase in CH4 oxidation rates and the loss of OTU richness due to tree growth (Fig. 4A and Table 2). In particular, the land use change revealed an increase in the proportions of USCα and cluster 5 pmoA sequences in soils while net CH4 sinks increased (R2 = 0.3032, P < 0.001) (Fig. 4B and Table 2). In other words, there was a negative relationship between the loss of methanotroph diversity (number of OTUs) and the increased dominance of members of the USCα and cluster 5 clades (R2 = 0.2019, P = 0.004) (see Fig. S3 in the supplemental material). The “ecosystem stability” was defined as a component of the amplitude of variation in plant respiration by Tilman et al. (35). We used the same logic for CH4 flux, which indicated that the standard deviation of net CH4 fluxes decreased as the CH4 sink rates increased in the forests, suggesting that the functional (ecosystem) stability improved with tree growth (P = 0.038) (Table 2).
Fig 4.
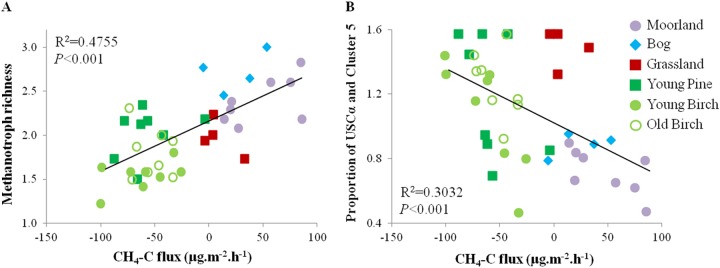
Relationships between methanotroph diversity and changes in CH4 flux associated with land use treatment (n = 4 replicates). (A) The methanotroph richness (or OTU richness) was calculated as the square root transformation of the number of T-RFs present in each sample (among the 15 most abundant T-RFs of the T-RFLP profiles, which constituted >94% coverage). (B) Proportions of USCα and cluster 5 microorganisms were calculated as the angular transformation (arcsine of the square root) of the ratio of the relative abundance of the T-RFs specific to Methylocapsa sp. (USCα/cluster 5; T-RFs Hha-32 and Hha-129) to the sum of the T-RFs specific to USCα/cluster 5 and the Methylocystaceae family (T-RF Hha-81). Refer to Table 1 for T-RF reference values.
Table 2.
Significance between land uses for net CH4 fluxes, methanotroph richness, proportions of USCα and cluster 5 members, and ecosystem stabilitya
| Habitat | CH4-C flux (μg · m−2 · h−1) | Methanotroph richness (no. of OTUs) | Proportion of USCα/cluster 5 | Stability |
|---|---|---|---|---|
| Moorland | 48.10α | 2.39αβ | 0.57α | 1.29αβ |
| Bog | 24.89α | 2.72α | 0.63αβ | 1.24αβ |
| Grassland | 9.26α | 1.98βγδ | 1.48γ | 0.73α |
| Young pine | −57.33β | 2.02βγ | 1.06βγ | 1.59β |
| Young birch | −61.87β | 1.54δ | 1.06αβγ | 1.52αβ |
| Old birch | −53.38β | 1.80γδ | 1.24βγ | 1.65β |
| P value | <0.001 | <0.001 | <0.001 | 0.038 |
Ecosystem stability was calculated as the square root transformation of the ratio of CH4 flux to its corresponding standard error (35). For each variable, land uses followed by different Greek letters (α, β, γ, δ) are statistically different according to multiple pairwise comparisons (α = 0.05). Tree growth was related to increased net CH4 sink, decreased methanotroph richness, increased abundance of USCα, and increased ecosystem stability.
Land use treatment did not consistently affect the abiotic properties of the soil (see Table S2 in the supplemental material). However, linear regression showed that some soil characteristics had a significant relationship with net CH4 fluxes (see Table S3 in the supplemental material). These abiotic factors were related mainly to soil structure (particle size, water-filled pore space [WFPS], and/or moisture), which overall helped with gas diffusivity. However, these changes were not consistently observed between sites (see Table S3 in the supplemental material).
DISCUSSION
Soil function and functional microflora are affected by land use treatment but not by temporal and spatial variations.
