

Abstract
Adenosine kinase (ADK; EC 2.7.1.20) is an evolutionarily conserved phosphotransferase that converts the purine ribonucleoside adenosine into 5′-adenosine-monophosphate. This enzymatic reaction plays a fundamental role in determining the tone of adenosine, which fulfills essential functions as a homeostatic and metabolic regulator in all living systems. Adenosine not only activates specific signaling pathways by activation of four types of adenosine receptors but it is also a primordial metabolite and regulator of biochemical enzyme reactions that couple to bioenergetic and epigenetic functions. By regulating adenosine, ADK can thus be identified as an upstream regulator of complex homeostatic and metabolic networks. Not surprisingly, ADK dysfunction is involved in several pathologies, including diabetes, epilepsy, and cancer. Consequently, ADK emerges as a rational therapeutic target, and adenosine-regulating drugs have been tested extensively. In recent attempts to improve specificity of treatment, localized therapies have been developed to augment adenosine signaling at sites of injury or pathology; those approaches include transplantation of stem cells with deletions of ADK or the use of gene therapy vectors to downregulate ADK expression. More recently, the first human mutations in ADK have been described, and novel findings suggest an unexpected role of ADK in a wider range of pathologies. ADK-regulating strategies thus represent innovative therapeutic opportunities to reconstruct network homeostasis in a multitude of conditions. This review will provide a comprehensive overview of the genetics, biochemistry, and pharmacology of ADK and will then focus on pathologies and therapeutic interventions. Challenges to translate ADK-based therapies into clinical use will be discussed critically.
I. Introduction
All living systems need efficient self-regulatory mechanisms to adjust metabolic demand to available energy sources. The purine ribonucleoside adenosine is the core partial structure of ATP and has been termed a “retaliatory metabolite” (Newby et al., 1985) in the sense that any drop in energy supplies and ATP lead to increased adenosine, which in turn provides negative feedback inhibition to reduce metabolic demand to save energy. Adenosine is not only part of the energy metabolites AMP, ADP, and ATP of the cell but also an integral component of RNA. In addition, it is part of several adenine-containing coenzymes such as NAD or FAD, part of second messenger systems such as cAMP, and is a central metabolite of biochemical pathways such as the transmethylation pathway. Given its tight link to the energy pool of the cell and to central biochemical reactions and messengers, it is not surprising that adenosine fulfills a key role as a metabolic regulator of energy homeostasis (Fredholm et al., 2011b). Adenosine thus controls important physiologic functions, such as blood supply, glucose homeostasis via interactions with both insulin and glucagon, and lipolysis (Hjemdahl and Fredholm, 1976; Fredholm and Sollevi, 1977). Under conditions of stress or distress adenosine levels rapidly rise, largely by breakdown of adenine nucleotides (Fredholm, 2007). Under those conditions adenosine exerts a multitude of protective functions on many different levels (Linden, 2005; Fredholm, 2007). Those include mechanisms to 1) increase oxygen supply or to decrease oxygen demand by regulation of blood flow, body temperature, and cell work; 2) induce tolerance to hypoxic damage by mechanisms of preconditioning; 3) regulate angiogenesis; and 4) regulate immune responses (Linden, 2005). Most of these physiologic functions of adenosine are mediated by four types of G-protein-coupled adenosine receptors (A1R, A2AR, A2BR, A3R) (Fredholm et al., 2000, 2001a, 2011a), although adenosine receptor independent functions of adenosine might also play a role (Fig. 1). In the following sections, I will discuss the existing literature on adenosine kinase (ADK) comprehensively and in detail. The extensive literature on adenosine and its receptors has been reviewed in several comprehensive review articles to which the reader is kindly referred (Camm and Garratt, 1991; Dunwiddie and Masino, 2001; Fredholm et al., 2005b, 2007, 2011a,b; Hasko et al., 2005; Jacobson and Gao, 2006; Fredholm, 2007, 2010; Sawynok, 2007; Cunha, 2008; Headrick and Lasley, 2009; Sebastiao and Ribeiro, 2009a; Stone et al., 2009; Burnstock et al., 2011). Therefore, the discussion of the general literature on adenosine and its receptors has been limited to selected and more recent articles and reviews.
Fig. 1.

Adenosine acts as a homeostatic network regulator via multiple adenosine receptor-dependent and -independent pathways.
A. Evolutionary Considerations
Adenine, the purine base of adenosine, might have played a role in prebiotic evolution. Importantly, adenine was shown to form nonenzymatically from hydrogen cyanide, a reaction that might have occurred on our primitive Earth (Oro, 1961). Therefore, it is most likely that adenine was already among the primordial compounds that played crucial roles in the origin of life on Earth (Miller and Urey, 1959a,b). Of note, the evolution of life started with self-replicating adenosine-containing RNA (“RNA world”) and not with deoxyadenosine-containing DNA, which evolved much later (Joyce, 2002). Three prerequisites for the origin of life have been suggested: energy (ATP), information (RNA), and membranes (Melendez-Hevia et al., 2008). Chemical evolution most likely led to the first proto-cells (Melendez-Hevia et al., 2008). It is tempting to speculate that in those first primitive organisms adenosine assumed a central position between energy and information. To construct a primordial self-regulatory system, a simple, rapid, and efficient method was needed to adjust metabolic demand to available energy supplies. Thus, if energy drops, ATP declines, adenosine increases, and it is this increase in adenosine that exerts a global inhibitory activity. Primordial regulatory systems needed to be simple and based on key biochemical and metabolic pathways. An early evolutionary appearance of adenosine as a key regulator of metabolism and energy homeostasis is supported by its ubiquitous involvement in many physiologic processes (Dunwiddie and Masino, 2001; Fredholm et al., 2011b). It is this primordial function as “master regulator” that is still maintained in all living systems today. More sophisticated regulatory systems as we know them today were added later, on different layers, to tune and fine-tune the system. It is logical that any disruption of this energy homeostasis-based primordial regulatory system has severe consequences for health and disease. Consequently, adenosine homeostasis needs to be kept under tight control.
B. Physiologic Role of Adenosine Kinase
Biochemically, adenosine can be formed by dephosphorylation of AMP via 5′-nucleotidase (EC 3.1.3.5) or by cleavage of S-adenosylhomocysteine (SAH) via SAH-hydrolase (EC 3.3.1.1). The major routes of adenosine removal are based on deamination to form inosine via adenosine deaminase (EC 3.5.4.4) or phosphorylation to AMP via adenosine kinase (ADK; EC 2.7.1.20). Importantly, 5′-nucleotidase and ADK are part of a highly active substrate cycle between adenosine and AMP, which enables a cell to rapidly respond to changes in the energy status; it has been shown that minor changes in ADK activity rapidly translate into major changes in the concentration of ambient adenosine (Bontemps et al., 1983, 1993a,b). Since levels of intracellular AMP, ADP, and ATP are high (millimolar range) and levels of adenosine are low (nanomolar range), any changes in the adenosine/AMP substrate cycle flow selectively effect the adenosine concentration without having major impact on the equilibrium of the phosphorylated compounds (Fredholm et al., 2005a; Boison et al., 2010). Several lines of evidence support the notion that ADK, which is a low-capacity and low-Km enzyme, is the primary enzyme for metabolic adenosine clearance under baseline conditions, with the goal to keep adenosine levels low (Boison et al., 2010). Thus, ADK expression levels are highest in those organs, in particular liver and placenta (Andres and Fox, 1979), which have the highest needs for metabolic adenosine clearance (Finkelstein and Martin, 1986). In contrast, ADA is a high-capacity and high-Km enzyme, which assists in metabolic adenosine clearance under conditions in which adenosine levels become excessive (e.g., due to pathologic activity) and the capacity of ADK is exceeded (Boison et al., 2010). Of note, ADK is an evolutionary ancient and highly conserved enzyme, which is directly related to bacterial ribokinases and fructokinases (Spychala et al., 1996; Park and Gupta, 2008). On the basis of these early evolutionary roots, it is not surprising that ADK has been identified in almost all living organisms that have been analyzed genetically, including microorganisms, yeasts, plants and animals, and in every tissue assayed.
II. Gene Structure and Transcription
The Adk gene at a remarkable size of 546 kb in humans and 390 kb in the mouse is, together with the human dystrophin gene (Tennyson et al., 1995), one of the largest genes known (Singh et al., 2001; Singh and Gupta, 2004). It has been located on chromosome 10q11-q24 in the human and on chromosome 14 A2-B in the mouse (Klobutcher et al., 1976; Samuelson and Farber, 1985). Although the size of the gene that encodes human ADK is 546-kb long, the coding sequence is only about 1.1 kb. Thereby, the human Adk gene has the highest intron/exon ratio of all known mammalian genes (Park and Gupta, 2012). Human Adk cDNAs encode proteins with sequence-derived molecular masses of 38.7 and 40.5 kDa, which differ in their N-terminal 21 amino acids (McNally et al., 1997).
A. Gene Structure and Homologies
The Adk gene has been characterized, cloned, and expressed from many species, including human (Singh et al., 1996, 2001; Spychala et al., 1996; McNally et al., 1997; Park et al., 2007), mouse (Singh et al., 1996; Boison et al., 2002b), rat (McNally et al., 1997), plants (Moffatt et al., 2000, 2002; Vanderpoorten et al., 2004), and human pathogens (Darling et al., 1999; Long et al., 2003). All known mammalian Adk genes have identical structures and comprise 11 relatively short exons (36 to 765 nucleotide range), which yield a coding sequence of ~1100 bp. In contrast, the intervening introns are huge and range from 4.2 to 128.6 kb in humans. The large size of the Adk gene seems to be a characteristic feature of amniotes (Park and Gupta, 2012), whereas the Adk genes in phylogenetically older eukaryotes, such as in fish or amphibians, are smaller in size (20 to 25 kb) (Singh et al., 2001; Singh and Gupta, 2004). Remarkably, the Adk coding sequence is highly conserved in evolution among vertebrate animals. Adk cDNA from Homo sapiens is 98% identical to Adk from Macaca mulatta, 88% identical to Bos taurus, 84% identical to Mus musculus, 83% identical to Rattus norvegicus, and even 76% identical to Xenopus sp. The Adk genes in the invertebrates Drosophila melanogaster and Cenorhabditis elegans diverge more in sequence similarities and are even smaller in size at 1.5 and 1.3 kb, respectively. The Adk gene of the plant Arabidopsis thaliana has only 10 small introns located within a 2.4-kb gene (Moffatt et al., 2000, 2002).
The enormous size of the Adk gene in amniotes, including human and mouse, is biologically intriguing. Its transcription alone should take about 4 hours (Tennyson et al., 1995). Therefore, it is unlikely that Adk-expression undergoes rapid regulatory changes at the transcriptional level. It is therefore more likely that the Adk gene drives the expression of a stable long-term product that might be subject to developmental regulation within the context of extended time spans. No additional genes have been identified within intronic Adk sequences of human and mouse.
B. Alternative Splice Variants
Two isoforms of ADK (ADK long or ADK-L, and ADK short or ADK-S) are present in mammalian cells (Juranka and Chan, 1985; Sahin et al., 1996, 2004; Sakowicz et al., 2001). Both isoforms are identical except for the amino acids encoded by their first exons (exon 1 and exon 1A) with exon 1A being located in the intron between exon 1 and 2. Differential splicing of the unique first exons with the remaining Adk exons gives rise to the two isoforms (Cui et al., 2011).
C. Alternative Promoter Use
Recent findings, based on the analysis of deletion mutants derived from cultured Chinese hamster cells and data mining of the human genome sequence, have identified two independent promoters driving the expression of each of the two isoforms (Singh and Gupta, 2004; Cui et al., 2011). The promoter driving the expression of ADK-L is bidirectional at least in human, hamster, and other mammals, and is linked in head-to-head orientation with the clathrin adaptor mu3A protein (Singh and Gupta, 2004), which is thought to be involved in protein sorting at the Golgi membrane (Drake et al., 2000). Recent blast searches of the human genome with the nucleotide sequence specific for ADK-S and its upstream noncoding region have identified a putative promoter region within the first intron of ADK-L and 350 bp upstream of the initiator codon of ADK-S. This putative promoter is located within a CpG island, and several transcription factor binding sites have been identified in its proximity (Cui et al., 2011). Although the functionality of this promoter region needs to be validated experimentally, this finding offers the intriguing possibility that each of the two isoforms of ADK is regulated independently at the transcriptional level. Independent transcriptional regulation might in turn suggest different physiologic functions of the two isoforms.
III. Biochemistry
First attempts at the biochemical characterization of ADK go back some 45 years and initially focused on mammalian tissue extracts or human tumor cells (Lindberg et al., 1967; Schnebli et al., 1967). The original interest in ADK was its tight link to nucleic acid metabolism as a salvage pathway for adenosine utilization. Despite the long interest in ADK and despite the wealth of biochemical information derived from modern technologies, the regulatory mechanisms that determine ADK activity, and hence adenosine homeostasis, still remain largely enigmatic. In the following, available information has been summarized and gaps of knowledge identified.
A. Catalytic Reaction
ADK is an ATP:adenosine 5′-phosphotransferase catalyzing the following phosphorylation reaction (Kornberg and Pricer, 1951): ATP + adenosine → ADP + AMP. This is an uncommon reaction type in which donor (ATP) and acceptor (adenosine) of the phosphoryl group share the same structural motif (adenine ring). ADK contains two catalytic sites: a high-affinity site, which binds adenosine and AMP selectively, and a site for ATP and ADP (Pelicano et al., 1997). These unique features of the ADK reaction complicated the interpretation of kinetic data and both a two-site ping-pong mechanism (Chang et al., 1983) and an ordered Bi-Bi mechanism (Henderson et al., 1972; Palella et al., 1980; Mimouni et al., 1994 ) have been proposed. Information obtained from the crystal structures of human (Mathews et al., 1998) and Toxoplasma gondii (Schumacher et al., 2000) ADK have confirmed an ordered Bi-Bi mechanism and a more detailed mechanism has recently been proposed (Park and Gupta, 2008). In a first step, inorganic phosphate or an activator compound binds to a conserved NXXE motif. Binding of an activator facilitates the binding of free Mg2+ and adenosine to the active site of ADK, causing a conformational change of the enzyme, which in turn increases the affinity for MgATP inducing the formation of an anion hole. This stabilizes the pentacovalent transition state, which is typical for an in-line SN2 displacement reaction. The magnesium ion plays a catalytic role and enhances the electrophilicity of the γ-phosphate of ATP, whereas the bound inorganic phosphate may increase the electrophilicity of its β-phosphate. This weakens the oxygen bridge between the two phosphate groups. At the same time the 5′-hydroxyl end of adenosine is deprotonated, attacking the positive center of the γ-phosphate. In a final step the γ-phosphate is transferred to adenosine, and products are released in the order of ADP and AMP (Park and Gupta, 2008).
B. Protein Structure
1. Isoforms
Catalytically active ADK exists as a monomer (Sen et al., 2006; Park and Gupta, 2008). Although monomer-stabilizing interaction partners have been identified in ADK from parasites (Sen et al., 2006), it remains to be determined whether similar interaction partners exist for mammalian ADK. Alternative promoter use and splicing (see above) yields two isoforms of mammalian ADK (Juranka and Chan, 1985; Singh et al., 1996; Spychala et al., 1996; McNally et al., 1997). Human Adk cDNAs encode proteins with sequence-derived molecular masses of 38.7 and 40.5 kDa, differing only in their N-terminal 21 amino acids (McNally et al., 1997). The long isoform of ADK, ADK-L, contains 21 additional N-terminal amino acids (MAAAEEEPKPKKLKVEAPQAL in human ADK-L), which replace four N-terminal amino acids of the short isoform ADK-S (MTSV in human ADK). Both isoforms are enzymatically functional and show no obvious differences in their kinetic properties (Sakowicz et al., 2001; Sahin et al., 2004).
2. Subcellular Localization
ADK is expressed in most organ systems of the mammalian body with highest expression levels in liver, pancreas, and placenta (Andres and Fox, 1979; Fedele et al., 2005; Cui et al., 2011). Founded on algorithms that predict subcellular localization based on sequence similarities, it was initially speculated that both isoforms of ADK are located in the cytoplasm (Nakai and Horton, 1999; Sakowicz et al., 2001). A recent study however, identified specific subcellular localizations of both isoforms of ADK (Cui et al., 2009). ADK-immunofluorescence analysis of cultured mammalian cells that expressed only ADK-L revealed only nuclear labeling, whereas cells that expressed both isoforms showed labeling in nucleus and cytoplasm (Cui et al., 2009). Transfection of cells with ADK-L or ADK-S carrying a C-terminal fusion with a c-myc epitope or a green fluorescent protein tag confirmed nuclear expression of ADK-L and cytoplasmic expression of ADK-S in vitro. Overexpression of an ADK-S transgene in an Adk-null background in the mouse revealed cytoplasmic localization of ADK-S (Fedele et al., 2005), a finding that was replicated by adeno-associated virus (AAV)-based overexpression of ADK-S in mouse brain (Shen et al., 2011; Theofilas et al., 2011).
Thus, independent lines of evidence from in vitro and in vivo studies show that ADK-S is located in the cytoplasm, whereas ADK-L is specific for the nucleus. The N-terminal sequence of ADK-L contains a cluster of conserved amino acids (PKPKKLKVE). When KK in this sequence was replaced by either AA or AD, nuclear localization of ADK was abolished; further fusion of this sequence to other proteins redirected their localization to the nucleus (Cui et al., 2009). These findings suggest that ADK-L contains a novel nuclear localization signal. The nuclear localization of ADK-L suggests a specific function for gene regulation (see below) (Studer et al., 2006). Interestingly, both isoforms of ADK are differentially expressed in a variety of mammalian tissues. Whereas both isoforms of ADK are prominently expressed in kidney, liver, lung, and pancreas, there is a predominance of ADK-L in brain, and ADK-S expression dominates in adrenal gland, spleen, and thymus; heart and muscle appear to express only ADK-S (Cui et al., 2011). The functional significance of isoform specific expression patterns in different organs has yet to be determined.
3. Crystal Structure
The crystal structure of ADK was first identified for human ADK (Mathews et al., 1998) and subsequently for the parasitic protozoan T. gondii (Cook et al., 2000; Schumacher et al., 2000). More recently, the crystal structures of several different eukaryotic and prokaryotic ADKs have been resolved (Reddy et al., 2007; Cassera et al., 2011; Kuettel et al., 2011). Identification of the crystal structure of ADK yielded important insights into the catalytic mechanism and provided information for the design of drugs acting on ADK. As outlined above, the enzymatically active form of ADK is a monomer. Crystallographic studies have identified a larger αβα three-layer sandwich domain with a smaller “lid.” The larger domain is composed of a central β-sheet with nine strands, which is flanked by 10 α-helices and provides the binding sites for the substrates adenosine and ATP (Mathews et al., 1998). The smaller domain is composed of a five-stranded mixed β-sheet flanked by two α-helices and forms a lid over the active site of the enzyme (Mathews et al., 1998). The two domains are connected by four peptide segments and adenosine binds in the cleft between those domains (Mathews et al., 1998). Studies from T. gondii, in which ADK was crystallized both as apo-enzyme and in its substrate-bound forms, revealed a major conformational change of the enzyme upon adenosine binding, reminiscent of opening and closing of the “lid” domain (Cook et al., 2000). In this model, the apo-enzyme is in the open confirmation with the adenosine-binding pocket exposed to the solvent environment. The substrate-bound form, in contrast, is in the closed confirmation, with the lid hiding the substrate binding pocket (Cook et al., 2000). This major conformational change is likely accomplished by a “GG-switch” composed of residues Gly68 and Gly69 (Cook et al., 2000). Although the sequences of Mycobacterium tuberculosis and human ADK are less than 20% identical, their overall structures, including the flexible lid, are similar (Mathews et al., 1998; Reddy et al., 2007). Remarkably, this structural similarity extends to bacterial ribokinases, suggesting an early evolutionary origin of ADK (Park et al., 2007; Park and Gupta, 2008, 2012).
4. Catalytic Site
Crystallography studies performed in different species (Mathews et al., 1998; Cook et al., 2000; Schumacher et al., 2000; Reddy et al., 2007; Cassera et al., 2011; Kuettel et al., 2011) uniformly revealed that the large domain of ADK contains the catalytic core, which is located at the domain interfaces, where adenosine binds in a deeply buried cavity and is covered by the smaller lid domain. The ATP binding site is located at an adjacent site in the large domain with the γ-phosphate group pointing near the 5′-end of the ribose moiety of adenosine. Binding of adenosine to the open apo-form of the enzyme induces a 30° rotation of the lid domain relative to the large domain. Thereby adenosine will be sequestered and formation of the ATP binding site in the large domain will be initiated at the same time. Local structural changes are induced by binding of ATP leading to the formation of an anion hole. Once ATP has bound, a closed conformation is achieved in which the small domain of ADK brings an evolutionary conserved catalytic arginine to the active site when adenine is bound (Schumacher et al., 2000). This catalytic arginine forms a hydrogen bond to the γ-phosphate of ATP and orientates the γ-phosphate into the catalytic position for a typical in-line SN2 displacement reaction (Schumacher et al., 2000).
5. Regulatory Site
The occurrence of substrate inhibition of ADK and a dual regulatory character of some adenosine analogs suggested the existence of an additional regulatory binding site for adenosine with a lower affinity for adenosine (Pelicano et al., 1997; Lin et al., 1988). Based on competition studies, this regulatory site was reported to differ from the catalytic site and might play a role under conditions of high adenosine production, such as during times of ischemia or seizures (Fisher and Newsholme, 1984; Hawkins and Bagnara, 1987; Lin et al., 1988). The existence of a second adenosine-binding site was further supported by chemical modification studies, which demonstrated that a highly active thiol group was essential for activity (Neudecker and Hartmann, 1972, 1978). Adenosine concentrations equivalent to the dissociation constant for the second binding site were shown to effectively protect the reactive thiol group from inactivation by 5,5′-dithio-bis(2-nitrobenzoic acid); consequently, this thiol group has been associated with the second regulatory binding site for adenosine that is different from the ATP binding site (Hawkins and Bagnara, 1987). The presence of two binding sites for adenosine has been confirmed in the crystal structure of human ADK (Mathews et al., 1998). The authors of this study identified a second adenosine-binding site at the ATP-binding site of the enzyme and concluded that substrate inhibition of ADK was due to competitive inhibition of ATP binding (Mathews et al., 1998). This conclusion, however, is not consistent with kinetic data from human placental ADK, which suggest that adenosine is a noncompetitive inhibitor of ATP binding (Palella et al., 1980), and with the 5,5′-dithio-bis(2-nitrobenzoic acid) inactivation studies, which suggest that the regulatory adenosine binding site is different from the ATP binding site (Hawkins and Bagnara, 1987).
6. Modeling Studies
Crystallography studies in combination with modeling studies for the binding of nucleoside and nonnucleoside inhibitors of ADK have revealed more details regarding the conformational changes of ADK. A semi-open conformation intermediate between open and closed, with a small lid-domain rotation of 12° degrees, was first described in T. gondii ADK (Zhang et al., 2007). In this model residues Gly143-X-X-Gly146 were suggested to be subject to torsional changes upon substrate binding, which together with a Gly68-Gly69 switch were predicted to induce a hinge bending of the lid domain. The authors of this study concluded that the intermediate conformation suggests that ATP binding is independent of adenosine binding. The possible existence of a semi-open conformation was subsequently confirmed in human ADK by modeling the binding of larger tubercidins, which were thought to stabilize the semi-open conformation (Bhutoria and Ghoshal, 2010). By use of an automated ligand-docking program with a genetic algorithm to explore the full range of ligand conformational flexibility and partial flexibility of the protein (Jones et al., 1997), it was shown that the semi-open conformation, resulting from a smaller degree of ligand-induced movement of the binding site, was sufficient to accommodate aryl compounds (Bhutoria and Ghoshal, 2010). Pharmacophore modeling suggested the existence of three distinct pharmacophoric elements for closed, semi-open, and open-state binders (Bhutoria and Ghoshal, 2010).
C. Kinetic Studies
Early kinetic studies on ADK were based on enzyme purified from ADK-rich tissues such as liver (Miller et al., 1979a,b; Yamada et al., 1981), placenta (Palella et al., 1980), brain (Yamada et al., 1980), or heart (Fisher and Newsholme, 1984). Remarkably, no major differences were observed in the kinetic properties among the different adenosine kinases (Yamada et al., 1982). The key properties of purified ADK can be summarized as follows: maximal enzyme activity is found at pH 6.5–6.8. Under those conditions ADK has an apparent Km for adenosine of 0.2–0.4 µM and an apparent Vmax of 2.2 µmol of AMP formed per minute per milligram of protein. In most tissues investigated, the Km values of ADK for adenosine were between one and two orders of magnitude lower than those of adenosine deaminase (ADA) (Arch and Newsholme, 1978; Phillips and Newsholme, 1979). On the basis of those kinetic data and studies with ADK and ADA inhibitors, it was concluded that under conditions that provide adequate oxygen and glucose, ADK plays a much greater role than ADA in regulating the extracellular concentration of adenosine. Only under conditions of increased energy depletion when adenosine formation is increased, ADA becomes important in regulating extracellular adenosine concentration (Lloyd and Fredholm, 1995). Furthermore, 5′-nucleotidase and ADK are simultaneously active in many tissues including liver or brain, so that a substrate cycle between AMP and adenosine results (Arch and Newsholme, 1978; Bontemps et al., 1983). The difference in Km values between ADK and ADA indicates that, via this substrate cycle, small changes in the activity of either ADK or 5′-nucleotidase produce rapid changes in adenosine concentration (Arch and Newsholme, 1978; Bontemps et al., 1983).
D. Transcriptional Activation of Adenosine Kinase
Few studies have addressed the transcriptional regulation of the Adk gene. ADK expression was found to be reduced in streptozotocin-induced diabetes mellitus in rats (Pawelczyk et al., 2000), whereas insulin was shown to restore Adk mRNA to normal levels within the first 7 hours of insulin treatment (Sakowicz and Pawelczyk, 2002). Mechanistic studies in splenocytes isolated from diabetic rats have shown a 3.9-fold increase in Adk mRNA 4 and 5 hours after the incubation of the cells with 10 nM insulin (Pawelczyk et al., 2003). Insulin-dependent activation of Adk transcription required activation of the mitogen-activated protein kinase (MAPK) pathway, since transcriptional activation of the Adk gene was blocked by the MAPK inhibitor PD98059 [2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one]. Insulin exposure also resulted in increased phosphorylation of ERK1/2 and Elk-1 and sustained elevation of c-Jun and c-Fos protein, whereas those changes could be prevented by incubating the cells with PD98059. The authors concluded that insulin activates Adk gene transcription via activation of the MAPK cascade and subsequent phosphorylation of Elk-1 and increased expression of c-fos and c-jun (Pawelczyk et al., 2003).
