

Summary
Intracellular parasitism has arisen only a few times during the long ancestry of protozoan parasites including in diverse groups such as microsporidians, kinetoplastids, and apicomplexans. Strategies used to gain entry differ widely from injection (e.g. microsporidians), active penetration of the host cell (e.g. Toxoplasma), recruitment of lysosomes to a plasma membrane wound (e.g. Trypanosoma cruzi), to host cell-mediated phagocytosis (e.g. Leishmania). The resulting range of intracellular niches is equally diverse ranging from cytosolic (e.g. T. cruzi) to residing within a nonfusigenic vacuole (e.g. Toxoplasma, Encephalitizoon) or a modified phagolysosome (e.g. Leishmania). These lifestyle choices influence access to nutrients, interaction with host cell signaling pathways, and detection by pathogen recognition systems. As such, intracellular life requires a repertoire of adaptations to assure entry-exit from the cell, as well as to thwart innate immune mechanisms and prevent clearance. Elucidating these pathways at the cellular and molecular level may identify key steps that can be targeted to reduce parasite survival or augment immunological responses and thereby prevent disease.
Keywords: Protozoan parasites, motility, cell invasion, protein secretion, cytoskeleton, signaling
Introduction
Protozoan parasites that infect humans are an extremely diverse collection of organisms that spans much of the eukaryotic tree of life (1) (Figure 1). Implicit from this diversity, parasitism as a life style evolved multiple times independently during eukaryotic history. Early branching groups such as Giardia and Trichomonas are members of the Parasbasala, reflecting their early divergence and degenerate and/or specialized mitochondria (2) (Figure 1). Entamoeba histolytica, the cause of amebic dysentery, also shows mitochondrial loss and extensive lateral gene transfer (3), but is phylogenetically more closely related to amoebas including Dictyostelium (Figure 1). Microsporidian parasites were also once thought to be early branching eukaryotes; however, phylogenetic comparisons, bolstered by whole genome sequencing, revealed that they are fungi (4) (Figure 1). Agents of human malaria, Plasmodium spp. belong to the Apicomplexa, which are most closely related to ciliates and dinoflagellates (5) (Figure 1). Other important apicomplexan parasites include Cryptosporidium spp., several species of which cause of water borne diarrheal disease in humans, and Toxoplasma gondii, which causes zoonotic infections and is primarily an opportunistic pathogen in humans. Finally, flagellated organisms related to Euglena are responsible for important human and animal diseases caused by Leishmania spp. and Trypanosoma spp. (6) (Figure 1). Some members of the cercozoa and hertokonts and chlorophyte algae are also parasitic on plants, invertebrates, or other protozoa, extending this adaptation to all the major branches of the eukaryotic tree of life (Figure 1). However, these later groups do not infect humans and hence are not considered further here.
Figure 1.

Phylogenetic relationships among the eight major groups of eukaryotes. Tree is based on consensus of molecular and morphological data. Protozoan parasite groups that commonly infect humans are denoted adjacent to the major branches they occupy. Modified with permission from (1).
The ancient nature of parasitism as a life style further implies that many of these organisms preyed on other early forms of life, prior to the appearance of higher vertebrates in evolution. Hence, while they may have co-adapted with humans throughout our existence, they also span a much broader slice of eukaryotic evolution and hence display greater diversity than their current hosts. Among the very diverse repertoire of parasitic “protists”, intracellular parasitism appears to have arisen only a few times independently. Interestingly, all protozoan parasites that have chosen the intracellular lifestyle are obligate, unlike many bacterial pathogens that have evolved as facultative intracellular parasites (7). The relative rarity of intracellular parasitism suggests there are substantial challenges to adopting this life style. Among these, the successful parasite must find a way to invade or enter host cells, and then escape endogenous mechanisms for clearing intracellular invaders, including induction of apoptosis (8), autophagy (see chpater by V. Deretic in this issue), and immunity related GTPases (9) and production of reactive oxygen-nitrogen intermediates (10), and lysosomal degradation. Intracellular parasites are also subject to surveillance by pattern recognition receptors such as TLRs (11) and intracellular senor systems such as NODs (12). If successful in avoiding these innate defenses, the parasite must then find access to nutrients, which may be limited compared to the comparatively rich environments of circulatory, mucosal, or the enteric systems. Although intracellular parasitism has evolved only a few times, it has been tremendously successful, as attested by the enormous expansion of species within the groups that have adopted this life style.
Here we consider the four major groups of obligate intracellular protozoan parasites that cause disease in humans (Table 1, Figure 2): microsporidians, using Encephalitzoon as an example, apicomplexan parasites focusing on Toxoplasma and Plasmodium, and the kinetoplastid parasites Leishmania spp, and T. cruzi. For each parasite, the invasion mechanisms and adaptations for intracellular survival are summarized with an emphasis on how these strategies influence immunological responses by the host.
Table 1.
| Encephalitozoon | Toxoplasma | Leishmania | Trypanosoma cruzi | |
|---|---|---|---|---|
| Invasive stages | Spore / sporoplasm | Tachyzoite Bradyzoite Sporozoite |
Promastigote | Trypomastigote |
| Intracellular stages | Meront | Tachyzoite Bradyzoite |
Amastigote | Amastigote |
| Cell types | Various, including macrophages | All nucleated, including macrophages | Primarily macrophages | Various; muscle, endothelial |
| Host receptors | GAGs, mannose receptors | GAGs, sialic acid | C1, C3, mannose scavenging | TGF-β, Bradykinin 2, G-protein coupled |
| Entry mechanism | Injection-induced invagination | Active penetration | Phagocytosis | Calcium-lysosomal Non-lysosomal |
| Host markers in the vacuole | Excludes endocytic and exocytic markers | Excludes endocytic and exocytic markers | EEA1, Rab5, Rab7, LAMP1 | LAMP1, PIP3, EEA1 |
| Niche | Nonfusigenic vacuole | Nonfusigenic vacuole | Delayed maturation, phagolysosome | Cytoplasmic |
| Secreted-surface virulence factors | Polar tube proteins | Rhoptry proteins ROPs, RONs Microneme proteins MICs |
Lysophosphoglycan LPG |
TcTox, LYT1, trans-sialidase, mucins, gp63 |
| Nutrient acquisition | Pores in vacuole membrane | Pores in vacuole membrane | Induced autophagy – phagolysosomal degradation | Direct access to cytosolic components |
Figure 2.
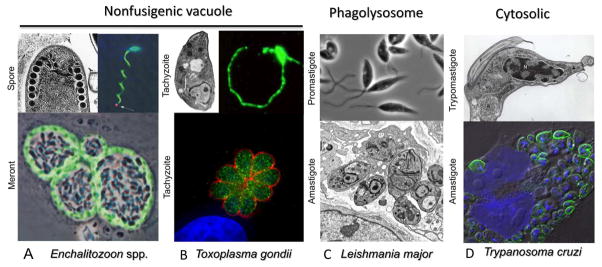
Summary of invasive forms and intracellular niches of major groups of intracellular protozoan parasites. A) Extracellular spore stages of E. intestinalis (left) showing coiled polar tube in cross-section. Image used with permission (216). Image at right shows discharge of sporoplasm (red) at the tip of polar tube (arrow), polar tube and spore wall stained with polyclonal serum (green), nuclei stained with DAPI (blue). Used with permission (28). Lower image shows intracellular meronts dividing within fibroblasts. Stained with mAb 6G2 that recognizes peripheral meronts (green), while sporoblasts occupy central region of the vacuole. Used with permission (28). B) Extracellular tachyzoites of T. gondii display polarized arrangement of organelles characteristic of apicomplexans (left upper). Image provided by W. Beatty. Gliding motility of tachyzoites in vitro as revealed by staining with the surface protein SGA1 (right image). Lower image shows intracellular replication of tachyzoites in a rosette. Cytoplasmic actin-like protein ALP1 (green) and surface antigen SAG1 (red) stained with specific antibodies, nuclei stained with DAPI (blue). Image used with permission (217). C) Extracellular promastigotes of L. major are flagellated, motile forms. Following invasion, the parasite differentiates into dividing amastigotes, which residue in a modified phagolysosome. Images provided by W. Beatty. D) Upper image shows extracellular trypomastigote of T. cruzi showing nucleus (N), kinetoplast (K) and flagellum (F). Image provided by S. Moreno. Lower image shows intracellular amastigotes dividing within cultured HeLa cells labeled with antibodies to T. cruzi PI-PLC (green), nuclei stained with DAPI (blue). Image modified with permission from (218).
Microsporidian parasites
There are more than 1,000 described species of microsporidian parasites, most of which are parasitic on invertebrates, including those that cause infestations of silkworm moths and honey bees; while others infect fish, where they cause economic loses. In contrast, microsporidian infections in humans are rare and typically associated with an immuncompromised state, although they also cause diarrheal disease in travelers and children in developing countries. Infection is spread by small environmentally resistant spores that are surrounded by double-layered wall comprised of an external exospore and internal enodospore (Figure 2A). Within the spore, a coiled polar tube filament is critical to the infection process (see below) and also contains an endomembrane system called the polaroplast and a posterior vacuole.
Intracellular parasitism by Encephalitizoon spp
Microsporidiosis is largely a zoonotic disease and humans are only an incidental host. Among the most common species infecting humans are Encephalitozoon cuniculi, E. hellem, E. intestinalis, and E. beneusi (13). Infections are transmitted by internalization of spores via the respiratory or gastrointestinal tracks. Persistent infections are characterized by diarrheal disease, although ocular and respiratory infections are also found in immunocompromised patients. Treatments with albendazole, a microtubule inhibitor, or fumagillin, a natural product produced by Aspergillus, are effective in controlling infection. Microsporidians were initially thought to be early branching eukaryotes based on the lack of mitochondria and simplified ribosome structure that resemble prokaryotic subunits. Subsequent molecular phylogenies revealed closer affinities to fungi, as first highlighted by examination of the tubulin encoding genes (4). Broader phylogenetic analyses from whole genome sequencing supported this revised classification and also revealed the remarkable genome reduction of this parasite (4). The complete genome sequence for E. cuniculi encodes just under 2,000 genes in 2.9 mb of total size (14). Introns, repeats and extragenic regions are all extremely short, and genes tend to be smaller than in other systems. As with other eukaryotic organisms once thought to predate the origin of mitochondria, remnant mitochondrial genes have also been found in microsporidians (15). Overall, the remarkable genome reduction of Encephalitozoon appears to be an adaptation to its obligate intracellular existence.
Mechanism of cell entry: ejection and induced uptake
Although all microsporidians produce environmentally resistant spores and propagate within their host cells, their mechanisms of entry and intracellular niches may vary. Most microsporidians are thought to develop directly within the host cytosol of their host cells, while a few genera are contained with a parasitophorous vacuole. Here we will focus on E. cuniculi as an example, since it represents the genus most commonly found in humans, and it has been better studied than most species. E. cuniculi infects a wide range of cell types (Table 1) and different mammalian hosts, where it is able to survive in macrophages as well as nonprofessional phagocytes (16).
During germination, microsporidian parasites eject themselves from the spore through a long polar filament that rapidly elongates into a tube (Figure 2A) (17). In response to environmental stimuli, which may include changes in pH, calcium, or other ions (18), polar filament extrusion is driven by changes in osmotic pressure from within the spore. During extrusion, the polar tube inverts as it grows from the distal tip, ultimately forming a hollow tube through which the sporoplasm migrates (19). The original plasma membrane is left behind and a new membrane is formed from the polaroplast, enveloping the sporoplasm that is released at the tip of the polar tube (20). Time-lapse video microscopy has captured this event in Nosema algerae and estimated that it occurs at ~100 μm / sec (21), making it one of the fastest motility events observed for small cells. Several proline rich proteins decorate the elongating tube and comprise the major polar tube proteins (PtP) (22). Germination has the potential to puncture the host cell plasma membrane directly and it is thought that many microsporidians simply pierce their host cell and inject the sporoplasm directly into the cell cytolsol.
The entry mechanism of Encephalitozoon is somewhat different as this parasite residues in a vacuole in the host cell, within which it replicates by binary fission, eventually differentiating to generate new spores (16). Although the compartment housing intracellular E. cuniculi was originally described to avoid fusion with lysosomal markers (23), later studies suggested that spore entry can also occur by phagocytosis, even in non-phagocytic cells (24). Consistent with this later entry mechanism, vacuoles formed by phagocytosis acquire endocytic and lysosomal markers, although this fate is not conducive to parasite survival (25). However, it was postulated that spores might hatch from within phagocytic vacuoles and deposit sporoplasms within the host cell cytosol, thereby escaping digestion (26). More recent studies have shown that although a considerable number of spores are ingested by phagocytosis, these do not appreciably contribute to productive infection (27). Examination of newly formed vacuoles containing E. cuniculi sporoplasms that enter by “invasion” reveals that they do not contain markers for the host cell plasma membrane nor do they acquire host endocytic markers such as transferrin receptor, EEA1 or LAMP1 (28). Hence, productive infection with E. cuniculi results in formation of a nonfusigenic vacuole (Table 1).
The use of fluorescent lipid probes reveals that host cell surface lipids are incorporated into the vacuole surrounding E. cuniculi shortly after its formation, indicating that the bulk of the lipid is likely derived by invagination of the plasma membrane (29). Both lipid raft associated and nonraft-associated domains of the plasma membrane were internalized into the membrane surrounding newly invaded parasites (29). Time-lapse video microscopy studies revealed that plasma membrane internalization is coincident with the extrusion of the sporoplasm (29). Hence, it appears that following extrusion from the polar tube, the sporoplasm induces uptake into the cell via a specialized invagination in the plasma membrane, perhaps induced by the tip of the polar tube (29). How this compartment gets internalized into the host cell remains undefined, although this process operates independently of host actin (27), indicating it is not a phagocytic event. Interaction of the spore with host cell membranes is facilitated by binding to glycosaminoglycans on the plasma membrane, an interaction that may be mediated by the spore wall protein EnP1 (30, 31). Given that spore discharge occurs outside of the cell prior to “invasion”, this interaction likely serves to indirectly enhance the probability of infection.