This study provides important correlative evidence that both soil function and associated functional microflora are stable over time and space. All sites showed increased CH4 consumption rates with afforestation, suggesting a tree growth-induced effect, which were directly linked to a shift in the methanotrophic communities. In addition, both rate of CH4 flux and community structure remained relatively stable throughout time and space. Our data demonstrate that a robust and consistent sampling procedure can overcome some variation in data which was previously assigned to temporal and spatial variations. We are confident in our data because previous studies demonstrated that our (laboratory-based) approach to CH4 flux measurements provides results that are very similar to in situ (field-based) measurements (25, 30). Our evidence is supported by previous findings which report a lack of effect of temperature on CH4 oxidation but an influence of soil physical structure (WFPS, soil moisture) (43, 44). Consistent and significant increases in CH4 sinks in forest soils indicate that afforestation can be effectively used to mitigate net CH4 fluxes. Upscaling of our data suggests that afforestation of 50% of all bogs and heathland in Scotland may offset about 5% of CH4 emissions from the farming sector (based on 2006 CH4 emission estimates [45]).
Our study provides correlative evidence that tree growth was associated with a change in CH4 flux which was related to a shift in the methanotrophic community structure. This finding was consistent with the different analytical techniques used in this study (T-RFLP, microarray, and PLFA-SIP). A strong effect of land use treatment on the methanotrophic community structure was detected. In particular, the site and type of land use treatment influenced the type of methanotrophs present. The soils under grassland and pine trees at Glensaugh were dominated mainly by members of USCα, which are type II methanotrophs involved in the oxidation of CH4 at atmospheric concentrations (37, 46). However, at the other sites (Bad à Cheo, Craggan, and Tulchan), members of the Methylocystaceae were relatively more dominant in the nonforested soils while USCα cells were characteristic of the forest soils. Methanotrophs of the USCα and cluster 5 clades were the main CH4 oxidizers, but members of cluster 5 had a low abundance in soils from forests. We found that tree growth, while improving CH4 sink, always favored USCα cells over type I methanotrophs (in New Zealand [25]) or over members of the Methylocystaceae family (in Scotland) (this study). Further analysis of the literature suggests that USCα-related species dominate forest ecosystems (25, 36, 38, 47, 48), which may explain the enhanced CH4 sink observed in forest soils worldwide. This further strengthens the link between change in functional microflora and improvement of soil function when tree growth occurred (see next section).
Overall, we did not detect any temporal variation due to seasonal change with regard to the methanotrophic community structure. There was some influence of the age of the birch forest at Craggan or, rather, of the methanotrophic community being slower to adapt to land use treatment at this site. Despite the comparatively weak effect of afforestation on community structure at Glensaugh, there was no spatial variation between sites with similar habitats following land use treatment (Table 3). In particular, our replicated moorland/birch woodland sites (Craggan/Tulchan) displayed a similar effect of tree invasion on methanotroph richness and relative abundance of USCα/cluster 5 cells (Table 3), thus suggesting a weak temporal and spatial variation. These results are supported by findings that methanotrophs lack spatial heterogeneity in agroecosystems (49, 50), which indicates that the effect of land use treatment on the soil microbial community is stronger and can be distinguished from temporal and spatial heterogeneity.
Table 3.
Spatial stability of the methanotroph communities between sites with similar land usesa
| Site | Habitat | Methanotroph richness | Proportion of USCα and cluster 5 |
|---|---|---|---|
| Bad à Cheo | Bog | α | αβ |
| Young pine | βγ | αβ | |
| Glensaugh | Grassland | γδ | γ |
| Young pine | γδε | γ | |
| Craggan | Moorland | αβγ | α |
| Young birch | δε | α | |
| Old birch | δε | βγ | |
| Tulchan | Moorland | αβ | α |
| Young birch | ε | γ | |
| Old birch | γδε | γ |
For each variable, land uses followed by different Greek letters (α, β, γ, δ, ε) are statistically different according to multiple pairwise comparisons (α = 0.05).
Macroecological theory can explain microbial control of net methane fluxes.