E. Transcriptional Repression of Adenosine Kinase
Hypoxia is known to lead to a rapid rise in adenosine—likely a homeostatic protective response of a tissue (Berne, 1963; Berne et al., 1974; Decking et al., 1997; Frenguelli et al., 2003). At least one transcriptional mechanism might contribute to this phenomenon. It was demonstrated in vitro that hypoxia induced in endothelial cells caused a robust repression (85% reduction) of Adk transcript levels. Transcription factor binding assays, hypoxia inducible factor 1-α (HIF-1α) loss- and gain-of-function studies, as well as abrogation of Adk transcriptional repression by ambient hypoxia in conditional HIF-1α mutant mice, demonstrated a definitive role of HIF-1α in the transcriptional repression of the Adk gene (Morote-Garcia et al., 2008).
F. Regulation by Metabolites
Uniquely, ADK activity is regulated by its own substrates and products, as well as by factors that reflect the energy state and health of a cell. Thus, ADK fulfills the role of a sensor for the energy state and bioenergetic equilibrium of a cell, and at the same time ADK acts as a switch determining ambient levels of the "retaliatory metabolite" adenosine to adjust metabolic demand to available energy supplies.
1. Adenosine
Surprisingly, ADK from several tissues is inhibited by its own substrate adenosine (Miller et al., 1979a; Fisher and Newsholme, 1984). The magnitude of substrate inhibition increases with rising concentrations of Mg2+ (Fisher and Newsholme, 1984). In human placental ADK, substrate inhibition was observed at adenosine concentrations greater than 2.5 µM at pH 7.4, with ATP and Mg2+ 0.2 mM, i.e., ~10 times higher than the Km of the enzyme for adenosine (Palella et al., 1980). Substrate inhibition was likewise found in ADK from human erythrocytes, where the degree of inhibition was found to be pH and Mg2+ dependent (Hawkins and Bagnara, 1987). In human liver, substrate inhibition was reported at significantly lower concentrations of adenosine (above 0.5 µM), indicating a rather narrow activity range of ADK in regard to ambient adenosine concentrations (Yamada et al., 1981). Similar adenosine concentrations for substrate inhibition were reported in rodent samples (Yamada et al., 1982; Fisher and Newsholme, 1984). Whereas physiologic adenosine concentrations in the range of 25–300 nM (Lonnroth et al., 1989) are not likely to affect ADK activity, substrate inhibition of ADK by higher concentrations of adenosine might be an important physiologic mechanism to potentiate endogenous adenosine responses under conditions of stress or distress, which can lead to micromolar concentrations of adenosine (Clark et al., 1997; Fredholm, 2007).
2. AMP
The activity of ADK also depends on the concentrations of AMP. It was found that AMP concentrations below 5 mM activated the enzyme, whereas concentrations above 5 mM inhibited the enzyme (Hawkins and Bagnara, 1987). Therefore, under physiologic concentrations of AMP in the range of 0.3 mM (Boesiger et al., 1994), ADK is expected to be activated by AMP, whereas only excessive AMP concentrations are likely to inhibit ADK, e.g., under conditions of severe energy stress, a meaningful physiologic response to augment adenosine signaling in stressful situations. Inhibition of ADK activity by higher concentrations of AMP was found to be competitive with respect to adenosine and noncompetitive with respect to ATP (Palella et al., 1980).
3. ADP
ADP was found to be a noncompetitive inhibitor with regard to adenosine and ATP (Palella et al., 1980; Rotllan and Miras Portugal, 1985; Mimouni et al., 1994). Hyperbolic inhibition was observed during noncompetitive inhibition of adenosine kinase by AMP and ADP (Palella et al., 1980).
4. ATP
ADK activity critically depends on available ATP levels in a Mg2+-dependent manner (Lindberg et al., 1967), whereas free ATP was found to inhibit ADK, the Mg2+-complexed form of ATP activated ADK (Palella et al., 1980; Rotllan and Miras Portugal, 1985). The Km of ADK for MgATP was determined as 75 µM (Palella et al., 1980). It needs to be mentioned that ATP can also be replaced by GTP as phosphate group donor (Miller et al., 1979b).
5. Magnesium
In most kinase reactions the true phosphate donating substrate is a complex of ATP4- and a divalent metal ion, typically Mg2+ forming MgATP2−, which then binds the enzyme. In agreement with this concept, a lack of Mg2+ in the medium resulted in lack of ADK activity, whereas maximal enzyme activity was achieved in the presence of Mg2+ at pH levels where ATP and Mg2+ existed primarily in the complexed, chelated form (Palella et al., 1980). The magnesium ion is thought to partly neutralize the negative charges on the phosphate groups of the nucleotide, which otherwise would prevent binding to the enzyme (Mildvan, 1987). After saturation of available ATP, a further increase in Mg2+ will result in free Mg2+. ADK activity increases further with increases in free Mg2+; however, once optimal activity levels have been reached, further increases in Mg2+ will inhibit the enzyme (Palella et al., 1980; Rotllan and Miras Portugal, 1985; Maj et al., 2002). This free, catalytic Mg2+ ion is thought to bind to the active site of the enzyme and induce the transition state of the reaction by increasing the electrophilicity of the µ-phosphorous atom of the nucleotide via its interaction with the oxygen atoms (Parducci et al., 2006). Furthermore, the free Mg2+ may optimize the spatial arrangement of the substrate’s functional groups (Rivas-Pardo et al., 2011).
6. Inorganic Phosphate
Interestingly, ADK displays a dependency on inorganic phosphate or other pentavalent ions as was first demonstrated in ADK isolated from Chinese hamster cells (Hao and Gupta, 1996). In those studies, the addition of inorganic phosphate, but also of arsenate or vanadate, increased the Vmax of the reaction and decreased the Km for adenosine. In contrast, these pentavalent ions did not change the Km for ATP. Dependency of the enzyme reaction on inorganic phosphate has been confirmed in ADK preparations derived from many different species (Maj et al., 2000, 2002; Park et al., 2006).
7. pH
Maximal activity of ADK derived from human placenta has been observed at pH 6.5 (Palella et al., 1980), whereas ADK from rat brain and human liver displayed a biphasic pH optimum with a sharp pH peak of activity at pH 5.5 and a broad peak of activity at pH 7.5–8.5 (Yamada et al., 1980,1981). A broad pH optimum in the pH 6–8 range was also reported for rat heart ADK (Fisher and Newsholme, 1984). In a more recent study it was shown that under more acidic conditions (pH 6.2) the presence of inorganic phosphate became a necessity for activation (Maj et al., 2000). The pH optimum at close to physiologic conditions implies that a drop in pH as occurs during or after injury is expected to inactivate ADK, thus contributing to an injury-induced surge of protective adenosine.
8. NO
Several studies have shown that nitric oxide (NO) induces the release of adenosine. Mechanistic studies performed on cultured neurons or hippocampal slices suggest that NO raises adenosine through inhibition of ADK (Rosenberg et al., 2000; Arrigoni and Rosenberg, 2006). However, it was not resolved whether inhibition of ADK was a direct effect of NO or an indirect effect caused by substrate inhibition.
G. Posttranslational Modifications
ADK does not seem to be a target for posttranslational modifications. A screen of a panel of protein kinases for their ability to phosphorylate recombinant mouse ADK yielded negative results (Sahin et al., 2004). Accordingly, ADK is most likely not an efficient substrate for PKA, PKC, PKG, CaMKII, CK1, CK2, MAPK, Cdk1, or Cdk5 (Sahin et al., 2004). Given the early evolutionary origin of ADK it might not seem too surprising that ADK is not regulated by mechanisms that evolved much later.
H. Protein-Protein Interactions
Protein-protein interactions might play important roles in the regulation of ADK activity. Seminal biochemical studies performed on ADK from the parasitic protozoan Leishmania donovani suggest a very attractive regulatory model, which is based on aggregation and disaggregation of the enzyme. With increasing concentrations, fully active L. donovani ADK formed soluble aggregates, resulting in inactivation of the enzyme. By using the aggregated inactive enzyme as the substrate, it was shown that a cyclophilin from L. donovani could induce complete disaggregation, leading to reactivation of the enzyme. It was further shown that the reactivating ability of cyclophilin remained unaffected even in the presence of cyclosporine A and macromolecular crowding agents. The reactivation occurred noncatalytically and was reversible (Chakraborty et al., 2002). The prevention of ADK aggregation by cyclophilin was shown to be mediated by an isomerase-independent chaperone function of cyclophilin (Chakraborty et al., 2004). It was further shown that ADP stabilized the aggregated form of ADK and that cyclophilin was able to disaggregate and activate ADK (Sen et al., 2006). Under conditions of cellular stress a rise in ADP is expected to stabilize the inactive aggregate of ADK and thereby promote a rise in adenosine, which in turn will suppress energy-consuming activities. On the other hand, a cyclophilin-based chaperone function may reactivate ADK any time, even under conditions of energy depletion. Whether mammalian ADK is regulated by a similar chaperone-based mechanism remains to be demonstrated. Intriguingly, it was shown that cyclosporine A and FK506 (tacrolimus) decreased ADK activity in T-lymphocytes (Spychala and Mitchell, 2002). Clinically, cyclosporine A and FK506 treatment led to a rise in plasma adenosine in kidney transplant recipients, suggesting that the resulting increase in plasma adenosine contributes to the immunosuppressive effects of these agents (Guieu et al., 1998).
I. Influence on Downstream Pathways
1. Adenosine Homeostasis
As outlined above, the concentration of adenosine in a tissue is mostly determined by the activities of adenosine-producing nucleotidases, by adenosine-producing transmethylation reactions, and by adenosine-removing ADK and ADA as well as by transmembrane transporters for adenosine (Boison et al., 2010). Extracellular adenosine flows into adenosine-metabolizing cells through equilibrative nucleoside transporters (Baldwin et al., 2004). Because those transport functions depend on the intracellular metabolic clearance rate of adenosine to maintain the inward flux of adenosine, the velocity of intracellular metabolic clearance of adenosine determines the rate of adenosine removal from the extracellular space. Thus, under steady-state conditions of adenosine production, the extracellular adenosine concentration is determined by the rate of intracellular adenosine clearance (Greene, 2011). ADK is the metabolic enzyme with the highest affinity for adenosine. Because of its low capacity, the rate of metabolic adenosine clearance—and therefore its extracellular concentration—seems to be largely dependent on the Vmax of ADK under physiologic conditions (Arch and Newsholme, 1978). Since ADK needs to bind both ATP and adenosine to transfer a phosphate from ATP to adenosine, resulting in the release of AMP and ADP, and since ADP might inactivate ADK by promoting its aggregated state, the velocity of the enzyme reaction and the resulting adenosine concentration in the extracellular space depend largely on the ATP/ADP ratio and the energy state of the tissue. Under physiologic conditions the homeostasis of adenosine is largely under the control of ADK, whose activity directly depends on the energy state of the cell. Adenosine affects several adenosine receptor dependent and independent pathways simultaneously (Fig. 1), as will be outlined in the following sections.
2. Adenosine Receptors
Adenosine activates four types of known G-protein-coupled adenosine receptors, which are designated as A1R, A2AR, A2BR, and A3R. The pharmacology and physiologic functions of the adenosine receptors have extensively been reviewed (Fredholm et al., 2000, 2001a, 2011a,b; Jacobson and Gao, 2006; Sebastiao and Ribeiro, 2009a; Stone et al., 2009) and only key functions will briefly be outlined below.
a. Adenosine A1 receptor
Activation of the A1R, which is coupled to pertussis toxin-sensitive Gi proteins, leads to inhibition of adenylyl cyclase activity (van Calker et al., 1979; Cooper et al., 1980) and to increased activity of phospholipase C (PLC) (Rogel et al., 2005; Tawfik et al., 2005). In the CNS as well as in the heart, A1R stimulation leads to activation of KATP channels and pertussis-toxin-sensitive K+ channels, whereas it leads to inhibition of Q-, P-, and N-type Ca2+ channels (Fredholm et al., 2001a, 2011a). In the heart, coupling to K+ channels mediates the bradycardia effects of adenosine (Belardinelli et al., 1995); whereas modulation of p44/42 extracellular signal-regulated protein kinase (ERK) signaling through A1R activation has been implicated mechanistically in the phenomenon of ischemic preconditioning (Reid et al., 2005).
b. Adenosine A2A receptor
In contrast, activation of the A2AR leads to an increase in adenylyl cyclase activity. In peripheral tissues the A2AR couples predominantly to GS proteins, whereas in striatum, a brain area that is particularly rich in A2ARs, the receptor couples predominantly to Golf, which likewise couples to adenylyl cyclase (Kull et al., 2000). A2AR activation was found to facilitate noradrenaline release and activation of the PLC and adenylyl cyclase pathways in tail arteries of the rat (Fresco et al., 2004). In addition, A2AR activation induced the formation of inositol phosphates, thus raising intracellular calcium and activating protein kinase C in COS-7 cells (Offermanns and Simon, 1995).
c. Adenosine A2B receptor
The A2BR couples positively to both adenylyl cyclase and PLC (Daly et al., 1983; Brackett and Daly, 1994; Peakman and Hill, 1994; Feoktistov and Biaggioni, 1997) and plays a major role in inflammation. A2BR activation was found to evoke interleukin-8 secretion via induction of inositol phosphate formation in a human mast cell line (Feoktistov and Biaggioni, 1995) and to mediate human chorionic vasoconstriction via activation of the arachidonic acid pathway (Donoso et al., 2005).
d. Adenosine A3 receptor
Activation of the A3R leads to inhibition of adenylyl cyclase (Zhou et al., 1992), stimulation of PLC (Abbracchio et al., 1995), and mobilization of calcium (Englert et al., 2002; Fossetta et al., 2003; Shneyvays et al., 2004, 2005). A3Rs can protect cardiomyocytes through activation of KATP channels (Tracey et al., 1998), and the anti-ischemic effect of A3R activation was found to be dependent on rhoA-phospholipase D1 signaling (Mozzicato et al., 2004). The A3R might also play a role in cancer and cell growth, as well as in cell differentiation, survival, and death, since the A3R couples to MAPK (Schulte and Fredholm, 2002, 2003) and since the WNT signaling pathway was found to contribute to A3R activation-dependent suppression of melanoma cells (Fishman et al., 2002). Furthermore, proliferation of human melanoma cells was found to be inhibited after A3R-dependent activation of the phosphatidylinositol 3-kinase-protein kinase B-ERK1/2 pathway (Merighi et al., 2005)
3. Adenosine Receptor-independent Pathways
The examples outlined above illustrate a multitude of AR-dependent pathways in multiple tissues and organ systems that directly depend on adenosine homeostasis. However, given the early evolutionary origin of adenosine and the relative late evolutionary appearance of the adenosine receptors (Burnstock and Verkhratsky, 2009; Fountain and Burnstock, 2009) it becomes plausible that adenosine might have additional, primordial functions that do not require ARs and that rely on biochemical and bioenergetic functions of adenosine.
a. Transmethylation
Adenosine is an obligatory end product of transmethylation reactions, which involve the transfer of a methyl group from S-adenosylmethionine (SAM) to a methyl group acceptor (e.g., ethanolamine or DNA) resulting in the formation of SAH, which in turn can be cleaved into adenosine and homocysteine by S-adenosylhomocysteine hydrolase when adenosine concentrations are kept low (Hoffman et al., 1979; Boison et al., 2002b; Mato et al., 2008) (Fig. 2). Importantly, transmethylation reactions can only be maintained when adenosine is constantly removed by ADK (Boison et al., 2002b; Mato et al., 2008). Therefore, it is not surprising that liver, the organ in which 80% of all mammalian transmethylation reactions take place, also has the highest expression levels of ADK (Yamada et al., 1981; Cui et al., 2011; Park and Gupta, 2012). Thus, transmethylation is not only a major source for adenosine (Lloyd et al., 1988; Deussen et al., 1989; Kroll et al., 1992) but 95% of the SAH-derived adenosine was found to be salvaged by ADK in isolated guinea pig heart preparations (Lloyd and Schrader, 1993). Conversely, if adenosine is not constantly removed by ADK, the thermodynamic equilibrium of the SAH hydrolase reaction favors the formation of SAH, which is a potent inhibitor of transmethylation reactions (Finkelstein and Martin, 1986; Finkelstein, 1998; Mato et al., 2008). Given the important role of ADK for metabolic clearance of SAH-derived adenosine, ADK is expected to be a regulator of transmethylation reactions. Indeed, the constitutive genetic disruption of the Adk gene in mice (Boison et al., 2002b) or in the plant Arabidopsis (Moffatt et al., 2002) provided the first direct evidence that ADK expression is a requirement for the maintenance of transmethylation. In mice, the deletion of ADK resulted in increased levels of SAH in the liver and microvesicular hepatic steatosis; all homozygous mutants developed steatotic liver and died within 14 days after birth (Boison et al., 2002b). Likewise, ADK-deficiency in Arabidopsis resulted in increased SAH and inhibition of SAM-dependent transmethylation reactions; affected plants were affected by reduced size and failure to elongate the primary shoot (Moffatt et al., 2002). More recently, six human patients with Adk mutations resulting in a functional ADK deficiency have been described (Bjursell et al., 2011). These human mutations resulted in disruption of the transmethylation pathway and the development of liver pathology and encephalopathy (Bjursell et al., 2011). SAM-dependent transmethylation reactions also determine the methylation status of CpG islands in promoter regions. Therefore, I hypothesized that the ADK/adenosine system might likewise determine the methylation status of DNA and thereby exert a novel function as epigenetic regulator (Williams-Karnesky et al., submitted manuscript). Infusion of adenosine or homocysteine into the hippocampus of rats induced global DNA hypomethylation, whereas the infusion of SAM induced hypermethylation of the DNA, demonstrating that the methylation status of DNA directly depended on the adenosine-sensitive transmethylation pathway. Importantly, blocking ADK with 5-iodotubercidin or genetic reduction of ADK expression resulted in hypomethylated DNA in the brain, whereas overexpression of either the cytoplasmic or the nuclear isoform of ADK resulted in increased DNA methylation in cultured cells, with the nuclear overexpression of ADK being more efficient in increasing the methylation status of the DNA (Williams-Karnesky et al., submitted manuscript). These data suggest a novel—likely adenosine receptor-independent—role of ADK in regulating the methylation status of DNA and thereby acting as an epigenetic regulator.
Fig. 2.

Transmethylation pathway. Adenosine is an obligatory end product of transmethylation reactions, including those catalyzed by DNA methyltransferases. If adenosine is not constantly removed by adenosine kinase, increased levels of adenosine drive the S-adenosylhomocysteine hydrolase reaction toward . SAH synthesis. SAH is a potent inhibitor of methyltransferases, which use SAM as methyl group (−CH3) donor.
b. Mitochondrial bioenergetics
As a "retaliatory metabolite" adenosine is directly linked to mitochondrial bioenergetics and energy homeostasis (Newby et al., 1985; Sommerschild and Kirkeboen, 2000; Peart and Headrick, 2007; Masino et al., 2009). It needs to be stressed that under basal conditions, levels of adenosine (∼100 nM in brain) are nearly 10,000-fold lower than ATP (Pazzagli et al., 1995; Delaney and Geiger, 1996). Therefore, even minor decreases in ATP levels can result in dramatic rises in adenosine levels. In line with this notion, adenosine levels increased as brain energy levels decreased following a variety of excitatory stimuli (Shepel et al., 2005). Interestingly, mitochondria are capable to release adenosine (Bukoski et al., 1983, 1986), and a mitochondrial adenosine-producing 5′-nucleotidase has been identified (Raatikainen et al., 1992). Because a concentration-dependent adenosine output from mitochondria by diffusion or facilitated diffusion has been suggested (Raatikainen et al., 1992), it is tempting to speculate that metabolic clearance of mitochondria-derived adenosine by cytoplasmic ADK drives mitochondrial adenosine production. Through this mechanism ADK could directly affect mitochondrial bioenergetics. In support of this notion hepatocyte mitochondria from ADK-knockout mice display a severe mitochondrial pathology (Boison et al., 2002b).
4. Nitric Oxide Metabolism
The interactions between adenosine and nitric oxide metabolism and signaling is a major topic and worth a dedicated review. However, relatively few studies have directly focused on the interactions between NO metabolism and ADK. Several studies have shown that pharmacological inhibition of ADK reduced lipopolysaccharide (LPS)-induced NO production and the induction of inducible NO synthase, most likely via an A2R-dependent mechanism (Lee et al., 2005; Petrov et al., 2005). On the other hand, as discussed above, NO triggers a rise in adenosine and subsequent inhibition of ADK by substrate inhibition (Rosenberg et al., 2000; Arrigoni and Rosenberg, 2006). This interrelationship between adenosine and NO homeostasis could be a self-limiting mechanism to terminate NO-dependent signaling.
IV. Pharmacology
A. Methods for Drug Development
Adenosine kinase inhibitors have received much attention in pharmaceutical drug development efforts during the late 1990s and early 2000s. Based on the rationale that ADK inhibitors would prevent the metabolic clearance of adenosine and thus potentiate the protective actions of endogenous adenosine, they were expected to augment adenosine signaling in a site- and event-specific manner and thus provide all the benefits of A1R activation, but with reduced potential for widespread systemic side effects (Kowaluk et al., 1998; Kowaluk and Jarvis, 2000). The primary applications for ADK inhibitors were considered to be anti-inflammatory, antinociceptive, and anticonvulsant therapy (Wiesner et al., 1999; Kowaluk and Jarvis, 2000; McGaraughty et al., 2001b, 2005). ADK inhibitor development was initially based on 5-iodo-7-β-d-ribofuranosyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (5-iodotubercidin, 5-ITU, Fig. 3) and 5′-amino-5′-deoxyadenosine as lead compounds (Cottam et al., 1993; Kowaluk et al., 1998; Wiesner et al., 1999), which were studied kinetically for inhibition of purified ADK activity (Cottam et al., 1993). A valuable strategy to modify and optimize existing lead molecules to improve their potency, bioavailability, or toxicity profile is based on fragmentation of existing leads and NMR-based screening of those fragments with the goal to identify suitable replacement of the fragments and incorporation of the newly identified fragments into the original scaffold (Hajduk et al., 2000). Structure-activity relationships and computational studies led to the identification of a wide range of ADK inhibitors (Cowart et al., 2001; Zheng et al., 2001; Gfesser et al., 2003; Perner et al., 2003; Ugarkar et al., 2003; Gomtsyan et al., 2004). A virtual screening approach led to the discovery of 2-aryloxazolopyrimidines as ADK inhibitors (Bauser et al., 2004). High throughput derivatization and liquid phase parallel synthesis of the 7-amino and the 2-aryl groups were subsequently used to generate highly potent derivatives (Fig. 3) (Bauser et al., 2004). To optimize ADK activity assays, capillary electrophoresis assays were developed, in which the enzymatic reaction was either performed in a test tube and subsequently injected into the capillary or in which the enzymatic reaction was directly performed in the capillary (Iqbal et al., 2006). The latter approach led to further sampling size reductions and increased throughput (Iqbal et al., 2006). To date, several classes of ADK inhibitors have been developed and characterized, which broadly fall into the categories of nucleoside and nonnucleoside ADK inhibitors.
Fig. 3.

Chemical structures of ADK’s endogenous substrate adenosine and of selected nucleoside and nonnucleoside ADK inhibitors. Numbers in black refer to the IC50 of the inhibitor in nanomolars for rat cytosolic ADK. For details and references, please refer to main text.
B. Nucleoside Adenosine Kinase Inhibitors
Nucleoside adenosine kinase inhibitors have a hydroxylated ribose or cyclopentane ring and an appended purine or pyrimidine heterocyclic base. The prototype of nucleoside ADK inhibitors is 5-iodotubercidin (Fig. 3), which is a derivative of adenosine in which the 5-aza group of the purine ring has been replaced by a carbon that is linked to an iodine moiety; those compounds compete with adenosine for binding to the enzyme (Kowaluk et al., 1998; Kowaluk and Jarvis, 2000; McGaraughty et al., 2001b, 2005). Development of ADK inhibitors was initially based on the generation of 5-iodotubercidin analogs with modifications of the 5′-group of the ribose moiety to include hydroxyl-, chloro-, azido-, deoxy-, amino-, or fluoro-groups in the 5′-position; however, none of those compounds exceeded the potency of 5-iodotubercidin (Cottam et al., 1993). The ADK inhibitors 5-iodotubercidin, 5′-amino-5′-deoxyadenosine, 5′-deoxy-5-iodotubercidin, as well as novel classes of ADK inhibitors such as 4-(N-phenylamino)-5-phenyl-7-(5′-deoxyribofuranosyl)pyrrolo[2,3-day]pyrimidine (GP683), were shown to inhibit seizures in the maximal electroshock (MES) model in rats (Wiesner et al., 1999). Among those pyrrolo[2,3-day]pyrimidine nucleoside analogs, the 5′-amino-5′-deoxy analogs of 5-bromo- and 5-iodotubercidin exhibited the highest potency and efficacy in the MES model (Ugarkar et al., 2000b). Although none of those compounds met a safety, efficacy, and side effect profile suitable for further drug development (Ugarkar et al., 2000a), substitution of the tubercidin molecule with aromatic rings at the N4 and C5 positions yielded highly potent ADK inhibitors with efficacy in the MES model and reduced side effects (Ugarkar et al., 2000a). Potency of nucleoside ADK inhibitors was significantly enhanced (e.g., 10-fold compared with 5′-deoxy-5′-aminoadenosine) in 6,8-disubstituted purine nucleosides (Bookser et al., 2005a). Since cytotoxicity was found to be due to phosphorylation at the 5′-position of the ribose base, l-xylofuranosyl analogs of tubercidin were synthesized, which could no longer be phosphorylated due to their altered stereochemical orientation; the lead compound GP790 of those α-l-lyxofuranosyl nucleosides displayed prominent anti-inflammatory activity in a rat paw swelling model (Ugarkar et al., 2003). Likewise, erythrofuranosyltubercidin analogs were resistant to phosphorylation, and the orally bioavailable lead compound GP3966 was shown to exhibit broad-spectrum analgesic properties in dogs (Boyer et al., 2005). Diaryltubercidins such as GP3269 were orally active in the rat formalin paw model; however, the utility of this compound class was limited due to poor water solubility. To improve water solubility while retaining ADK inhibition potency a new compound class was generated by replacing the hydrophobic C4-phenylamino substituent with a hydrophilic glycinamide group. Although drugs from this compound class showed strong oral efficacy in pain models in the rat and marmoset monkey (ED50 estimated at 0.9 mg/kg) without evidence of side effects such as ataxia, sedation, or emesis, one compound caused lethal toxicity in the rat formalin paw model. Therefore, work on this series of compounds was discontinued (Bookser et al., 2005b).