Intracellular niche and nutrient access
Comparison of the genomes of microsporidian parasites reveals a reduced reliance on metabolic pathways involved in biosynthesis, with an expansion of transporters designed to capture host metabolites. Although E. cuniculi has a core carbon pathway consisting of glycolysis and a pentose phosphate shunt (14), both pathways are absent in E. benusi, which lacks a system for generating ATP from glycolysis (32). Microsporidians also lack functional mitochondria and hence also do not rely on electron transport to generate energy. Microsporidian genomes encode ATP transporters, which are thought to salvage ATP from the host cell cytosol (33). Intriguingly, E. cuniculi resides in a vacuole that contains pores that allow diffusion of host molecules into the lumen of the vacuole (29). Although the molecular identity of these pores is unknown, they allow passage of small molecules such as ATP, amino acids, vitamins, and sugars from the host cytosol to the proximity of the parasite. Within this semi-permeable compartment, the parasite divides by binary fission, and eventually differentiates into sporoplast cells that give rise to new spores (17). The process of egress is thought to occur by release of spores from terminally infected cells, although no details are available about cellular pathways that might govern this.
Host-pathogen interactions in vivo
Although little is known about the direct affects of microsporidian infection on induction or evasion of immune mechanisms at a cellular level, the control of infection on a broader level has been studied in the murine model infected with E. cuniculi. Inbred mice strains differ in susceptibility, but all strains of mice mount a potent Th1 response (34). Immune protection is largely dependent on T cell-mediated immunity that requires the production of IL-12 and IFN-γ (35, 36). CD8+ T cells are critical both for producing cytokines such as IFN-γ and for direct killing of infected targets, whereas iNOs is not essential for control of the acute infection (37). In contrast, CD4+ T cells appear to be dispensable for induction of immunity (38). Instead, γδ T cells are important for producing IFN-γ and inducing CD8+ T cell immunity (39). Following oral challenge, dendritic cells are critical to development of immunity mediated by intraepithelial lymphocytes (40). In the human system, disseminated disease is characterized by monocyte infiltration. In vitro infection of monocyte-derived macrophages with E. cuniculi is mediated by TLR2, upregulating secretion of TNF-α and IL-8, as well as numerous chemokines, and recruiting additional host cells (41, 42). Innate immune responses to microsporidia in natural insect hosts, and model organisms like Drosophila, consist of common pathways for melanization and antimicrobial peptides, as well as transcriptionally unique pathways (16). Microspordia are common parasites of insects and as such they have likely evolved elaborate ways of thwarting innate immunity, although these remain largely unexplored.
Apicomplexans
The phylum Apicomplexa is a diverse collection of more than 5,000 species, most of which are parasitic on invertebrates (43). Early branching members include gregarines, which parasitize the enteric tract of various invertebrates, especially worms and insects (44). Apicomplexans are unified by an apical complex consisting of a polarized microtubule organizing center called the conoid, and a cluster of apical secretory organelles (45). The also contain submembrane alveolar sacs, consistent with their relationship with ciliates and inclusion in the Alveolata grouping of eukaryotes (Figure 1). Apicomplexans rely on a unique form of substrate-dependent motility referred to as gliding, which propels them across the substrate and which supports active penetration of host cells (46) (Figure 2B).
Given the ancient ancestry of the Apicomplexa, it is thought that parasitism on mammals evolved late, and likely occurred multiple times (44, 47). The Apicomplexa includes enteric parasites such as Cryptosporidium, two species of which infect humans, causing of waterborne diarrheal disease (48). Cryptosporidiosis is temporarily debilitating in healthy adults, but much more severe in immunocompromised hosts or in children, especially in the developing world (48). Other enteric species include Eimeria spp. and related coccidian parasites some of which cause severe disease in livestock, but which only rarely infect humans (i.e. Isospora, Cyclospora,). A number of members of the phylum specialize in infecting blood cells, including Babesia spp. that cause disease in animals and sometimes humans (49). More notable are Plasmodium spp., which cause malaria, the most severe human infections among this group (50), and the related rodent malarias that serve as model systems. Thieleria spp. also cause blood-borne infections that result in serious disease in cattle in Africa, but instead of infecting red blood cells, they parasitizes leukocytes, immortalizing them in the process (51). The final group is the tissue-cyst forming coccidian, which have both an enteric form and a tissue form, typically involved in chronic infection and transmission (52). Among this group, Toxoplasma gondii is the most wide spread, being a truly equal opportunity pathogen; infecting all types of nucleated cells in all forms of warm-blooded vertebrates. Toxoplasma has become the model for this phylum due to the ease of use, genetic tools, and excellent animal models (53). We will briefly consider the general features of intracellular parasitism that have been obtained from studies with T. gondii, summarizing key similarities and differences with other apicomplexan parasites.
Intracellular parasitism by Toxoplasma gondii
Toxoplasma gondii commonly infects a variety of wild and domestic animals and is a zoonotic infection, humans being accidental hosts that play a limited role in transmission (53). The acute infection is characterized by fast growing forms called tachyzoites, which lyse their host cells within 24–48 hrs to release large numbers of progeny. In response to immune pressure, the parasite differentiates into a slow growing form called a bradyzoite, which resides within an intracellular cyst. Tissue cysts normally occur in long-lived cells such as muscle, endothelial, or neuronal cells. Ingestion of the tissue cysts by members of the cat family results in sexual development within intestinal epithelial cells, culminating in the shedding of oocysts that undergo meiosis in the environment to form eight haploid sporozoites. Oral ingestion of oocysts by a wide variety of hosts leads to acute infection. T. gondii is unique among the tissue-forming coccidian in propagating between intermediate hosts due following ingestion of tissue cysts that lead to infection in carnivorous or omnivorous animals (54). This adaptation may account for the highly clonal populations structure of T. gondii that exists in North America and Europe (55). Humans can become infected by ingesting oocysts that can contaminate food or water, or by ingesting tissue cysts in undercooked meat. Treatment of acute infections with sulfa drugs combined with pyramethamine is capable of controlling active proliferation, yet will not eliminate tissue cysts (56). Toxoplasmosis is primarily a problem of immunocompromised patients, although severe disease can also occur due to congenital infection or due to ocular infection in otherwise healthy adults (57).
Apicomplexan parasites lack cilia and flagella (except for microgametes) and navigate through their environments using a unique form of substrate-dependent motility called gliding (46) (Figure 2B). Sporozoites of all apicomplexans exhibit gliding including many gregarines (58). Gliding has been best studied in T. gondii tachyzoites, which undergo circular and helical gliding in vitro, only the later of which leads to productive cell invasion (59). Gliding relies on the translocation of adhesins from the anterior pole, where they are secreted, to the posterior pole in a process akin to a conveyor belt (46). The force for translocation is provided by a small myosin motor anchored in the inner membrane complex (60). Within the space between the inner and outer membranes polymerization of short actin filaments is essential for gliding motility (61). The interaction between the cytoplasmic domain of adhesins and the actin filaments is provided by the F-actin binding protein aldolase, which also plays an important role in energy production through glycolysis (62). A variety of surface adhesins likely contribute to motility in T. gondii, although the best studied is the surface protein microneme protein 2 (MIC2), which plays an essential role in gliding (63). Following translocation along the surface, adhesins are shed by intramembrane proteolysis via a family of rhomboid proteases (64). Recent evidence reveals that ROM4 functions to trim surface proteins and hence maintains a gradient of adhesins from the front to the back that is important for efficient gliding motility (65).
Gliding motility of Plasmodium sporozoites also generates circular and helical patterns and requires the MIC-2 orthologue called TRAP, a microneme protein that links to actin filaments via aldolase, and is thought to rely on a similar small myosin motor that is conserved in apicomplexans (66, 67). Gliding motility in Plasmodium sporozoites is critical for invasion of the salivary gland in the mosquito. Gliding motility is also important for ookinete migration across the midgut epithelium in the mosquito and this requires CTRAP, an orthologue of TRAP (68). Finally, gliding motility of sporozoites is important in the vertebrate host to exit the skin and ultimately infect liver hepatocytes (68). Although merozoites of Plasmodium do not display gliding motility, they nonetheless rely on actin-myosin based motility for penetration of the red blood cells (69). Additional details on the cell biology of Plasmodium infection and immunological control can be found in the accompanying chapter by K. Matuschewski.
Mechanisms of cell entry: active invasion propelled by parasite motility
A combination of pharmacological inhibition with cytochalasin D and genetic evaluation of resistance mutants, demonstrated that cell invasion by T. gondii requires polymerization of F-actin by the parasite, while it is largely independent of the host cell cytoskeleton (70). Since cytochalasin D resistant mutants in the parasite regain both gliding motility and cell invasion in the presence of drug, it was concluded that gliding motility provides the force for cell invasion (70). This mode of active invasion consistent with the lack of response in calcium signaling, membrane ruffling, phosphorylation of tyrosine residues, or actin rearrangement in the host cell during invasion (71). More recent reports have suggested a role for actin polymerization in the host cell based on a variety of chemical or genetic treatments that disrupt actin assembly (72). All these manipulations also affect the steady state F-actin pool, hence it is equally possible that these findings reflect a requirement for a rigid host cell cytoskeleton for parasite attachment, rather than active polymerization. Sporozoites and merozoites of Plasmodium are also thought to invade cells actively using this same process, based largely on inhibition by cytochalasin D, although similar genetic confirmation of the role of F-actin in the parasite has not been conducted in this system. Sporozoites of Plasmodium also exhibit cell traversal behavior, being able to successively migrate through multiple cells before finally invading into a membrane bound compartment (68). Initially thought to be important for liver stage invasion, it now seems apparent that the main role of cell traversal is for sporozoites that have been deposited in the skin by mosquito bite, to escape from the dermis and enter into capillaries (73).
Penetration of T. gondii into the host cell results in formation of a membrane bound vacuole that is primarily derived by invagination of the host cell plasma membrane as shown both by electrophysiological recordings and surface membrane lipid labeling (74). In the process of entry, the parasite excludes the majority of host cell surface proteins, both through interactions with the underlying cytoskeleton, and through differences in anchoring in the plasma membrane that influence fluidity (75, 76). Similar sorting has been described during invasion of merozoites into red blood cells (77). In the case of T. gondii, sorting excludes many determinants that would otherwise target the vacuole for endocytic processing (78, 79). Ultimately, T. gondii resides in a vacuole that is nonfusigenic and which resists acquisition of markers from both the endocytic and exocytic systems, while being enveloped by host ER and mitochondria (Figure 2B, Table 1). Intracellular T. gondii parasites are cut off from the extracellular environment and the host endomembrane system and likely gain access to host metabolites via small pores in the membrane of the vacuole, which allow free diffusion of molecules ~ 1600 daltons (80)
Apicomplexan parasites are highly specialized for regulated protein secretion, which is tightly coupled with motility and cell invasion (81). As mentioned above, motility relies on proteins in the micronemes (MIC), apical secretory organelles that are discharged in response to elevated cytoplasmic calcium (82). Microneme discharge occurs by fusion of the secretory vesicles with the cell surface at the apical end of the cell, transferring proteins such as MIC2 onto the surface membrane. Micronemal proteins contain a variety of adhesive domains including type I thrombospondin repeats, integrin A domains, apple domains, EGF repeats, Sushi domains, and fibronectin type II and III domains (83). These domains are found in different combinations on a wide range of proteins in the Apicomplexa and they are thought to mediate binding to the substratum and host cell. In the majority of cases, the host receptors recognized by these proteins is unknown, although several bind to generic surface molecules such as glycosaminoglycans and sialic acid. Release of microneme proteins is tightly coupled to elevated calcium, and this signal is mediated by the calcium-dependent kinase protein 1 (CDPK1) (84), which is a member of a family of plant like kinases found in apicomplexans (85). In addition to controlling release of MIC2, this pathway controls release of other microneme proteins such as AMA1, which is essential for invasion in both T. gondii (86) and Plasmodium (87) and also implicated in red cell invasion by Babesia (88).
The second wave of protein secretion during invasion discharges rhoptries, which inject their contents into the host cell at the point of invasion (89). The precise mechanism of this transfer is unknown, but it may result from small breaks in the plasma membrane that subsequently reseal. This mechanism introduces effector rhoptry proteins (ROP) that are destined to interact with host cell targets, directly into the cytosol. As described further below, several of these secretory ROP proteins mediate important events in pathogenesis and immune evasion. A second important role for ROP proteins is the formation of a tight junction between the host cell and parasite plasma membrane that defines the point of invasion (90). During penetration into the host cell, the parasite squeezes through a noticeable construction that defines a tight junction between the host cell and parasite. This interface has been referred to as the moving junction, although it remains fixed with respect to the substrate and the parasite moves past the junction to enter the host cell. Rhoptry neck proteins (RONs) comprise the junction and form a specific interface to facilitate invasion (91, 92). Within the junctional complex, RON2, 4, and 5 interact with the microneme adhesin AMA1, thereby forming a bridge from the parasite surface to the host membrane. At least one of the proteins, RON2, is thought to be a transmembrane protein, while several others are detected on the cytoplasmic face of the host cell (92, 93). The ability of the parasite to insert its own receptor into the membrane may account for the very broad host range of T. gondii. A number of the RON proteins are conserved in apicomplexans and a similar complex is thought to be important in Plasmodium invasion of red blood cells (94).
The process of exit from the host cell has been studied in T. gondii and Plasmodium and despite the similarity in their invasion pathways, the events that define egress are quite different (95, 96). At the end of the lytic cycle for T. gondii, the parasite activates motility and emerges from the host cell by rupturing the vacuole and breaching the host cell plasma membrane. Egress in T. gondii is similar to invasion in that it relies on elevated cytoplasmic calcium, microneme secretion, and motility. However, egress is topologically opposite of invasion, implying that any host receptors involved must be different. Elevation in intracellular calcium is triggered by a cADPR pathway, mediated by accumulation of the plant hormone abscisic acid (97). Elevated calcium activates microneme secretion of a membrane lytic protein called perforin-like protein (TgPLP1), which is important for causing the initial breach in the vacuolar membrane (98). Secretion of microneme proteins is essential for egress as shown by the finding that suppression of CDPK1 blocks egress (84). In the absence of these signal-dependent pathways, parasites are also able to rupture cells mechanically, although this process is much less efficient.