The lack of ecological theory to explain the microbial regulation of ecosystem functions hampers the interpretation of the consequences of microbial shifts at the functional level and is hindered further by the lack of microbial data in predictive models. Here we provide correlative evidence that ecological theories developed for macroorganisms, such as plants and animals (macroecology), can be used to explain microbial regulation of biogeochemical cycles. Our data suggest that the microbial regulation of the CH4 flux may be explained by the selection theory and illustrate that members of USCα are key species in atmospheric CH4 sink globally (25, 46–48). Contrastingly, previous studies reported that microbial diversity (initial community evenness and increased species richness) enhances functional predictability and stability of ecosystems (35, 51–53). However, our results indicate a decrease in the methanotroph diversity (OTU richness) when CH4 consumption increased (Fig. 4A). This discrepancy was able to be explained because we studied systems with large-scale land use changes whereas other studies either manipulated species richness and abundance in the laboratory (52, 53) or used agricultural fields (35) which are highly disturbed due to management practices such as plowing, irrigation, and fertilizer application (51). Additionally, the focus of our study was CH4 consumption, which is restricted to a selected group of microbial communities. Biodiversity offers a buffer (insurance hypothesis) to maintain an ecosystem process when environmental changes occur and is due to the functional redundancy of the organisms involved (54, 55). Although this principle can be applied theoretically to any organisms (6), the situation for specific physiological groups of microbes, such as methanotrophs, is unusual because functional redundancy may not exist as such and because methanotrophy is a process limited to selected microbial taxa. Thus, a change in environmental conditions, such as land use treatment, can affect only the competition between certain methanotrophs, which will then affect species richness and evenness. Our data support the importance of the soil methanotrophic community structure as a regulator of CH4 flux during land use change. Specifically, the proportions of USCα and cluster 5 microorganisms were associated with the net CH4 oxidation rates of a habitat. Moreover, the decrease in methanotroph diversity was associated with an increase of the dominance of the USCα/cluster 5 cluster (see Fig. S3A in the supplemental material). This suggests that, in the systems we investigated, there was a niche-specific distribution of USCα/cluster 5 cells induced by land use change. Nonetheless, we argue that even though it is possible that ecosystem functioning can be explained by the selection theory, maintaining biodiversity should remain a key goal in conservation policy, as natural or anthropogenic disturbance may create new niches, and if these new niches remain unoccupied due to a lack of physiological diversity, it may have serious consequences for ecosystem functioning.
Although other studies investigated the relationship between land use change and CH4 oxidation rates (56) or between land use change and the methanotrophic communities involved (26), this is the first time that a relationship linking land use change with a change in CH4 oxidation rates and community structure and activity has been characterized over time and space. Our data suggest that the proportions of USCα and cluster 5 methanotrophs in the habitats investigated influence whether a soil is a source or sink for CH4. Especially in the moorland/birch forest sites (Craggan and Tulchan) (Fig. 4B and Table 1), the switch between net CH4 emissions and sinks occurred when the relative abundance of USCα/cluster 5 was about the same as that of Methylocystaceae (50/50), although the transition was slower in the young birch in Craggan. Data analysis from previous works suggests that a similar threshold was observed in New Zealand (25) except that USCα/cluster 5 cells had to constitute ∼80% of the total methanotrophs to observe significantly higher CH4 consumption rates. Nevertheless, the improvement of CH4 sinks was concomitant with a change in the methanotrophic community structure in all sites, therefore indicating that microbes were driving the ecosystem function rather than being a consequence of changes in soil properties.