C. Nonnucleoside Adenosine Kinase Inhibitors
Nonnucleoside ADK inhibitors lack ribose or cyclopentane rings and are either built on pyridopyrimidine cores or on alkynylpyrimidine cores, which were shown to reduce pain and inflammation in a variety of animal models (Cowart et al., 2001; Zheng et al., 2001; Gfesser et al., 2003; Gomtsyan et al., 2002, 2004; Gomtsyan and Lee, 2004). A virtual screening approach led to the discovery of a different class of nonnucleoside ADK inhibitors based on 2-aryl oxazolo-pyrimidines, which were further optimized to yield a variety of highly potent derivatives (Fig. 3) (Bauser et al., 2004). Earlier classes of nonnucleoside ADK inhibitors tended to cause locomotor side effects, a problem that was remedied by introducing polar 7-substituents of pyridopyrimidine derivatives (Zheng et al., 2003). Improved analgesic properties were achieved by the introduction of 5,6,7-trisubstituted 4-aminopyrido[2,3-day]pyrimidines as a novel class of nonnucleoside ADK inhibitors (Perner et al., 2003). In contrast, 6,7-disubstituted 4-aminopyrido[2,3-day]pyrimidines displayed only modest potency to inhibit ADK in intact cells (Perner et al., 2005). From the class of 4-amino-5,7-disubstituted pyridopyrimidines, which had been considered for clinical drug development, 5-(3-bromophenyl)-7-(6-morpholin-4-4-ylpyridin-3-yl)pyrido[2,3-day]pyrimidin-4-ylamine (ABT-702) has most widely been studied. ABT-702 (Fig. 3) was shown to have an EC50 of 1.7 nM and was equally effective on long and short isoforms of ADK from different organs and species (Jarvis et al., 2000). It was shown to be orally active and efficacious in reducing acute somatic nociception (ED50: 65 µmol/kg p.o.) in the mouse hot-plate assay. It also dose-dependently reduced nociception in the phenyl-p-quinone-induced abdominal constriction assay (Jarvis et al., 2000) and was shown to be efficacious in a wide range of pain- and inflammation-related tests, including carrageenan-induced thermal hyperalgesia, the formalin test of persistent pain, and models of nerve injury-induced and diabetic neuropathic pain. Therapeutic effects were reversed by blocking adenosine receptors, indicating that the therapeutic effects were based on a rise in adenosine (Kowaluk et al., 2000; Suzuki et al., 2001). Although 4-amino-5,7-disubstituted pyridopyrimidines were characterized as potent ADK inhibitors, compounds with a nitrogen atom in position C7 of the heterocyclic ring, however, were shown to have mutagenic properties in the Ames assay (Matulenko et al., 2005).
D. Pronucleotides
A series of 6-(het)aryl-7-deazapurine pronucleotides was recently synthetized and shown to exhibit cytostatic activity. Interestingly, several of these pronucleotides strongly inhibited human ADK; however, the mechanistic implications of this finding have not been investigated further (Spacilova et al., 2010).
E. Substrates of Adenosine Kinase
A different pharmacological application makes use of the capability of ADK to phosphorylate nucleoside-based prodrugs into their active derivatives. This strategy has been employed to develop potential anticancer drugs. Thus, it was found that the proapoptotic effects of N6-substituted derivatives of adenosine are related to their intracellular conversion into corresponding mononucleotides by ADK (Mlejnek and Dolezel, 2005). Vidarabine (9-β-d-ribofuranosyladenine or AraA) is an analog of adenosine containing d-arabinose instead of d-ribose and was originally considered as an anticancer drug (LePage et al., 1973). However, AraA also exhibits antiviral activity (Bryson et al., 1974) and was the first antiviral nucleoside to be licensed for the treatment of herpes virus infections in humans (Whitley et al., 1976). AraA needs to be phosphorylated to its 5′-triphosphate to be effective as an inhibitor of herpes virus replication (Balzarini and De Clercq, 1990). ADK converts AraA to its 5′-monophosphate, which is then further converted to its antiviral and cytotoxic 5′-triphosphate derivative (Chan and Juranka, 1981; Chan and Guttman, 1985).
V. Physiology and Pathophysiology
A. Lessons from Genetically Modified Organisms
Whereas important insights into the biochemistry of ADK were gained from mutant cell lines, the complex physiologic and pathophysiological roles of ADK were largely derived from genetic manipulations of ADK. Of note are genetic manipulations in mice and in the plant A. thaliana (mouse-ear cress). Together these studies demonstrate that ADK expression needs to be tightly controlled to maintain normal physiologic function. Studies on ADK from parasites will be discussed in a later section of this review.
1. Constitutive Deletion of Adenosine Kinase
A homozygous constitutive disruption of the Adk-gene was first accomplished in the mouse via a standard gene targeting approach (Boison et al., 2002b). Homozygous mutants were characterized by early postnatal mortality. Three causes of death were identified, as follows. 1) Mutant pups were affected by deficits in thermoregulation. When separated from their mothers at a room temperature of 22°C the body temperature of Adk−/− mutants dropped to 24°C within 15.6 minutes in contrast to wild-type littermates, which took 26.3 minutes to reach the same temperature (Boison et al., 2002b). Adenosine is known to regulate thermoregulation through A1R- and A2AR-dependent mechanisms (Jonzon et al., 1986; Zarrindast and Heidari, 1993; Fredholm et al., 2011b) and cooler pups were more likely to be culled by their mothers than normothermic littermates. 2) Mutant pups developed intermittent periods of apnea up to two times per hour and up to 20 seconds in duration, which contributed to lethal outcome during the first days after birth (Boison et al., 2002b). Periods of apnea in the mutant pups is consistent with increased activation of adenosine receptors in brain stem, which contribute to the control of respiratory function (Aoki et al., 2004; Wilson et al., 2004). 3) From postnatal day 4 onward, Adk−/− mutants developed microvesicular hepatic steatosis and failed to thrive as evidenced by significantly reduced weight gain and early death: 35% of the mutants died within the first 4 days of life, 53% between postnatal day 5 and 8, and only 12% survived up to 14 days. At postnatal day 7 a brightly colored yellow liver (Fig. 4) could visually be detected beneath the skin (Boison et al., 2002b). Metabolite analysis from liver samples revealed 2.3-fold elevated SAH and SAM and a 35% decrease in ATP in the homozygotes, whereas heterozygous mutants appeared to be normal. Increased SAH and SAM indicate disruption of the transmethylation pathway and demonstrate that constant removal of adenosine by ADK is necessary for the maintenance of transmethylation reactions (Boison et al., 2002b). Since liver is both the organ with the highest expression levels of ADK and the organ in which 80% of all transmethylation reactions take place, it is tempting to conclude that liver ADK plays a major role in the maintenance of transmethylation (Boison et al., 2002b). Because of the prominent liver pathology and early death of most mutants, further insight into the role of ADK in other organ systems could not be derived.
Fig. 4.
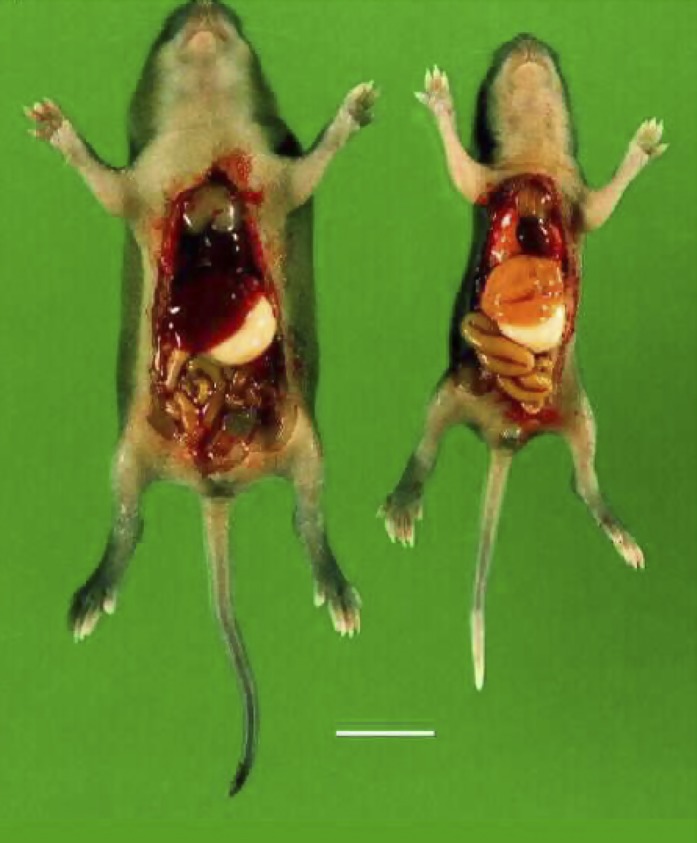
Genetic disruption of ADK leads to hepatic steatosis. Wild-type mouse (left) and Adk−/− mouse (right) prepared at postnatal day 7. Note the reduced body size of the mutant and the yellow discoloration of the liver. Scale bar: 1 cm.
Interestingly, a genetic disruption of the Adk gene in A. thaliana led to a remarkably similar phenotype (Moffatt et al., 2002). Affected plants were characterized by major developmental abnormalities, including small growth with rounded, wavy leaves and a compact, bushy appearance. Importantly, the lack of adenosine salvage in the ADK-deficient plants led to elevated SAH and resulted in the inhibition of SAM-dependent transmethylation reactions. The authors of this study concluded that adenosine must be steadily removed by ADK to prevent feedback inhibition of SAH hydrolase and maintain SAM utilization and recycling (Moffatt et al., 2002).
2. Transgenic Overexpression of Adenosine Kinase
To investigate the role of ADK in the control of brain activity, a mouse model was developed containing a loxP-flanked Adk transgene encoding the short cytoplasmic isoform of ADK under the control of a human ubiquitin promoter within the Adk−/− background (Adk-tg) (Fedele et al., 2005). Adk-tg mice displayed increased brain ADK activity and constitutive overexpression of transgenic ADK throughout the brain, with particularly high levels in hippocampal pyramidal neurons. Consequently, the Adk-tg mice were characterized by various abnormalities in brain function. Brain ADK expression levels were found to 1) critically affect the basal concentration of ambient adenosine as evaluated by microelectrode biosensors, 2) determine the degree of tonic adenosine-dependent synaptic inhibition and hippocampal plasticity, 3) modulate the age-dependent effects of brain derived neurotrophic factor on hippocampal synaptic transmission, and 4) influence GABAA receptor-mediated currents in CA3 pyramidal neurons (Diógenes et al., 2012). Physiologically, overexpression of ADK in the brain of Adk-tg mice resulted in frequent electrographic seizures at a rate of about four seizures per hour (Fedele et al., 2005; Li et al., 2007a, 2008b). Furthermore, the animals displayed increased susceptibility to stroke- or seizure- induced neuronal cell death (Pignataro et al., 2007a; Li et al., 2008a,b; Shen et al., 2011), indicating that overexpression of ADK, resulting in a decreased concentration of endogenous adenosine, rendered the brain more vulnerable to seizures and to neuronal cell death. Behaviorally, Adk-tg mice were resistant to amphetamine induced hyperlocomotion (Yee et al., 2007; Shen et al., 2012) and displayed severe learning deficits in the Morris water maze task and in Pavlovian conditioning (Yee et al., 2007). Adenosine is known to be an important regulator of sleep physiology (Bjorness and Greene, 2009; Huang et al., 2011; Porkka-Heiskanen and Kalinchuk, 2011; Schmitt et al., 2012). Consequently, disruption of adenosine homeostasis by overexpression of ADK altered sleep physiology, with Adk-tg mice being awake more than 58 minutes more per day than wild-type mice and spending significantly less time in rapid eye movement (REM) sleep (Palchykova et al., 2010). In addition, ADK expression in brain stem might play an important role in addictive behavior, since morphine withdrawal behavior was significantly diminished in Adk-tg mice (Wu et al., 2013). Together, these data suggest that ADK expression in the brain is crucial for the regulation of a multitude of behaviors that depend on maintenance of adenosine homeostasis.
3. Brain-Specific Alterations of Adenosine Kinase Expression in Mice
In a first attempt to gain region-specific insights into the role of ADK expression on brain function, an Emx1-Cre transgene (Iwasato et al., 2004) was bred into the Adk-tg line to delete the loxP-flanked Adk-tg gene within the entire dorsal telencephalon. The resulting fb-Adk-def mice were characterized by a forebrain-selective reduction of ADK expression (Li et al., 2008b), increased levels of adenosine in the cerebral cortex (Shen et al., 2011), and resistance to acute seizures, after the excitotoxin kainic acid was injected into the amygdala (Li et al., 2008b). The animals were also resistant to seizure- or stroke-induced neuronal cell loss, indicating a strong neuroprotective effect of raised adenosine levels in the cortex (Li et al., 2008b; Shen et al., 2011). Importantly, fb-Adk-def mice were also resistant to the development of epilepsy in a mouse model of intra-amygdaloid kainic acid-induced epileptogenesis, suggesting for the first time that adenosine might have antiepileptogenic properties (Li et al., 2008b). Behaviorally, fb-Adk-def mice showed profound impairment in spatial working memory and enhanced motor responses to N-methyl-d-aspartate receptor blockade (Singer et al., 2012). More work is needed to identify the role of ADK in specific brain areas, and new lines of conditional Adk-mutants are needed to address pertinent region-specific questions.
B. Adenosine Kinase Mutations in Humans
Six human patients have been described recently with mutations in Adk that prevent the expression of functional protein (Bjursell et al., 2011). All mutations caused disruptions in the methionine cycle resulting in hypermethioninemia, inhibition of transmethylation, and severe liver pathology reminiscent to changes found in Adk−/− mice (Bjursell et al., 2011). In the neonatal period, affected infants failed to thrive and the children were affected by severe developmental delay and encephalopathy. Epileptic seizures developed in all six children with an age of onset between 10 and 35 months (Bjursell et al., 2011). This seizure phenotype is not consistent with the general anticonvulsant role of ADK reduction. However, developmental or epigenetic effects contributing to this seizure phenotype cannot be excluded and warrant further investigation. One girl died during sleep at the age of 10 years and 9 months (Bjursell et al., 2011), an event that might be related to sudden unexpected death in epilepsy (SUDEP) and insufficiencies in metabolic adenosine clearance (Shen et al., 2010). This human condition validates results obtained from transgenic animals and demonstrates that ADK is a crucial enzyme for the maintenance of normal body functions.
C. Human Neuropathology
In the adult brain, ADK is predominantly expressed in astrocytes (Studer et al., 2006). Many neurologic conditions are associated with inflammatory processes and the development of astrogliosis, which is a macroglial response characterized by astroglial cell proliferation and hypertrophy (Pekny and Nilsson, 2005). As demonstrated in different animal models of neurologic disease, overexpression of ADK appears to be a general response to astroglial activation (Boison, 2012b; Boison et al., 2010). These findings prompted the investigation of specimens surgically resected from the human brain and of human post mortem samples. Importantly, ADK was significantly overexpressed in surgically resected tissue from patients with mesial temporal lobe epilepsy (Aronica et al., 2011; Masino et al., 2011). In addition, ADK was found to be overexpressed in human astrocytic tumors and related to tumor-associated epilepsy (de Groot et al., 2012). These histopathological findings demonstrate an association of overexpression of ADK with human epilepsy and support data from transgenic animals showing a tight link between ADK expression levels and seizure susceptibility.
D. Role of Adenosine Kinase in Brain Development
ADK expression undergoes a remarkable shift during early postnatal brain development in rodents (Fig. 5) (Studer et al., 2006). After birth, ADK expression is largely limited to the expression of the long isoform in the nuclei of neurons. During the first 14 days of postnatal brain development ADK expression gradually shifts from neurons to astrocytes and from expression of the long nuclear isoform to the short cytoplasmic isoform (Studer et al., 2006). By postnatal day 21, the brain shows the adult expression pattern of ADK, with ADK expression largely restricted to the cytoplasmic isoform (Fedele et al., 2005) and to expression in astrocytes (Studer et al., 2006). The only neurons in the adult brain that maintain high expression levels of ADK are neurons from the olfactory bulb, whereas dentate granular neurons maintain a low level expression of the nuclear isoform of ADK into adulthood (Gouder et al., 2004; Studer et al., 2006). During early postnatal development of the hippocampal formation, the nuclear expression of neuronal ADK is gradually phased out as the cells mature (Fig. 5) (Studer et al., 2006). Since ADK is a metabolic clearance enzyme necessary for the maintenance of transmethylation reactions, it is tempting to speculate that the expression of the nuclear isoform of ADK in immature or developing neurons might be implicated in epigenetic functions based on interaction with DNA methylation pathways. This transient neuronal expression profile of ADK could therefore play important roles for brain plasticity and development.
Fig. 5.

ADK expression changes during early postnatal brain development of the mouse. Top, first row: ADK immunohistochemistry (brown) shows strong ADK labeling in cell bodies of CA1 pyramidal cells at P4 and P8 but not at P14. Arrow pairs denote outer and inner boundaries of stratum pyramidale. Top, second row: Confocal imaging of immunofluorescence for ADK (red) and the neuronal marker NeuN (green) shows colocalization of ADK and NeuN in CA1 pyramidal cell bodies at P4 and P8 (yellow). Black scale bar: 75 µm; white scale bar: 12 µm. Bottom: Corresponding immunohistochemical characterization of the CA3 area, which loses neuronal ADK expression earlier than CA1.
E. Role of Adenosine Kinase in Specific Organ Systems and Pathologies
ADK controls specific organ functions through a combination of adenosine receptor-dependent and -independent mechanisms. Any change in adenosine homeostasis will affect activation of all adenosine receptors simultaneously and the resulting effects on organ function are more likely based on changes in network homeostasis rather than a specific receptor subtype. If not mentioned otherwise, the net effects described below are based on a combination of adenosine receptor-dependent and -independent mechanisms.
1. Liver
Liver is the organ with the highest expression levels of ADK (Fedele et al., 2005; Cui et al., 2011) and the organ in which 80% of all transmethylation reactions of the human body take place (Mato et al., 2008). The biochemistry of transmethylation reactions has been discussed at an earlier place in this review. Importantly, ADK expression in liver is a requirement to maintain the metabolic clearance of adenosine and thus the flow of transmethylation reactions. As discussed above, disruption of ADK expression has dire consequences for the liver: hepatic steatosis develops, a pathology shared between Adk−/− mice and human patients with ADK deficiency (Boison et al., 2002a; Bjursell et al., 2011). Thus, ADK deficiency in the liver is likely to affect the availability of many methylated compounds, such as choline or related metabolites in fat metabolism.
2. Pancreas
In pancreas, the nuclear isoform of ADK is specifically expressed in β-cells, whereas α-cells and fibroblasts express exclusively the cytoplasmic isoform of ADK (Annes et al., 2012). These findings suggest a specific role of nuclear ADK for β-cell function. Using a lentiviral approach to infect cultured β-cells with an RNAi targeted to ADK, it was shown in mixed cultures containing infected, uninfected, and control-infected cells that both types of control cells exhibited the same basal proliferation rate, whereas cells that received the ADK-directed siRNA demonstrated a 2.5-fold increase in their proliferation rate (Annes et al., 2012). These findings demonstrate that the nuclear isoform of ADK attenuates the proliferation of β-cells in a cell-autonomous manner.
3. Heart
Adenosine exerts a variety of cardioprotective effects, which are largely based on the activation of A1Rs (Hedqvist and Fredholm, 1979). Those cardioprotective effects include protection against ischemia/reperfusion injury (Mubagwa and Flameng, 2001; Peart and Headrick, 2007), reduction of oxidative stress (Narayan et al., 2001; Reichelt et al., 2009), and attenuation of hypertrophy and heart failure (Liao et al., 2003; Lu et al., 2008; Xu et al., 2008). Since the antihypertrophic effects of adenosine cannot completely be abrogated by genetic deletion or pharmacological blockade of the adenosine receptors (Lu et al., 2008; Fassett et al., 2011), the intracellular metabolism of adenosine in cardiomycytes might play a critical role. Interestingly, cardiomyocytes preferentially express the nuclear isoform of ADK (Fassett et al., 2011), indicating an intracellular function of adenosine in cardiomyocyte physiology. In line with this notion, blockade of ADK with either RNAi or ADK inhibitors (iodotubercidin or ABT-702) completely reversed the antihypertrophic effects of external adenosine or its analog 2-chloroadenosine (Fassett et al., 2011). These results support an inhibitory role of ADK on cell growth of cardiomyocytes. Analysis of cell signaling pathways identified Raf-dependent signaling to the mTOR/p70Sk complex as an important contributor to cardiomyocyte hypertrophy, which can be disrupted by adenosine through a mechanism dependent on ADK (Fassett et al., 2011). However, whether this interaction with the mTOR pathway is direct or indirect, e.g., via changes in DNA methylation, has not been identified. In conclusion, as in β-cells, these studies suggest an important role of nuclear ADK in the regulation of cell proliferation.
4. Brain
Endogenous adenosine has long been known to regulate excitability within the brain (Dunwiddie, 1980; Dunwiddie et al., 1981). Consequently, dysregulation of ADK expression and resulting disruption of adenosine homeostasis is implicated in a wide range of neurologic and neuropsychiatric pathologies. Although developmental changes in ADK expression have been documented in the developing brain (Studer et al., 2006), functional implications of ADK expression have only been studied in the adult brain to date.
a. Cell type specificity of ADK expression
In the adult brain, ADK expression is largely restricted to astrocytes (Studer et al., 2006). As mentioned earlier, notable exceptions are neurons from the olfactory bulb, which maintain high levels of ADK expression into adulthood (Gouder et al., 2004). In addition, the cell bodies and nuclei of dentate granular neurons maintain low levels of nuclear ADK expression (Li et al., 2008b). The functional implications of neuronal ADK expression in the adult brain remain to be determined.
b. Isoform specificity of ADK expression
In the adult brain, expression of the short cytoplasmic isoform of ADK dominates quantitatively as determined in Western blots, which separate the two isoforms (Cui et al., 2009; Fedele et al., 2005). Cytoplasmic ADK spreads throughout the astroglial network and gives the impression of a ubiquitous ADK background (Gouder et al., 2004; Studer et al., 2006). Widespread distribution of ADK immunoreactivity is in line with regulation of the adenosine concentration in brain tissue. Indeed, engineered changes in cytoplasmic ADK expression in mouse brain were shown to be sufficient to alter the tissue concentration of adenosine (Shen et al., 2011). Conversely, the nuclear isoform of ADK shows distinct expression in the nuclei of astrocytes and to a lesser degree in dentate granular cell neurons (Studer et al., 2006; Li et al., 2008b), whereas neurons from the olfactory bulb have high expression levels of both isoforms of ADK. Interestingly, the nuclear expression of ADK is seen in cell types that maintain plastic behavior into adulthood, such as astrocytes or cells from the granular cell layer of the dentate gyrus, whereas terminally differentiated cells, such as most neurons, lack ADK expression. The nuclear expression of ADK is thus consistent with cell-autonomous effects of adenosine, which might be related to an epigenetic role of ADK as regulator of DNA methylation. A possible epigenetic role of nuclear ADK would be consistent with the expression of nuclear ADK in plastic cell types and findings from β-cells of the pancreas and from cardiomyocytes, in which ADK was shown to have prominent effects on cell proliferation (Fassett et al., 2011; Annes et al., 2012). This is an exciting possibility that warrants further investigation.
c. Epilepsy
Epilepsy is a chronic seizure disorder that affects about 1% of the population. It is widely believed that most forms of epilepsy are acquired and result from a precipitating injury, which can be a traumatic injury to the brain, a stroke or period of hypoxia, a viral infection, or febrile seizure (Pitkanen and Lukasiuk, 2011; Vezzani et al., 2011; Aronica and Vezzani, 2012). Importantly, inflammatory processes as well as microglial and astroglial activation play important roles in the development of epilepsy. In particular, reactive gliosis, a fairly common morphologic and biochemical conversion of astrocytes into a pathologically hyperactive state is a pathologic hallmark of epilepsy. Since astrocytes form complex astroglial networks (Giaume et al., 2010), any disruption of astrocyte function in epilepsy, such as structural, biochemical, and metabolic changes, is expected to disrupt network homeostasis within the brain on a global scale. Synaptic levels of adenosine are largely controlled by an astrocytic sink for adenosine, which is based on astroglial expression of ADK (Boison et al., 2010). Although neurons, which mostly lack ADK, can constitute a major source for the direct release of adenosine (Lovatt et al., 2012), astrocytes can release ATP as the metabolic precursor of adenosine (Pascual et al., 2005). Reuptake of adenosine into the astrocyte is mediated via two types of equilibrative nucleoside transporters and driven by metabolic clearance of adenosine via phosphorylation into AMP by ADK (Fig. 6). Following an insult to the brain, astrocytic ADK expression undergoes a biphasic response: acute downregulation of the enzyme within hours as an acute neuroprotective response (Gouder et al., 2004; Pignataro et al., 2008) is followed by astrogliosis and associated overexpression of ADK within days or weeks (Gouder et al., 2004; Li et al., 2007a, 2012). Consequently, ADK was found to be upregulated and causing adenosine deficiency in epileptogenic sclerotic tissue in a variety of rodent models of epilepsy (Fig. 7) as well as in human specimens resected from patients with temporal lobe epilepsy and hippocampal sclerosis (Li et al., 2008b; Aronica et al., 2011). By use of a mouse model of CA3-selective astrogliosis it was shown that spontaneous recurrent seizures were both temporally and spatially related to the astrogliotic focus and to the area of overexpressed ADK. Since similar seizures were triggered by transgenic overexpression of ADK in brain, increased metabolic clearance of adenosine via increased expression of ADK in astrocytes is a likely contributing mechanism for seizure generation in epilepsy (Li et al., 2008b). In addition, homeostatic functions of the adenosine system appear to play a crucial role in epileptogenesis. Both transgenic animals with forebrain-selective reduction of ADK, as well as recipients of adenosine releasing stem cell-derived infrahippocampal grafts, showed a significant attenuation of astrogliosis following a kainic acid-induced status epilepticus, failed to increase ADK expression, and most importantly, did not develop any spontaneous seizures following an adequate trigger for epileptogenesis (Li et al., 2008b). These findings indicate that astroglial ADK is a promising target for the prediction and prevention of seizures in epilepsy
Fig. 6.

Astrocytes constitute a sink for the metabolic clearance of adenosine in the brain. Whereas neurons are capable of releasing adenosine directly, astrocytes can release ATP via vesicular release and/or by direct release through hemichannels (h-ch). Extracellular ATP is rapidly degraded into adenosine (ADO) by a series of ectonucleotidases. Adenosine can also be released directly via equilibrative nucleoside transporters (nt). Intracellular adenosine levels are largely controlled by adenosine kinase, which phosphorylates adenosine into AMP. Small changes in adenosine kinase activity rapidly translate into major changes in adenosine. Intracellular astrocytic adenosine kinase is considered to be a metabolic reuptake system for adenosine. Only selected mechanisms and pathways are shown; for details please refer to main text.
Fig. 7.