Egress in Plasmodium-infected red blood cells relies much less on calcium but rather is triggered by a cascade of proteases that are active within the vacuole (95). Independent of proteolysis, there is evidence that the parasite induces poration of the host cell membrane and that swelling leads to vacuole rupture (99). A variety of studies reveal that egress is also dependent on proteases and is blocked by protease inhibitors, principally those that target cysteine proteinases (95). Consistent with this, disruption of a gene encoding a cysteine proteinase that is expressed in the insect cell stages, blocks egress of sporozoites from the oocyst within the midgut of mosquitoes (100). Paralogs of this gene expressed in red cells stages encode proteins known as SERAs, and the maturation of SERA5 by the subtilisin 1 protease has been linked to egress of merozoites from red blood cells (101). More recently, a calcium-dependent step was identified by the genetic knockdown of CPDK5 (102), which blocks egress at a step downstream of protease activation. Cysteine proteinases are also implicated in egress of malaria merosomes from infected hepatocytes, a process that involves induced cell death and shedding of merozoites packaged in membrane blebs that are discharged into the liver sinusoids (103). Finally, recent studies indicate that activation of host cell calpain is important in release of both malaria merozoites from red blood cells and T. gondii tachyzoites from host cells (104).
Intracellular niche, nutrient access, immune detection and evasion
Toxoplasma is unique in being able to actively penetrate both phagocytic and nonphagocytic cells and set up residence in a nonfusigenic vacuole. However, the intracellular fate of T. gondii is dependent on the active mechanism of entry and antibody opsonized parasites are engulfed by phagocytosis and rapidly undergo endosome-lysosome fusion, resulting in death of the parasite (79). Following activation, macrophages and other cell types are also able to curtail the survival of intracellular T. gondii using a variety of mechanisms including increase reactive oxygen radicals (105), nitric oxide production (106), nutrient limitation (107), and finally vacuole destruction mediated by immunity related GTPases (IRGs) (9). The IRG proteins are strongly upregulated following IFN-γ exposure and they are normally found on endomembranes or reside in the cytosol (108). Following infection, IRGs are recruited to the pathogen-containing vacuole where they oligomerize and destroy the vacuole membrane, resulting in digestion of the pathogen (9). IRGs have been implicated in resistance to a wide variety of intracellular bacterial pathogens as well as protozoans (108). Interestingly, delivery of the IRG proteins to Toxoplasma-containing vacuoles also relies on Atg5, although this appears to operate by a mechanism that is independent of the normal autophagy pathway (109). Not surprisingly, T. gondii has also evolved mechanisms to resist destruction and virulent strains of the parasite avoid clearance by the IRG pathway (110, 111), although the mechanism of this escape is presently unknown.
Infection with T. gondii has been shown to inhibit a wide range of host cell signaling pathways involved innate and adaptive immune signaling (112). For example, depending on different cell types, infection has been shown to block or activate NFκB signaling, a pathway that controls both cell survival and inflammatory responses. Infection with T. gondii has also been shown to block STAT1-based signaling and generally disrupt IFN-γ signaling (113). Infection also blocks apoptosis thorough both cell death activating receptors and endogenous pathways (114). Although the mechanisms responsible for these altered signaling events remain unknown, parasite-specific molecules likely induce them as a means of disrupting host cell immune responses.
Initial clues about the potential for the parasite to alter gene expression came from microarray studies, which identified a variety of metabolite pathways as being upregulated (115). A number of these pro-parasite genes were found to be under the regulation of HIF1, a transcription factor that responds to low oxygen. Additionally, EGR2 (116) and AP-1 (117) transcription factors are upregulated by T. gondii infection and these may play a role in growth promotion or survival. More recent approaches have taken advantage of strain polymorphisms between the three major lineages of T. gondii that are commonly found in North America and Europe (55). Type I strains are acutely virulent in the mouse model and this difference is mediated by a small number of genetic differences between types II and III, which are relatively avirulent (118). Generation of a set of linkage maps for T. gondii and the availability of genetic crosses paved the way for genetic analysis of such complex traits (119). Using this approach, alterations in host cell gene transcription induced in a strain-specific manner were used to map a polymorphic ROP kinase called ROP16 that alters host transcription (120). Interesting, ROP16 is discharged into the host cell and accumulates in the host cell nucleus (120). ROP16 expression in types I and III is associated with prolonged phosphorylation of STAT3 and STAT6 transcription factors, thus enhancing IL4 and IL6 production while down regulating IL-12 production (120). This pathway partially explains a previously described phenotype that distinguishes type II strains as strong inducers of IL-12, from types I and III, which are quiescent (121). Consistent with this, recent studies have shown that ROP16 directly phosphorylates STAT3 (122) and STAT6 (123). Other polymorphic kinases also contribute to overcoming host control, for example transgenic expression of ROP18 from either type I (124) or II (125) is able to greatly enhance the virulence of type III strains, which normally under express this polymorphic kinase. ROP18 is secreted into the host cell cytosol at the time of invasion and is targeted to the surface of the parasite-containing vacuole. ROP18 is also conserved in a variety of virulent lineages from South America, suggesting it may be responsible for the success of these lineage in the wild (126). The target(s) of ROP18 remain unknown, but it is associated with survival and rapid expansion in vivo, although this difference is not the result of intrinsic growth rates.
Host-pathogen interactions in vivo
Control of toxoplasmosis in the murine model critically depends on IL-12, TNF-α, and IFN-γ, which collectively arm hematopoietic and nonhematopoietic cells with potent microbicidal activities (127). The importance of innate and adaptive responses that drive the Th1 response, and of counter regulation by Th2 pathways, in various animal models of toxoplasmosis are considered in detail elsewhere in this volume (Liesenfeld chapter). Although T. gondii can infect all nucleated cells, the macrophage is key to the intracellular infection. Inflammatory monocytes are recruited to the site of infection and are critical for control in the mouse model (128, 129). Inflammatory monocytes are defined by the presence of surface markers for CCR2 that responds to the chemokine CCL2, allowing homing to sites of inflammation (130). Recruitment of inflammatory monocytes in the lamina propria is critical for control of oral infection, and these cells also produce IL-12, TNF-α, and NO, which are implicated in control of intracellular parasites (130).
Gliding is critical for movement across the substrate and also for penetration of tissues. For example, tachyzoites of T. gondii are capable of penetrating though intestinal epithelial cells, and entering the submucosa, and disseminating widely in vivo, including migration across the blood-brain and placental barriers (131). Following infection in the submucosal, infected monocytes and dendritic cells may also disseminate the parasite to deeper tissues, including the CNS (132). Infection of host cells with T. gondii also influences their migration behavior, acting as a second mechanism for dissemination. Infection of dendritic cells and macrophages with T. gondii greatly enhances active migratory behavior and facilitates infection of the CNS (133). The extent to which strains use active migration vs. enhanced host cell motility is dependent on the genotype of the parasite (133).
Other apicomplexans
Although the features of apicomplexan gliding motility and cell invasion are largely preserved in related apicomplexan parasites (58), they differ some what among other members. For example, sporozoites of Cryptosporidium exhibit gliding motility and actin-dependent cell entry (134). However, following initial contact during which the parasite becomes enveloped in the host cell membrane (134), the host actin cytoskeleton undergoes elaborate remodeling to form a unique pedestal that supports intracellular development at the apex of the cell (135). Invasion is associated with rapid phosphorylation of tyrosine residues in host cells and within 30 min, elaborate accumulations of F-actin are detected at the host-parasite interface (136). Actin assembly involves activation of CDC42 and recruitment of N-WASp and Arp2/3 ((135), consistent with known pathways for actin assembly. The activation of host signaling and cytoskeletal rearrangement contrasts sharply with what is seen for T. gondii. Cryptosporidium also differs from other apicomplexans in that occupies a unique niche that lies outwith the cytosol, but encased within a parasite-containing vacuole and surrounded by the host cell plasma membrane. These differences may reflect an independent event leading to intracellular parasitism, consistent with its deep phylogenic branching (44). Cryptosporidium lacks de novo pathways for synthesizing many key metabolites and hence is highly dependent on nutrients obtained from the host cell. Nutrients are likely acquired through a membranous interface called the feeder organelle that forms beneath the unique parasite containing vacuole. Within the intestinal epithelium, the parasite undergoes several rounds of development, egress, and reinvasion (i.e. merogony, gametocyte formation, fertilization) before shedding oocysts into the lumen of the intestine, although the processes governing these steps are poorly understood.
Even more divergent is the entry mechanism of Thieleria, which invades and replicates within lymphocytes. Sporozoites of Theileria are nonmotile, bind to cells with various orientations, and enter by a process that involves zippering of the host membrane around the parasite (137). Entry is independent of the actin cytoskeleton and does not involve formation of a moving junction and protein secretion also appears to play little role in this process (137). Although this process differs from that described for other apicomplexans, the mechanism of entry remains largely uncharacterized, due in part to the difficulty in working with this organism. Following this unusual method of entry, the vacuole membrane dissolves and the parasite lies free within the cytosol. Recruitment of host cell microtubules and activation of IκK kinase assures both the propagation of the parasite during host cell division, and is thought to prevent apoptosis of the transformed cell (51).
Kinetoplastids
The kinetoplastidae are a group of parasitic protozoa that are unified by the presence of a flagellum in at least one life cycle stage, an unusual expanded mitochondrial DNA called the kinetoplast, and the presence of morphological variants through the life cycle. They are most closely related to euglenoids and are parasitic on plants, insects, and in some cases vertebrates. Intracellular parasitism appears to have developed several times independently within this group. All members of the Leishmania group, which are otherwise related to parasites of insects such as Crithidia, Herpetomonas, etc., are obligate intracellular parasites in their vertebrate hosts. Their widespread abundance, and variety of clinical presentations in humans, gives evidence to the success of the intracellular life style. Separately, intracellular parasitism arose in T. cruzi, a member of the trypanosomatids, which are largely extracellular parasites of a range of different species including reptiles, birds, and mammals. Not surprisingly, the mechanisms of host cell entry and intracellular niches of these two organisms differ greatly.
Intracellular parasitism by Leishmania
Leishmania parasites are widespread in the old and new world where they are transmitted by a variety of species of sand flies from two major genera Phlebotomus and Lutzomyia, respectively. Within the insect host, the parasite lives as a motile promastigote form (Figure 2C), which attaches to the midgut wall to avoid being expelled with the blood meal. Differentiation into nondividing metacyclic forms is associated with detachment from the midgut and migration forward in the digestive tract. During feeding, metacyclic promastimgotes are injected into the mammalian host where they enter macrophages and differentiate into non-flagelated amastigotes (Figure 2C). Amastigotes replicate and persist intracellularly, providing a reservoir for transmission. Different species of Leishmania are responsible for a spectrum of human diseases from cutaneous forms caused by L. major, L. tropica, and L. mexicana to the more sever mucocutaneous disease caused by L. braziliensis brasiliensis and finally the most severe form, visceral disease caused by L. donovani. Although cutaneous forms are largely self-healing and lead to lasting immunity, visceral forms of the disease are often fatal and difficult to treat. Leishmania parasites provide excellent models to study cellular immunity to intracellular parasites, as discussed further by the chapter by N. Glaichenhaus in this volume.
Mechanism of cell entry: phagocytosis
Entry of promastigotes into macrophages occurs following serum opsonization via complement receptor 1 (CR1) or 3 (CR3) and occurs by a RhoA-dependent process of phagocytosis (138). Promastigotes do not replicate in macrophages, but rather differentiate into amastigotes, loosing the flagellum and repressing expression of surface molecules in the process. During this differentiation process, promastigotes arrest the maturation of the phagosome, which shows delayed or reduced recruitment of late endosome lysosome markers such as rab7 and LAMP1 (139) (Table 1). Arrested phagosomes are also characterized by over-coating with host actin, associated polymerization factors such as Arp 2/3, Nck, and WASP (140), and recruitment of a variety of host GTPases involved in actin polymerization (141). Additional remodeling of the phagosome is associated with alteration in lipid rafts and with impaired assembly of the NADPH oxidase complex (142). Uptake of virulent metacyclic promastigotes into murine macrophages has been associated with entry into caveolae, and this pathway is important for avoiding early lysosome fusion (143). Additionally, uptake of metacyclic promastigotes by human macrophages is associated with avoidance of mannose receptor positive compartments and subsequent delayed lysosome fusion (144).
Similar to promastigotes, uptake of amastigotes occurs by a classical phagocytic process that can be influenced by opsonization with different serum components ((145) (Table 1). Uptake of uncoated parasites is mediated by Rho and Cdc42, while IgG-coated parasites evoke uptake via a Rac1-dependent process (146). Vacuoles formed by uptake of amastigotes readily undergo fusion and acquire markers associated phagosome maturation into phagolysosomes. The vacuole is positive for the H+ ATPase and becomes strongly acidic and contains hydrolyses as well as markers such as rab7, LAMP1, and LAMP2 (139). Amastigotes are resistant to this hydrolytic environment and replicate with the phagolysosome (Table 1). Not all vacuoles occupied by Leishmania spp. are the same; small tight fitting vacuoles characterize L. major and L. donovani, while L mexicana and L. amazonensis occupy large spacious vacuoles. In response to infection, upregulation of Beige LYST, a regulator of lysosome size, restrains the growth of vacuoles containing L. amazonensis, thereby acting to enhance oxidative stress and limit intracellular growth (147). Regardless of the vacuole size preferred by a given species of Leishmania, replication of the parasite eventually leads to cell lysis and release of amastigotes that infect other cells, although little is known about the factors that govern the process of egress.
The ability of Leishmania to modulate phagosome maturation depends on an abundant surface glycolipid called lypophosphoglycan or LGP, which is a member of a family of phosphoglycans. This group of molecules shares a repeating structure of disaccharides of Gal (1,4) Man (α1PO4 – 6) that is not found in the vertebrate host. LPG contains a core backbone of repeating disaccharides linked to the membrane by a singly acylated phosphoinositol anchor. In addition to this core backbone, LPGs from different species of Leishmania differ in side change modifications ranging for highly branched structures in L. major and L. tropica to the simpler structure of L. donovani (148). Differences in the side chain repeats are thought to control attachment to the fly midgut as shown by in vitro binding experiments, competition experiments where soluble LPG will displace parasite binding, and using LPG-deficient mutants that are rapidly cleared from flies (148). The diversity of LPG side chains is also thought to influence vector compatibility and hence control transmission (148).