Herein we provide important evidence of regulation of CH4 flux by methanotrophic bacteria. Some of our results should be interpreted with caution because this study has some limitations. (i) We provide correlative evidence that macroecology theory may explain CH4 flux in soils. However, unlike plants and animals, the species concept is not well defined for microbial communities. Consequently, OTUs obtained from T-RFLP and microarray analyses may be at the taxonomic level, which is not exactly comparable to plant and animal species. Additionally, both molecular approaches used here are based on PCR amplification, which is well known to introduce bias in data and have some implications in data interpretations. (ii) We measured ecosystem stability by variance of CH4 flux without distinguishing rates of CH4 production and consumption, and therefore, our data are a proxy to actual ecosystem stability. (iii) The nature of our study and conclusions that are drawn are correlative only. For achieving a mechanistic link between diversity and CH4 flux, manipulative experiments, such as diversity dilution (52), may be needed. (iv) There are some other processes (e.g., anaerobic CH4 consumption) and microbial groups (e.g., verrumicrobial methanotrophs) which were not accounted for in this work. These factors may also have played a role in the total CH4 flux. (v) Our spatial and temporal data are limited. To confirm the conclusions from this study, future works should address all the above-mentioned issues by explicitly considering many more land use types, management practices, more-frequent flux measurements, and statistically designed spatial measurements.
In conclusion, this study provides experimental evidence for the increase and stability of CH4 sink after afforestation, which was correlated with a shift in methanotrophic communities. The establishment of forest was accompanied with a decrease in methanotroph richness correlated to an increase in dominance of members of the USCα, while ecosystem stability improved. This study illustrates the microbial regulation of biogeochemical cycles by using CH4 flux as an example. This supports the growing demand for explicit consideration of the inclusion of microbial data in predictive models studying the effects of global changes.
Supplementary Material
ACKNOWLEDGMENTS
This study was financed by the Macaulay Development Trust. Work in the B.K.S. laboratory is supported by funding from the Australian Research Council (DP130104841), Grain Research and Development Corporation, and Cotton Research and Development Corporation.
We thank Barry Thornton and William Tottey for their technical assistance with PLFA and methanogen community analyses, respectively. We are grateful to Lucinda Robertson, Richard Gwatkin, and Luca Giaramida for their help during field sampling.
Footnotes
Published ahead of print 26 April 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00095-13.
REFERENCES
- 1. Gans J, Wolinsky M, Dunbar J. 2005. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309:1387–1390 [DOI] [PubMed] [Google Scholar]
- 2. Harris J. 2009. Soil microbial communities and restoration ecology: facilitators or followers? Science 325:573–574 [DOI] [PubMed] [Google Scholar]
- 3. Isbell F, Calcagno V, Hector A, Connolly J, Harpole WS, Reich PB, Scherer-Lorenzen M, Schmid B, Tilman D, van Ruijven J, Weigelt A, Wilsey BJ, Zavaleta ES, Loreau M. 2011. High plant diversity is needed to maintain ecosystem services. Nature 477:199–202 [DOI] [PubMed] [Google Scholar]
- 4. Reich PB, Tilman D, Naeem S, Ellsworth DS, Knops J, Craine J, Wedin D, Trost J. 2004. Species and functional group diversity independently influence biomass accumulation and its response to CO2 and N. Proc. Natl. Acad. Sci. U. S. A. 101:10101–10106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reich PB, Tilman D, Isbell F, Mueller K, Hobbie SE, Flynn DF, Eisenhauer N. 2012. Impacts of biodiversity loss escalate through time as redundancy fades. Science 336:589–592 [DOI] [PubMed] [Google Scholar]