Astrogliosis and overexpression of ADK in a mouse model of temporal lobe epilepsy. (A and B) Brains from kainic acid (KA)-treated mice were taken at 4 weeks after either intrahippocampal KA or saline injection. Transverse brain sections of the KA-injected brain hemisphere were stained for ADK-immunoreactivity. Note prominent overexpression of astrogliosis in association with spontaneous seizures activity in B. (C and D) Colocalization of ADK and GFAP immunofluorescence, as seen by confocal laser scanning microscopy. Transverse brain sections of a KA-injected animal taken 4 weeks after the injection and those from a naive control animal were double stained for ADK (red) and the astrocyte marker GFAP (green). Optical sections were digitized at high magnification and superimposed for display. (C) Dentate gyrus of a control animal. Note that the cell bodies of individual astrocytes (green processes) are stained for ADK (red). (D) Dentate gyrus of a KA-injected animal. Note the massive gliosis characterized by the swelling of cell bodies, the enlargement of astrocytic processes, and the expansion of ADK-immunoreactivity into the processes (colocalization of ADK and GFAP, yellow). sp, Stratum pyramidale; sml, stratum moleculare; sg, stratum granulosum.
d. Traumatic brain injury
Astrogliosis and associated overexpression of ADK has also been identified in a rat model of severe traumatic brain injury (TBI) induced by a lateral fluid percussion injury. Interestingly, the injured animals developed epileptiform bursts associated with astrogliosis and overexpression of ADK well before the development of clinical epilepsy (Lusardi et al., 2012).
e. Central apnea
Adenosine homeostasis in the brain stem is implicated in the regulation of respiratory function by A1 and A2ARs (Fredholm, 1984; Lagercrantz et al., 1984). Remarkably, suppression of respiratory function is a major cause of death following a severe TBI, and high levels of adenosine in the cerebrospinal fluid were associated with acute lethal outcome in human victims of a severe TBI (Clark et al., 1997). Consequently, a combination of an excessive injury-related surge in adenosine, in combination with deficiencies in metabolic clearance of brain stem adenosine by ADK, would constitute a major risk factor for the development of lethal apnea. This hypothesis was tested in a rat model of severe TBI with an acute mortality rate of 46.7%. As expected, the acute mortality was found to be related to prolonged apnea. To determine whether excessive adenosine receptor activation contributed to lethal outcome, a subset of rats was treated with a 25 mg/kg concentration of the nonselective adenosine receptor antagonist caffeine intraperitoneally within 1 minute of the injury. Importantly, a single acute injection of caffeine was shown to completely prevent TBI-induced mortality when given immediately following the TBI, demonstrating that excessive adenosine contributed to lethal outcome (Lusardi et al., 2012). As in TBI, sudden unexpected death in epilepsy (SUDEP) has been associated with respiratory suppression (Langan, 2000; So, 2008). To address the hypothesis whether seizure-induced adenosine release, in combination with deficient metabolic adenosine clearance, might be a sufficient cause for SUDEP, seizures in mice were triggered in combination with pharmacologically (combination of ADK and adenosine deaminase inhibitor) induced deficiency in metabolic adenosine clearance. The combination of impaired adenosine clearance with kainic acid-induced seizures triggered sudden death in all animals. However, caffeine, when given after seizure onset, significantly increased the survival time in affected animals (Shen et al., 2010). Together, the TBI and SUDEP studies suggest that the capacity for metabolic adenosine clearance in brain stem by ADK might critically determine vulnerability to lethal apnea. In line with these findings, it is important to note that sudden infant death syndrome (SIDS) is a condition frequently characterized by lethal respiratory suppression and familial occurrence (Oren et al., 1987; Harper et al., 2000). Interestingly, a retrospective study has demonstrated the incidence of hepatic steatosis in about 10% of a total of 418 SIDS cases (Boles et al., 1998). Given the occurrence of apnea and hepatic steatosis in Adk−/− mice, it is tempting to speculate that inborn deficiencies in ADK might contribute at least to a subset of SIDS cases, a possibility that warrants further investigation.
f. Stroke
ADK expression levels critically determine the brain’s vulnerability to the effects of a stroke. Thus, transgenic overexpression of ADK in Adk-tg mice led to a 3-fold increase in infarct volume compared with wild-type control animals, when exposed to 15 minutes of middle cerebral artery occlusion (MCAO) followed by 24 hours of reperfusion, whereas all Adk-tg mice died following 60 minutes of MCAO, a condition in which wild-type animals routinely survive (Pignataro et al., 2007a). In contrast, transgenic fb-Adk-def mice with increased ADK expression in striatum (164%) and reduced ADK expression in cortical forebrain (65%) demonstrate increased striatal infarct volume (126%) but reduced cortical infarct volume (27%) after 60 minutes of MCAO and 23 hours of reperfusion compared with wild-type controls. These findings indicate that ADK expression levels in the CNS determine cerebral injury levels by regulating the availability of adenosine activating the neuroprotective function of the A1R (Shen et al., 2011). These findings were further corroborated using an adeno associated virus-based strategy to modify ADK expression in astrocytes. Mice receiving intrastriatal injections of virus that carried either Adk-sense or -antisense constructs to overexpress or knockdown ADK in vivo were characterized by increased (126%) or decreased (51%) infarct volume, respectively, when subjected to MCAO (Shen et al., 2011). Together, these data define ADK as a possible therapeutic target for modulating the degree of stroke-induced brain injury.
Ischemic preconditioning is a phenomenon in which tolerance to injury develops based on the experience of a preceding noninjurious challenge (Dirnagl et al., 2009). Thus, mice subjected to 15 minutes of MCAO followed 72 hours later by 60 minutes of MCAO display robust protection of the affected brain hemisphere (Stenzel-Poore et al., 2003). Because of its neuroprotective capabilities, adenosine is a logical candidate to mediate ischemic tolerance (Williams-Karnesky and Stenzel-Poore, 2009). To investigate whether adenosine might play a role in protecting the hippocampus after focal ischemia, Adk-tg mice were subjected to transient MCAO. Although the hippocampus of wild-type mice was consistently spared from injury after 60 minutes of MCAO, hippocampal injury became evident in Adk-tg mice after only 15 minutes of MCAO. To determine whether downregulation of endogenous ADK might qualify as a candidate mechanism mediating endogenous neuroprotection, ADK expression in wild-type mice was evaluated various time points after an MCAO. Although ADK expression was found to be reduced brain wide up to 1 day following 60 minutes of MCAO (Fig. 8), a significant reduction of ADK expression was also found in the ipsilateral hippocampus after 15 minutes of MCAO and 3 hours of reperfusion (Pignataro et al., 2008). Moreover, abrogation of lipopolysaccharide (LPS)-induced ischemic preconditioning in Adk-tg mice indicated that ADK activity negatively regulates LPS-induced tolerance to stroke (Shen et al., 2011). Thus, transient downregulation of hippocampal ADK after a stroke might be an endogenous neuroprotective mechanism of the brain.
Fig. 8.

Downregulation of ADK in a mouse model of cerebral stroke. Brains from mice were taken 3 hours after 60 minutes of middle cerebral artery occlusion (right) or a sham surgery and stained for ADK immunoreactivity. Images show part of the striatum ipsilateral to the stroke.
g. Sleep
Sleep and the intensity of sleep are enhanced by adenosine and its receptor agonists, whereas antagonists such as caffeine or theophylline induce wakefulness (Huang et al., 2011; Porkka-Heiskanen and Kalinchuk, 2011). In rodents, adenosine metabolic enzymes, including ADK, undergo diurnal changes, with higher enzymatic activities usually observed during the active period of the animals (Alanko et al., 2003; Mackiewicz et al., 2003). In line with a role of ADK in sleep modulation, sleep was found to be profoundly altered in Adk-tg mice (Palchykova et al., 2010). The mutant animals displayed a profound reduction in electroencephalogram power in low frequencies in all vigilance states and a reduction in the 6–11 Hz theta activity in REM sleep and in waking. Adk-tg mice also slept about 1 hour less per day compared with wild-type control mice, and REM sleep duration was reduced by 20%. In Adk-tg mice, the effects of sleep deprivation on slow-wave activity and energy were significantly reduced (Palchykova et al., 2010). These findings are in line with pharmacological data, in which the ADK inhibitor ABT-702 caused a significant shift in the slow-wave sleep and REM sleep ratio in rats (Radek et al., 2004). Together, these data demonstrate that levels of ADK activity in the brain critically determine important parameters of sleep physiology.
h. Cognition
As an upstream regulator of major neurotransmitter systems, including glutamatergic neurotransmission (Sebastiao and Ribeiro, 2009b; Ribeiro and Sebastiao, 2010; Diógenes et al., 2012), adenosine is a prime candidate for the modulation of cognitive processes. Importantly, transgenic overexpression of ADK in the brain of mice (Adk-tg mice) caused prominent cognitive impairment on several levels (Yee et al., 2007; Singer et al., 2012). The motor stimulant effect of MK-801 was potentiated in Adk-tg mice suggesting N-methyl-d-aspartate receptor hypofunction (Yee et al., 2007). In line with this finding Adk-tg mice displayed severe learning deficits in the domains of reference memory, working memory, and associative learning (Yee et al., 2007). The link between overexpression of ADK and cognitive impairment might be of pathologic relevance for neurologic conditions in which overexpression of ADK has either been confirmed (epilepsy) or suspected (Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis) (Boison, 2007; Boison et al., 2010). Interestingly, these conditions share 1) astrogliosis as a histopathological hallmark (Schiffer et al., 1996; Blumcke et al., 1999; Renkawek et al., 1999; Ala et al., 2000; Yamanaka et al., 2008) and 2) cognitive impairment as comorbidity or key pathologic feature (Palop and Mucke, 2009; Aarsland and Kurz, 2010; Rusina et al., 2010; Bell et al., 2011). Thus, astrogliosis-associated overexpression of ADK might be causally involved in the development of cognitive comorbidities spanning a wide range of neurologic conditions.
i. Schizophrenia
The adenosine hypothesis of schizophrenia postulates that hypofunction of adenosine signaling may contribute to the pathophysiology of schizophrenia (Boison et al., 2012) and constitutes a novel concept to integrate the dopaminergic hyperfunction (Seeman, 1987) and the glutamatergic hypofunction (Gordon, 2010) hypotheses of schizophrenia. Presynaptically, adenosine regulates the release of both glutamate and dopamine largely via A1Rs (Thompson et al., 1993; Wu and Saggau, 1997), whereas the output of glutamatergic and dopaminergic neurotransmission might be regulated postsynaptically by the proposed heterodimerization of A2A receptors with glutamate or dopamine receptors (Franco et al., 2007; Fuxe et al., 2007, 2010). Although this interaction remains hypothetical, it provides an attractive mechanism whereby adenosine could act as an upstream regulator of glutamatergic and dopaminergic neurotransmission. Consequently, any disruption in adenosine homeostasis would lead to imbalances in both glutamatergic and dopaminergic neurotransmission. In line with this notion, adenosine-deficient Adk-tg mice show altered locomotor behavior in response to both dopaminergic (amphetamine) and glutamatergic (MK-801) stimulants (Yee et al., 2007; Shen et al., 2012). In addition, Adk-tg mice display attentional deficits, which are a characteristic hallmark of schizophrenia (Yee et al., 2007; Shen et al., 2012). If adenosine deficiency is implicated in the expression of symptoms that are of relevance for schizophrenia, then therapeutic adenosine augmentation should be beneficial for the treatment of schizophrenia. Indeed, blockade of ADK by ABT-702 exerted potent antipsychotic-like activity in wild-type mice, whereas local cell-based adenosine augmentation in striatum restored responsiveness to amphetamine in Adk-tg mice, whereas the same manipulation of the hippocampus reversed the working memory deficits of the mutants (Shen et al., 2012). These findings suggest that ADK plays a critical role as upstream regulator of several molecular pathways implicated in the pathophysiology of schizophrenia.
5. Cochlea
Adenosine plays important roles in the auditory system, in particular in protecting the cochlea from oxidative stress (Vlajkovic et al., 2009). An otoprotective role of adenosine is supported by findings showing that A1R activation can prevent cochlear injury caused by acoustic trauma or by ototoxic drugs (Vlajkovic et al., 2009). Therefore, adenosine metabolic enzymes, such as ADK, have emerged as attractive targets for controlling oxidative stress in the cochlea (Vlajkovic et al., 2009). In the adult cochlea of the rat, ADK immunoreactivity was mostly localized to the nuclear or perinuclear region of spiral ganglion neurons, to lateral wall tissue, and to epithelial cells lining the scala media (Vlajkovic et al., 2010). Like in the brain, ADK expression was subject to highly coordinated expression changes during early postnatal development of the cochlea (Vlajkovic et al., 2010), implicating a putative role of ADK in the regulation of developmental processes. Therapeutically, the chronic application of the ADK inhibitor ABT-702 (1.5 mg/kg twice per week for 3 or 6 months) was shown to attenuate the development of age-related hearing loss in C57Bl/6 mice (Vlajkovic et al., 2011). At the age of 9 months, when nontreated control mice exhibited significant loss of hair cells and hearing capability, ABT-702-treated mice showed better hearing thresholds in auditory brain stem responses as reflected in lower threshold shifts at 10 and 16 kHz. Importantly, the treated animals were also characterized by increased hair cell survival in the apical cochlea (Vlajkovic et al., 2011). Although ADK inhibition was shown to be a promising therapeutic approach for the attenuation of age-related hearing loss, ABT-702 treatment was not able to prevent the development of sound-induced hearing loss (Vlajkovic et al., 2010).
6. Diabetes
In diabetes mellitus, adenosine homeostasis is critically altered in several tissues. Thus it was shown that the cytosolic activity (Vmax) of ADK was decreased by 40 to 50% in kidney, heart, and liver of rats, in which diabetes mellitus was induced by streptozotocin (Pawelczyk et al., 2000). In line with these findings, Adk transcript levels were found to be reduced by up to 50% in the same organs and as early as 24 hours following the induction of diabetes (Pawelczyk et al., 2000). Reduced ADK expression and reversed transport of adenosine from cells into the extracellular space were also found to be associated with suppressed proliferation of diabetic T lymphocytes, an effect linked to excessive A2AR stimulation (Sakowicz-Burkiewicz et al., 2006). In the brain of diabetic rats, binding densities of the neuroprotective A1 receptors were found to be reduced by 36%, whereas those of the facilitatory A2aR increased by 83% in total hippocampal membranes (Duarte et al., 2006). Thus, increased adenosine signaling in the diabetic brain in combination with a shift in adenosine receptor expression patterns might be an explanation for the development of diabetic encephalopathy and the protective effects of caffeine (Duarte et al., 2007,2009). Conversely, insulin-treatment was found to restore expression and activity levels of Adk transcripts and ADK protein, respectively (Sakowicz-Burkiewicz et al., 2006). Restoration of ADK expression by insulin in rat lymphocytes was mediated by activation of the mitogen-activated protein kinase pathway (Pawelczyk et al., 2003). As already discussed above, the nuclear isoform of ADK plays a key role in regulating the proliferation of β-cells in the pancreas. In particular, the therapeutic inhibition of nuclear ADK in β-cells might constitute a promising therapeutic avenue to increase the number of insulin-producing cells in diabetic conditions, in which glucose-based mechanisms in the control of β-cell replication fail (Porat et al., 2011; Annes et al., 2012).
7. Arthritis
Homeostasis of adenosine receptor signaling is of crucial importance in the regulation of inflammation and the release of proinflammatory cytokines (Hasko et al., 2008; Cronstein, 2010; Ernst et al., 2010). The A2A and A3Rs in particular play key roles in the regulation of inflammatory pathways in a variety of conditions including arthritis (Morello et al., 2006; Hasko et al., 2008). While A2A and A3 receptors were shown to be upregulated in patients with rheumatoid arthritis (Varani et al., 2009, 2010, 2011), adenosine was shown to suppress elevated levels of the proinflammatory cytokines TNF-α and IL-1β in patients with rheumatoid arthritis (Forrest et al., 2005; Varani et al., 2010). It is now well accepted that adenosine exerts potent anti-inflammatory effects via activation of A2A and A3 receptors. Therefore, A2A and A3 receptor agonists (Yan et al., 2003; Akkari et al., 2006; Flogel et al., 2012) and ADK inhibitors (Cronstein et al., 1995; Boyle et al., 2001) constitute rational therapeutic strategies for the treatment of arthritis.
8. Colitis
Ulcerative colitis is an inflammatory bowel disease (IBD) that causes long-lasting inflammation in part of the digestive tract. As discussed above, the homoeostasis of adenosine receptor signaling is also of critical significance for the chronic inflammatory reactions in IBD (Hasko et al., 2008). Limited oxygen availability and inflammation in mucosal membranes lead to increased production of adenosine from degradation of ATP and ADP; increased expression of adenosine receptors, in particular the A2B receptor; reduced uptake of extracellular adenosine, and decreased metabolic clearance of adenosine (Eltzschig et al., 2009). Recent data demonstrate that enhanced adenosine signaling via the A2BR attenuated mucosal inflammation, permeability, and tissue injury during intestinal ischemia or experimental colitis, whereas suppression of macrophage activation was also involved in the beneficial effects of A2BR activation (Eltzschig et al., 2009; Hasko et al., 2009). Therefore, therapeutic modulation of adenosine signaling appears to be a rational approach for the treatment of IBD.
9. Cancer
The role of the adenosine/ADK regulatory system in cancer may depend on the type of cancer. Several studies report a cytotoxic role of extracellular adenosine. Thus, extracellular adenosine was shown to induce apoptosis in MCF-7 human breast cancer cells, an effect that was shown to be intracellular and independent of adenosine receptor activation (Tsuchiya et al., 2012). Mechanistically, it was shown that adenosine promoted the translocation of apoptosis-inducing factor-homologous mitochondrion-associated inducer of death from the cytosol into the nucleus (Tsuchiya et al., 2012). An earlier study suggested that the cytotoxic effects of adenosine on breast cancer cells might be based on conversion of adenosine to AMP (ADK dependent) followed by activation of nucleoside kinase and activation of the mitochondrial/intrinsic apoptotic pathway (Hashemi et al., 2005). An intracellular adenosine receptor-independent apoptotic effect on astrocytoma cells was found to depend on intracellular activation of an adenosine analog (Ceruti et al., 2000). A cytotoxic role for adenosine was also demonstrated in human gastric cancer cells, with a mechanism based on conversion of adenosine to AMP and activation of the intrinsic apoptotic pathway through AMP kinase activation (Saitoh et al., 2004). In addition, A3 receptor activation might be of benefit for the treatment of colorectal cancer with clinical trials on the way (Yan et al., 2003). In line with a cytotoxic role of adenosine, Adk-gene expression was found to be significantly higher in human patient-derived colorectal cancer tissue than in healthy control tissue (Giglioni et al., 2008; Vannoni et al., 2004a,b), suggesting that either more efficient metabolic clearance of cytotoxic adenosine might provide an advantage for tumor cells or that a general increase in purine metabolic enzymes might permit accelerated purine metabolism to support the growth of cancerous tissue. In contrast to those findings, ADK activity was found to be reduced in hepatoma cells, suggesting that increased adenosine might provide a selective advantage for hepatic cancers. Although increased adenosine has been linked to cytotoxic and apoptotic effects in several cancer types, it needs to be stressed that increased levels of adenosine also inhibit immune responses and inflammatory responses and stimulate angiogenesis, effects that might benefit tumor growth on a physiologic level. More work needs to be done to fully understand the role of adenosine in cancer biology, in particular regarding epigenetic ramifications of adenosine regulation, and to investigate whether ADK might constitute a therapeutic target for the treatment of cancer.
VI. Therapeutic Applications of Adenosine Kinase-Based Interventions
A. Strategies to Alter Adenosine Kinase Activity
1. Pharmacology
Pharmacological approaches to harness the therapeutic potential of adenosine augmentation are based on nucleoside and nonnucleoside ADK inhibitors, which have been discussed in preceding sections. The major advantage of this approach, in contrast to more selective adenosine receptor agonists, is that ADK inhibitors can potentiate an endogenous stress response of the body and potentiate the actions of endogenous adenosine in a site- and event-specific manner (Kowaluk et al., 1998; Britton et al., 1999; Wiesner et al., 1999; Kowaluk and Jarvis, 2000). An additional advantage of ADK inhibitors is the rise of the tissue concentration of adenosine, which will not only lead to the increased activation of all subtypes of adenosine receptors but also to adenosine receptor-independent effects, including epigenetic changes due to interference of adenosine with DNA methylation. Therefore, in contrast to receptor-specific ligands, ADK inhibitors are capable of affecting complex networks synergistically on multiple different levels, taking advantage of the multimodal activity of an endogenous homeostatic regulator of network function. However, therapeutic adenosine augmentation by systemic ADK inhibition might not be a viable therapeutic option due to liver toxicity (Boison et al., 2002b) and the occurrence of brain hemorrhage in some of the preclinical studies (McGaraughty et al., 2005). Therefore, localized or focal therapeutic approaches might be better suited to harness the therapeutic potential of adenosine in a more refined way.
2. Gene Therapy
One strategy to targeted local or even cell-type selective adenosine augmentation is gene therapy. In contrast to conventional gene therapies in which a transgene is added, the therapeutic goal here is to use gene therapy to reduce expression of the endogenous Adk gene. This can best be achieved using antisense approaches (Boison, 2010) to knock down gene expression. Two studies have used this approach to knock down ADK expression in models of seizures and stroke (Shen et al., 2011; Theofilas et al., 2011). Both studies are based on the same AAV8-based vector, which expresses an Adk cDNA in antisense orientation under the control of an astrocyte specific gfaABC1D promoter (Lee et al., 2008). When injected into the hippocampus of Adk-tg mice with spontaneous electrographic seizures, recipients of the Adk antisense virus had a substantial unilateral decrease in seizure activity ipsilateral to the virus injection site with 0.6 ± 0.6 seizures/hour compared with 5.8 ± 0.5 seizures/hour on the contralateral (noninjected) side (Theofilas et al., 2011). Similarly, injection of the antisense virus into the striatum of mice decreased their infarct volume to 51% of control, when these animals were subjected to 60 minutes of MCAO to model a stroke (Shen et al., 2011). Together, these studies constitute a proof of principle that a gene therapy targeting ADK, restricted to a specific brain area (hippocampus or striatum) and to a specific cell type (astrocyte), can have potent therapeutic effects based on augmenting the anticonvulsive and neuroprotective properties of adenosine. More work needs to be done to evaluate whether anti-ADK gene therapies are effective in clinically relevant models of temporal lobe epilepsy.
3. Cell Therapy
A different approach for the local augmentation of adenosine signaling is to first delete the Adk gene in cultured cells to induce therapeutic adenosine release and then to transplant the cells into a host to therapeutically exploit locally enhanced levels of adenosine. The first successful cell therapy approach was achieved in baby hamster kidney (BHK) cells in which the Adk gene had been disrupted by a combination of chemical mutagenesis and selection for ADK deficiency; importantly, disruption of the Adk gene was more effective in inducing cellular adenosine release than disruption of the adenosine deaminase gene (Huber et al., 2001). When encapsulated into semipermeable polymer fibers and transplanted into the ventricular system of epileptic rats that were kindled in the hippocampus, ADK-deficient BHK cells releasing about 40 ng adenosine per 105 cells per day almost completely suppressed any seizures in an A1R-dependent manner, whereas animals receiving control implants with wild-type cells continued to display their preimplantation seizure behavior (Huber et al., 2001). Unfortunately, seizure suppression was limited to 2 weeks due to the reduced longevity of the encapsulated cells. To develop a more versatile cell-based system for seizure control, both alleles of the Adk gene were disrupted in mouse embryonic stem (ES) cells by homologous recombination with a gene targeting construct; the Adk−/− ES cells yielded glial populations with an adenosine release of up to 40 ng/105 cells/h comparable to the amounts of adenosine released from BHK cells (Fedele et al., 2004). When differentiated into neural precursor cells and grafted into the infrahippocampal fissure of rats, the Adk−/− cell grafts profoundly suppressed kindling epileptogenesis (Li et al., 2007b). More importantly, when grafted into the infrahippocampal fissure of mice 24 hours after a status epilepticus, the same cells prevented the development of epilepsy (Li et al., 2008b). Recipients of the Adk−/− cells were characterized by attenuated astrogliosis, almost normal ADK expression levels, and complete lack of any seizures, whereas recipients of wild-type cells, or sham-treated control animals developed astrogliosis with overexpressed ADK as well as spontaneous electrographic seizures at a rate of about 4 seizures/hour (Li et al., 2008b). In a different approach, the same cells were transplanted into the striatum of mice 7 days prior to the onset of a stroke, modeled by 60 minutes of MCAO. After 23 hours of reperfusion, recipients of the Adk−/− cells were characterized by a significant reduction in infarct volume. Neuroprotection was strongest in adenosine-releasing glial precursor cell recipients, which were characterized by an 85% reduction of the infarct area. Graft-mediated neuroprotection correlated with a significant improvement of general and focal neurologic scores (Pignataro et al., 2007b). In an attempt to engineer human stem cells for therapeutic adenosine release, human mesenchymal stem cells were infected with a lentivirus engineered to express a micro RNA directed against Adk. This RNAi approach resulted in a reduction of ADK to 20% of its normal levels and triggered the release of about 1 ng adenosine/105 cells/h (Ren et al., 2007). When transplanted into the infrahippocampal fissure of mice, these implants reduced acute seizure-induced cell death (Ren et al., 2007) and led to a partial suppression of epileptogenesis (Li et al., 2009). This partial therapeutic effect is most likely due to the 40 times lower amounts of adenosine released by those cells compared with the engineered ES cells that completely lacked any ADK expression. Together, these reports demonstrate that disruption of ADK expression in cells is a promising therapeutic strategy to augment adenosine signaling at a local site within the brain, with potent therapeutic effects resulting in neuroprotection, seizure suppression, and, ultimately, prevention of epileptogenesis.
4. Ketogenic Diet
A high-fat, low-carbohydrate ketogenic diet is a metabolic intervention that provides effective seizure control in many forms of pharmacoresistant epilepsy, particularly in children (Neal et al., 2008; Yellen, 2008; Freeman, 2009; Kossoff and Rho, 2009; Kossoff et al., 2009). Despite its clinical use for over 80 years, the mechanisms underlying the therapeutic actions of a ketogenic diet have remained enigmatic. A ketogenic diet forces the brain to use ketones instead of glucose as primary energy source, and it is those metabolic changes that are thought to underlie the therapeutic effects of this type of dietary intervention (Bough et al., 2006; Kalapos, 2007; Ma et al., 2007; Bough, 2008; Yellen, 2008). A large body of evidence supports the notion that a ketogenic diet leads to increased adenosine signaling in the brain (Masino and Geiger, 2008, 2009; Masino et al., 2009, 2012). Indeed, it was recently shown that a ketogenic diet reduced the expression of ADK in mice (Masino et al., 2011). In support of increased adenosine signaling, a ketogenic diet suppressed seizures in adenosine deficient Adk-tg mice, but not in A1R-deficient mice, demonstrating that functional A1R activation is necessary for the antiepileptic effects of the diet (Masino et al., 2011). Apart from seizure control, ketogenic diets have also been shown to be beneficial in experimental paradigms of pain and inflammation, a therapeutic outcome compatible with increased adenosine signaling (Ruskin et al., 2009).
5. Transcriptional Repression
In an innate response to hypoxia, vasculature reacts with a rise in adenosine (Berne, 1963; Berne et al., 1974). Although functional inhibition of ADK in response to hypoxia has been reported previously (Decking et al., 1997), a recent study reported transcriptional repression of the Adk gene resulting in a 85% reduction of the endothelial Adk transcript (Morote-Garcia et al., 2008). It was further shown that transcriptional repression of the Adk gene was dependent on hypoxia inducible factor 1-α (HIF-1α) and that repression of ADK led to an attenuation of vascular leakage in vitro and in vivo (Morote-Garcia et al., 2008). This transcriptional mechanism appears to be an evolutionary conserved strategy to directly couple adenosine homeostasis to a system that can sense an environmental condition of critical importance for the bioenergetic equilibrium of a cell.