Mutants in LPG biosynthesis have been useful for probing the role of these components in intracellular survival and virulence. Lpg1−/− parasites lack the ability to elongate the LPG core, yet have normal levels of other PGs. In contrast, lpg2−/− mutants are unable to elongate the core repeats on LPG and other PGs. Lpg2−/− L. major parasites fail to survive in macrophages and are unable to delay phagolysosomal maturation (149). This affect was attributed to LPG based on add-back experiments and the similar phenotype of Lpg1−/− mutants (150, 151). Consistent with this, amastigotes, which lack LPG, are unable to delay phagosome maturation and rapidly acquire rab7 and LAMP markers (139). Recent studies suggest that LPG expression is associated with vacuoles that avoid recruitment of synaptotagmin V, thereby avoiding delivery of the H+ ATPase responsible for acidification (152). The susceptibility of Lpg1−/− or Lpg2−/− mutants to clearance in macrophages suggests that the delay in maturation is necessary to protect promastigotes from clearance, whereas amastigotes are much more resistant to the hydrolytic environment of the phagolysosomal system. However, the same susceptibility in Lpg- parasites is not observed for L. mexicana (153), a finding that may relate to the larger vacuole size, which has been postulated to provide some protection from oxidative stress. Additionally, the role of LPG1 in survival within dendritic cells has not been determined in either species.
Intracellular niche, nutrient access, immune detection and evasion
Although the phagolysosome environment of the replicating amastigote would appear to be a hostile environment, it does provide ready access to nutrients that are being degraded by the host cell. Leishmania mexicana has further enhanced this process by inducing an autophagy like pathway in infected cells, thereby shunting cytosolic proteins for degradation and making them available to parasites within the phagolysosome (154). Like many intracellular pathogens, Leishmania is dependent on iron it acquires from the host. One of the main processes for limiting access of iron to intravacuolar pathogens is the NRAMP1 protein, which acts to pump Fe2+ iron from the phagosome lumen to the cytosol. NRAMP plays an important role in controlling intracellular growth of Leishmania. In order to obtain iron, Leishmania relies on a ZIP family iron transporter called LIT1, which is expressed by amastigotes and upregulated during intracellular growth (155). LIT1 encodes an enzyme with iron reductase activity that is expressed on the parasite surface, converting Fe3+ to Fe2+ iron that is transported for cellular utilization (155). LIT1 expression is essential for intracellular parasite survival and virulence, demonstrating the importance of this iron salvage pathway.
As discussed above, the major entry pathways of metacyclic promastigotes via CR1 and CR3 assures minimal respiratory burst. Additionally, LGP is thought to scavenge oxygen radicals, inhibit PKC, and suppress NO production by macrophages (156). The vacuole surrounding intracellular Leishmania contains MHC class II molecules that would seem to facilitate presentation of antigens. Although antigen presentation occurs following infection with promastigotes, the process is inhibited following infection with amastigotes (139, 156). Inhibition of IFN-γ mediated upregulation of MHCII and iNOS is blocked by L. donovani, which disruptes nuclear translocation of STAT1α (157). Leishmania are also potent inhibitors of IL-12 production by macrophages in vitro and in vivo (156); as this cytokine plays a pivotal role in Th1 responses, this activity is likely extremely important in persistence in vivo.
Host-pathogen interactions during in vivo infection
During primary infection in vivo, Leishmania recruits neutrophils to the site of inoculation following a sandfly bite and survival within these cells is important for productive infection as shown by depletion studies (158). Neutrophils are normally equipped to dispose of microbes (159), yet Leishmania promastigotes are able to establish vacuoles that avoid lysosome fusion and provide a protective environment for survival, if not replication (160). During the first week post inoculation, the proportion of parasites found in neutrophils drops and parasites are increasingly seen in macrophages, which are the normal resident cell (158). Type I interferon receptors are important in regulating the recruitment of both neutrophils and monocytes to the site of infection and in their absence, mice develop attenuated lesions (161). Leishmania infection also induces recruitment of CCR2+ CD11b+ Lyc6+ inflammatory monocytes to the site of infection and these cells are important for control of acute infection (162). Within tissues, it has been suggested that Leishmania persists in safe cells, those that support growth but are less able to clear parasites, perhaps due to more restricted microbicidial capacity (163). Induction of iNOS is considered to be an important control mechanism, and hence parasites may infect Langherhan cells (164) in the skin during primary infection, or fibroblasts (165) during chronic infection, as a means of escaping this effector mechanism (163).
Intracellular parasitism by T. cruzi
Trypanosoma cruzi is an exclusively new world kinetoplastid parasite, being found throughout Central and South America in a wide range of mammalian hosts, and also extending into the southern United States (166). Transmission to the vertebrate host occurs by triatome insect vectors, which pass metacyclic trypomastigotes in their feces following feeding (Figure 2D, Table 1). Infection occurs by entry of the trypomastigotes into the bite, possibly facilitated by rubbing the wound, or by oral ingestion of contaminated food, for example fruit juices that can contain triatome feces. Following entry into the vertebrate host, trypomastigotes are adapted for entry into macrophages were they differentiation into nonmotile, replicative forms called amastigotes (Figure 2D, Table 1). T. cruzi is an obligate intracellular parasite in the vertebrate host and replication takes place in the cytosol, eventually liberating trypomastigotes that can reinvade cells. Uptake of trypomastigotes by tritome bugs results in replication as epimastigotes in the insect, again giving rise to infectious metacylic trypomastigotes that can be transmitted to other vertebrates. T. cruzi is genetically very diverse and two major groups have been described; Tc1 is largely sylvatic while TcII has an urban distribution and is most often associated with human disease (167). In humans, T. cruzi infection is typified by a short-lived acute phase, an intermediate phase where infection is not easily detected, and finally a chronic phase associated with disease that occurs in 10–30% individuals, often only manifest decades later. The disease if referred to as Chagas disease, named after Carlos Chagas who first described the organism in 1909 while working in Brazil. The major symptoms include cardiomyopathy and megasyndromes of the digestive system, both causing significant morbidity and potentially leading to mortality. T. cruzi infections are also a significant cause of disease in immunocompromised patients, including those with HIV/AIDS. Treatment of acute phase infections is limited to two drugs nifurtitox and benznidizole, neither of which is effective in eliminating chronic infection (168). With increasing immigration and travel, blood transfusion from donors with long-term chronic infections pose a risk of transmission to populations not exposed to natural transmission routes (169).
Mechanism of cell entry: lysosome-dependent vs. -independent uptake
T. cruzi is capable of invading both phagocytic cells such as macrophages and dendritic cells and a wide range of nonphagocytic cells. One of the key steps important for entry is the recognition and binding to host cell surface receptors and a diverse array of T. cruzi surface proteins participate in this process (170). Among the principle parasite surface proteins involved in host cell binding and invasion are the gp63 proteases, the gp85 transialidase family, and the mucins and mucin-like proteins (Table 1). Transialidases play an important role in transferring sialic acid from host proteins to mucins, since T. cruzi is unable to synthesize sialic acid. Thus modified, mucins participate in cell attachment, entry, and survival. A variety of other interactions have also been implicated in cell attachment and invasion including TGF-β, the nerve growth factor receptor TrkA, and bradykinnin 1 receptors, and the extent to which these are utilized may depend on the particular strain of T. cruzi (170)(Table 1).
Entry of T. cruzi trypomastigotes into phagocytic or nonphagocytic cells differs from endocytosis/phagocytosis in that it occurs independently of the host actin cytoskeleton. In the case of trypomastigotes, entry of most but not all strains is enhanced by depolymerization of host actin filaments with cytochalasin D. In contrast to trypomastigotes, entry of amastigotes into HeLa cells requires actin microfilaments and also relies on the small GTPase Rac1 (171). Amastigote invasion is calcium-dependent and relies on cAMP signaling (171). Uptake of both amastigotes and promastigotes is sensitive to inhibition by dynasore, a compound that disrupts the GTPase activity of dynamin, which participates in fission of endocytic vesicles as they pinch-off from the plasma membrane (172).
Two major pathways have been described for trypomastigote entry into mammalian cells (Table 1). The first, involves activation of calcium signaling in the host cell and the recruitment of host cell lysosomes to the site of entry, where they fuse with the vacuole formed by the entering parasite (173, 174). The extremely active motility of the parasite likely contributes to this by causing local breaks in the plasma membrane and hence lysosomal recruitment is triggered as part of the wound healing response, akin to what happens in mammalian cells that have been damaged (175). Consistent with this, synaptogamin VII (Syt VII) mediates lysosomal recruitment during T. cruzi infection and during membrane resealing by calcium-mediated exocytosis (176, 177). Recent evidence suggests that autophagy contributes to this pathway as disruption of Atg5 or beclin greatly reduces the number of parasites that enter into cells via the lysosomal recruitment pathway (178).
A second pathway for entry does not rely on lysosome recruitment, but rather occurs by actin-independent invagination of the plasma membrane in a class I PI-kinase-dependent manner (179). Parasites engaging this pathway only intercept lysosomal markers at a later stage. Consistent with these two pathways being functional distinct, inhibition of Syt VII only partially reduces parasite entry (180). As well, the lysosomal recruitment pathway is dependent on class III PI-3 kinases that are inhibited by wortmanin, while the alternative pathway relies on class I PI-3 kinase (180). Although both pathways operate simultaneously in cells, it has been suggested that the early lysosomal recruitment pathway is more efficient. Under conditions where parasites selectively enter by the non-lysosomal pathway (i.e. in the presence of wortmanin to inhibit class III PI-3 kinases), they are not retained by the cell but rather are extruded (181). Similar results have been shown for treatment with cytochalasin D, which inhibits the delivery of parasites to mature to a lysosomal compartments (182). Hence, acquisition of the lysosomal niche is key to parasite survival, regardless of which initial entry pathway is utilized.
During host cell invasion, there is evidence that the parasite directly activates calcium signaling in host cells through a variety of pathways (Table 1). Generation of kinnins released by the action of the parasite protease cruzipain (gp63) results in bradykinnin receptor 2 (B2R)-mediated signaling and activation of PLC to release calcium from the ER (183). Trypomastigotes secrete a soluble factor that interacts with host cell surface G-protein coupled receptors, which in turn triggers calcium release via a PLC-dependent pathway that activates IP3 channels (184). This pathway relies on oligopeptidase B, which is a cytosolic enzyme in the parasite that processes an unknown ligand that is released from the cell (185, 186). Additionally, cAMP dependent calcium-regulated exocytosis contributes to lysosome exocytosis and T. cruzi invasion (187). Collectively these pathways for calcium activation contribute to localized turnover of the actin cytoskeleton, enhanced lysosomal fusion with the plasma membrane, and perhaps other cellular pathways involved in parasite uptake.
Intracellular niche, nutrient access, immune detection and evasion
Following engulfment into lysosome-rich compartments, the parasite undergoes differentiation into the nonflagellated amastigote form that will undergo division. Prior to completing this developmental switch, it ruptures the parasitophorous vacuolar membrane and takes up residence directly in the host cell cytosol (180, 188). Escape is mediated by several molecules including a protein with hemolysin activity that cross-reacts with complement component C9, called TxTOX (189) (Table 1). The gene encoding TcTOX activity has never been identified, but it may correspond to a factor called lytic factor LYT1, which is critical for vacuole rupture (190), as both proteins react with antibodies to C9. Vacuole escape is important for intracellular replication as null mutants in the gene encoding LYT1 show reduced infectivity in vitro and in vivo (191). Cell surface transialidase also contributes to vacuole rupture by processing glycoconjugates on host lysosomal proteins including LAMP1 and LAMP2 (192, 193). Once liberated in the cytosol, the parasite has ready access to small metabolites that serve as nutrients (i.e. sugars, amino acids, vitamins, nucleobases). Little is known about the nutrient uptake pathways of intracellular T. cruzi, although the parasite contains amino acid and sugar transporters (194). Within the host cell cytosol, amastigotes reportedly undergo a precise 9 rounds of mitotic replication, before reverting to non-dividing trypomastigotes that are released by host cell lysis (188). Although the factors governing egress are not known, the precise number of cellular divisions that are choreographed to development, suggests that the process is highly regulated.
Infection with T. cruzi in the murine model evokes a strong type 1 cytokine response and control of infection is dependent on IL-12, IFN-γ, and iNOS (195). CD8+ T cells responses are critical to control and a surprisingly large fraction of antigen-specific T cells are generated to transialidases (196). Innate responses are trigged both by the MyD88 and TRIF pathways that serve as adaptors for toll-like receptors (TLRs). Important pattern recognition receptors include TLR2, which responds to GPI anchors on parasite proteins (197, 198). Additionally, TLR9, which responds to CpG methylated DNA is important for control of infection (199, 200). Together TLR2 and TLR9 combine to induce expression of iNOS, IL-12, and TNF-α from dendritic cells and macrophages, thus contributing to parasite control. The induction of type I interferon also contributes to control of infection as shown by the increased susceptibility of MyD88−/− TRIF−/− mice compared to those lacking MyD88 alone (201). Cell-autonomous control of intracellular parasites is mediated by the IRG family of GTPases, and mice lacking LRG-47 (i.e. Irgm1) have enhanced susceptibility to infection with T. cruzi (202). Type I interferons are also produced in vivo in an intradermal challenge model, and this pathway relies primarily on direct feedback as shown by the lowered responsiveness of IFNAR−/− mice (203). Despite this robust response, absence of type I interferon does not influence development of T. cruzi reactive CD8+ T cells in the murine model (204).
TLR-independent mechanisms also contribute to innate immunity. For example induction of type I interferon has also been described to result from activation of TBK1 that acts on IRF3 (205), although the cytosolic sensor involved has not been identified. Additionally NOD-like receptor 1 (Nod1) is required for control of T. cruzi infection mediated through NFκB activation (206). NOD proteins normally respond to bacterial cell wall products and the parasite molecule sensed by this pathway is also not known. Finally, elevated calcium signals described above can activate NFATc1, which is critical for IFN-γ production and dendritic cell maturation (207).