- 6. Loreau M, Hector A. 2001. Partitioning selection and complementarity in biodiversity experiments. Nature 412:72–76 [DOI] [PubMed] [Google Scholar]
- 7. Martin P. 1992. Exe—a climatically sensitive model to study climate change and CO2 enhancement effects on forests. Aust. J. Bot. 40:717–735 [Google Scholar]
- 8. Cramer W, Bondeau A, Woodward FI, Prentice IC, Betts RA, Brovkin V, Cox PM, Fisher V, Foley JA, Friend AD, Kucharik C, Lomas MR, Ramankutty N, Sitch S, Smith B, White A, Young-Molling C. 2001. Global response of terrestrial ecosystem structure and function to CO2 and climate change: results from six dynamic global vegetation models. Glob. Change Biol. 7:357–373 [Google Scholar]
- 9. Gottfried M, Pauli H, Grabherr G. 1998. Prediction of vegetation patterns at the limits of plant life: a new view of the alpine-nival ecotone. Arctic Alp. Res. 30:207–221 [Google Scholar]
- 10. Box EO, Crumpacker DW, Hardin ED. 1999. Predicted effects of climatic change on distribution of ecologically important native tree and shrub species in Florida. Clim. Change 41:213–248 [Google Scholar]
- 11. Ferrier S, Guisan A. 2006. Spatial modelling of biodiversity at the community level. J. Appl. Ecol. 43:393–404 [Google Scholar]
- 12. Davidson EA, Janssens IA. 2006. Temperature sensitivity of soil carbon decomposition and feedbacks to climate change. Nature 440:165–173 [DOI] [PubMed] [Google Scholar]
- 13. He R, Wooller MJ, Pohlman JW, Quensen J, Tiedje JM, Leigh MB. 2012. Shifts in identity and activity of methanotrophs in Arctic lake sediments in response to temperature changes. Appl. Environ. Microbiol. 78:4715–4723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pace NR. 1997. A molecular view of microbial diversity and the biosphere. Science 276:734–740 [DOI] [PubMed] [Google Scholar]
- 15. Amann RI, Ludwig W, Schleifer K-H. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Levy PE, Gray A, Leeson SR, Gaiawyn J, Kelly MPC, Cooper MDA, Dinsmore KJ, Jones SK, Sheppard LJ. 2011. Quantification of uncertainty in trace gas fluxes measured by the static chamber method. Eur. J. Soil Sci. 62:811–821 [Google Scholar]
- 17. Matthias AD, Blackmer AM, Bremner JM. 1980. A simple chamber technique for field measurement of emissions of nitrous oxide from soils. J. Environ. Qual. 9:251–256 [Google Scholar]
- 18. McDonald IR, Bodrossy L, Chen Y, Murrell JC. 2008. Molecular ecology techniques for the study of aerobic methanotrophs. Appl. Environ. Microbiol. 74:1305–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dedysh SN, Liesack W, Khmelenina VN, Suzina NE, Trotsenko YA, Semrau JD, Bares AM, Panikov NS, Tiedje JM. 2000. Methylocella palustris gen. nov., sp. nov., a new methane-oxidizing acidophilic bacterium from peat bags, representing a novel subtype of serine-pathway methanotrophs. Int. J. Syst. Evol. Microbiol. 50:955–969 [DOI] [PubMed] [Google Scholar]
- 20. Dunfield PF, Khmelenina VN, Suzina NE, Trotsenko YA, Dedysh SN. 2003. Methylocella silvestris sp. nov., a novel methanotroph isolated from an acidic forest cambisol. Int. J. Syst. Evol. Microbiol. 53:1231–1239 [DOI] [PubMed] [Google Scholar]
- 21. Vorobev AV, Baani M, Doronina NV, Brady AL, Liesack W, Dunfield PF, Dedysh SN. 2010. Methyloferula stellata gen nov., sp. nov., an acidophilic, obligately methanotrophic bacterium possessing only a soluble methane monooxygenase. Int. J. Syst. Evol. Microbiol. 61:2456–2463 [DOI] [PubMed] [Google Scholar]
- 22. Chen Y, Dumont MG, Cebron A, Murrell JC. 2007. Identification of active methanotrophs in a landfill cover soil through detection of expression of 16S rRNA and functional genes. Environ. Microbiol. 9:2855–2869 [DOI] [PubMed] [Google Scholar]
- 23. Bourne DG, McDonald IR, Murrell JC. 2001. Comparison of pmoA PCR primer sets as tools for investigating methanotroph diversity in three Danish soils. Appl. Environ. Microbiol. 67:3802–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luton PE, Wayne JM, Sharp RJ, Riley PW. 2002. The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology 148:3521–3530 [DOI] [PubMed] [Google Scholar]