B. Applications in Preclinical Studies
On the basis of the rationale outlined in preceding sections, therapeutic adenosine augmentation is of value in a variety of pathogenic conditions. Importantly, therapeutic adenosine augmentation is uniquely suited to synergistically modify disrupted networks via an endogenous upstream regulator through activation of four adenosine receptor-dependent pathways, but also through additional epigenetic and bioenergetic mechanisms. In the following preclinical examples, therapeutic gain through ADK manipulation is illustrated.
1. Diabetes
One approach in diabetes therapy is promotion of β-cell replication, which normally is under the control of glucose (Porat et al., 2011). However, it is the responsiveness of β-cell replication to excess glucose, which fails in the diabetic condition. Therefore, promotion of β-cell replication independent of glucose constitutes an important therapeutic goal. By using a small-molecule screening platform to identify molecules that increase β-cell replication, Annes and colleagues (2012) recently identified a class of ADK inhibitors that specifically promoted β-cell replication in a cell type-selective manner. ADK inhibitor-dependent β-cell replication was blocked by the phosphoinositide kinase inhibitor wortmannin and the mammalian target of rapamycin (mTOR) inhibitor rapamycin, suggesting involvement of the phosphoinositide kinase/mTOR pathway. In line with this finding, ADK inhibition resulted in the increased phosphorylation status of ribosomal protein S6, a downstream target of the mTOR pathway. Intriguingly, mTOR is a cytoplasmic and nuclear kinase (Zhang et al., 2002), which might be of relevance for the presence of nuclear ADK in β-cells. In vivo, the ADK inhibitor ABT-702 resulted in a robust increase in β-cell proliferation, and this effect was shown to be specific to β-cells since the replication rate of exocrine cells or that of hepatocytes was not altered by ABT-702 (Annes et al., 2012). Although ADK inhibitors show promising potential in increasing β-cell proliferation, and thus the number of insulin secreting cells, it remains to be demonstrated whether ADK inhibitors can improve responsiveness to glucose in a model of diabetes.
2. Epilepsy
Adenosine augmentation therapies make rational therapeutic use of an endogenous anticonvulsant and neuroprotectant of the brain with the potential to not only suppress seizures, but also to prevent epileptogenesis (Boison, 2009, 2012a). ADK inhibitors are capable of raising the levels of endogenous adenosine and, as originally proposed, to potentiate an endogenous adenosine response, such as the well-documented seizure-induced adenosine release, in a site- and event-specific manner (Kowaluk et al., 1998; Kowaluk and Jarvis, 2000; McGaraughty et al., 2001b, 2005). Since pathologic overexpression of ADK has been demonstrated within epileptogenic brain areas (Gouder et al., 2004; Li et al., 2008b; Aronica et al., 2011; Boison, 2012b) and since overexpression of ADK alone can trigger electrographic seizures (Etherington et al., 2009; Li et al., 2008b, 2012; Theofilas et al., 2011), the scientific rationale for the use of ADK inhibitors in epilepsy therapy is strong. Importantly, the ADK inhibitor 5-ITU was shown to suppress seizures in mice that were resistant to conventional antiepileptic drugs (Gouder et al., 2004), suggesting that ADK inhibitors might be effective in pharmacoresistant epilepsy. Moreover, acute seizures induced by injection of bicuculline into the prepiriform cortex of rats were shown to be blocked by subsequent injection of the ADK inhibitors 5′-amino-5′-deoxyadenosine or 5-ITU, but not by the ADA inhibitor 2′-deoxycoformycin, suggesting that the anti-ictogenic activity of ADK inhibition is superior to ADA inhibition (Zhang et al., 1993). Among a wide spectrum of ADK inhibitors that have subsequently been developed for seizure control (Ugarkar et al., 2000a,b), GP-3269 showed enhanced oral bioavailability (60%) and extended plasma half-life (>4 hour), attenuation of the seizure response in the rat maximum electroshock (MES) and kindling models, and lack of profound cardiovascular side effects (Erion et al., 1997; McGaraughty et al., 2005). Despite an improved cardiovascular side effect profile, the brain wide therapeutic modulation of ADK might not be a therapeutic option due to psychiatric and cognitive effects (Yee et al., 2007; Boison et al., 2012; Shen et al., 2012). As discussed in preceding sections, in vivo or ex vivo gene therapies targeting ADK may provide a promising alternative to restrict therapeutic effects to an identifiable epileptogenic focal area with demonstrated adenosine dysfunction. Thus, infrahippocampal grafts of cells engineered to lack Adk were shown not only to suppress seizures but also to prevent epileptogenesis in a variety of experimental paradigms in which an epileptic state was created in rats or mice by repeated suprathreshold electrical stimulation (kindling) or by status epilepticus-induced brain injury (Huber et al., 2001; Li et al., 2007b, 2008b, 2009; Ren et al., 2007).
3. Pain
Several states of pathologic pain, in particular neuropathic pain, appear to share common mechanisms with epilepsy and, not surprisingly, antiepileptic drugs are frequently highly effective in the treatment of chronic pain (Horga de la Parte and Horga, 2006; Malawska and Kulig, 2008). As in epilepsy, adenosine provides potent inhibition to hyperexcitable neuronal circuits resulting in profound antinociceptive effects of adenosine (Lynch et al., 2003; Sawynok and Liu, 2003). Consequently, ADK inhibitors have been considered as a very attractive target for the treatment of various pain states, and proof of feasibility studies with prototypes of ADK inhibitors have demonstrated efficacy in several animal models of nociception (Keil and DeLander, 1994; Poon and Sawynok, 1995, 1998; Sawynok and Liu, 2003). Unfortunately, short half lives in vivo, poor bioavailability, lack of pharmacological selectivity, and potential to form cytotoxic metabolites limited further preclinical testing of those prototype inhibitors (Cottam et al., 1993; Wiesner et al., 1999; Ugarkar et al., 2000a). Therefore, most subsequent studies have focused on the structurally novel nucleoside (A-134974), nonnucleoside (ABT-702), and carbocyclic (A-286501) ADK inhibitors (Fig. 3). These compounds were shown to be orally active and to alleviate acute nociception, neuropathic allodynia, chemogenic nociception, and inflammatory thermal hyperalgesia (Kowaluk et al., 2000; McGaraughty et al., 2001a; Jarvis et al., 2002b). Importantly, all drugs selectively attenuated inflammatory hyperalgesia selectively in the inflamed hindpaw consistent with the notion that ADK inhibitors increase adenosine concentration preferentially at sites of injury or trauma. These drugs also showed a remarkable improvement in their therapeutic window compared with adenosine receptor agonists. Although a 10- to 16-fold separation between ED50 values for motor depressant and antihyperalgesic actions was noted for the newer ADK inhibitors, there was significantly less separation for directly acting adenosine receptor agonists, with the largest effect ratio being only 4.3 (Jarvis et al., 2002a). Likewise, the cardiovascular side effect profile of the ADK inhibitors was improved compared with direct adenosine receptor agonists (Kowaluk et al., 2000; Jarvis et al., 2002b). The antinociceptive effects of ADK inhibitors are primarily based on a spinal site of action. Intrathecal administration of A-134974 was more effective (ED50 = 6 nmol) than intracerebroventricular (ED50 = 100 nmol) or intraplanar (ED50 > 300 nmol) injection in its antihyperalgesic effects, whereas supraspinal activity of the drug was associated with motor depressant effects (McGaraughty et al., 2001a). As in epilepsy, the challenge to implement ADK-based therapies is restriction of treatment to the pathogenetic area responsible for pain generation. Restriction of treatment to a spinal site, e.g., by anti-ADK gene therapy, might provide local benefit without side effects.
4. Inflammation
As outlined in a preceding section, adenosine is an endogenous anti-inflammatory agent. Consequently, ADK inhibitors hold promise for the treatment of a large spectrum of inflammatory conditions. Thus, ADK inhibitors were shown to provide benefit in a rat model of pleuritis (Cottam et al., 1993) and in inflammatory pain models in the rat (Poon and Sawynok, 1998; Kowaluk et al., 2000; McGaraughty et al., 2001a; Suzuki et al., 2001). The ADK inhibitor GP-515 dose-dependently inhibited carrageenan-induced rat paw swelling and reduced cutaneous neutrophil invasion and vascular leakage in a rat skin lesion model; the latter effects were shown to be A2 receptor dependent (Rosengren et al., 1995). Furthermore, GP-515 affected carrageenan-induced inflammation in air pouches induced in BALB/c mice. Importantly, adenosine concentrations in pouch exudates were found to be increased. The anti-inflammatory effects of GP-515 were abrogated after the injection of ADA into the pouch, indicating that enhanced adenosine signaling induced by ADK inhibition was responsible for the therapeutic effect. GP-515 also reduced leukocyte counts and TNFα concentrations in the exudate (Cronstein et al., 1995). Suppression of TNFα production by GP-515 was further demonstrated in LPS-stimulated peripheral blood mononuclear cells (Eigler et al., 2000). Subsequently, GP-515 was shown to improve clinical and histologic outcome in a murine model of dextran sulfate sodium-induced colitis, a well-accepted model of inflammatory bowel disease (Siegmund et al., 2001). In addition, colon shortening, an indirect parameter for the degree of inflammation, was reduced as was the weight of the spleen. Mechanistically, GP-515 suppressed interferon-γ synthesis in LPS-induced splenocytes isolated from the colitis mice. In addition, CD69 expression, a marker for immune activation, was found to be reduced in the GP-515-treated colitis mice (Siegmund et al., 2001).
5. Cerebral Stroke
To capitalize on the neuroprotective potential of endogenous adenosine, three independent studies demonstrated a protective effect of ADK inhibitors on the infarct volume in a rat focal ischemia model (Miller et al., 1996; Jiang et al., 1997; Tatlisumak et al., 1998). When given 30 minutes before the onset of middle cerebral artery occlusion, 5′-deoxy-5-iodotubercidin reduced the infarct volume by 34 to 57% depending on the experimental paradigm and dose used (Miller et al., 1996; Jiang et al., 1997). More importantly, 5′-deoxy-5-iodotubercidin and GP-683 reduced the infarct volume up to 44% when given 30 to 360 minutes after the MCAO (Miller et al., 1996; Jiang et al., 1997; Tatlisumak et al., 1998). However, GP-683 treatment at a dose of 2 mg/kg was associated with a nonstatistically significant increase in mortality (Tatlisumak et al., 1998). Surprisingly, 5-iodotubercidin failed to protect against cerebral ischemic injury in gerbils in a temporary bilateral carotid artery occlusion model (Phillis and Smith-Barbour, 1993). In line with the neuroprotective effect of ADK inhibition, focal treatment strategies were realized by stem cell therapy and gene therapy (Pignataro et al., 2007b; Shen et al., 2011). Thus, a focal intrastriatal implant of ADK-deficient ES cell-derived glial progenitor cells led to an 85% reduction of the infarct volume in mice, when the cells were transplanted 7 days prior to 60 minutes of MCAO (Pignataro et al., 2007b). Likewise, an AAV-based gene therapy virus engineered to knock down ADK in astrocytes was shown to reduce infarct volume in mice by 51% when injected 4 weeks prior to the artery occlusion (Shen et al., 2011). Although preventative in nature and therefore not translatable to a clinical stroke scenario, these studies demonstrate that glial interventions and local ADK treatment approaches can provide significant neuroprotective effects in a mouse focal ischemia model.
6. Hearing Loss
Only two studies tested the otoprotective potential of the ADK inhibitor ABT-702. Although ABT-702 failed to restore hearing thresholds after exposure to traumatic noise (Vlajkovic et al., 2010), the chronic treatment with ABT-702 (1.5 mg/kg intraperitoneally twice a week) attenuated hair cell loss and age-related hearing loss in C57BL/6 mice (Vlajkovic et al., 2011). It remains to be determined why ABT-702 had an opposite outcome in those studies.
7. Schizophrenia
The adenosine hypothesis of schizophrenia predicts a deficiency of endogenous adenosine signaling in schizophrenia, which would synergistically affect dopaminergic and glutamatergic neurotransmission (Boison et al., 2012). In line with this hypothesis, adenosine-deficient Adk-tg mice display altered locomotor responses to both glutamatergic and dopaminergic psychostimulants. In addition, these animals display a wide spectrum of phenotypes in the affective and cognitive domains (Yee et al., 2007; Boison et al., 2012; Shen et al., 2012). If adenosine deficiency is implicated in the pathophysiology of schizophrenia, then ADK inhibition should provide benefit. Indeed, ABT-702 was recently shown to exhibit antipsychotic-like activity in a prepulse inhibition (PPI) paradigm in mice (Shen et al., 2012), which is a widely accepted measure of sensorimotor gating deficits believed to underlie sensory flooding and cognitive fragmentation in schizophrenia (Braff et al., 2001a,b). PPI disruption in rodents can be induced with the dopaminergic agonist apomorphine and is a well-established model of schizophrenia with predictive validity for antipsychotic drugs (Swerdlow et al., 2008). Furthermore, enhancement of basal PPI is considered to be a marker of antipsychotic action, since antipsychotics increase PPI in drug-naive animals (Singer et al., 2009). A recent dose response analysis (2.5, 5, and 10 mg/kg ABT-702) revealed that ABT-702 increased PPI in drug-naive animals independent of dose. In addition, ABT-702 at a dose of 5 mg/kg was found to be effective in reversing the PPI-disruptive effect of apomorphine (2 mg/kg). Together, these data demonstrated that ADK inhibition enhanced basal PPI in drug naive wild-type mice and exerted antipsychotic-like efficacy in a pharmacologically induced animal model of schizophrenia (Shen et al., 2012).
8. Cardioprotection
Adenosine is not only a potent neuroprotectant in the brain but also a powerful cardioprotectant, but the underlying protective mechanisms might be different (Przyklenk and Whittaker, 2005; Peart and Headrick, 2007; Gomes et al., 2011; McIntosh and Lasley, 2012). Whereas adenosine homeostasis in the brain is largely under the control of ADK (Pak et al., 1994; Boison et al., 2010), in the rabbit heart ADA inhibition was found to have a more profound adenosine augmenting effect than ADK inhibition (Manthei et al., 1998). In an initial study, ADK inhibition with 5-ITU or ADA inhibition with erythro-9-(2-hydroxy-3-nonyl)adenine, but not a combination of both drugs, was shown to improve functional recovery in the ischemic-reperfused mouse heart (Peart et al., 2001). Interestingly, ADK inhibition was shown to attenuate the cardioprotective effects of exogenous adenosine, suggesting that cardioprotection involves purine salvage through ADK (Peart et al., 2002). A mitochondrial ATP-sensitive K+ channel blocker was shown to abrogate the cardioprotective effects of adenosine when coinfused with 5-ITU, demonstrating that conversion of adenosine to AMP, and thus ADK activity, might play a critical role in the cardioprotective mechanisms of adenosine (Peart et al., 2003). Together, these data suggest that both ADA or ADK inhibition can limit injury during ischemia-reperfusion via adenosine receptor activation. However, cardioprotection via either enzyme inhibitor appears to require an alternative purine-salvage pathway to be functional, and this pathway was reduced in aged hearts, which are increasingly susceptible to ischemic damage (Willems and Headrick, 2005). Subsequently, those findings were reproduced in a rat model of myocardial infarction (Peart and Gross, 2005). It was shown that the ADK inhibitor GP-515 induced vascular endothelial growth factor expression in cultured rat myocardial myoblasts, an effect that was completely blocked by the addition of ADA, which, when given alone, led to a decrease in baseline vascular endothelial growth factor expression (Gu et al., 2000). Since ADA—in contrast to brain—is highly expressed in the heart (Barankiewicz et al., 1997), ADK inhibitors might not be useful adenosine augmenting agents to promote cardioprotection. The interaction between ADA and ADK in adenosine metabolism of the heart might also be a reason why ADK inhibitors (for CNS applications) have fewer cardiac side effects than direct adenosine receptor agonists (Kowaluk et al., 2000). More recently, it was shown that the ADK inhibitors 5-ITU and ABT-702 as well as RNAi directed against Adk prevented the antihypertrophic effects of adenosine on cardiomyocytes, suggesting that ADK activity is needed to provide those antihypertrophic cardioprotective effects (Fassett et al., 2011). In conclusion, given the antihypertrophic role of nuclear ADK in cardiomyocytes and given the complexity of interactions between ADK and ADA in regulating cardiac adenosine homeostasis, ADK inhibition might not be a useful approach for cardioprotection.
9. Sepsis
Since ADK inhibitors are potent anti-inflammatory agents, they may also be of therapeutic benefit in septic shock. The ADK inhibitor GP-515 significantly decreased mortality in two models of septic shock induced by either lethal i.v. injection of endotoxin or induction of bacterial peritonitis (Firestein et al., 1994). It was shown that the protective effect of GP-515 was adenosine receptor dependent and that decreased neutrophil accumulation in the lungs and reduced TNFα levels in plasma were involved (Firestein et al., 1994). It was further shown that ADK inhibition prevented hypoxia-induced vascular leakage (Morote-Garcia et al., 2008). Since only few studies have addressed ADK as potential target for the treatment of sepsis, more work is needed to judge therapeutic usefulness.
10. Cartilage Protection
The anti-inflammatory properties of ADK inhibitors are likewise of therapeutic value in cartilage protection. The ADK inhibitor ITU was shown to attenuate cartilage damage induced by either IL-1β or by LPS in an in vitro cartilage explant model; depending on the model system used, ADK inhibition inhibited glycosaminoglycan release, prostaglandin E2 release, or NO production (Petrov et al., 2005) and was shown to be more effective in the prevention of NO formation than ADA inhibition (Tesch et al., 2002). Similarly, ABT-702 significantly decreased cartilage destruction in a rat adjuvant arthritis model, a therapeutic effect that was associated with suppression of collagenase and stromelysin gene expression (Boyle et al., 2001).
C. New Therapeutic Concepts and Future Trends
The examples presented above demonstrate a general therapeutic utility of ADK inhibitors in augmenting the multiple beneficial cytoprotective and anti-inflammatory effects of endogenous adenosine. However, despite very promising results, in particular in the areas of inflammation, pain, and epilepsy, clinical ADK inhibitor development has largely been abandoned since around 2005 due to risks of toxicity and intolerable side effects. Since then, as outlined in earlier sections of this review, our knowledge on the role and function of endogenous ADK has increased tremendously leading to the re-definition of ADK as a promising therapeutic target. New solutions to challenging hurdles in ADK-based therapy development and newly defined functions of endogenous ADK will lead to a new era of ADK-based therapeutics. Several new trends and novel functions of ADK are noteworthy (Fig. 9):
Fig. 9.

Emerging new roles of adenosine kinase. Summary of key findings; for details please refer to main text.
Local therapies: The identification of ADK overexpression in many neuropathological conditions with causal implications in pathogenesis and pathophysiology (Boison, 2008; Boison et al., 2010) redefines ADK as a rational therapeutic target. The realization that ADK dysfunction is restricted to certain brain areas (e.g., hippocampus) and cell types (astrocytes) provides the therapeutic rationale to develop region- and cell type-specific therapeutic interventions with the goal to normalize ADK function within an affected brain area. This goal could most effectively be achieved via gene therapies targeting ADK in a region- and cell type-specific manner (Boison, 2009). Local therapeutic interventions would also circumvent challenges and side effects, which hampered the clinical development of systemic ADK inhibitors.
Adenosine receptor-independent mechanisms: Historically, ADK inhibitor development has aimed at achieving increased adenosine receptor activation as a consequence of an increased concentration of endogenous adenosine (Kowaluk et al., 1998; Kowaluk and Jarvis, 2000; McGaraughty et al., 2005). However, a recent study suggests a novel use of ADK inhibitors that is independent of increased adenosine receptor activation. Importantly, ADK inhibitors were shown to promote the proliferation of β-cells in the pancreas through an adenosine receptor-independent mechanism (Annes et al., 2012).
Role of nuclear isoform of ADK: Tissue levels of adenosine and the concentration of extracellular adenosine, which determine the degree of adenosine receptor activation, are thought to depend on the cytoplasmic isoform of ADK (Studer et al., 2006; Shen et al., 2011). However, the nuclear isoform of ADK might play key roles in the regulation of cell proliferation, as demonstrated in β-cells of the pancreas and in cardiomyocytes (Fassett et al., 2011; Annes et al., 2012). On the basis of a tightly controlled developmental expression profile of ADK in neonatal rodent brain, a role of the nuclear isoform of ADK in the control of neurogenesis can also be postulated (Studer et al., 2006). Obviously, the nuclear isoform of ADK might play an important role in cancer biology. Thus, nuclear ADK emerges as a novel therapeutic target to capitalize on specific nuclear functions of ADK such as interaction with the mTOR pathway (Fassett et al., 2011; Annes et al., 2012). However, it will be a challenge to develop novel therapeutics to selectively target the nuclear isoform of ADK.
Epigenetic role of ADK: Adenosine is an obligatory end product of transmethylation reactions, including DNA methylation (Finkelstein and Martin, 1986; Mato et al., 2008). If adenosine is not constantly removed by ADK, adenosine accumulates and reverses the direction of the SAH-hydrolase reaction, resulting in increased levels of SAH (Moffatt et al., 2002; Boison et al., 2002b). SAH in turn is known to inhibit DNA methyltransferases through substrate inhibition (James et al., 2002). Since direct DNA methyltransferase inhibitors, such as azacytidine and decitabine are highly toxic (Weisman et al., 1985; Yogelzang et al., 1997), ADK inhibitors might find new uses as DNA regulating agents, e.g., in cancer therapy.
Regulation of cell proliferation: Through a combination of epigenetic and additional adenosine receptor-independent mechanisms, such as interaction with the mTOR pathway, ADK regulating agents might find new uses as therapeutics to affect cell proliferation. The use of ADK inhibitors to stimulate β-cell proliferation is one promising step in this direction.
VII. Implications for Human Pathogens
Prokaryotic and eukaryotic microorganisms usually have distinct adaptations in their nucleoside and nucleotide metabolism that differ from their hosts. Those differences can be exploited to develop antiparasitic drugs, which are specific for the parasites but do not affect the host. In particular, purine salvage pathways play important roles for parasites, which frequently lack pathways for the de novo synthesis of purines. Parasitic ADK therefore plays important roles in adenosine salvage, a feature that can be exploited therapeutically. In the following sections parasitic ADK and therapeutic avenues will briefly be discussed.
A. Mycobacterium tuberculosis
Mycobacterium tuberculosis is a pathogenic bacterial species and the causative agent of most cases of tuberculosis. M. tuberculosis Adk was the first bacterial ADK to be cloned and characterized and shown to exhibit a >12-fold enhanced affinity for the antimycobacterial prodrug 2-methyladenosine compared with human ADK (Long et al., 2003). Recent crystallographic data from recombinant M. tuberculosis ADK and structural differences to human ADK are expected to yield information guiding the design of more potent and selective antimycobacterial agents (Wang et al., 2005; Reddy et al., 2007). Identification of structure-activity relationships allowed the identification of highly selective substrates (i.e., prodrugs of toxic metabolites), such as 2-aza-adenosine, 8-aza-9-deaza-adenosine, 2-fluoro-adenosine, carbocyclic-adenosine, 8-aza-carbocyclic-adenosine, or 9-[α-l-lyxofuranosyl]-adenine and potent inhibitors of purine salvage, such as N1-benzyl-adenosine, 2-fluoro-adenosine, 6-cyclopentyloxy-purine riboside, 7-iodo-7-deaza-adenosine, or 5′-amino-5′-deoxy-adenosine (Long and Parker, 2006; Long et al., 2008). In particular, a new class of halogenated 3-deaza-adenosine analogs was shown to be 10-fold better substrates for M. tuberculosis ADK compared with human ADK, a finding that may lead to a new class of antitubercular agents (Long et al., 2007).
B. Trypanosoma brucei
Trypanosoma brucei is an extracellular eukaryotic parasite that causes sleeping sickness. It lacks de novo purine synthesis and therefore depends on adenosine taken up from the host’s blood by high affinity transporters (Vodnala et al., 2008). Although T. brucei ADK is not essential for the survival of the parasite (Luscher et al., 2007), the combination of the high affinity of T. brucei ADK for adenosine and the existence of efficient adenosine transporters result in a strong purine salvage system in T. brucei, which, potentially, should render the parasite more sensitive than mammalian cells to antimetabolites such as AraA that need to be phosphorylated by ADK to be transformed into their cytotoxic form. Indeed, it was recently shown that AraA inhibited parasite proliferation in an ADK-dependent manner by affecting nucleotide levels and inhibition of nucleic acid synthesis (Vodnala et al., 2008).
C. Leishmania donovanii
Leishmania donovanii is a purine-auxotrophic parasitic protozoan that causes visceral leishmaniasis, also known as kala-azar. The stage-specific differential activity pattern of the parasitic ADK made the enzyme an attractive target for chemotherapeutic intervention. L. donovanii ADK can be inhibited by very low concentrations of the adenosine analogs tubercidin and 6-methylmercaptopurine riboside, whereas ADK-deficient promastigotes were shown to survive and grow in the presence of 20 µM tubercidin (Iovannisci and Ullman, 1984; Datta et al., 1987). The biochemistry of L. donovanii ADK has been studied intensively, and regulatory mechanisms governing the activity of L. donovanii ADK might be instructive to understand ADK regulation in more complex mammalian systems, however, distinct differences also exist. Similar to mammalian ADK, L. donovanii has a kinetic mechanism of a sequential Bi-Bi reaction, with AMP and ADP acting as enzyme regulators in vivo (Bhaumik and Datta, 1988); however, immunologically, the enzyme was found to be distinct from mammalian ADK (Bhaumik and Datta, 1989). In contrast to mammalian ADK, the protozoan enzyme binds adenosine exclusively through the catalytic site and is therefore not inhibited by its own substrate at high adenosine concentrations (Bhaumik and Datta, 1992). L. donovanii ADK enzyme is an aggregation-prone protein and its activity is regulated by an aggregation-disaggregation cycle, in which a L. donovanii cyclophilin disaggregates the aggregated form and thus stabilizes the active form of the enzyme (Chakraborty et al., 2002), whereas the aggregated inactive form was found to be stabilized by ADP (Sen et al., 2006). Stress-induced translocation of the cyclophilin from the lumen of the endoplasmic reticulum to the cytosol was found to be implicated in the regulation of ADK activity (Sen et al., 2007). To date, possible protein interaction partners have not been identified for mammalian ADKs; however, regulation of ADK by an aggregation-disaggregation cycle is an intriguing mechanism allowing ADK to respond rapidly and reversibly to changes in environmental conditions.