To provide insight into host pathways that might be selectively altered by T. cruzi, transcriptional profiling studies have been conducted using host microarrays to examine changes in gene expression in several cell types following in vitro infection (208). These studies reveal the strong type I interferon pathway described above and associated genes controlled by this pathway (208). Additionally, they reveal that pathways controlling cell proliferation, amino acid catabolism, and wound healing responses are altered by infection (208). For example, the down regulation of genes involved in branch chain amino acid catabolism presumably makes these essential nutrients more available to the parasite. Unexpectedly, these studies also revealed a block in cytokinesis, suggesting that T. cruzi arrests the cell cycle to prolong its intracellular survival (208). Alterations in pathways that promote cell survival were also observed in microarray studies and separate studies reveal that intracellular T. cruzi activates Akt as a mechanism to prevent apoptosis (209).
Host-pathogen interactions during in vivo infection
Although extensive in vitro studies have characterized cellular entry mechanisms and the intracellular niche, less is known about how T. cruzi finds appropriate cells and survives in them in vivo. Following introduction into the tissue, trypomastigotes likely infect fibroblasts, mesenchymal and tissue macrophages in the local environment. Recruitment of inflammatory monocytes and CD8+ T cells expressing CCR2 is important for control of infection as shown by the increased susceptibility of mice deficient in the chemokine Ccl2−/− (210). The mechanism by which the parasite exits the local lesion and migrates to tissues such as the heart and skeletal muscle remains unresolved. However, during the first 300 days in the murine model, parasites can be detected in apdipose tissue where they are associated with increased levels of cytokines and chemokines (211). Within this reservoir, increased inflammation may result from suppression of adiponectin, and this process may contribute to cardiomyopathy seen during chronic T. cruzi infection. In infected mice, recruitment of CD68+ macrophages to the heart is associated with production of arginase I, reminiscent of type II or alternatively activated macrophages, which may provide a safe haven for parasite replication in spite of strong Th1 responses (212).
The heart and smooth muscle of the gut are the primary sites of pathology during chronic disease, leading to the suggestion that T. cruzi has tissue tropism, although the molecular basis for this is unknown. Nonetheless, distinct genetic types have been detected within different tissues from the same patient, suggesting a genetic basis for tissue tropism (213). Different parasite genotypes have also been shown to exhibit preferential infection of enteric vs. cardiac tissues in the mouse, supporting the idea that genetic differences may control this predisposition (214). Alternatively, differences in the tissue distribution of different genetic types in patients might result from different routes of transmission. Oral infection occurs across the mucosa and metacyclic trypomastigotes infect mucosal epithelial cells through interactions with gp82, which binds to mucin and triggers calcium signaling in the host cell (215). The variable susceptibility of the parasite surface molecule called gp90 to pepsin treatment may accentuate oral infection, thus influencing pathogenicity by this route (215).
Conclusions
Defining the cellular niches of intracellular parasites is important for several reasons including identifying possible unique adaptations that might define new therapeutic targets, maximizing the efficiency of antibiotic or drug delivery, and identifying parasite molecules that are recognized by cellular immune responses, such as those sensed by innate recognitions systems. What we have learned from a few laboratory strains of the example species summarized above, now needs to be extended to other more clinically relevant isolates, and to interactions with human cells, in order to define to what extent they define conserved vs. unique adaptations. Recent expansion of genetic tools, use of model organisms, and live imaging techniques have the potential to inform us about important host-parasite interactions that occur in vivo. Defining tissue tropisms and the cellular interactions that occur during acute and persistent infections in vivo will ultimately be important for understanding pathogenesis and for improved methods for disease control.
Acknowledgments
I am grateful to members of my laboratory who have contributed to the studies described here and colleagues who contributed the images used here and provided helpful suggestions and comments. Supported by grants from the NIH.
References
- 1.Baldauf SL. The deep roots of eukaryotes. Science. 2003;300:1703–1706. doi: 10.1126/science.1085544. [DOI] [PubMed] [Google Scholar]
- 2.Shiflett AM, Johnson PJ. Mitochondrion-related organelles in eukaryotic protists. Annu Rev Microbiol. 2010 doi: 10.1146/annurev.micro.62.081307.162826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loftus B, et al. The genome of the protist parasite Entamoeba histolytica. Nature. 2005;433:865–868. doi: 10.1038/nature03291. [DOI] [PubMed] [Google Scholar]
- 4.Keeling PJ, Fast NM. Microsporidia: biology and evolution of highly reduced intracellular parasites. Annu Rev Microbiol. 2002;56:93–116. doi: 10.1146/annurev.micro.56.012302.160854. [DOI] [PubMed] [Google Scholar]
- 5.Escalante AA, Ayala FJ. Evolutionary origin of Plasmodium and other Apicomplexa based on rRNA genes. Proc Natl Acad Sci USA. 1995;92:5793. doi: 10.1073/pnas.92.13.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simpson AG, Stevens JR, Lukes J. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 2006;22:168–174. doi: 10.1016/j.pt.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Haas A. Everybody has a home of their own - “The phagosome zoo”. In: Schaible UE, Haas A, editors. Intracellular Niches of Microbes. Weinheim: Wiley VCH; 2009. pp. 159–190. [Google Scholar]
- 8.Kennedy AD, DeLeo FR. Neutrophil apoptosis and the resolution of infection. Immunol Res. 2009;43:25–61. doi: 10.1007/s12026-008-8049-6. [DOI] [PubMed] [Google Scholar]
- 9.Martens S, Howard JC. The interferon-inducible GTPases. Annu Rev Cell Dev Biol. 2006;22:559–589. doi: 10.1146/annurev.cellbio.22.010305.104619. [DOI] [PubMed] [Google Scholar]
- 10.Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeda K, Akira S. Toll receptors and pathogen resistance. Cellular Mirobiology. 2003;5:143–153. doi: 10.1046/j.1462-5822.2003.00264.x. [DOI] [PubMed] [Google Scholar]
- 12.Inohara, Chamaillard, McDonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 13.Didier ES, Didier PJ, Snowden KF, Shadduck JA. Microsporidiosis in mammals. Microb Infect. 2000;2:709–720. doi: 10.1016/s1286-4579(00)00354-3. [DOI] [PubMed] [Google Scholar]
- 14.Katinka MD, et al. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature. 2001;414:450–453. doi: 10.1038/35106579. [DOI] [PubMed] [Google Scholar]
- 15.Keeling PJ, Fast NM, Law JS, Williams BA, Slamovits CH. Comparative genomics of microsporidia. Folia Parasitol (Praha) 2005;52:8–14. doi: 10.14411/fp.2005.002. [DOI] [PubMed] [Google Scholar]
- 16.Texier C, Vidau C, Vigues B, El Alaoui H, Delbac F. Microsporidia: a model for minimal parasite894 host interactions. Curr Opin Microbiol. 2010 doi: 10.1016/j.mib.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Bigliardi E, Sacchi L. Cell biology and invasion of the microsporidia. Microb Infect. 2001;3:373–379. doi: 10.1016/s1286-4579(01)01393-4. [DOI] [PubMed] [Google Scholar]
- 18.Pleshinger J, Weidner E. The microsporidian spore invasion tube. IV. Discharge activation begins with pH-triggered Ca2+ influx. J Cell Biol. 1985;100:1834–1838. doi: 10.1083/jcb.100.6.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weidner E. The microsporidian spore invasion tube. III. Tube extrusion and assembly. J Cell Biol. 1982;93:976–979. doi: 10.1083/jcb.93.3.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weidner E, Byrd W, Scarborough A, Pleshinger J, DS Microsporidian spore discharge and the transfer of polaroplast organelle membrane into the plasma membrane. J Protozool. 1984;31:195–198. [Google Scholar]
- 21.Frixione E, Ruiz L, Cerbon J, Undeen AH. Germination of Nosema algerae (Microspora) spores: conditional inhibition by D2O, ethanol and Hg2+ suggests dependence of water influx upon membrane hydration and specific transmembrane pathways. J Eukaryot Microbiol. 1997;44:109–116. doi: 10.1111/j.1550-7408.1997.tb05946.x. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Weiss LM. The microsporidian polar tube: a highly specialised invasion organelle. Int J Parasitol. 2005;35:941–953. doi: 10.1016/j.ijpara.2005.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weidner E. Interactions between Encephalitozoon cuniculi and macrophages. Parasitophorous vacuole growth and absence of lysosomal fusion. Z Parasitenk Parasitol Res. 1975;47:109. doi: 10.1007/BF00418060. [DOI] [PubMed] [Google Scholar]
- 24.Couzinet S, Cejas E, Schittny J, Deplazes P, Weber R, Zimmerli S. Phagocytic uptake of Encephalitozoon cuniculi by nonprofessional phagocytes. Infect Immun. 2000;68:6939–6945. doi: 10.1128/iai.68.12.6939-6945.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leitch GJ, Ward TL, Shaw AP, Newman G. Apical spore phagocytosis is not a significant route of infection of differentiated enterocytes by Encephalitozoon intestinalis. Infect Immun. 2005;73:7697–7704. doi: 10.1128/IAI.73.11.7697-7704.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franzen C, Muller A, Hartmann P, Salzberger B. Cell invasion and intracellular fate of Encephalitozoon cuniculi (Microsporidia) Parasitol. 205;130:285–292. doi: 10.1017/s003118200400633x. [DOI] [PubMed] [Google Scholar]
- 27.Orlik J, Bottcher K, Gross U, Bohne W. Germination of phagocytosed E. cuniculi spores does not significantly contribute to parasitophorous vacuole formation in J774 cells. Parasitol Res. 2010;106:753–755. doi: 10.1007/s00436-010-1736-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fasshauer V, Gross U, Bohne W. The parasitophorous vacuole membrane of Encephalitozoon cuniculi lacks host cell membrane proteins immediately after invasion. Eukaryot Cell. 2005;4:221–224. doi: 10.1128/EC.4.1.221-224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ronnebaumer K, Gross U, Bohne W. The nascent parasitophorous vacuole membrane of Encephalitozoon cuniculi is formed by host cell lipids and contains pores which allow nutrient uptake. Eukaryot Cell. 2008;7:1001–1008. doi: 10.1128/EC.00004-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayman JR, Southern TR, Nash TE. Role of sulfated glycans in adherence of the microsporidian Encephalitozoon intestinalis to host cells in vitro. Infect Immun. 2005;73:841–848. doi: 10.1128/IAI.73.2.841-848.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Southern TR, Jolly CE, Lester ME, Hayman JR. EnP1, a microsporidian spore wall protein that enables spores to adhere to and infect host cells in vitro. Eukaryot Cell. 2007;6:1354–1362. doi: 10.1128/EC.00113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keeling PJ, et al. The reduced genome of the parasitic microsporidian Enterocytozoon bieneusi lacks genes for core carbon metabolism. Genome Biol Evol. 2010;2:304–309. doi: 10.1093/gbe/evq022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsaousis AD, Kunji ER, Goldberg AV, Lucocq JM, Hirt RP, Embley TM. A novel route for ATP acquisition by the remnant mitochondria of Encephalitozoon cuniculi. Nature. 2008;453:553–556. doi: 10.1038/nature06903. [DOI] [PubMed] [Google Scholar]
- 34.Niederkorn JY, Shadduck JA, Schmidt EC. Susceptibility of selected inbred strains of mice to Encephalitozoon cuniculi. J Infect Dis. 1981;144:249–253. doi: 10.1093/infdis/144.3.249. [DOI] [PubMed] [Google Scholar]
- 35.Khan IA, Moretto M. Role of gamma interferon in cellular immune response against murine Encephalitozoon cuniculi infection. Infect Immun. 1999;67:1887–1893. doi: 10.1128/iai.67.4.1887-1893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moretto MM, Lawlor EM, Khan IA. Lack of interleukin-12 in p40-deficient mice leads to poor CD8+ T-cell immunity against Encephalitozoon cuniculi infection. Infect Immun. 2010;78:2505–2511. doi: 10.1128/IAI.00753-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan IA, Schwartzman JD, Kasper LH, Moretto M. CD8+ CTLs are essential for protective immunity against Encephalitozoon cuniculi infection. J Immunol. 1999;162:6086–6091. [PubMed] [Google Scholar]
- 38.Moretto M, Casciotti L, Durell B, Khan IA. Lack of CD4+ T cells does not affect induction of CD8+ T-cell immunity against Encephalitozoon cuniculi infection. Infect Immun. 2000;68:6223–6232. doi: 10.1128/iai.68.11.6223-6232.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moretto M, Durell B, Schwartzman JD, Khan IA. Gamma delta T cell-deficient mice have a down951 regulated CD8+ T cell immune response against Encephalitozoon cuniculi infection. J Immunol. 2001;166:7389–7397. doi: 10.4049/jimmunol.166.12.7389. [DOI] [PubMed] [Google Scholar]