- 25. Nazaries L, Tate KR, Ross DJ, Singh J, Dando J, Saggar S, Baggs EM, Millard P, Murrell JC, Singh BK. 2011. Response of methanotrophic communities to afforestation and reforestation in New Zealand. ISME J. 5:1832–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh BK, Tate KR, Kolipaka G, Hedley CB, Macdonald CA, Millard P, Murrell JC. 2007. Effect of afforestation and reforestation of pastures on the activity and population dynamics of methanotrophic bacteria. Appl. Environ. Microbiol. 73:5153–5161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stralis-Pavese N, Sessitsch A, Weilharter A, Reichenauer T, Riesing J, Csontos J, Murrell JC, Bodrossy L. 2004. Optimization of diagnostic microarray for application in analysing landfill methanotroph communities under different plant covers. Environ. Microbiol. 6:347–363 [DOI] [PubMed] [Google Scholar]
- 28. Stralis-Pavese N, Abell GCJ, Sessitsch A, Bodrossy L. 2011. Analysis of methanotroph community composition using a pmoA-based microbial diagnostic microarray. Nat. Protoc. 6:609–624 [DOI] [PubMed] [Google Scholar]
- 29. Bodrossy L, Stralis-Pavese N, Murrell JC, Radajewski S, Weilharter A, Sessitsch A. 2003. Development and validation of a diagnostic microbial microarray for methanotrophs. Environ. Microbiol. 5:566–582 [DOI] [PubMed] [Google Scholar]
- 30. Tate KR, Ross DJ, Saggar S, Hedley CB, Dando J, Singh BK, Lambie SM. 2007. Methane uptake in soils from Pinus radiata plantations, a reverting shrubland and adjacent pastures: effects of land-use change, and soil texture, water and mineral nitrogen. Soil Biol. Biochem. 39:1437–1449 [Google Scholar]
- 31. Frostegård Å, Tunlid A, Bååth E. 1993. Phospholipid fatty-acid composition, biomass, and activity of microbial communities from two soil types experimentally exposed to different heavy-metals. Appl. Environ. Microbiol. 59:3605–3617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thornton B, Zhang ZL, Mayes RW, Hogberg MN, Midwood AJ. 2011. Can gas chromatography combustion isotope ratio mass spectrometry be used to quantify organic compound abundance? Rapid Commun. Mass Spectrom. 25:2433–2438 [DOI] [PubMed] [Google Scholar]
- 33. Culman SW, Bukowski R, Gauch HG, Cadillo-Quiroz H, Buckley DH. 2009. T-REX: software for the processing and analysis of T-RFLP data. BMC Bioinformatics 10:171–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Culman SW, Gauch HG, Blackwood CB, Thies JE. 2008. Analysis of T-RFLP data using analysis of variance and ordination methods: a comparative study. J. Microbiol. Methods 75:55–63 [DOI] [PubMed] [Google Scholar]
- 35. Tilman D, Reich PB, Knops JM. 2006. Biodiversity and ecosystem stability in a decade-long grassland experiment. Nature 441:629–632 [DOI] [PubMed] [Google Scholar]
- 36. Singh BK, Tate KR, Ross DJ, Singh J, Dando J, Thomas N, Millard P, Murrell JC. 2009. Soil methane oxidation and methanotroph responses to afforestation of pastures with Pinus radiata stands. Soil Biol. Biochem. 41:2196–2205 [Google Scholar]
- 37. Holmes AJ, Roslev P, McDonald IR, Iversen N, Henriksen K, Murrell JC. 1999. Characterization of methanotrophic bacterial populations in soils showing atmospheric methane uptake. Appl. Environ. Microbiol. 65:3312–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Knief C, Vanitchung S, Harvey NW, Conrad R, Dunfield PF, Chidthaisong A. 2005. Diversity of methanotrophic bacteria in tropical upland soils under different land uses. Appl. Environ. Microbiol. 71:3826–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gauch HG. 2006. Statistical analysis of yield trials by AMMI and GGE. Crop Sci. 46:1488–1500 [Google Scholar]