D. T. gondii
T. gondii is a purine auxotroph intracellular parasitic protozoan that causes toxoplasmosis in humans. ADK-mediated phosphorylation of salvaged adenosine provides the major route of purine acquisition of the parasite. Consequently, T. gondii ADK represents a promising target for the rational design of antiparasitic compounds. Enzyme inhibition was observed with the purine nucleoside analogs AraA, 4-nitrobenzylthioinosine, N6-(p-methoxybenzoyl)adenosine, tubercidin, and iodotubercidin; ADK-deficient mutants of T. gondii were resistant to these drugs (Iltzsch et al., 1995; Darling et al., 1999; Sullivan et al., 1999). Recombinant expression and crystallographic analysis of T. gondii ADK revealed structural differences compared with human ADK, resulting in a major change in the orientation of the two domains and changes in substrate binding properties; these structural differences might form the basis for the rational design of ADK inhibitors that are selective for T. gondii ADK (Cook et al., 2000; Recacha et al., 2000; Schumacher et al., 2000; Zhang et al., 2006, 2007). 6-Benzylthioinosine was identified as a prototype subversive substrate for T. gondii ADK and used as a lead to develop a new class of compounds shown to be selectively toxic to the parasites but not their host (Yadav et al., 2004; Rais et al., 2005). In particular, due to their increased internal flexibility, the 7-deaza-6-benzylthioinosine analogs were shown to exhibit improved binding to the hydrophobic pocket of the T. gondii enzyme (Kim et al., 2008). Consequently, those agents showed a selective antitoxoplasmic effect in wild-type parasites, whereas ADK-deficient mutants were resistant to the drug (Al Safarjalani et al., 2008, 2010). A different approach was taken in identifying short inhibitory RNAs or double-stranded RNAs targeting the expression of T. gondii Adk gene, an approach that might eventually lead to the development of novel therapeutics or vaccines (Yu et al., 2008, 2009).
E. Cryptosporidium parvum
Cryptosporidium parvum is one of several protozoan species that cause cryptosporidiosis, a parasitic disease of the mammalian intestinal tract. C. parvum is a purine auxotroph and the sole route for purine salvage by the parasite is ADK. Overexpression and purification of recombinant C. parvum has recently been achieved, and initial data suggest that 4-nitro-6-benzylthioinosine, a compound with therapeutic promise against the related parasite T. gondii, also inhibits C. parvum ADK (Galazka et al., 2006).
F. Anopheles gambia and Plasmodium falciparum
Anopheles gambia is the most common vector for the transmission of Plasmodium falciparum in Africa, a purine auxotroph parasite that causes malaria. P. falciparum does not have ADK by itself and therefore requires access to host- or vector-derived purines to survive. A. gambia ADK was recently cloned, expressed, and characterized; remarkably, A. gambia ADK has the highest affinity for adenosine (Km = 8.1 nM) of any known ADKs. The ability to salvage adenosine by ADK separates the insect host from the parasite and provides a rationale for metabolic and inhibitor design studies to investigate targets in host-parasite interactions (Cassera et al., 2011).
VIII. Conclusions and Outlook
Adenosine is a ubiquitous energy metabolite that fulfills many beneficial functions in most organ systems. Early attempts to harness the therapeutic potential of adenosine were based on the development of adenosine receptor-specific ligands. However, because of the widespread distribution of the receptors, systemic side effects precluded the clinical development of many of the most potent drugs. A breakthrough came with the realization that ADK inhibitors could potentiate the beneficial effects of endogenous adenosine in a site- and event-specific manner and thereby prevent widespread side effects of systemic adenosine receptor drugs. Promising preclinical data in the areas of inflammation, pain, and epilepsy led to intense drug development efforts, mostly between 1995 and 2005. According to a press release from 1996, GP-3269 is the only ADK inhibitor studied in phase I studies in humans, but data have not been disclosed. Subsequently, limitations of ADK inhibitors became evident, and drug development efforts have largely been stalled. Limitations included liver toxicity, and, according to a preliminary report, the development of GP-3966 was halted due to CNS hemorrhage in rats and dogs. The advent of new molecular tools and the report of unexpected basic research findings during that past 10 years have resulted in a recent surge of interest in ADK. An almost-abandoned old target has been completely reinvented and is likely to lead to unprecedented new therapeutic opportunities in many different areas (Table 1). Of note are:
TABLE 1.
Therapeutic opportunities of ADK manipulation: summary of major findings and new trends
| Goal | Method | Outcome |
|---|---|---|
| Adenosine augmentation | Systemic ADK inhibitors | Seizure suppression |
| Antinociception | ||
| Anti-inflammatory action | ||
| Neuroprotection | ||
| Attenuation of age-related hearing loss | ||
| Antipsychotic-like activity | ||
| Adenosine augmentation | Ketogenic diet | Seizure suppression |
| Adenosine augmentation | Adk−/− stem cell transplantation | Neuroprotection |
| Seizure suppression | ||
| Prevention of epilepsy | ||
| Adenosine augmentation | Anti-ADK gene therapy | Neuroprotection |
| Seizure suppression | ||
| Cell proliferation | ADK inhibitors | Stimulation of β-cell proliferation |
| Parasitology | Prodrug activation via ADK | Parasite toxicity |
Overexpression of ADK has been identified in several pathologies, in which resulting adenosine deficiency plays a key role in pathophysiological mechanisms. The identification of pathologic ADK overexpression provides a strong scientific rationale to target ADK therapeutically.
New developments suggest that ADK might be exploitable therapeutically by antisense gene therapy, which might be a unique strategy to target ADK in a region- and cell type-specific manner.
Novel roles of ADK, in particular of the nuclear isoform of ADK, have been identified, which could be exploited to modulate cell proliferation therapeutically with ADK regulating agents.
A possible role of ADK in regulating epigenetic functions of the cell is likely to lead to novel applications of ADK-regulating agents as "epigenetic medicines."
Exciting developments in the area of bacteriology and parasitology suggest that ADK in purine auxotroph organisms differs significantly enough from human ADK to allow therapeutic exploitation of those differences to develop new classes of antiparasitic drugs.
Perhaps the most exciting future therapeutic perspective is the availability of ADK modulating agents that can be used to regulate the availability of the homeostatic bioenergetic network regulator adenosine. In targeting ADK, the unique opportunity exists to modulate an entire network not only based on activation of multiple adenosine receptor-dependent pathways but also based on adenosine receptor-independent biochemical, bioenergetic, and epigenetic mechanisms. This multimodal approach is a novel therapeutic concept that differs from conventional pathway-centric drug development efforts. The realization that adenosine homeostasis—and consequently network function—is disrupted in many pathologic conditions offers the unique therapeutic opportunity to use ADK-regulating agents to reconstruct or restore network homeostasis in disease. Those novel concepts may indeed redefine ADK as a therapeutic target and may offer new hopes to finding cures for intractable diseases.
Acknowledgments
The author thanks Sue Crawford for proofreading the manuscript.
Abbreviations
- A-134974
N7-[(1′R,2′S,3′R,4′S)-2′,3′-dihydroxy-4′-aminocyclopentyl]-4-amino-5-iodopyrrolopyrimidine
- A-286501
N7-((1′R,2′S,3′R,4′S)-2′,3′-dihydroxy-4′-amino-cyclopentyl)-4-amino-5-bromo-pyrrolo[2,3-a]pyrimidine
- AAV
adeno-associated virus
- ABT-702
4-amino-5-(3-bromophenyl)-7-(6-morpholino-pyridin-3-yl)pyrido[2,3-d]pyrimidine
- ADA
adenosine deaminase
- ADK
adenosine kinase
- ADK-L
long isoform of ADK
- ADK-S
short isoform of ADK
- A1R
adenosine A1 receptor
- A2AR
adenosine A2A receptor
- A2BR
adenosine A2B receptor
- A3R
adenosine A3 receptor
- AraA
9-β-d-ribofuranosyladenine
- bp
base pair
- BHK
baby hamster kidney
- CNS
central nervous system
- ERK
extracellular signal-regulated kinase
- ES
embryonic stem
- GP-515
4-amino-1-(5-amino-5-deoxy-1-β-d-ribofuranosyl)-3-bromo-pyrazol[3,4-d] pyrimidine
- GP-3269
7-(5-deoxy-β-D-ribofuranosyl)-N-(4-fluorophenyl)-5-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine
- 5-ITU
5-iodotubercidin
- GP-3966
4-N-(4-fluorophenyl)amino-5-phenyl-7-(β-D-erythrofuranosyl) pyrrolo[2,3-d]pyrimidine
- HIF
hypoxia inducible factor
- IBD
inflammatory bowel disease
- 5-ITU
5-iodotubercidin
- kb
kilobase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MCAO
middle cerebral artery occlusion
- MES
maximal electroshock
- MK-801
dizocilpine
- mTOR
mammalian target of rapamycin
- PD98059
2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one
- PK
protein kinase
- PLC
phospholipase C
- PPI
prepulse inhibition
- REM
rapid eye movement
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- SIDS
sudden infant death syndrome
- SUDEP
sudden unexpected death in epilepsy
- TBI
traumatic brain injury
- TNF-α
tumor necrosis factor α
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Boison.
Footnotes
This work was funded by the National Institutes of Health National Institute of Neurologic Disorders and Stroke [Grants NS061844, NS065957]; the National Institutes of Health National Institute of Mental Health [Grant MH083973]; the US Department of the Army (USAMRMC–PRMRP) [Contract W81XWH-12-1-0283]; the Parkinson’s Northwest Group; and the Legacy Hospital Foundations.
References
- Aarsland D, Kurz MW. (2010) The epidemiology of dementia associated with Parkinson’s disease. Brain Pathol 20:633–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbracchio MP, Brambilla R, Ceruti S, Kim HO, von Lubitz DK, Jacobson KA, Cattabeni F. (1995) G protein-dependent activation of phospholipase C by adenosine A3 receptors in rat brain. Mol Pharmacol 48:1038–1045 [PubMed] [Google Scholar]
- Akkari R, Burbiel JC, Hockemeyer J, Müller CE. (2006) Recent progress in the development of adenosine receptor ligands as antiinflammatory drugs. Curr Top Med Chem 6:1375–1399 [DOI] [PubMed] [Google Scholar]
- Al Safarjalani ON, Rais RH, Kim YA, Chu CK, Naguib FN, el Kouni MH. (2008) 7-Deaza-6-benzylthioinosine analogues as subversive substrate of Toxoplasma gondii adenosine kinase: activities and selective toxicities. Biochem Pharmacol 76:958–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Safarjalani ON, Rais RH, Kim YA, Chu CK, Naguib FN, El Kouni MH. (2010) Carbocyclic 6-benzylthioinosine analogues as subversive substrates of Toxoplasma gondii adenosine kinase: biological activities and selective toxicities. Biochem Pharmacol 80:955–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ala TA, Beh GO, Frey WH., 2nd (2000) Pure hippocampal sclerosis: a rare cause of dementia mimicking Alzheimer’s disease. Neurology 54:843–848 [DOI] [PubMed] [Google Scholar]
- Alanko L, Heiskanen S, Stenberg D, Porkka-Heiskanen T. (2003) Adenosine kinase and 5′-nucleotidase activity after prolonged wakefulness in the cortex and the basal forebrain of rat. Neurochem Int 42:449–454 [DOI] [PubMed] [Google Scholar]
- Andres CM, Fox IH. (1979) Purification and properties of human placental adenosine kinase. J Biol Chem 254:11388–11393 [PubMed] [Google Scholar]
- Annes JP, Ryu JH, Lam K, Carolan PJ, Utz K, Hollister-Lock J, Arvanites AC, Rubin LL, Weir G, Melton DA. (2012) Adenosine kinase inhibition selectively promotes rodent and porcine islet β-cell replication. Proc Natl Acad Sci USA 109:3915–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Yamada E, Endoh T, Suzuki T. (2004) Multiple actions of extracellular ATP and adenosine on calcium currents mediated by various purinoceptors in neurons of nucleus tractus solitarius. Neurosci Res 50:245–255 [DOI] [PubMed] [Google Scholar]
- Arch JR, Newsholme EA. (1978) Activities and some properties of 5′-nucleotidase, adenosine kinase and adenosine deaminase in tissues from vertebrates and invertebrates in relation to the control of the concentration and the physiological role of adenosine. Biochem J 174:965–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Ravizza T, Zurolo E, Vezzani A. (2012) Astrocyte immune responses in epilepsy. Glia 60:1258–1268 [DOI] [PubMed] [Google Scholar]
- Aronica E, Zurolo E, Iyer A, de Groot M, Anink J, Carbonell C, van Vliet EA, Baayen JC, Boison D, Gorter JA. (2011) Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia 52:1645–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni E, Rosenberg PA. (2006) Nitric oxide-induced adenosine inhibition of hippocampal synaptic transmission depends on adenosine kinase inhibition and is cyclic GMP independent. Eur J Neurosci 24:2471–2480 [DOI] [PubMed] [Google Scholar]
- Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD. (2004) The equilibrative nucleoside transporter family, SLC29. Pflugers Arch 447:735–743 [DOI] [PubMed] [Google Scholar]
- Balzarini J, De Clercq E. (1990) 9-Beta-D-arabinofuranosyladenine 5′-monophosphate (araAMP) is converted directly to its antivirally active 5′-triphosphate form by 5-phosphoribosyl-1-pyrophosphate (PRPP) synthetase. Biochem Biophys Res Commun 173:781–787 [DOI] [PubMed] [Google Scholar]
- Barankiewicz J, Danks AM, Abushanab E, Makings L, Wiemann T, Wallis RA, Pragnacharyulu PVP, Fox A, Marangos PJ. (1997) Regulation of adenosine concentration and cytoprotective effects of novel reversible adenosine deaminase inhibitors. J Pharmacol Exp Ther 283:1230–1238 [PubMed] [Google Scholar]
- Bauser M, Delapierre G, Hauswald M, Flessner T, D’Urso D, Hermann A, Beyreuther B, De Vry J, Spreyer P, Reissmüller E, et al. (2004) Discovery and optimization of 2-aryl oxazolo-pyrimidines as adenosine kinase inhibitors using liquid phase parallel synthesis. Bioorg Med Chem Lett 14:1997–2000 [DOI] [PubMed] [Google Scholar]
- Belardinelli L, Shryock JC, Song Y, Wang D, Srinivas M. (1995) Ionic basis of the electrophysiological actions of adenosine on cardiomyocytes. FASEB J 9:359–365 [DOI] [PubMed] [Google Scholar]
- Bell B, Lin JJ, Seidenberg M, Hermann B. (2011) The neurobiology of cognitive disorders in temporal lobe epilepsy. Nat Rev Neurol 7:154–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berne RM. (1963) Cardiac nucleotides in hypoxia: possible role in regulation of coronary blood flow. Am J Physiol 204:317–322 [DOI] [PubMed] [Google Scholar]
- Berne RM, Rubio R, Curnish RR. (1974) Release of adenosine from ischaemic brain: effect on cerebral vascular resistance and incorporation into cerebral adenine nucleotides. Circ Res 35:262–271 [Google Scholar]
- Bhaumik D, Datta AK. (1988) Reaction kinetics and inhibition of adenosine kinase from Leishmania donovani. Mol Biochem Parasitol 28:181–187 [DOI] [PubMed] [Google Scholar]
- Bhaumik D, Datta AK. (1989) Immunochemical and catalytic characteristics of adenosine kinase from Leishmania donovani. J Biol Chem 264:4356–4361 [PubMed] [Google Scholar]
- Bhaumik D, Datta AK. (1992) Active site thiol(s) in Leishmania donovani adenosine kinase: comparison with hamster enzyme and evidence for the absence of regulatory adenosine binding site. Mol Biochem Parasitol 52:29–38 [DOI] [PubMed] [Google Scholar]
- Bhutoria S, Ghoshal N. (2010) Deciphering ligand dependent degree of binding site closure and its implication in inhibitor design: A modeling study on human adenosine kinase. J Mol Graph Model 28:577–591 [DOI] [PubMed] [Google Scholar]
- Bjorness TE, Greene RW. (2009) Adenosine and sleep. Curr Neuropharmacol 7:238–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjursell MK, Blom HJ, Cayuela JA, Engvall ML, Lesko N, Balasubramaniam S, Brandberg G, Halldin M, Falkenberg M, Jakobs C, et al. (2011) Adenosine kinase deficiency disrupts the methionine cycle and causes hypermethioninemia, encephalopathy, and abnormal liver function. Am J Hum Genet 89:507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blümcke I, Zuschratter W, Schewe JC, Suter B, Lie AA, Riederer BM, Meyer B, Schramm J, Elger CE, Wiestler OD. (1999) Cellular pathology of hilar neurons in Ammon’s horn sclerosis. J Comp Neurol 414:437–453 [DOI] [PubMed] [Google Scholar]
- Boesiger P, Buchli R, Meier D, Steinmann B, Gitzelmann R. (1994) Changes of liver metabolite concentrations in adults with disorders of fructose metabolism after intravenous fructose by 31P magnetic resonance spectroscopy. Pediatr Res 36:436–440 [DOI] [PubMed] [Google Scholar]
- Boison D. (2007) Adenosine as a modulator of brain activity. Drug News Perspect 20:607–611 [DOI] [PubMed] [Google Scholar]
- Boison D. (2008) The adenosine kinase hypothesis of epileptogenesis. Prog Neurobiol 84:249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. (2009) Adenosine augmentation therapies (AATs) for epilepsy: prospect of cell and gene therapies. Epilepsy Res 85:131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. (2010) Inhibitory RNA in epilepsy: research tools and therapeutic perspectives. Epilepsia 51:1659–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. (2012a) Adenosine augmentation therapy for epilepsy, in Jasper's Basic Mechanisms of the Epilepsies (Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV. eds) pp 1150–1160, Oxford University Press, Oxford [Google Scholar]
- Boison D. (2012b) Adenosine dysfunction in epilepsy. Glia 60:1234–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Chen JF, Fredholm BB. (2010) Adenosine signaling and function in glial cells. Cell Death Differ 17:1071–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Huber A, Padrun V, Déglon N, Aebischer P, Möhler H. (2002a) Seizure suppression by adenosine-releasing cells is independent of seizure frequency. Epilepsia 43:788–796 [DOI] [PubMed] [Google Scholar]
- Boison D, Scheurer L, Zumsteg V, Rülicke T, Litynski P, Fowler B, Brandner S, Mohler H. (2002b) Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci USA 99:6985–6990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Singer P, Shen HY, Feldon J, Yee BK. (2012) Adenosine hypothesis of schizophrenia—opportunities for pharmacotherapy. Neuropharmacology 62:1527–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles RG, Buck EA, Blitzer MG, Platt MS, Cowan TM, Martin SK, Yoon H, Madsen JA, Reyes-Mugica M, Rinaldo P. (1998) Retrospective biochemical screening of fatty acid oxidation disorders in postmortem livers of 418 cases of sudden death in the first year of life. J Pediatr 132:924–933 [DOI] [PubMed] [Google Scholar]
- Bontemps F, Mimouni M, Van den Berghe G. (1993a) Phosphorylation of adenosine in anoxic hepatocytes by an exchange reaction catalysed by adenosine kinase. Biochem J 290:679–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontemps F, Van den Berghe G, Hers HG. (1983) Evidence for a substrate cycle between AMP and adenosine in isolated hepatocytes. Proc Natl Acad Sci USA 80:2829–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontemps F, Vincent MF, Van den Berghe G. (1993b) Mechanisms of elevation of adenosine levels in anoxic hepatocytes. Biochem J 290:671–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookser BC, Matelich MC, Ollis K, Ugarkar BG. (2005a) Adenosine kinase inhibitors. 4. 6,8-Disubstituted purine nucleoside derivatives. Synthesis, conformation, and enzyme inhibition. J Med Chem 48:3389–3399 [DOI] [PubMed] [Google Scholar]
- Bookser BC, Ugarkar BG, Matelich MC, Lemus RH, Allan M, Tsuchiya M, Nakane M, Nagahisa A, Wiesner JB, Erion MD. (2005b) Adenosine kinase inhibitors. 6. Synthesis, water solubility, and antinociceptive activity of 5-phenyl-7-(5-deoxy-beta-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidines substituted at C4 with glycinamides and related compounds. J Med Chem 48:7808–7820 [DOI] [PubMed] [Google Scholar]
- Bough K. (2008) Energy metabolism as part of the anticonvulsant mechanism of the ketogenic diet. Epilepsia 49 (Suppl 8):91–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, Shaw R, Smith Y, Geiger JD, Dingledine RJ. (2006) Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol 60:223–235 [DOI] [PubMed] [Google Scholar]
- Boyer SH, Ugarkar BG, Solbach J, Kopcho J, Matelich MC, Ollis K, Gomez-Galeno JE, Mendonca R, Tsuchiya M, Nagahisa A, et al. (2005) Adenosine kinase inhibitors. 5. Synthesis, enzyme inhibition, and analgesic activity of diaryl-erythro-furanosyltubercidin analogues. J Med Chem 48:6430–6441 [DOI] [PubMed] [Google Scholar]
- Boyle DL, Kowaluk EA, Jarvis MF, Lee CH, Bhagwat SS, Williams M, Firestein GS. (2001) Anti-inflammatory effects of ABT-702, a novel non-nucleoside adenosine kinase inhibitor, in rat adjuvant arthritis. J Pharmacol Exp Ther 296:495–500 [PubMed] [Google Scholar]
- Brackett LE, Daly JW. (1994) Functional characterization of the A2b adenosine receptor in NIH 3T3 fibroblasts. Biochem Pharmacol 47:801–814 [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Light GA, Sprock J, Perry W, Cadenhead KS, Swerdlow NR. (2001a) Impact of prepulse characteristics on the detection of sensorimotor gating deficits in schizophrenia. Schizophr Res 49:171–178 [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. (2001b) Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 156:234–258 [DOI] [PubMed] [Google Scholar]
- Britton DR, Mikusa J, Lee CH, Jarvis MF, Williams M, Kowaluk EA. (1999) Site and event specific increase of striatal adenosine release by adenosine kinase inhibition in rats. Neurosci Lett 266:93–96 [DOI] [PubMed] [Google Scholar]
- Bryson Y, Connor JD, Sweetman L, Carey S, Stuckey MA, Buchanan R. (1974) Determination of plaque inhibitory activity of adenine arabinoside (9-beta-D-arabinofuranosyladenine) for herpesviruses using an adenosine deaminase inhibitor. Antimicrob Agents Chemother 6:98–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukoski RD, Sparks HV, Mela-Riker LM. (1986) A role for mitochondria in myocardial adenosine production. Adv Exp Med Biol 194:157–167 [DOI] [PubMed] [Google Scholar]
- Bukoski RD, Sparks HV, Mela LM. (1983) Rat heart mitochondria release adenosine. Biochem Biophys Res Commun 113:990–995 [DOI] [PubMed] [Google Scholar]
- Burnstock G, Fredholm BB, Verkhratsky A. (2011) Adenosine and ATP receptors in the brain. Curr Top Med Chem 11:973–1011 [DOI] [PubMed] [Google Scholar]
- Burnstock G, Verkhratsky A. (2009) Evolutionary origins of the purinergic signalling system. Acta Physiol (Oxf) 195:415–447 [DOI] [PubMed] [Google Scholar]
- Camm AJ, Garratt CJ. (1991) Adenosine and supraventricular tachycardia. N Engl J Med 325:1621–1629 [DOI] [PubMed] [Google Scholar]
- Cassera MB, Ho MC, Merino EF, Burgos ES, Rinaldo-Matthis A, Almo SC, Schramm VL. (2011) A high-affinity adenosine kinase from Anopheles gambiae. Biochemistry 50:1885–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceruti S, Franceschi C, Barbieri D, Malorni W, Camurri A, Giammarioli AM, Ambrosini A, Racagni G, Cattabeni F, Abbracchio MP. (2000) Apoptosis induced by 2-chloro-adenosine and 2-chloro-2′-deoxy-adenosine in a human astrocytoma cell line: differential mechanisms and possible clinical relevance. J Neurosci Res 60:388–400 [DOI] [PubMed] [Google Scholar]
- Chakraborty A, Das I, Datta R, Sen B, Bhattacharyya D, Mandal C, Datta AK. (2002) A single-domain cyclophilin from Leishmania donovani reactivates soluble aggregates of adenosine kinase by isomerase-independent chaperone function. J Biol Chem 277:47451–47460 [DOI] [PubMed] [Google Scholar]
- Chakraborty A, Sen B, Datta R, Datta AK. (2004) Isomerase-independent chaperone function of cyclophilin ensures aggregation prevention of adenosine kinase both in vitro and under in vivo conditions. Biochemistry 43:11862–11872 [DOI] [PubMed] [Google Scholar]
- Chan VL, Guttman S. (1985) Codominant and recessive 9-beta-D-arabinofuranosyladenine-resistant mutations of baby hamster cells. Mutat Res 149:141–146 [DOI] [PubMed] [Google Scholar]
- Chan VL, Juranka P. (1981) Isolation and preliminary characterization of 9-β-d-arabinofuranosyladenine-resistant mutants of baby hamster cells. Somatic Cell Genet 7:147–160 [DOI] [PubMed] [Google Scholar]
- Chang CH, Cha S, Brockman RW, Bennett LL., Jr (1983) Kinetic studies of adenosine kinase from L1210 cells: a model enzyme with a two-site ping-pong mechanism. Biochemistry 22:600–611 [DOI] [PubMed] [Google Scholar]
- Clark RS, Carcillo JA, Kochanek PM, Obrist WD, Jackson EK, Mi Z, Wisneiwski SR, Bell MJ, Marion DW. (1997) Cerebrospinal fluid adenosine concentration and uncoupling of cerebral blood flow and oxidative metabolism after severe head injury in humans. Neurosurgery 41:1284–1292, discussion 1292–1293 [DOI] [PubMed] [Google Scholar]
- Cook WJ, DeLucas LJ, Chattopadhyay D. (2000) Crystal structure of adenosine kinase from Toxoplasma gondii at 1.8 A resolution. Protein Sci 9:704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DM, Londos C, Rodbell M. (1980) Adenosine receptor-mediated inhibition of rat cerebral cortical adenylate cyclase by a GTP-dependent process. Mol Pharmacol 18:598–601 [PubMed] [Google Scholar]
- Cottam HB, Wasson DB, Shih HC, Raychaudhuri A, Di Pasquale G, Carson DA. (1993) New adenosine kinase inhibitors with oral antiinflammatory activity: synthesis and biological evaluation. J Med Chem 36:3424–3430 [DOI] [PubMed] [Google Scholar]