- 40.Moretto MM, Weiss LM, Combe CL, Khan IA. IFN-gamma-producing dendritic cells are important for priming of gut intraepithelial lymphocyte response against intracellular parasitic infection. J Immunol. 2007;179:2485–2492. doi: 10.4049/jimmunol.179.4.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fischer J, West J, Agochukwu N, Suire C, Hale-Donze H. Induction of host chemotactic response by Encephalitozoon spp. Infect Immun. 2007;75:1619–1625. doi: 10.1128/IAI.01535-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischer J, Suire C, Hale-Donze H. Toll-like receptor 2 recognition of the microsporidia Encephalitozoon spp. induces nuclear translocation of NF-kappaB and subsequent inflammatory responses. Infect Immun. 2008;76:4737–4744. doi: 10.1128/IAI.00733-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levine ND. The Protozoan Phylum Apicomplexa. Boca Raton: CRC Press; 1988. [Google Scholar]
- 44.Leander BS. Marine gregarines: evolutionary prelude to the apicomplexan radiation? Trends Parasitol. 2008;24:60–67. doi: 10.1016/j.pt.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 45.Morrissette NS, Sibley LD. Cytoskeleton of apicomplexan parasites. Microbiol Mol Biol Rev. 2002;66:21–38. doi: 10.1128/MMBR.66.1.21-38.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sibley LD. Invasion strategies of intracellular parasites. Science. 2004;304:248–253. doi: 10.1126/science.1094717. [DOI] [PubMed] [Google Scholar]
- 47.Barta JR. Phylogenetic analysis of the class sporozoea (Phylum Apicomplexan Levine 1970): evidence for the independent evolution of heteroxenous life cycles. J Parasitol. 1989;75:195–206. [PubMed] [Google Scholar]
- 48.Tzipori S, Widmer G. A hundred-year retrospective on cryptosporidiosis. Trends Parasitol. 2008;24:184–189. doi: 10.1016/j.pt.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Homer MJ, Aguilar-Delfin I, Telford SR, 3rd, Krause PJ, Persing DH. Babesiosis. Clin Microbiol Rev. 2000;13:451–469. doi: 10.1128/cmr.13.3.451-469.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller LH, Good MF, Milon G. Malaria pathogenesis. Science. 1994;264:1878–1883. doi: 10.1126/science.8009217. [DOI] [PubMed] [Google Scholar]
- 51.Dobbelaere DA, Kuenzi P. The strategies of the Theileria parasite: a new twist in host-pathogen interactions. Curr Opin Immunol. 2004;16:524–530. doi: 10.1016/j.coi.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 52.Dubey JP. Toxoplasma, Hammondia, Besniotia, Sarcocystis, and other tissue cyst-forming coccidia of man and animals. In: Kreier JP, editor. Parasitic Protozoa. New York: Academic Press; 1977. pp. 101–237. [Google Scholar]
- 53.Dubey JP. Toxoplasmosis of animals and humans. Boca Raton: CRC Press; 2010. [Google Scholar]
- 54.Su C, Evans D, Cole RH, Kissinger JC, Ajioka JW, Sibley LD. Recent expansion of Toxoplasma through enhanced oral transmission. Science. 2003;299:414–416. doi: 10.1126/science.1078035. [DOI] [PubMed] [Google Scholar]
- 55.Sibley LD, Ajioka JW. Population structure of Toxoplasma gondii: Clonal expansion driven by infrequent recombination and selective sweeps. Ann Rev Microbiol. 2008;62:329–351. doi: 10.1146/annurev.micro.62.081307.162925. [DOI] [PubMed] [Google Scholar]
- 56.McCabe RE. Antitoxoplasma chemotherapy. In: Joynson DHM, Wreghitt TG, editors. Toxoplasmosis: a comprehensive clinical guide. Cambridge: Cambridge Univ. Press; 2001. pp. 319–359. [Google Scholar]
- 57.Joynson DH, Wreghitt TJ. Toxoplasmosis: A comprehensive clinical guide. Cambridge University Press; 2001. [Google Scholar]
- 58.Heintzelman MB. Cellular and molecular mechanics of gliding locomotion in eukaryotes. Int Rev Cytol. 2006;251:79–129. doi: 10.1016/S0074-7696(06)51003-4. [DOI] [PubMed] [Google Scholar]
- 59.Håkansson S, Morisaki H, Heuser JE, Sibley LD. Time-lapse video microscopy of gliding motility in Toxoplasma gondii reveals a novel, biphasic mechanism of cell locomotion. Mol Biol Cell. 1999;10:3539–3547. doi: 10.1091/mbc.10.11.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meissner M, Schluter D, Soldati D. Role of Toxoplasma gondii myosin A in powering parasite gliding and host cell invasion. Science. 2002;298:837–840. doi: 10.1126/science.1074553. [DOI] [PubMed] [Google Scholar]
- 61.Sahoo N, Beatty WL, Heuser JE, Sept D, Sibley LD. Unusual kinetic and structural properties control rapid assembly and turnover of actin in the parasite Toxoplasma gondii. Mol Biol Cell. 2006;17:895–906. doi: 10.1091/mbc.E05-06-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Starnes GL, Coincon M, Sygusch J, Sibley LD. Aldolase is essential for energy production and bridging adhesin-actin cytoskeletal interactions during parasite invasion of host cells. Cell Host Microbe. 2009;5:353–364. doi: 10.1016/j.chom.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huynh MH, Carruthers VB. Toxoplasma MIC2 is a major determinant of invasion and virulence. PloS Pathog. 2006;2:753–762. doi: 10.1371/journal.ppat.0020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dowse T, Soldati D. Rhomboid-like proteins in Apicomplexa: phylogeny and nomenclature. Trends Parastiol. 2005;35:747–756. doi: 10.1016/j.pt.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 65.Buguliskis JS, Brossier F, Shuman J, Sibley LD. Rhomboid 4 (ROM4) affects the processing of surface adhesins and facilitates host cell invasion by Toxoplasma gondii. PLoS Pathog. 2010;6:e1000858. doi: 10.1371/journal.ppat.1000858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ménard R. Gliding motility and cell invasion by Apicomplexa: insights from the Plasmodium sporozoite. Cell Micro. 2001;3:63–73. doi: 10.1046/j.1462-5822.2001.00097.x. [DOI] [PubMed] [Google Scholar]
- 67.Soldati D, Meissner M. Toxoplasma as a novel system for motility. Curr Opin Cell Biol. 2004;16:32–40. doi: 10.1016/j.ceb.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 68.Tardieux I, Menard R. Migration of Apicomplexa across biological barriers: the Toxoplasma and Plasmodium rides. Traffic. 2008;9:627–635. doi: 10.1111/j.1600-0854.2008.00703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baum J, Papenfuss AT, Baum B, Speed TP, Cowman AF. Regulation of apicomplexan actin-based motility. Nat Rev Microbiol. 2006;4:621–628. doi: 10.1038/nrmicro1465. [DOI] [PubMed] [Google Scholar]
- 70.Dobrowolski JM, Sibley LD. Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell. 1996;84:933–939. doi: 10.1016/s0092-8674(00)81071-5. [DOI] [PubMed] [Google Scholar]
- 71.Morisaki JH, Heuser JE, Sibley LD. Invasion of Toxoplasma gondii occurs by active penetration of the host cell. J Cell Sci. 1995;108:2457–2464. doi: 10.1242/jcs.108.6.2457. [DOI] [PubMed] [Google Scholar]
- 72.Gonzalez V, et al. Host cell entry by apicomplexa parasites requires actin polymerization in the host cell. Cell Host Microbe. 2009;5:259–272. doi: 10.1016/j.chom.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 73.Amino R, et al. Host cell traversal is important for progression of the malaria parasite through the dermis to the liver. Cell Host Microbe. 2008;3:88–96. doi: 10.1016/j.chom.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 74.Suss-Toby E, Zimmerberg J, Ward GE. Toxoplasma invasion: The parasitophorous vacuole is formed from host cell plasma membrane and pinches off via a fusion pore. Proc Natl Acad Sci USA. 1996;93:8413–8418. doi: 10.1073/pnas.93.16.8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Charron AJ, Sibley LD. Molecular partitioning during host cell penetration by Toxoplasma gondii. Traffic. 2004;5:855–867. doi: 10.1111/j.1600-0854.2004.00228.x. [DOI] [PubMed] [Google Scholar]
- 76.Mordue DG, Desai N, Dustin M, Sibley LD. Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. J Exp Med. 1999;190:1783–1792. doi: 10.1084/jem.190.12.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Murphy SC, et al. Erythrocyte detergent-resistant membrane proteins: their characterization and selective uptake during malarial infection. Blood. 2004;103:1920–1928. doi: 10.1182/blood-2003-09-3165. [DOI] [PubMed] [Google Scholar]
- 78.Mordue D, Håkansson S, Niesman I, Sibley LD. Toxoplasma gondii resides in a vacuole that avoids fusion with host cell endocytic and exocytic vesicular trafficking pathways. Exp Parasitol. 1999;92:87–99. doi: 10.1006/expr.1999.4412. [DOI] [PubMed] [Google Scholar]
- 79.Mordue DG, Sibley LD. Intracellular fate of vacuoles containing Toxoplasma gondii is determined at the time of formation and depends on the mechanism of entry. J Immunol. 1997;159:4452–4459. [PubMed] [Google Scholar]
- 80.Schwab JC, Beckers CJM, Joiner KA. The parasitophorous vacuole membrane surrounding intracellular Toxoplasma gondii functions as a molecular sieve. Proc Natl Acad Sci USA. 1994;91:509–513. doi: 10.1073/pnas.91.2.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carruthers VB, Sibley LD. Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur J Cell Biol. 1997;73:114–123. [PubMed] [Google Scholar]
- 82.Carruthers VB, Tomley FM. Microneme proteins in apicomplexans. Subcell Biochem. 2008;47:33–45. doi: 10.1007/978-0-387-78267-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anantharaman V, Iyer LM, Balaji S, Aravind L. Adhesion molecules and other secreted host1051 interaction determinants in Apicomplexa: insights from comparative genomics. Int Rev Cytol. 2007;262:1–74. doi: 10.1016/S0074-7696(07)62001-4. [DOI] [PubMed] [Google Scholar]
- 84.Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, Sibley LD. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature. 2010;465:359–362. doi: 10.1038/nature09022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Billker O, Lourido S, Sibley LD. Calcium-dependent signaling and kinases in apicomplexan parasites. Cell Host Microbe. 2009;5:612– 622. doi: 10.1016/j.chom.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mital J, Meissner M, Soldati D, Ward GE. Conditional expression of Toxoplasma gondii apical membrane antigen-1 (TgAMA1) demonstrates that TgAMA1 plays a critical role in host cell invasion. Mol Biol Cell. 2005;16:4341– 4349. doi: 10.1091/mbc.E05-04-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leykauf K, et al. Protein kinase a dependent phosphorylation of apical membrane antigen 1 plays an important role in erythrocyte invasion by the malaria parasite. PLoS Pathog. 2010;6:e1000941. doi: 10.1371/journal.ppat.1000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Montero E, Rodriguez M, Oksov Y, Lobo CA. Babesia divergens apical membrane antigen 1 and its interaction with the human red blood cell. Infect Immun. 2009;77:4783–4793. doi: 10.1128/IAI.00969-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Håkansson S, Charron AJ, Sibley LD. Toxoplasma evacuoles: a two-step process of secretion and fusion forms the parasitophorous vacuole. EMBO J. 2001;20:3132–3144. doi: 10.1093/emboj/20.12.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bradley PJ, Sibley LD. Rhoptries: an arsenal of secreted virulence factors. Curr Opin Microbiol. 2007;10:582–587. doi: 10.1016/j.mib.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alexander DL, Mital J, Ward GE, Bradley PJ, Boothroyd JC. Identification of the moving junction complex of Toxoplasma gondii: a collaboration between distinct secretory organelles. PLoS Pathog. 2005;1:137–149. doi: 10.1371/journal.ppat.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Besteiro S, Michelin A, Poncet J, Dubremetz J, Lebrun M. Export of a Toxoplasma gondii rhoptry neck protein complex at the host cell membrane to form the moving junction during invasion. PLoS Path. 2009;5:e1000309. doi: 10.1371/journal.ppat.1000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Straub KW, Cheng SJ, Sohn CS, Bradley PJ. Novel components of the apicomplexan moving junction reveal conserved and coccidia-restricted elements. Cell Microbiol. 2009;11:590–603. doi: 10.1111/j.1462-5822.2008.01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Richard D, et al. Interaction between Plasmodium falciparum apical membrane antigen 1 and the rhoptry neck protein complex defines a key step in the erythrocyte invasion process of malaria parasites. J Biol Chem. 2010;285:14812–14822. doi: 10.1074/jbc.M109.080770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Blackman MJ. Malaria proteases and host cell egress: an emerging casade. Cell Microbiol. 2008;10:1925–1934. doi: 10.1111/j.1462-5822.2008.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roiko MS, Carruthers VB. New roles for perforins and proteases in apicomplexan egress. Cell Microbiol. 2009;11:1444–1452. doi: 10.1111/j.1462-5822.2009.01357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nagamune K, Hicks LM, Fux B, Broissier F, Chini EN, Sibley LD. Abscisic acid controls calcium1086 dependent egress and development in Toxoplasma gondii. Nature. 2008;451:207–211. doi: 10.1038/nature06478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kafsack BF, Pena JD, Coppens I, Ravindran S, Boothroyd JC, Carruthers VB. Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science. 2009;323:530–533. doi: 10.1126/science.1165740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Glushakova S, Humphrey G, Leikina E, Balaban A, Miller J, Zimmerberg J. New stages in the program of malaria parasite egress imaged in normal and sickle erythrocytes. Curr Biol. 2010;20:1117–1121. doi: 10.1016/j.cub.2010.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aly AS, Matuschewski K. A malarial cysteine protease is necessary for Plasmodium sporozoite egress from oocysts. J Exp Med. 2005;202:225–230. doi: 10.1084/jem.20050545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yeoh S, et al. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell. 2007;131:1072–1083. doi: 10.1016/j.cell.2007.10.049. [DOI] [PubMed] [Google Scholar]