- 40. Chen Y, Dumont MG, McNamara NP, Chamberlain PM, Bodrossy L, Stralis-Pavese N, Murrell JC. 2008. Diversity of the active methanotrophic community in acidic peatlands as assessed by mRNA and SIP-PLFA analyses. Environ. Microbiol. 10:446–459 [DOI] [PubMed] [Google Scholar]
- 41. Bowman JP, Sly LI, Nichols PD, Hayward AC. 1993. Revised taxonomy of the methanotrophs: description of Methylobacter gen. nov., emendation of Methylococcus, validation of Methylosinus and Methylocystis species, and a proposal that the family Methylococcaceae includes only the group I methanotrophs. Int. J. Syst. Evol. Microbiol. 43:735–753 [Google Scholar]
- 42. Bodelier PLE, Gillisen MJB, Hordijk K, Damste JSS, Rijpstra WIC, Geenevasen JAJ, Dunfield PF. 2009. A reanalysis of phospholipid fatty acids as ecological biomarkers for methanotrophic bacteria. ISME J. 3:606–617 [DOI] [PubMed] [Google Scholar]
- 43. Smith KA, Ball T, Conen F, Dobbie KE, Massheder J, Rey A. 2003. Exchange of greenhouse gases between soil and atmosphere: interactions of soil physical factors and biological processes. Eur. J. Soil Sci. 54:779–791 [Google Scholar]
- 44. Ball BC, Smith KA, Klemedtsson L, Brumme R, Sitaula BK, Hansen S, Priemé A, MacDonald J, Horgan GW. 1997. The influence of soil gas transport properties on methane oxidation in a selection of Northern European soils. J. Geophys. Res. Atmos. 102:23309–23317 [Google Scholar]
- 45. Choudrie SL, Jackson J, Watterson JD, Murrells T, Passant N, Thomson A, Cardenas L, Leech A, Mobbs DC, Thistlethwaite G. 2008. UK greenhouse gas inventory 1990 to 2006: annual report for submission under the Framework Convention on Climate Change. AEA Technology plc, Didcot, Oxfordshire, United Kingdom [Google Scholar]
- 46. Knief C, Lipski A, Dunfield PF. 2003. Diversity and activity of methanotrophic bacteria in different upland soils. Appl. Environ. Microbiol. 69:6703–6714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dörr N, Glaser B, Kolb S. 2010. Methanotrophic communities in Brazilian ferralsols from naturally forested, afforested, and agricultural sites. Appl. Environ. Microbiol. 76:1307–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kolb S. 2009. The quest for atmospheric methane oxidizers in forest soils. Environ. Microbiol. Rep. 1:336–346 [DOI] [PubMed] [Google Scholar]
- 49. Krause S, Lüke C, Frenzel P. 2009. Spatial heterogeneity of methanotrophs: a geostatistical analysis of pmoA-based T-RFLP patterns in a paddy soil. Environ. Microbiol. Rep. 1:393–397 [DOI] [PubMed] [Google Scholar]
- 50. Shrestha PM, Kammann C, Lenhart K, Dam B, Liesack W. 2012. Linking activity, composition and seasonal dynamics of atmospheric methane oxidizers in a meadow soil. ISME J. 6:1115–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Levine UY, Teal TK, Robertson GP, Schmidt TM. 2011. Agriculture's impact on microbial diversity and associated fluxes of carbon dioxide and methane. ISME J. 5:1683–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McGrady-Steed J, Harris PM, Morin PJ. 1997. Biodiversity regulates ecosystem predictability. Nature 390:162–165 [Google Scholar]
- 53. Wittebolle L, Marzorati M, Clement L, Balloi A, Daffonchio D, Heylen K, De Vos P, Verstraete W, Boon N. 2009. Initial community evenness favours functionality under selective stress. Nature 458:623–626 [DOI] [PubMed] [Google Scholar]
- 54. Naeem S, Li SB. 1997. Biodiversity enhances ecosystem reliability. Nature 390:507–509 [Google Scholar]
- 55. Yachi S, Loreau M. 2007. Does complementary resource use enhance ecosystem functioning? A model of light competition in plant communities. Ecol. Lett. 10:54–62 [DOI] [PubMed] [Google Scholar]
- 56. Smith KA, Dobbie KE, Ball BC, Bakken LR, Sitaula BK, Hansen S, Brumme R, Borken W, Christensen S, Priemé A, Fowler D, MacDonald JA, Skiba U, Klemedtsson L, Kasimir-Klemedtsson A, Degórska A, Orlanski P. 2000. Oxidation of atmospheric methane in Northern European soils, comparison with other ecosystems, and uncertainties in the global terrestrial sink. Glob. Change Biol. 6:791–803 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.