- Cowart M, Lee CH, Gfesser GA, Bayburt EK, Bhagwat SS, Stewart AO, Yu H, Kohlhaas KL, McGaraughty S, Wismer CT, et al. (2001) Structure-activity studies of 5-substituted pyridopyrimidines as adenosine kinase inhibitors. Bioorg Med Chem Lett 11:83–86 [DOI] [PubMed] [Google Scholar]
- Cronstein B. (2010) How does methotrexate suppress inflammation? Clin Exp Rheumatol 28(5, Suppl 61)S21–S23 [PubMed] [Google Scholar]
- Cronstein BN, Naime D, Firestein G. (1995) The antiinflammatory effects of an adenosine kinase inhibitor are mediated by adenosine. Arthritis Rheum 38:1040–1045 [DOI] [PubMed] [Google Scholar]
- Cui XA, Agarwal T, Singh B, Gupta RS. (2011) Molecular characterization of Chinese hamster cells mutants affected in adenosine kinase and showing novel genetic and biochemical characteristics. BMC Biochem 12:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui XA, Singh B, Park J, Gupta RS. (2009) Subcellular localization of adenosine kinase in mammalian cells: The long isoform of AdK is localized in the nucleus. Biochem Biophys Res Commun 388:46–50 [DOI] [PubMed] [Google Scholar]
- Cunha RA. (2008) Different cellular sources and different roles of adenosine: A1 receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A2A receptor-mediated facilitation of plasticity. Neurochem Int 52:65–72 [DOI] [PubMed] [Google Scholar]
- Daly JW, Butts-Lamb P, Padgett W. (1983) Subclasses of adenosine receptors in the central nervous system: interaction with caffeine and related methylxanthines. Cell Mol Neurobiol 3:69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling JA, Sullivan WJ, Jr, Carter D, Ullman B, Roos DS. (1999) Recombinant expression, purification, and characterization of Toxoplasma gondii adenosine kinase. Mol Biochem Parasitol 103:15–23 [DOI] [PubMed] [Google Scholar]
- Datta AK, Bhaumik D, Chatterjee R. (1987) Isolation and characterization of adenosine kinase from Leishmania donovani. J Biol Chem 262:5515–5521 [PubMed] [Google Scholar]
- de Groot M, Iyer A, Zurolo E, Anink J, Heimans JJ, Boison D, Reijneveld JC, Aronica E. (2012) Overexpression of ADK in human astrocytic tumors and peritumoral tissue is related to tumor-associated epilepsy. Epilepsia 53:58–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decking UK, Schlieper G, Kroll K, Schrader J. (1997) Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res 81:154–164 [DOI] [PubMed] [Google Scholar]
- Delaney SM, Geiger JD. (1996) Brain regional levels of adenosine and adenosine nucleotides in rats killed by high-energy focused microwave irradiation. J Neurosci Methods 64:151–156 [DOI] [PubMed] [Google Scholar]
- Deussen A, Lloyd HG, Schrader J. (1989) Contribution of S-adenosylhomocysteine to cardiac adenosine formation. J Mol Cell Cardiol 21:773–782 [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Becker K, Meisel A. (2009) Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol 8:398–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diógenes MJ, Neves-Tomé R, Fucile S, Martinello K, Scianni M, Theofilas P, Lopatár J, Ribeiro JA, Maggi L, Frenguelli BG, et al. (2012) Homeostatic control of synaptic activity by endogenous adenosine is mediated by adenosine kinase. Cereb Cortex. DOI:10.1093/cercor/bhs284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoso MV, López R, Miranda R, Briones R, Huidobro-Toro JP. (2005) A2B adenosine receptor mediates human chorionic vasoconstriction and signals through arachidonic acid cascade. Am J Physiol Heart Circ Physiol 288:H2439–H2449 [DOI] [PubMed] [Google Scholar]
- Drake MT, Zhu Y, Kornfeld S. (2000) The assembly of AP-3 adaptor complex-containing clathrin-coated vesicles on synthetic liposomes. Mol Biol Cell 11:3723–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte JM, Carvalho RA, Cunha RA, Gruetter R. (2009) Caffeine consumption attenuates neurochemical modifications in the hippocampus of streptozotocin-induced diabetic rats. J Neurochem 111:368–379 [DOI] [PubMed] [Google Scholar]
- Duarte JM, Oliveira CR, Ambrósio AF, Cunha RA. (2006) Modification of adenosine A1 and A2A receptor density in the hippocampus of streptozotocin-induced diabetic rats. Neurochem Int 48:144–150 [DOI] [PubMed] [Google Scholar]
- Duarte JM, Oses JP, Rodrigues RJ, Cunha RA. (2007) Modification of purinergic signaling in the hippocampus of streptozotocin-induced diabetic rats. Neuroscience 149:382–391 [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV. (1980) Endogenously released adenosine regulates excitability in the in vitro hippocampus. Epilepsia 21:541–548 [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Hoffer BJ, Fredholm BB. (1981) Alkylxanthines elevate hippocampal excitability. Evidence for a role of endogenous adenosine. Naunyn Schmiedebergs Arch Pharmacol 316:326–330 [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24:31–55 [DOI] [PubMed] [Google Scholar]
- Eigler A, Matschke V, Hartmann G, Erhardt S, Boyle D, Firestein GS, Endres S. (2000) Suppression of TNF-alpha production in human mononuclear cells by an adenosine kinase inhibitor. J Leukoc Biol 68:97–103 [PubMed] [Google Scholar]
- Eltzschig HK, Rivera-Nieves J, Colgan SP. (2009) Targeting the A2B adenosine receptor during gastrointestinal ischemia and inflammation. Expert Opin Ther Targets 13:1267–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englert M, Quitterer U, Klotz KN. (2002) Effector coupling of stably transfected human A3 adenosine receptors in CHO cells. Biochem Pharmacol 64:61–65 [DOI] [PubMed] [Google Scholar]
- Erion MD, Ugarkar BG, Dare J, Castellino AJ, Fujitaki JM, Dixon R, Appleman JR, Wiesner JB. (1997) Design, synthesis and anticonvulsant activity of the potent adenosine kinase inhibitor Gp3269. Nucleosides Nucleotides 16:1013–1021 [Google Scholar]
- Ernst PB, Garrison JC, Thompson LF. (2010) Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J Immunol 185:1993–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherington LA, Patterson GE, Meechan L, Boison D, Irving AJ, Dale N, Frenguelli BG. (2009) Astrocytic adenosine kinase regulates basal synaptic adenosine levels and seizure activity but not activity-dependent adenosine release in the hippocampus. Neuropharmacology 56:429–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassett JT, Hu X, Xu X, Lu Z, Zhang P, Chen Y, Bache RJ. (2011) Adenosine kinase regulation of cardiomyocyte hypertrophy. Am J Physiol Heart Circ Physiol 300:H1722–H1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele DE, Gouder N, Güttinger M, Gabernet L, Scheurer L, Rülicke T, Crestani F, Boison D. (2005) Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain 128:2383–2395 [DOI] [PubMed] [Google Scholar]
- Fedele DE, Koch P, Scheurer L, Simpson EM, Möhler H, Brüstle O, Boison D. (2004) Engineering embryonic stem cell derived glia for adenosine delivery. Neurosci Lett 370:160–165 [DOI] [PubMed] [Google Scholar]
- Feoktistov I, Biaggioni I. (1995) Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J Clin Invest 96:1979–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistov I, Biaggioni I. (1997) Adenosine A2B receptors. Pharmacol Rev 49:381–402 [PubMed] [Google Scholar]
- Finkelstein JD. (1998) The metabolism of homocysteine: pathways and regulation. Eur J Pediatr 157 (Suppl 2):S40–S44 [DOI] [PubMed] [Google Scholar]
- Finkelstein JD, Martin JJ. (1986) Methionine metabolism in mammals. Adaptation to methionine excess. J Biol Chem 261:1582–1587 [PubMed] [Google Scholar]
- Firestein GS, Boyle D, Bullough DA, Gruber HE, Sajjadi FG, Montag A, Sambol B, Mullane KM. (1994) Protective effect of an adenosine kinase inhibitor in septic shock. J Immunol 152:5853–5859 [PubMed] [Google Scholar]
- Fisher MN, Newsholme EA. (1984) Properties of rat heart adenosine kinase. Biochem J 221:521–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman P, Madi L, Bar-Yehuda S, Barer F, Del Valle L, Khalili K. (2002) Evidence for involvement of Wnt signaling pathway in IB-MECA mediated suppression of melanoma cells. Oncogene 21:4060–4064 [DOI] [PubMed] [Google Scholar]
- Flogel U, Burghoff S, van Lent PL, Temme S, Galbarz L, Ding Z, El-Tayeb A, Huels S, Bonner F, Borg N, Jacoby C, Muller CE, van den Berg WB, and Schrader J (2012) Selective activation of adenosine A2A receptors on immune cells by a CD73-dependent prodrug suppresses joint inflammation in experimental rheumatoid arthritis. Sci Transl Med 4:146ra108. [DOI] [PubMed]
- Forrest CM, Harman G, McMillan RB, Stoy N, Stone TW, Darlington LG. (2005) Modulation of cytokine release by purine receptors in patients with rheumatoid arthritis. Clin Exp Rheumatol 23:89–92 [PubMed] [Google Scholar]
- Fossetta J, Jackson J, Deno G, Fan X, Du XK, Bober L, Soudé-Bermejo A, de Bouteiller O, Caux C, Lunn C, et al. (2003) Pharmacological analysis of calcium responses mediated by the human A3 adenosine receptor in monocyte-derived dendritic cells and recombinant cells. Mol Pharmacol 63:342–350 [DOI] [PubMed] [Google Scholar]
- Fountain SJ, Burnstock G. (2009) An evolutionary history of P2X receptors. Purinergic Signal 5:269–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Lluis C, Canela EI, Mallol J, Agnati L, Casadó V, Ciruela F, Ferré S, Fuxe K. (2007) Receptor-receptor interactions involving adenosine A1 or dopamine D1 receptors and accessory proteins. J Neural Transm 114:93–104 [DOI] [PubMed] [Google Scholar]
- Fredholm BB. (1984) Effects of methylxanthines on skeletal muscle and on respiration. Prog Clin Biol Res 158:365–375 [PubMed] [Google Scholar]
- Fredholm BB. (2007) Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ 14:1315–1323 [DOI] [PubMed] [Google Scholar]
- Fredholm BB. (2010) Adenosine receptors as drug targets. Exp Cell Res 316:1284–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W. (2000) Structure and function of adenosine receptors and their genes. Naunyn Schmiedebergs Arch Pharmacol 362:364–374 [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. (2005a) Adenosine and brain function. Int Rev Neurobiol 63:191–270 [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Masino SA, Vaugeois JM. (2005b) Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu Rev Pharmacol Toxicol 45:385–412 [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Chern Y, Franco R, Sitkovsky M. (2007) Aspects of the general biology of adenosine A2A signaling. Prog Neurobiol 83:263–276 [DOI] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. (2001a) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53:527–552 [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. (2011a) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev 63:1–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Irenius E, Kull B, Schulte G. (2001b) Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem Pharmacol 61:443–448 [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Johansson S, Wang YQ. (2011b) Adenosine and the regulation of metabolism and body temperature. Adv Pharmacol 61:77–94 [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Sollevi A. (1977) Antilipolytic effect of adenosine in dog adipose tissue in situ. Acta Physiol Scand 99:254–256 [DOI] [PubMed] [Google Scholar]
- Freeman JM. (2009) Seizures, EEG events, and the ketogenic diet. Epilepsia 50:329–330 [DOI] [PubMed] [Google Scholar]
- Frenguelli BG, Llaudet E, Dale N. (2003) High-resolution real-time recording with microelectrode biosensors reveals novel aspects of adenosine release during hypoxia in rat hippocampal slices. J Neurochem 86:1506–1515 [DOI] [PubMed] [Google Scholar]
- Fresco P, Diniz C, Gonçalves J. (2004) Facilitation of noradrenaline release by activation of adenosine A(2A) receptors triggers both phospholipase C and adenylate cyclase pathways in rat tail artery. Cardiovasc Res 63:739–746 [DOI] [PubMed] [Google Scholar]
- Fuxe K, Ferré S, Genedani S, Franco R, Agnati LF. (2007) Adenosine receptor-dopamine receptor interactions in the basal ganglia and their relevance for brain function. Physiol Behav 92:210–217 [DOI] [PubMed] [Google Scholar]
- Fuxe K, Marcellino D, Leo G, Agnati LF. (2010) Molecular integration via allosteric interactions in receptor heteromers. A working hypothesis. Curr Opin Pharmacol 10:14–22 [DOI] [PubMed] [Google Scholar]
- Galazka J, Striepen B, Ullman B. (2006) Adenosine kinase from Cryptosporidium parvum. Mol Biochem Parasitol 149:223–230 [DOI] [PubMed] [Google Scholar]
- Gfesser GA, Bayburt EK, Cowart M, DiDomenico S, Gomtsyan A, Lee CH, Stewart AO, Jarvis MF, Kowaluk EA, Bhagwat SS. (2003) Synthesis and structure-activity relationships of 5-heteroatom-substituted pyridopyrimidines as adenosine kinase inhibitors. Eur J Med Chem 38:245–252 [DOI] [PubMed] [Google Scholar]
- Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N. (2010) Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci 11:87–99 [DOI] [PubMed] [Google Scholar]
- Giglioni S, Leoncini R, Aceto E, Chessa A, Civitelli S, Bernini A, Tanzini G, Carraro F, Pucci A, Vannoni D. (2008) Adenosine kinase gene expression in human colorectal cancer. Nucleosides Nucleotides Nucleic Acids 27:750–754 [DOI] [PubMed] [Google Scholar]
- Gomes CV, Kaster MP, Tomé AR, Agostinho PM, Cunha RA. (2011) Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim Biophys Acta 1808:1380–1399 [DOI] [PubMed] [Google Scholar]
- Gomtsyan A, Didomenico S, Lee CH, Matulenko MA, Kim K, Kowaluk EA, Wismer CT, Mikusa J, Yu H, Kohlhaas K, et al. (2002) Design, synthesis, and structure-activity relationship of 6-alkynylpyrimidines as potent adenosine kinase inhibitors. J Med Chem 45:3639–3648 [DOI] [PubMed] [Google Scholar]
- Gomtsyan A, Didomenico S, Lee CH, Stewart AO, Bhagwat SS, Kowaluk EA, Jarvis MF. (2004) Synthesis and biological evaluation of pteridine and pyrazolopyrimidine based adenosine kinase inhibitors. Bioorg Med Chem Lett 14:4165–4168 [DOI] [PubMed] [Google Scholar]
- Gomtsyan A, Lee CH. (2004) Nonnucleoside inhibitors of adenosine kinase. Curr Pharm Des 10:1093–1103 [DOI] [PubMed] [Google Scholar]
- Gordon JA. (2010) Testing the glutamate hypothesis of schizophrenia. Nat Neurosci 13:2–4 [DOI] [PubMed] [Google Scholar]
- Gouder N, Scheurer L, Fritschy J-M, Boison D. (2004) Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci 24:692–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene RW. (2011) Adenosine: front and center in linking nutrition and metabolism to neuronal activity. J Clin Invest 121:2548–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu JW, Ito BR, Sartin A, Frascogna N, Moore M, Adair TH. (2000) Inhibition of adenosine kinase induces expression of VEGF mRNA and protein in myocardial myoblasts. Am J Physiol Heart Circ Physiol 279:H2116–H2123 [DOI] [PubMed] [Google Scholar]
- Guieu R, Dussol B, Devaux C, Sampol J, Brunet P, Rochat H, Bechis G, Berland YF. (1998) Interactions between cyclosporine A and adenosine in kidney transplant recipients. Kidney Int 53:200–204 [DOI] [PubMed] [Google Scholar]
- Hajduk PJ, Gomtsyan A, Didomenico S, Cowart M, Bayburt EK, Solomon L, Severin J, Smith R, Walter K, Holzman TF, et al. (2000) Design of adenosine kinase inhibitors from the NMR-based screening of fragments. J Med Chem 43:4781–4786 [DOI] [PubMed] [Google Scholar]
- Hao W, Gupta RS. (1996) Pentavalent ions dependency of mammalian adenosine kinase. Biochem Mol Biol Int 38:889–899 [PubMed] [Google Scholar]
- Harper RM, Kinney HC, Fleming PJ, Thach BT. (2000) Sleep influences on homeostatic functions: implications for sudden infant death syndrome. Respir Physiol 119:123–132 [DOI] [PubMed] [Google Scholar]
- Hashemi M, Karami-Tehrani F, Ghavami S, Maddika S, Los M. (2005) Adenosine and deoxyadenosine induces apoptosis in oestrogen receptor-positive and -negative human breast cancer cells via the intrinsic pathway. Cell Prolif 38:269–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskó G, Csóka B, Németh ZH, Vizi ES, Pacher P. (2009) A(2B) adenosine receptors in immunity and inflammation. Trends Immunol 30:263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskó G, Linden J, Cronstein B, Pacher P. (2008) Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 7:759–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskó G, Pacher P, Vizi ES, Illes P. (2005) Adenosine receptor signaling in the brain immune system. Trends Pharmacol Sci 26:511–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins CF, Bagnara AS. (1987) Adenosine kinase from human erythrocytes: kinetic studies and characterization of adenosine binding sites. Biochemistry 26:1982–1987 [DOI] [PubMed] [Google Scholar]
- Headrick JP, Lasley RD. (2009) Adenosine receptors and reperfusion injury of the heart. Handb Exp Pharmacol 193:189–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedqvist P, Fredholm BB. (1979) Inhibitory effect of adenosine on adrenergic neuroeffector transmission in the rabbit heart. Acta Physiol Scand 105:120–122 [DOI] [PubMed] [Google Scholar]
- Henderson JF, Mikoshiba A, Chu SY, Caldwell IC. (1972) Kinetic studies of adenosine kinase from Ehrlich ascites tumor cells. J Biol Chem 247:1972–1975 [PubMed] [Google Scholar]
- Hjemdahl P, Fredholm BB. (1976) Cyclic AMP-dependent and independent inhibition of lipolysis by adenosine and decreased pH. Acta Physiol Scand 96:170–179 [DOI] [PubMed] [Google Scholar]
- Hoffman DR, Cornatzer WE, Duerre JA. (1979) Relationship between tissue levels of S-adenosylmethionine, S-adenylhomocysteine, and transmethylation reactions. Can J Biochem 57:56–65 [DOI] [PubMed] [Google Scholar]
- Horga de la Parte JF, Horga A. (2006) [Pregabalin: new therapeutic contributions of calcium channel alpha2delta protein ligands on epilepsy and neuropathic pain]. Rev Neurol 42:223–237 [PubMed] [Google Scholar]
- Huang ZL, Urade Y, Hayaishi O. (2011) The role of adenosine in the regulation of sleep. Curr Top Med Chem 11:1047–1057 [DOI] [PubMed] [Google Scholar]
- Huber A, Padrun V, Déglon N, Aebischer P, Möhler H, Boison D. (2001) Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. Proc Natl Acad Sci USA 98:7611–7616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iltzsch MH, Uber SS, Tankersley KO, el Kouni MH. (1995) Structure-activity relationship for the binding of nucleoside ligands to adenosine kinase from Toxoplasma gondii. Biochem Pharmacol 49:1501–1512 [DOI] [PubMed] [Google Scholar]
- Iovannisci DM, Ullman B. (1984) Characterization of a mutant Leishmania donovani deficient in adenosine kinase activity. Mol Biochem Parasitol 12:139–151 [DOI] [PubMed] [Google Scholar]
- Iqbal J, Burbiel JC, Müller CE. (2006) Development of off-line and on-line capillary electrophoresis methods for the screening and characterization of adenosine kinase inhibitors and substrates. Electrophoresis 27:2505–2517 [DOI] [PubMed] [Google Scholar]
- Iwasato T, Nomura R, Ando R, Ikeda T, Tanaka M, Itohara S. (2004) Dorsal telencephalon-specific expression of Cre recombinase in PAC transgenic mice. Genesis 38:130–138 [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Gao ZG. (2006) Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 5:247–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SJ, Melnyk S, Pogribna M, Pogribny IP, Caudill MA. (2002) Elevation in S-adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine-related pathology. J Nutr 132(8, Suppl)2361S–2366S [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Mikusa J, Chu KL, Wismer CT, Honore P, Kowaluk EA, McGaraughty S. (2002a) Comparison of the ability of adenosine kinase inhibitors and adenosine receptor agonists to attenuate thermal hyperalgesia and reduce motor performance in rats. Pharmacol Biochem Behav 73:573–581 [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Yu H, Kohlhaas K, Alexander K, Lee CH, Jiang M, Bhagwat SS, Williams M, Kowaluk EA. (2000) ABT-702 (4-amino-5-(3-bromophenyl)-7-(6-morpholinopyridin-3-yl)pyrido[2, 3-d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti-inflammatory properties: I. In vitro characterization and acute antinociceptive effects in the mouse. J Pharmacol Exp Ther 295:1156–1164 [PubMed] [Google Scholar]
- Jarvis MF, Yu H, McGaraughty S, Wismer CT, Mikusa J, Zhu C, Chu K, Kohlhaas K, Cowart M, Lee CH, et al. (2002b) Analgesic and anti-inflammatory effects of A-286501, a novel orally active adenosine kinase inhibitor. Pain 96:107–118 [DOI] [PubMed] [Google Scholar]
- Jiang N, Kowaluk EA, Lee CH, Mazdiyasni H, Chopp M. (1997) Adenosine kinase inhibition protects brain against transient focal ischemia in rats. Eur J Pharmacol 320:131–137 [DOI] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. (1997) Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267:727–748 [DOI] [PubMed] [Google Scholar]
- Jonzon B, Bergquist A, Li YO, Fredholm BB. (1986) Effects of adenosine and two stable adenosine analogues on blood pressure, heart rate and colonic temperature in the rat. Acta Physiol Scand 126:491–498 [DOI] [PubMed] [Google Scholar]
- Joyce GF. (2002) The antiquity of RNA-based evolution. Nature 418:214–221 [DOI] [PubMed] [Google Scholar]
- Juranka P, Chan V-L. (1985) Analysis of adenosine kinase mutants of baby hamster kidney cells using affinity-purified antibody. J Biol Chem 260:7738–7743 [PubMed] [Google Scholar]
- Kalapos MP. (2007) Possible mechanism for the effect of ketogenic diet in cases of uncontrolled seizures. The reconsideration of acetone theory. Med Hypotheses 68:1382–1388 [DOI] [PubMed] [Google Scholar]
- Keil GJ, 2nd, DeLander GE. (1994) Adenosine kinase and adenosine deaminase inhibition modulate spinal adenosine- and opioid agonist-induced antinociception in mice. Eur J Pharmacol 271:37–46 [DOI] [PubMed] [Google Scholar]
- Kim YA, Sharon A, Chu CK, Rais RH, Al Safarjalani ON, Naguib FN, el Kouni MH. (2008) Structure-activity relationships of 7-deaza-6-benzylthioinosine analogues as ligands of Toxoplasma gondii adenosine kinase. J Med Chem 51:3934–3945 [DOI] [PubMed] [Google Scholar]
- Klobutcher LA, Nichols EA, Kucherlapati RS, Ruddle FH. (1976) Assignment of the gene for human adenosine kinase to chromosome 10 using a somatic cell hybrid clone panel. Cytogenet Cell Genet 16:171–174 [DOI] [PubMed] [Google Scholar]
- Kornberg A, Pricer WE., Jr (1951) Enzymatic phosphorylation of adenosine and 2,6-diaminopurine riboside. J Biol Chem 193:481–495 [PubMed] [Google Scholar]
- Kossoff EH, Rho JM. (2009) Ketogenic diets: evidence for short- and long-term efficacy. Neurotherapeutics 6:406–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossoff EH, Zupec-Kania BA, Rho JM. (2009) Ketogenic diets: an update for child neurologists. J Child Neurol 24:979–988 [DOI] [PubMed] [Google Scholar]
- Kowaluk EA, Bhagwat SS, Jarvis MF. (1998) Adenosine kinase inhibitors. Curr Pharm Des 4:403–416 [PubMed] [Google Scholar]
- Kowaluk EA, Jarvis MF. (2000) Therapeutic potential of adenosine kinase inhibitors. Expert Opin Investig Drugs 9:551–564 [DOI] [PubMed] [Google Scholar]
- Kowaluk EA, Mikusa J, Wismer CT, Zhu CZ, Schweitzer E, Lynch JJ, Lee CH, Jiang M, Bhagwat SS, Gomtsyan A, et al. (2000) ABT-702 (4-amino-5-(3-bromophenyl)-7-(6-morpholino-pyridin- 3-yl)pyrido[2,3-d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti-inflammatory properties. II. In vivo characterization in the rat. J Pharmacol Exp Ther 295:1165–1174 [PubMed] [Google Scholar]
- Kroll K, Deussen A, Sweet IR. (1992) Comprehensive model of transport and metabolism of adenosine and S-adenosylhomocysteine in the guinea pig heart. Circ Res 71:590–604 [DOI] [PubMed] [Google Scholar]
- Kuettel S, Greenwald J, Kostrewa D, Ahmed S, Scapozza L, Perozzo R. (2011) Crystal structures of T. b. rhodesiense adenosine kinase complexed with inhibitor and activator: implications for catalysis and hyperactivation. PLoS Negl Trop Dis 5:e1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kull B, Svenningsson P, Fredholm BB. (2000) Adenosine A(2A) receptors are colocalized with and activate g(olf) in rat striatum. Mol Pharmacol 58:771–777 [DOI] [PubMed] [Google Scholar]
- Lagercrantz H, Yamamoto Y, Fredholm BB, Prabhakar NR, von Euler C. (1984) Adenosine analogues depress ventilation in rabbit neonates. Theophylline stimulation of respiration via adenosine receptors? Pediatr Res 18:387–390 [DOI] [PubMed] [Google Scholar]
- Langan Y. (2000) Sudden unexpected death in epilepsy (SUDEP): risk factors and case control studies. Seizure 9:179–183 [DOI] [PubMed] [Google Scholar]
- Lee JK, Won JS, Singh AK, Singh I. (2005) Adenosine kinase inhibitor attenuates the expression of inducible nitric oxide synthase in glial cells. Neuropharmacology 48:151–160 [DOI] [PubMed] [Google Scholar]
- Lee Y, Messing A, Su M, Brenner M. (2008) GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia 56:481–493 [DOI] [PubMed] [Google Scholar]
- LePage GA, Khaliq A, Gottlieb JA. (1973) Studies of 9-beta-D-arabinofuranosyladenine in man. Drug Metab Dispos 1:756–759 [PubMed] [Google Scholar]
- Li T, Lan JQ, Boison D. (2008a) Uncoupling of astrogliosis from epileptogenesis in adenosine kinase (ADK) transgenic mice. Neuron Glia Biol 4:91–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Lytle N, Lan J-Q, Sandau US, Boison D. (2012) Local disruption of glial adenosine homeostasis in mice associates with focal electrographic seizures: a first step in epileptogenesis? Glia 60:83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Quan Lan J, Fredholm BB, Simon RP, Boison D. (2007a) Adenosine dysfunction in astrogliosis: cause for seizure generation? Neuron Glia Biol 3:353–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Ren G, Kaplan DL, Boison D. (2009) Human mesenchymal stem cell grafts engineered to release adenosine reduce chronic seizures in a mouse model of CA3-selective epileptogenesis. Epilepsy Res 84:238–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D. (2008b) Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest 118:571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A, Segschneider M, Simon RP, Brüstle O, Boison D. (2007b) Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain 130:1276–1288 [DOI] [PubMed] [Google Scholar]