- 102.Dvorin JD, et al. A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science. 2010;328:910–912. doi: 10.1126/science.1188191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sturm A, et al. Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science. 2006;313:1287–1290. doi: 10.1126/science.1129720. [DOI] [PubMed] [Google Scholar]
- 104.Chandramohanadas R, et al. Apicomplexan parasites co-opt host calpains to facilitate their escape from infected cells. Science. 2009;324:794–797. doi: 10.1126/science.1171085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wilson CB, Tsai V, Remington JS. Failure to trigger the oxidative burst of normal macrophages. J Exp Med. 1980;151:328–346. doi: 10.1084/jem.151.2.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Adams LB, Hibbs JB, Taintor RR, Krahenbuhl JL. Microbiostatic effect of murine-activated macrophages for Toxoplasma gondii: Role for synthesis of inorganic nitrogen oxides from L-arginine. J Immunol. 1990;144:2725–2729. [PubMed] [Google Scholar]
- 107.Pfefferkorn ER. Interferon-γ blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host to degrade tryptophan. Proc Natl Acad Sci USA. 1984;81:908–912. doi: 10.1073/pnas.81.3.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Taylor GA, Feng CG, Sher A. Control of IFN-γ mediated host resistance to intracellular pathogens by immunity-related GTPases (p47 GTPases) Microb Infect. 2007;9:1644–1651. doi: 10.1016/j.micinf.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 109.Zhao Z, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Khaminets A, et al. Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell Microbiol. 2010;12:939–961. doi: 10.1111/j.1462-5822.2010.01443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhao Y, Ferguson DJ, Wilson DC, Howard JC, Sibley LD, Yap GS. Virulent Toxoplasma gondii evade immunity-related GTPas-mediated parasite vacuole disruption within primed macrophages. J Immunol. 2009;182:3775–3781. doi: 10.4049/jimmunol.0804190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Leng J, Butcher BA, Denkers EY. Dysregulation of macrophage signal transduction by Toxoplasma gondii: past progress and recent advances. Parasite Immunol. 2009;31:717–728. doi: 10.1111/j.1365-3024.2009.01122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim SK, Fouts AE, Boothroyd JC. Toxoplasma gondii dysregulates IFN-γ inducible gene expression in human fiboblasts: insights from a genome-wide transcriptional profiling. J Immunol. 2007;178:5154–5165. doi: 10.4049/jimmunol.178.8.5154. [DOI] [PubMed] [Google Scholar]
- 114.Hippe D, Gais A, Gross U, Luder CG. Modulation of caspase activation by Toxoplasma gondii. Methods Mol Biol. 2009;470:275–288. doi: 10.1007/978-1-59745-204-5_19. [DOI] [PubMed] [Google Scholar]
- 115.Blader I, Manger ID, Boothroyd JC. Microarray analysis reveals previously unknown changes in Toxoplasma gondii infected human cells. J Biol Chem. 2001;276:24223–24231. doi: 10.1074/jbc.M100951200. [DOI] [PubMed] [Google Scholar]
- 116.Phelps ED, Sweeney KR, Blader IJ. Toxoplasma gondii rhoptry discharge correlates with activation of the early growth response 2 host cell transcription factor. Infect Immun. 2008;76:4703–4712. doi: 10.1128/IAI.01447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Molestina RE, Sinai AP. Host and parasite-derived IKK activities direct distinct temporal phases of NF-kappaB activation and target gene expression following Toxoplasma gondii infection. J Cell Sci. 2005;118:5785–5796. doi: 10.1242/jcs.02709. [DOI] [PubMed] [Google Scholar]
- 118.Su C, Howe DK, Dubey JP, Ajioka JW, Sibley LD. Identification of quantitative trait loci controlling acute virulence in Toxoplasma gondii. Proc Natl Acad Sci (USA) 2002;99:10753–10758. doi: 10.1073/pnas.172117099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Khan A, et al. Composite genome map and recombination parameters derived from three archetypal lineages of Toxoplasma gondii. Nuc Acids Res. 2005;33:2980–2992. doi: 10.1093/nar/gki604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Saeij JPJ, Coller S, Boyle JP, Jerome ME, White ME, Boothroyd JC. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature. 2007;445:324–327. doi: 10.1038/nature05395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Robben PM, Mordue DG, Truscott SM, Takeda K, Akira S, Sibley LD. Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. J Immunol. 2004;172:3686–3694. doi: 10.4049/jimmunol.172.6.3686. [DOI] [PubMed] [Google Scholar]
- 122.Yamamoto M, et al. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J Exp Med. 2009;206:2747–2760. doi: 10.1084/jem.20091703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ong YC, Reese ML, Boothroyd JC. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J Biol Chem. 2010 doi: 10.1074/jbc.M110.112359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Taylor S, et al. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science. 2006;314:1776–1780. doi: 10.1126/science.1133643. [DOI] [PubMed] [Google Scholar]
- 125.Saeij JPJ, et al. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science. 2006;314:1780–1783. doi: 10.1126/science.1133690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Khan A, Taylor S, Ajioka JW, Rosenthal BM, Sibley LD. Selection at a single locus leads to widespread expansion of Toxoplasma gondii lineages that are virulence in mice. PLoS Genetics. 2009;5:e1000404. doi: 10.1371/journal.pgen.1000404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yap GS, Sher A. Cell-mediated immunity to Toxoplasma gondii: initiation, regulation and effector function. Immunobiol. 1999;201:240–247. doi: 10.1016/S0171-2985(99)80064-3. [DOI] [PubMed] [Google Scholar]
- 128.Mordue DG, Sibley LD. A novel population of Gr-1+ activated macrophages induced during acute toxoplasmosis. J Leukoc Biol. 2003;74:1015–1025. doi: 10.1189/jlb.0403164. [DOI] [PubMed] [Google Scholar]
- 129.Robben PR, LaRegina M, Kuziel WA, Sibley LD. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J Exp Med. 2005;201:1761–1769. doi: 10.1084/jem.20050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dunay IR, et al. Gr1+ inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity. 2008;29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Barragan A, Sibley LD. Migration of Toxoplasma gondii across biological barriers. Trends Microbiol. 2003;11:426– 430. doi: 10.1016/s0966-842x(03)00205-1. [DOI] [PubMed] [Google Scholar]
- 132.Courret N, Darche S, Songio P, Milon G, Buzoni-Gatel D, Tardieux I. CD11c and CD11b expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood. 2005;107:309–316. doi: 10.1182/blood-2005-02-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lambert H, Barragan A. Modelling parasite dissemination: host cell subversion and immune evasion by Toxoplasma gondii. Cell Microbiol. 2010;12:292–300. doi: 10.1111/j.1462-5822.2009.01417.x. [DOI] [PubMed] [Google Scholar]
- 134.Wetzel DM, Schmidt J, Kuhlenschmidt M, Dubey JP, Sibley LD. Gliding motility leads to active cellular invasion by Cryptosporidium parvum sporozoites. Infect Immun. 2005;73:5379–5387. doi: 10.1128/IAI.73.9.5379-5387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Borowski H, Clode PL, Thompson RC. Active invasion and/or encapsulation? A reappraisal of host-cell parasitism by Cryptosporidium. Trends Parasitol. 2008;24:509–516. doi: 10.1016/j.pt.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 136.Forney JR, DeWald DB, Yang S, Speer CA, Healey MC. A role for host phosphoinositide 3-kinase and cytoskeletal remodeling during Cryptosporidium parvum infection. Infect Immun. 1999;67:844–852. doi: 10.1128/iai.67.2.844-852.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shaw MK. Cell invasion by Theileria sporozoites. Trends Parasitol. 2003;19:2–6. doi: 10.1016/s1471-4922(02)00015-6. [DOI] [PubMed] [Google Scholar]
- 138.Lodge R, Descoteaux A. Modulation of phagolysosome biogenesis by the lipophosphoglycan of Leishmania. Clin Immunol. 2005;114:256–265. doi: 10.1016/j.clim.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 139.Lodge R, Descoteaux A. Leishmania invasion and phagosome biogenesis. Subcell Biochem. 2008;47:174–181. doi: 10.1007/978-0-387-78267-6_14. [DOI] [PubMed] [Google Scholar]
- 140.Holm A, Tejle K, Magnusson KE, Descoteaux A, Rasmusson B. Leishmania donovani lipophosphoglycan causes periphagosomal actin accumulation: correlation with impaired translocation of PKC-alpha and defective phagosome maturation. Cell Microbiol. 2001;3:439–447. doi: 10.1046/j.1462-5822.2001.00127.x. [DOI] [PubMed] [Google Scholar]
- 141.Lodge R, Descoteaux A. Leishmania donovani promastigotes induce periphagosomal F-actin accumulation through retention of the GTPase Cdc42. Cell Microbiol. 2005;7:1647–1658. doi: 10.1111/j.1462-5822.2005.00582.x. [DOI] [PubMed] [Google Scholar]
- 142.Lodge R, Diallo TO, Descoteaux A. Leishmania donovani lipophosphoglycan blocks NADPH oxidase assembly at the phagosome membrane. Cell Microbiol. 2006;8:1922–1931. doi: 10.1111/j.1462-5822.2006.00758.x. [DOI] [PubMed] [Google Scholar]
- 143.Rodriguez NE, Gaur U, Wilson ME. Role of caveolae in Leishmania chagasi phagocytosis and intracellular survival in macrophages. Cell Microbiol. 2006;8:1106–1120. doi: 10.1111/j.1462-5822.2006.00695.x. [DOI] [PubMed] [Google Scholar]
- 144.Ueno N, Bratt CL, Rodriguez NE, Wilson ME. Differences in human macrophage receptor usage, lysosomal fusion kinetics and survival between logarithmic and metacyclic Leishmania infantum chagasi promastigotes. Cell Microbiol. 2009;11:1827–1841. doi: 10.1111/j.1462-5822.2009.01374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 146.Lodge R, Descoteaux A. Phagocytosis of Leishmania donovani amastigotes is Rac1 dependent and occurs in the absence of NADPH oxidase activation. Eur J Immunol. 2006;36:2735–2744. doi: 10.1002/eji.200636089. [DOI] [PubMed] [Google Scholar]
- 147.Wilson J, et al. Control of parasitophorous vacuole expansion by LYST/Beige restricts the intracellular growth of Leishmania amazonensis. PLoS Pathog. 2008;4:e1000179. doi: 10.1371/journal.ppat.1000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sacks DL. Leishmania-sand fly interactions controlling species-specific vector competence. Cell Microbiol. 2001;3:189–196. doi: 10.1046/j.1462-5822.2001.00115.x. [DOI] [PubMed] [Google Scholar]
- 149.Desjardins M, Descoteaux A. Inhibition of phagolysosomal biogenesis by the Leishmania lipophosphoglycan. J Exp Med. 1997;185:2061–2068. doi: 10.1084/jem.185.12.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Scianimanico S, Desrosiers M, Dermine JF, Meresse S, Descoteaux A, Desjardins M. Impaired recruitment of the small GTPase rab7 correlates with the inhibition of phagosome maturation by Leishmania donovani promastigotes. Cell Microbiol. 1999;1:19–32. doi: 10.1046/j.1462-5822.1999.00002.x. [DOI] [PubMed] [Google Scholar]
- 151.Dermine JF, Scianimanico S, Prive C, Descoteaux A, Desjardins M. Leishmania promastigotes require lipophosphoglycan to actively modulate the fusion properties of phagosomes at an early step of phagocytosis. Cell Microbiol. 2000;2:115–126. doi: 10.1046/j.1462-5822.2000.00037.x. [DOI] [PubMed] [Google Scholar]
- 152.Vinet AF, Fukuda M, Turco SJ, Descoteaux A. The Leishmania donovani lipophosphoglycan excludes the vesicular proton-ATPase from phagosomes by impairing the recruitment of synaptotagmin V. PLoS Pathog. 2009;5:e1000628. doi: 10.1371/journal.ppat.1000628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ilg T, Demar M, Harbecke D. Phosphoglycan repeat-deficient Leishmania mexicana parasites remain infectious within monocytes. J Immunol. 2001;276:4988–4997. doi: 10.1074/jbc.M008030200. [DOI] [PubMed] [Google Scholar]
- 154.Schaible UE, Schlesinger PH, Steinberg TH, Mangel WF, Kobayashi T, Russell DG. Parasitophorous vacuoles of Leishmania mexicana acquire macrophages form the host cell cytosol via two independent routes. J Cell Sci. 1999;112:681–693. doi: 10.1242/jcs.112.5.681. [DOI] [PubMed] [Google Scholar]
- 155.Huynh C, Sacks DL, Andrews NW. A Leishmania amazonensis ZIP family iron transporter is essential for parasite replication within macrophage phagolysosomes. J Exp Med. 2006;203:2363–2375. doi: 10.1084/jem.20060559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Alexander J, Satoskar AR, Russell DG. Leishmania species: models of intracellular parasitism. J Cell Sci. 1999;112:2993–3002. doi: 10.1242/jcs.112.18.2993. [DOI] [PubMed] [Google Scholar]
- 157.Matte C, Descoteaux A. Leishmania donovani amastigotes impair IFN-gamma-induced STAT1-alpha nuclear translocation by blocking the interaction between STAT1-alpha and importin-alpha5. Infect Immun. 2010 doi: 10.1128/IAI.00046-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Peters NC, Sacks DL. The impact of vector-mediated neutrophil recruitment on cutaneous leishmaniasis. Cell Microbiol. 2009;11:1290–1296. doi: 10.1111/j.1462-5822.2009.01348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gueirard P, Laplante A, Rondeau C, Milon G, Desjardins M. Trafficking of Leishmania donovani promastigotes in non-lytic compartments in neutrophils enables the subsequent transfer of parasites to macrophages. Cell Microbiol. 2008;10:100–111. doi: 10.1111/j.1462-5822.2007.01018.x. [DOI] [PubMed] [Google Scholar]
- 161.Xin L, et al. Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J Immunol. 2010;184:7047–7056. doi: 10.4049/jimmunol.0903273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Conrad SM, Strauss-Ayali D, Field AE, Mack M, Mosser DM. Leishmania-derived murine monocyte chemoattractant protein 1 enhances the recruitment of a restrictive population of CC chemokine receptor 2-positive macrophages. Infect Immun. 2007;75:653–665. doi: 10.1128/IAI.01314-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bogdan C. Mechanisms and consequences of persistence of intracellular pathogens: leishmaniasis as an example. Cell Microbiol. 2008;10:1221–1234. doi: 10.1111/j.1462-5822.2008.01146.x. [DOI] [PubMed] [Google Scholar]
- 164.Blank C, Bogdan C, Bauer C, Erb K, Moll H. Murine epidermal Langerhans cells do not express inducible nitric oxide synthase. Eur J Immunol. 1996;26:792–796. doi: 10.1002/eji.1830260410. [DOI] [PubMed] [Google Scholar]
- 165.Bogdan C, Donhauser N, Doring R, Rollinghoff M, Diefenbach A, Rittig MG. Fibroblasts as host cells in latent leishmaniosis. J Exp Med. 2000;191:2121–2130. doi: 10.1084/jem.191.12.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Milei J, Guerri-Guttenberg RA, Grana DR, Storino R. Prognostic impact of Chagas disease in the United States. Am Heart J. 2009;157:22–29. doi: 10.1016/j.ahj.2008.08.024. [DOI] [PubMed] [Google Scholar]
- 167.Macedo AM, Machado CR, Oliveira RP, Pena SD. Trypanosoma cruzi: genetic structure of populations and relevance of genetic variability to the pathogenesis of chagas disease. Mem Inst Oswaldo Cruz. 2004;99:1–12. doi: 10.1590/s0074-02762004000100001. [DOI] [PubMed] [Google Scholar]
- 168.Lescure FX, et al. Chagas disease: changes in knowledge and management. Lancet Infect Dis. 2010;10:556–570. doi: 10.1016/S1473-3099(10)70098-0. [DOI] [PubMed] [Google Scholar]
- 169.Bern C, Montgomery SP, Katz L, Caglioti S, Stramer SL. Chagas disease and the US blood supply. Curr Opin Infect Dis. 2008;21:476–482. doi: 10.1097/QCO.0b013e32830ef5b6. [DOI] [PubMed] [Google Scholar]
- 170.Epting C, Coates BM, Engman DM. Molecular mechanisms of host cell invasion by Trypanosoma cruzi. Exp Parasitol. 2010 doi: 10.1016/j.exppara.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mortara RA, et al. Host cell actin remodeling in response to Trypanosoma cruzi: trypomastigote versus amastigote entry. Subcell Biochem. 2008;47:101–109. doi: 10.1007/978-0-387-78267-6_8. [DOI] [PubMed] [Google Scholar]
- 172.Barrias ES, Reignault LC, De Souza W, Carvalho TM. Dynasore, a dynamin inhibitor, inhibits Trypanosoma cruzi entry into peritoneal macrophages. PLoS One. 2010;5:e7764. doi: 10.1371/journal.pone.0007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rodriguez A, Samoff E, Rioult MG, Chung A, Andrews NW. Host cell invasion by trypanosomes requires lysosomes and microtubule/kinesin-mediated transport. J Cell Biol. 1996;134:349–362. doi: 10.1083/jcb.134.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tardieux I, et al. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell. 1992;71:1117–1130. doi: 10.1016/s0092-8674(05)80061-3. [DOI] [PubMed] [Google Scholar]
- 175.McNeil PL, Kirchhausen T. An emergency response team for membrane repair. Nat Rev Mol Cell Biol. 2005;6:499–505. doi: 10.1038/nrm1665. [DOI] [PubMed] [Google Scholar]
- 176.Caler EV, Chakrabarti S, Fowler KT, Rao S, Andrews NW. The exocytosis-regulatory protein synaptotagmin VII mediates cell invasion by Trypanosoma cruzi. J Exp Med. 2001;193:1097–1104. doi: 10.1084/jem.193.9.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106:157–169. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- 178.Romano PS, Arboit MA, Vazquez CL, Colombo MI. The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy. 2009;5:6–18. doi: 10.4161/auto.5.1.7160. [DOI] [PubMed] [Google Scholar]
- 179.Woolsey AM, Sunwoo L, Petersen CA, Brachmann SM, Cantley LC, Burleigh BA. Novel PI 3-kinase-dependent mechanisms of trypanosome invasion and vacuole maturation. J Cell Sci. 2003;116:3611–3622. doi: 10.1242/jcs.00666. [DOI] [PubMed] [Google Scholar]
- 180.Mott GA, Burleigh BA. The role of host cell lysosomes in Trypanosoma cruzi invasion. Subcell Biochem. 2008;47:165–173. doi: 10.1007/978-0-387-78267-6_13. [DOI] [PubMed] [Google Scholar]
- 181.Andrade LO, Andrews NW. Lysosomal fusion is essential for the retention of Trypanosoma cruzi inside host cells. J Exp Med. 2004;200:1135–1143. doi: 10.1084/jem.20041408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Woolsey AM, Burleigh BA. Host cell actin polymerization is required for cellular retention of Trypanosoma cruzi and early association with endosomal/lysosomal compartments. Cell Microbiol. 2004;6:829–838. doi: 10.1111/j.1462-5822.2004.00405.x. [DOI] [PubMed] [Google Scholar]
- 183.Scharfstein J, et al. Host cell invasion by Trypanosoma cruzi is potentiated by activation of bradykinin B (2) receptors. J Exp Med. 2000;192:1289–1300. doi: 10.1084/jem.192.9.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Rodriguez A, Rioult MG, Ora A, Andrews NW. A trypanosome-soluble factor induces IP3 formation, intracellular Ca2+ mobilization and microfilament rearrangement in host cells. J Cell Biol. 1995;129:1263–1273. doi: 10.1083/jcb.129.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Burleigh BA, Caler EV, Webster P, Andrews NW. A cytosolic serine endopeptidase from Trypanosoma cruzi is required for the generation of Ca2+ signaling in mammalian cells. J Cell Biol. 1997;136:609–620. doi: 10.1083/jcb.136.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Caler EV, Vaena de Avalos S, Haynes PA, Andrews NW, Burleigh BA. Oligopeptidase B1298 dependent signaling mediates host cell invasion by Trypanosoma cruzi. EMBO J. 1998;17:4975–4986. doi: 10.1093/emboj/17.17.4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rodriguez A, Martinez I, Chung A, Berlot CH, Andrews NW. cAMP regulates Ca2+-dependent exocytosis of lysosomes and lysosome-mediated cell invasion by trypanosomes. J Biol Chem. 1999;274:16754–16759. doi: 10.1074/jbc.274.24.16754. [DOI] [PubMed] [Google Scholar]
- 188.Andrade LO, Andrews NW. The Trypanosoma cruzi - host - cell interplay: location, invasion, retention. Nat Rev Microbiol. 2005;3:819–823. doi: 10.1038/nrmicro1249. [DOI] [PubMed] [Google Scholar]
- 189.Andrews NW, Abrams CK, Slatin SL, Griffiths G. A T. cruzi secreted protein immunologically related to the complement component C9: evidence for membrane pore-forming activity at low pH. Cell. 1990;61:1277–1287. doi: 10.1016/0092-8674(90)90692-8. [DOI] [PubMed] [Google Scholar]
- 190.Manning-Cela R, Cortes A, Gonzalez-Rey E, Van Voorhis WC, Swindle J, Gonzalez A. LYT1 protein is required for efficient in vitro infection by Trypanosoma cruzi. Infect Immun. 2001;69:3916–3923. doi: 10.1128/IAI.69.6.3916-3923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zago MP, Barrio AB, Cardozo RM, Duffy T, Schijman AG, Basombrio MA. Impairment of infectivity and immunoprotective effect of a LYT1 null mutant of Trypanosoma cruzi. Infect Immun. 2008;76:443–451. doi: 10.1128/IAI.00400-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Rubin-de-Celis SS, Uemura H, Yoshida N, Schenkman S. Expression of trypomastigote trans1315 sialidase in metacyclic forms of Trypanosoma cruzi increases parasite escape from its parasitophorous vacuole. Cell Microbiol. 2006;8:1888–1898. doi: 10.1111/j.1462-5822.2006.00755.x. [DOI] [PubMed] [Google Scholar]
- 193.Hall BF, Webster P, Ma AK, Joiner KA, Andrews NW. Desialylation of lysosomal membrane glycoproteins by Trypanosoma cruzi: a role for the surface neuraminidase in facilitating parasite entry into the host cell cytoplasm. J Exp Med. 1992;176:313–325. doi: 10.1084/jem.176.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Silber AM, Colli W, Ulrich H, Alves MJ, Pereira CA. Amino acid metabolic routes in Trypanosoma cruzi: possible therapeutic targets against Chagas’ disease. Curr Drug Targets Infect Disord. 2005;5:53–64. doi: 10.2174/1568005053174636. [DOI] [PubMed] [Google Scholar]
- 195.Kayama H, Takeda K. The innate immune response to Trypanosoma cruzi infection. Microb Infect. 2010;12:511–517. doi: 10.1016/j.micinf.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 196.Tarleton RL. Immune system recognition of Trypanosoma cruzi. Curr Opin Immunol. 2007;19:430–434. doi: 10.1016/j.coi.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 197.Almeida IC, et al. Highly purified glycosylphosphatidylinositols from Trypanosoma cruzi are potent proinflammatory agents. EMBO J. 2000;19:1476–1485. doi: 10.1093/emboj/19.7.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Campos MA, et al. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J Immunol. 2001;167:416–423. doi: 10.4049/jimmunol.167.1.416. [DOI] [PubMed] [Google Scholar]
- 199.Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol. 2006;177:3515–3519. doi: 10.4049/jimmunol.177.6.3515. [DOI] [PubMed] [Google Scholar]
- 200.Shoda LK, et al. DNA from protozoan parasites Babesia bovis, Trypanosoma cruzi, and T. brucei is mitogenic for B lymphocytes and stimulates macrophage expression of interleukin-12, tumor necrosis factor alpha, and nitric oxide. Infect Immun. 2001;69:2162–2171. doi: 10.1128/IAI.69.4.2162-2171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Koga R, et al. TLR-dependent induction of IFN-beta mediates host defense against Trypanosoma cruzi. J Immunol. 2006;177:7059–7066. doi: 10.4049/jimmunol.177.10.7059. [DOI] [PubMed] [Google Scholar]
- 202.Santiago HC, et al. Mice deficient in LRG-47 display enhanced susceptibility to Trypanosoma cruzi infection associated with defective hemopoiesis and intracellular control of parasite growth. J Immunol. 2005;175:8165–8172. doi: 10.4049/jimmunol.175.12.8165. [DOI] [PubMed] [Google Scholar]
- 203.Chessler AD, Unnikrishnan M, Bei AK, Daily JP, Burleigh BA. Trypanosoma cruzi triggers an early type I IFN response in vivo at the site of intradermal infection. J Immunol. 2009;182:2288–2296. doi: 10.4049/jimmunol.0800621. [DOI] [PubMed] [Google Scholar]
- 204.Martin DL, Murali-Krishna K, Tarleton RL. Generation of Trypanosoma cruzi-specific CD8+ T-cell immunity is unaffected by the absence of type I interferon signaling. Infect Immun. 2010;78:3154–3159. doi: 10.1128/IAI.00275-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chessler AD, Ferreira LR, Chang TH, Fitzgerald KA, Burleigh BA. A novel IFN regulatory factor 3-dependent pathway activated by trypanosomes triggers IFN-beta in macrophages and fibroblasts. J Immunol. 2008;181:7917–7924. doi: 10.4049/jimmunol.181.11.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Silva GK, et al. Cutting edge: nucleotide-binding oligomerization domain 1-dependent responses account for murine resistance against Trypanosoma cruzi infection. J Immunol. 2010;184:1148–1152. doi: 10.4049/jimmunol.0902254. [DOI] [PubMed] [Google Scholar]
- 207.Kayama H, et al. NFATc1 mediates Toll-like receptor-independent innate immune responses during Trypanosoma cruzi infection. PLoS Pathog. 2009;5:e1000514. doi: 10.1371/journal.ppat.1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Costales JA, Daily JP, Burleigh BA. Cytokine-dependent and-independent gene expression changes and cell cycle block revealed in Trypanosoma cruzi-infected host cells by comparative mRNA profiling. BMC Genomics. 2009;10:252. doi: 10.1186/1471-2164-10-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chuenkova MV, PereiraPerrin M. Trypanosoma cruzi targets Akt in host cells as an intracellular antiapoptotic strategy. Sci Signal. 2009;2:ra74. doi: 10.1126/scisignal.2000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Paiva CN, et al. CCL2/MCP-1 controls parasite burden, cell infiltration, and mononuclear activation during acute Trypanosoma cruzi infection. J Leukoc Biol. 2009;86:1239–1246. doi: 10.1189/jlb.0309187. [DOI] [PubMed] [Google Scholar]
- 211.Nagajyothi F, et al. Chagas disease, adipose tissue and the metabolic syndrome. Mem Inst Oswaldo Cruz. 2009;104 (Suppl 1):219–225. doi: 10.1590/s0074-02762009000900028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Cuervo H, Pineda MA, Aoki MP, Gea S, Fresno M, Girones N. Inducible nitric oxide synthase and arginase expression in heart tissue during acute Trypanosoma cruzi infection in mice: arginase I is expressed in infiltrating CD68+ macrophages. J Infect Dis. 2008;197:1772–1782. doi: 10.1086/529527. [DOI] [PubMed] [Google Scholar]
- 213.Vago AR, et al. Kinetoplast DNA signatures of Trypanosoma cruzi strains obtained directly from infected tissues. Am J Pathol. 1996;149:2153–2159. [PMC free article] [PubMed] [Google Scholar]
- 214.Andrade LO, Machado CR, Chiari E, Pena SD, Macedo AM. Differential tissue distribution of diverse clones of Trypanosoma cruzi in infected mice. Mol Biochem Parasitol. 1999;100:163–172. doi: 10.1016/s0166-6851(99)90035-x. [DOI] [PubMed] [Google Scholar]
- 215.Yoshida N. Molecular mechanisms of Trypanosoma cruzi infection by oral route. Mem Inst Oswaldo Cruz. 2009;104 (Suppl 1):101–107. doi: 10.1590/s0074-02762009000900015. [DOI] [PubMed] [Google Scholar]
- 216.Kolter D, Orenstein JM. Clinical syndromes associated with microsporidosis. In: Wittner M, Weiss LM, editors. Microsporidia and Microsporidiosis. Washington, DC: ASM Press; 1999. pp. 258–292. [Google Scholar]
- 217.Gordon JL, Beatty WL, Sibley LD. A novel actin-related protein is associated with daughter cell formation in Toxoplasma. Euk Cell. 2008;7:1500–1512. doi: 10.1128/EC.00064-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Martins VD, Galizzi M, Salto ML, Docampo R, Moreno SN. Developmental expression of a Trypanosoma cruzi phosphoinositide-specific phospholipase C in amastigotes and stimulation of host phosphoinositide hydrolysis. Infect Immun. 2010;113:134–138. doi: 10.1128/IAI.00473-10. [DOI] [PMC free article] [PubMed] [Google Scholar]