- Liao Y, Takashima S, Asano Y, Asakura M, Ogai A, Shintani Y, Minamino T, Asanuma H, Sanada S, Kim J, et al. (2003) Activation of adenosine A1 receptor attenuates cardiac hypertrophy and prevents heart failure in murine left ventricular pressure-overload model. Circ Res 93:759–766 [DOI] [PubMed] [Google Scholar]
- Lin BB, Hurley MC, Fox IH. (1988) Regulation of adenosine kinase by adenosine analogs. Mol Pharmacol 34:501–505 [PubMed] [Google Scholar]
- Lindberg B, Klenow H, Hansen K. (1967) Some properties of partially purified mammalian adenosine kinase. J Biol Chem 242:350–356 [PubMed] [Google Scholar]
- Linden J. (2005) Adenosine in tissue protection and tissue regeneration. Mol Pharmacol 67:1385–1387 [DOI] [PubMed] [Google Scholar]
- Lloyd HG, Deussen A, Wuppermann H, Schrader J. (1988) The transmethylation pathway as a source for adenosine in the isolated guinea-pig heart. Biochem J 252:489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd HG, Fredholm BB. (1995) Involvement of adenosine deaminase and adenosine kinase in regulating extracellular adenosine concentration in rat hippocampal slices. Neurochem Int 26:387–395 [DOI] [PubMed] [Google Scholar]
- Lloyd HG, Schrader J. (1993) Adenosine metabolism in the guinea pig heart: the role of cytosolic S-adenosyl-L-homocysteine hydrolase, 5′-nucleotidase and adenosine kinase. Eur Heart J 14 (Suppl I):27–33 [PubMed] [Google Scholar]
- Long MC, Allan PW, Luo MZ, Liu MC, Sartorelli AC, Parker WB. (2007) Evaluation of 3-deaza-adenosine analogues as ligands for adenosine kinase and inhibitors of Mycobacterium tuberculosis growth. J Antimicrob Chemother 59:118–121 [DOI] [PubMed] [Google Scholar]
- Long MC, Escuyer V, Parker WB. (2003) Identification and characterization of a unique adenosine kinase from Mycobacterium tuberculosis. J Bacteriol 185:6548–6555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MC, Parker WB. (2006) Structure-activity relationship for nucleoside analogs as inhibitors or substrates of adenosine kinase from Mycobacterium tuberculosis. I. Modifications to the adenine moiety. Biochem Pharmacol 71:1671–1682 [DOI] [PubMed] [Google Scholar]
- Long MC, Shaddix SC, Moukha-Chafiq O, Maddry JA, Nagy L, Parker WB. (2008) Structure-activity relationship for adenosine kinase from Mycobacterium tuberculosis II. Modifications to the ribofuranosyl moiety. Biochem Pharmacol 75:1588–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lönnroth P, Jansson PA, Fredholm BB, Smith U. (1989) Microdialysis of intercellular adenosine concentration in subcutaneous tissue in humans. Am J Physiol 256:E250–E255 [DOI] [PubMed] [Google Scholar]
- Lovatt D, Xu Q, Liu W, Takano T, Smith NA, Schnermann J, Tieu K, Nedergaard M. (2012) Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity. Proc Natl Acad Sci USA 109:6265–6270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Fassett J, Xu X, Hu X, Zhu G, French J, Zhang P, Schnermann J, Bache RJ, Chen Y. (2008) Adenosine A3 receptor deficiency exerts unanticipated protective effects on the pressure-overloaded left ventricle. Circulation 118:1713–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusardi TA, Lytle NK, Szybala C, Boison D. (2012) Caffeine prevents acute mortality after TBI in rats without increased morbidity. Exp Neurol 234:161–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher A, Onal P, Schweingruber AM, Mäser P. (2007) Adenosine kinase of Trypanosoma brucei and its role in susceptibility to adenosine antimetabolites. Antimicrob Agents Chemother 51:3895–3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch ME, Clark AJ, Sawynok J. (2003) Intravenous adenosine alleviates neuropathic pain: a double blind placebo controlled crossover trial using an enriched enrolment design. Pain 103:111–117 [DOI] [PubMed] [Google Scholar]
- Ma W, Berg J, Yellen G. (2007) Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci 27:3618–3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackiewicz M, Nikonova EV, Zimmerman JE, Galante RJ, Zhang L, Cater JR, Geiger JD, Pack AI. (2003) Enzymes of adenosine metabolism in the brain: diurnal rhythm and the effect of sleep deprivation. J Neurochem 85:348–357 [DOI] [PubMed] [Google Scholar]
- Maj M, Singh B, Gupta RS. (2000) The influence of inorganic phosphate on the activity of adenosine kinase. Biochim Biophys Acta 1476:33–42 [DOI] [PubMed] [Google Scholar]
- Maj MC, Singh B, Gupta RS. (2002) Pentavalent ions dependency is a conserved property of adenosine kinase from diverse sources: identification of a novel motif implicated in phosphate and magnesium ion binding and substrate inhibition. Biochemistry 41:4059–4069 [DOI] [PubMed] [Google Scholar]
- Malawska B, Kulig K. (2008) Brivaracetam: a new drug in development for epilepsy and neuropathic pain. Expert Opin Investig Drugs 17:361–369 [DOI] [PubMed] [Google Scholar]
- Manthei SA, Reiling CM, Van Wylen DG. (1998) Dual cardiac microdialysis to assess drug-induced changes in interstitial purine metabolites: adenosine deaminase inhibition versus adenosine kinase inhibition. Cardiovasc Res 37:171–178 [DOI] [PubMed] [Google Scholar]
- Masino SA, Geiger JD. (2008) Are purines mediators of the anticonvulsant/neuroprotective effects of ketogenic diets? Trends Neurosci 31:273–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Geiger JD. (2009) The ketogenic diet and epilepsy: is adenosine the missing link? Epilepsia 50:332–333 [DOI] [PubMed] [Google Scholar]
- Masino SA, Kawamura M, Jr, Ruskin DN, Geiger JD, Boison D. (2012) Purines and neuronal excitability: links to the ketogenic diet. Epilepsy Res 100:229–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Kawamura M, Wasser CD, Pomeroy LT, Ruskin DN. (2009) Adenosine, ketogenic diet and epilepsy: the emerging therapeutic relationship between metabolism and brain activity. Curr Neuropharmacol 7:257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Li T, Theofilas P, Sandau US, Ruskin DN, Fredholm BB, Geiger JD, Aronica E, Boison D. (2011) A ketogenic diet suppresses seizures in mice through adenosine A₁ receptors. J Clin Invest 121:2679–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews II, Erion MD, Ealick SE. (1998) Structure of human adenosine kinase at 1.5 A resolution. Biochemistry 37:15607–15620 [DOI] [PubMed] [Google Scholar]
- Mato JM, Martínez-Chantar ML, Lu SC. (2008) Methionine metabolism and liver disease. Annu Rev Nutr 28:273–293 [DOI] [PubMed] [Google Scholar]
- Matulenko MA, Lee CH, Jiang M, Frey RR, Cowart MD, Bayburt EK, Didomenico S, Gfesser GA, Gomtsyan A, Zheng GZ, et al. (2005) 5-(3-Bromophenyl)-7-(6-morpholin-4-ylpyridin-3-yl)pyrido[2,3-d]pyrimidin-4-ylamine: structure-activity relationships of 7-substituted heteroaryl analogs as non-nucleoside adenosine kinase inhibitors. Bioorg Med Chem 13:3705–3720 [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Chu KL, Wismer CT, Mikusa J, Zhu CZ, Cowart M, Kowaluk EA, Jarvis MF. (2001a) Effects of A-134974, a novel adenosine kinase inhibitor, on carrageenan-induced inflammatory hyperalgesia and locomotor activity in rats: evaluation of the sites of action. J Pharmacol Exp Ther 296:501–509 [PubMed] [Google Scholar]
- McGaraughty S, Cowart M, Jarvis MF. (2001b) Recent developments in the discovery of novel adenosine kinase inhibitors: mechanism of action and therapeutic potential. CNS Drug Rev 7:415–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaraughty S, Cowart M, Jarvis MF, Berman RF. (2005) Anticonvulsant and antinociceptive actions of novel adenosine kinase inhibitors. Curr Top Med Chem 5:43–58 [DOI] [PubMed] [Google Scholar]
- McIntosh VJ, Lasley RD. (2012) Adenosine receptor-mediated cardioprotection: are all 4 subtypes required or redundant? J Cardiovasc Pharmacol Ther 17:21–33 [DOI] [PubMed] [Google Scholar]
- McNally T, Helfrich RJ, Cowart M, Dorwin SA, Meuth JL, Idler KB, Klute KA, Simmer RL, Kowaluk EA, Halbert DN. (1997) Cloning and expression of the adenosine kinase gene from rat and human tissues. Biochem Biophys Res Commun 231:645–650 [DOI] [PubMed] [Google Scholar]
- Meléndez-Hevia E, Montero-Gómez N, Montero F. (2008) From prebiotic chemistry to cellular metabolism—the chemical evolution of metabolism before Darwinian natural selection. J Theor Biol 252:505–519 [DOI] [PubMed] [Google Scholar]
- Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Borea PA. (2005) A3 adenosine receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase/Akt-dependent inhibition of the extracellular signal-regulated kinase 1/2 phosphorylation in A375 human melanoma cells. J Biol Chem 280:19516–19526 [DOI] [PubMed] [Google Scholar]
- Mildvan AS. (1987) Role of magnesium and other divalent cations in ATP-utilizing enzymes. Magnesium 6:28–33 [PubMed] [Google Scholar]
- Miller LP, Jelovich LA, Yao LP, DaRe J, Ugarkar B, Foster AC. (1996) Pre- and peristroke treatment with the adenosine kinase inhibitor, 5′-deoxyiodotubercidin, significantly reduces infarct volume after temporary occlusion of the middle cerebral artery in rats. Neurosci Lett 220:73–76 [DOI] [PubMed] [Google Scholar]
- Miller RL, Adamczyk DL, Miller WH. (1979a) Adenosine kinase from rabbit liver. I. Purification by affinity chromatography and properties. J Biol Chem 254:2339–2345 [PubMed] [Google Scholar]
- Miller RL, Adamczyk DL, Miller WH, Koszalka GW, Rideout JL, Beacham LM, 3rd, Chao EY, Haggerty JJ, Krenitsky TA, Elion GB. (1979b) Adenosine kinase from rabbit liver. II. Substrate and inhibitor specificity. J Biol Chem 254:2346–2352 [PubMed] [Google Scholar]
- Miller SL, Urey HC. (1959a) Organic compound synthesis on the primitive earth. Science 130:245–251 [DOI] [PubMed] [Google Scholar]
- Miller SL, Urey HC. (1959b) Origin of Life. Science 130:1622–1624 [DOI] [PubMed] [Google Scholar]
- Mimouni M, Bontemps F, Van den Berghe G. (1994) Kinetic studies of rat liver adenosine kinase. Explanation of exchange reaction between adenosine and AMP. J Biol Chem 269:17820–17825 [PubMed] [Google Scholar]
- Mlejnek P, Dolezel P. (2005) Apoptosis induced by N6-substituted derivatives of adenosine is related to intracellular accumulation of corresponding mononucleotides in HL-60 cells. Toxicol In Vitro 19:985–990 [DOI] [PubMed] [Google Scholar]
- Moffatt BA, Stevens YY, Allen MS, Snider JD, Pereira LA, Todorova MI, Summers PS, Weretilnyk EA, Martin-McCaffrey L, Wagner C. (2002) Adenosine kinase deficiency is associated with developmental abnormalities and reduced transmethylation. Plant Physiol 128:812–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt BA, Wang L, Allen MS, Stevens YY, Qin W, Snider J, von Schwartzenberg K. (2000) Adenosine kinase of Arabidopsis. Kinetic properties and gene expression. Plant Physiol 124:1775–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello S, Ito K, Yamamura S, Lee KY, Jazrawi E, Desouza P, Barnes P, Cicala C, Adcock IM. (2006) IL-1 beta and TNF-alpha regulation of the adenosine receptor (A2A) expression: differential requirement for NF-kappa B binding to the proximal promoter. J Immunol 177:7173–7183 [DOI] [PubMed] [Google Scholar]
- Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. (2008) HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 111:5571–5580 [DOI] [PubMed] [Google Scholar]
- Mozzicato S, Joshi BV, Jacobson KA, Liang BT. (2004) Role of direct RhoA-phospholipase D1 interaction in mediating adenosine-induced protection from cardiac ischemia. FASEB J 18:406–408 [DOI] [PubMed] [Google Scholar]
- Mubagwa K, Flameng W. (2001) Adenosine, adenosine receptors and myocardial protection: an updated overview. Cardiovasc Res 52:25–39 [DOI] [PubMed] [Google Scholar]
- Nakai K, Horton P. (1999) PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci 24:34–36 [DOI] [PubMed] [Google Scholar]
- Narayan P, Mentzer RM, Jr, Lasley RD. (2001) Adenosine A1 receptor activation reduces reactive oxygen species and attenuates stunning in ventricular myocytes. J Mol Cell Cardiol 33:121–129 [DOI] [PubMed] [Google Scholar]
- Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, Whitney A, Cross JH. (2008) The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol 7:500–506 [DOI] [PubMed] [Google Scholar]
- Neudecker TJ, Hartmann GR. (1972) Involvement of -SH and -S-S-groups in the stabilisation of adenosine kinase. Hoppe Seylers Z Physiol Chem 353:1553. [PubMed] [Google Scholar]
- Neudecker TJ, Hartmann GR. (1978) On the modification of adenosine kinase by thiols. Hoppe Seylers Z Physiol Chem 359:1771–1776 [DOI] [PubMed] [Google Scholar]
- Newby AC, Worku Y, Holmquist CA. (1985) Adenosine formation. Evidence for a direct biochemical link with energy metabolism. Adv Myocardiol 6:273–284 [PubMed] [Google Scholar]
- Offermanns S, Simon MI. (1995) G alpha 15 and G alpha 16 couple a wide variety of receptors to phospholipase C. J Biol Chem 270:15175–15180 [DOI] [PubMed] [Google Scholar]
- Oren J, Kelly DH, Shannon DC. (1987) Familial occurrence of sudden infant death syndrome and apnea of infancy. Pediatrics 80:355–358 [PubMed] [Google Scholar]
- Oro J. (1961) Mechanism of synthesis of adenine from hydrogen cyanide under possible primitive earth conditions. Nature 191:1193–1194 [DOI] [PubMed] [Google Scholar]
- Pak MA, Haas HL, Decking UKM, Schrader J. (1994) Inhibition of adenosine kinase increases endogenous adenosine and depresses neuronal activity in hippocampal slices. Neuropharmacology 33:1049–1053 [DOI] [PubMed] [Google Scholar]
- Palchykova S, Winsky-Sommerer R, Shen H-Y, Boison D, Gerling A, Tobler I. (2010) Manipulation of adenosine kinase affects sleep regulation in mice. J Neurosci 30:13157–13165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palella TD, Andres CM, Fox IH. (1980) Human placental adenosine kinase. Kinetic mechanism and inhibition. J Biol Chem 255:5264–5269 [PubMed] [Google Scholar]
- Palop JJ, Mucke L. (2009) Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol 66:435–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parducci RE, Cabrera R, Baez M, Guixé V. (2006) Evidence for a catalytic Mg2+ ion and effect of phosphate on the activity of Escherichia coli phosphofructokinase-2: regulatory properties of a ribokinase family member. Biochemistry 45:9291–9299 [DOI] [PubMed] [Google Scholar]
- Park J, Gupta RS. (2008) Adenosine kinase and ribokinase—the RK family of proteins. Cell Mol Life Sci 65:2875–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Gupta RS. (2012) Adenosine metabolism, adenosine kinase, and evolution, in Adenosine: a key link between metabolism and central nervous system activity (Boison D, Masino MA. eds) pp 23–54, Springer, New York [Google Scholar]
- Park J, Singh B, Gupta RS. (2006) Inhibition of adenosine kinase by phosphonate and bisphosphonate derivatives. Mol Cell Biochem 283:11–21 [DOI] [PubMed] [Google Scholar]
- Park J, van Koeverden P, Singh B, Gupta RS. (2007) Identification and characterization of human ribokinase and comparison of its properties with E. coli ribokinase and human adenosine kinase. FEBS Lett 581:3211–3216 [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. (2005) Astrocytic purinergic signaling coordinates synaptic networks. Science 310:113–116 [DOI] [PubMed] [Google Scholar]
- Pawelczyk T, Sakowicz M, Podgorska M, Szczepanska-Konkel M. (2003) Insulin induces expression of adenosine kinase gene in rat lymphocytes by signaling through the mitogen-activated protein kinase pathway. Exp Cell Res 286:152–163 [DOI] [PubMed] [Google Scholar]
- Pawelczyk T, Sakowicz M, Szczepanska-Konkel M, Angielski S. (2000) Decreased expression of adenosine kinase in streptozotocin-induced diabetes mellitus rats. Arch Biochem Biophys 375:1–6 [DOI] [PubMed] [Google Scholar]
- Pazzagli M, Corsi C, Fratti S, Pedata F, Pepeu G. (1995) Regulation of extracellular adenosine levels in the striatum of aging rats. Brain Res 684:103–106 [DOI] [PubMed] [Google Scholar]
- Peakman MC, Hill SJ. (1994) Adenosine A2B-receptor-mediated cyclic AMP accumulation in primary rat astrocytes. Br J Pharmacol 111:191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peart J, Flood A, Linden J, Matherne GP, Headrick JP. (2002) Adenosine-mediated cardioprotection in ischemic-reperfused mouse heart. J Cardiovasc Pharmacol 39:117–129 [DOI] [PubMed] [Google Scholar]
- Peart J, Matherne GP, Cerniway RJ, Headrick JP. (2001) Cardioprotection with adenosine metabolism inhibitors in ischemic-reperfused mouse heart. Cardiovasc Res 52:120–129 [DOI] [PubMed] [Google Scholar]
- Peart J, Willems L, Headrick JP. (2003) Receptor and non-receptor-dependent mechanisms of cardioprotection with adenosine. Am J Physiol Heart Circ Physiol 284:H519–H527 [DOI] [PubMed] [Google Scholar]
- Peart JN, Gross GJ. (2005) Cardioprotection following adenosine kinase inhibition in rat hearts. Basic Res Cardiol 100:328–336 [DOI] [PubMed] [Google Scholar]
- Peart JN, Headrick JP. (2007) Adenosinergic cardioprotection: multiple receptors, multiple pathways. Pharmacol Ther 114:208–221 [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. (2005) Astrocyte activation and reactive gliosis. Glia 50:427–434 [DOI] [PubMed] [Google Scholar]
- Pelicano H, Maury G, Elalaoui A, Shafiee M, Imbach JL, Goody RS, Divita G. (1997) Study of the substrate-binding properties of bovine liver adenosine kinase and inhibition by fluorescent nucleoside analogues. Eur J Biochem 248:930–937 [DOI] [PubMed] [Google Scholar]
- Perner RJ, Gu YG, Lee CH, Bayburt EK, McKie J, Alexander KM, Kohlhaas KL, Wismer CT, Mikusa J, Jarvis MF, et al. (2003) 5,6,7-trisubstituted 4-aminopyrido[2,3-d]pyrimidines as novel inhibitors of adenosine kinase. J Med Chem 46:5249–5257 [DOI] [PubMed] [Google Scholar]
- Perner RJ, Lee CH, Jiang M, Gu YG, Didomenico S, Bayburt EK, Alexander KM, Kohlhaas KL, Jarvis MF, Kowaluk EL, et al. (2005) Synthesis and biological evaluation of 6,7-disubstituted 4-aminopyrido[2,3-d]pyrimidines as adenosine kinase inhibitors. Bioorg Med Chem Lett 15:2803–2807 [DOI] [PubMed] [Google Scholar]
- Petrov R, MacDonald MH, Tesch AM, Benton HP. (2005) Inhibition of adenosine kinase attenuates interleukin-1- and lipopolysaccharide-induced alterations in articular cartilage metabolism. Osteoarthritis Cartilage 13:250–257 [DOI] [PubMed] [Google Scholar]
- Phillips E, Newsholme EA. (1979) Maximum activities, properties and distribution of 5′ nucleotidase, adenosine kinase and adenosine deaminase in rat and human brain. J Neurochem 33:553–558 [DOI] [PubMed] [Google Scholar]
- Phillis JW, Smith-Barbour M. (1993) The adenosine kinase inhibitor, 5-iodotubercidin, is not protective against cerebral ischemic injury in the gerbil. Life Sci 53:497–502 [DOI] [PubMed] [Google Scholar]
- Pignataro G, Maysami S, Studer FE, Wilz A, Simon RP, Boison D. (2008) Downregulation of hippocampal adenosine kinase after focal ischemia as potential endogenous neuroprotective mechanism. J Cereb Blood Flow Metab 28:17–23 [DOI] [PubMed] [Google Scholar]
- Pignataro G, Simon RP, Boison D. (2007a) Transgenic overexpression of adenosine kinase aggravates cell death in ischemia. J Cereb Blood Flow Metab 27:1–5 [DOI] [PubMed] [Google Scholar]
- Pignataro G, Studer FE, Wilz A, Simon RP, Boison D. (2007b) Neuroprotection in ischemic mouse brain induced by stem cell-derived brain implants. J Cereb Blood Flow Metab 27:919–927 [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Lukasiuk K. (2011) Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol 10:173–186 [DOI] [PubMed] [Google Scholar]
- Poon A, Sawynok J. (1995) Antinociception by adenosine analogs and an adenosine kinase inhibitor: dependence on formalin concentration. Eur J Pharmacol 286:177–184 [DOI] [PubMed] [Google Scholar]
- Poon A, Sawynok J. (1998) Antinociception by adenosine analogs and inhibitors of adenosine metabolism in an inflammatory thermal hyperalgesia model in the rat. Pain 74:235–245 [DOI] [PubMed] [Google Scholar]
- Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, Stolovich-Rain M, Dadon D, Granot Z, Ben-Hur V, White P, et al. (2011) Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab 13:440–449 [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Kalinchuk AV. (2011) Adenosine, energy metabolism and sleep homeostasis. Sleep Med Rev 15:123–135 [DOI] [PubMed] [Google Scholar]
- Przyklenk K, Whittaker P. (2005) Cardioprotection with adenosine: ‘a riddle wrapped in a mystery’. Br J Pharmacol 145:699–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raatikainen MJ, Peuhkurinen KJ, Kiviluoma KT, Hiltunen JK, Hassinen IE. (1992) 5′-Nucleotidase activity and adenosine production in rat liver mitochondria. Biochim Biophys Acta 1099:238–246 [DOI] [PubMed] [Google Scholar]
- Radek RJ, Decker MW, Jarvis MF. (2004) The adenosine kinase inhibitor ABT-702 augments EEG slow waves in rats. Brain Res 1026:74–83 [DOI] [PubMed] [Google Scholar]
- Rais RH, Al Safarjalani ON, Yadav V, Guarcello V, Kirk M, Chu CK, Naguib FN, el Kouni MH. (2005) 6-Benzylthioinosine analogues as subversive substrate of Toxoplasma gondii adenosine kinase: activities and selective toxicities. Biochem Pharmacol 69:1409–1419 [DOI] [PubMed] [Google Scholar]
- Recacha R, Talalaev A, DeLucas LJ, Chattopadhyay D. (2000) Toxoplasma gondii adenosine kinase: expression, purification, characterization, crystallization and preliminary crystallographic analysis. Acta Crystallogr D Biol Crystallogr 56:76–78 [DOI] [PubMed] [Google Scholar]
- Reddy MC, Palaninathan SK, Shetty ND, Owen JL, Watson MD, Sacchettini JC. (2007) High resolution crystal structures of Mycobacterium tuberculosis adenosine kinase: insights into the mechanism and specificity of this novel prokaryotic enzyme. J Biol Chem 282:27334–27342 [DOI] [PubMed] [Google Scholar]
- Reichelt ME, Shanu A, Willems L, Witting PK, Ellis NA, Blackburn MR, Headrick JP. (2009) Endogenous adenosine selectively modulates oxidant stress via the A1 receptor in ischemic hearts. Antioxid Redox Signal 11:2641–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid EA, Kristo G, Yoshimura Y, Ballard-Croft C, Keith BJ, Mentzer RM, Jr, Lasley RD. (2005) In vivo adenosine receptor preconditioning reduces myocardial infarct size via subcellular ERK signaling. Am J Physiol Heart Circ Physiol 288:H2253–H2259 [DOI] [PubMed] [Google Scholar]
- Ren G, Li T, Lan JQ, Wilz A, Simon RP, Boison D. (2007) Lentiviral RNAi-induced downregulation of adenosine kinase in human mesenchymal stem cell grafts: a novel perspective for seizure control. Exp Neurol 208:26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkawek K, Stege GJ, Bosman GJ. (1999) Dementia, gliosis and expression of the small heat shock proteins hsp27 and alpha B-crystallin in Parkinson’s disease. Neuroreport 10:2273–2276 [DOI] [PubMed] [Google Scholar]
- Ribeiro JA, Sebastião AM. (2010) Modulation and metamodulation of synapses by adenosine. Acta Physiol (Oxf) 199:161–169 [DOI] [PubMed] [Google Scholar]
- Rivas-Pardo JA, Caniuguir A, Wilson CA, Babul J, Guixé V. (2011) Divalent metal cation requirements of phosphofructokinase-2 from E. coli. Evidence for a high affinity binding site for Mn2+. Arch Biochem Biophys 505:60–66 [DOI] [PubMed] [Google Scholar]
- Rogel A, Bromberg Y, Sperling O, Zoref-Shani E. (2005) Phospholipase C is involved in the adenosine-activated signal transduction pathway conferring protection against iodoacetic acid-induced injury in primary rat neuronal cultures. Neurosci Lett 373:218–221 [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Li Y, Le M, Zhang Y. (2000) Nitric oxide-stimulated increase in extracellular adenosine accumulation in rat forebrain neurons in culture is associated with ATP hydrolysis and inhibition of adenosine kinase activity. J Neurosci 20:6294–6301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosengren S, Bong GW, Firestein GS. (1995) Anti-inflammatory effects of an adenosine kinase inhibitor. Decreased neutrophil accumulation and vascular leakage. J Immunol 154:5444–5451 [PubMed] [Google Scholar]
- Rotllan P, Miras Portugal MT. (1985) Adenosine kinase from bovine adrenal medulla. Eur J Biochem 151:365–371 [DOI] [PubMed] [Google Scholar]
- Rusina R, Ridzon P, Kulist'ák P, Keller O, Bartos A, Buncová M, Fialová L, Koukolík F, Matej R. (2010) Relationship between ALS and the degree of cognitive impairment, markers of neurodegeneration and predictors for poor outcome. A prospective study. Eur J Neurol 17:23–30 [DOI] [PubMed] [Google Scholar]
- Ruskin DN, Kawamura M, Masino SA. (2009) Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PLoS ONE 4:e8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin B, Kansy JW, Nairn AC, Spychala J, Ealick SE, Fienberg AA, Greene RW, Bibb JA. (2004) Molecular characterization of recombinant mouse adenosine kinase and evaluation as a target for protein phosphorylation. Eur J Biochem 271:3547–3555 [DOI] [PubMed] [Google Scholar]
- Saitoh M, Nagai K, Nakagawa K, Yamamura T, Yamamoto S, Nishizaki T. (2004) Adenosine induces apoptosis in the human gastric cancer cells via an intrinsic pathway relevant to activation of AMP-activated protein kinase. Biochem Pharmacol 67:2005–2011 [DOI] [PubMed] [Google Scholar]
- Sakowicz-Burkiewicz M, Kocbuch K, Grden M, Szutowicz A, Pawelczyk T. (2006) Diabetes-induced decrease of adenosine kinase expression impairs the proliferation potential of diabetic rat T lymphocytes. Immunology 118:402–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakowicz M, Grdeń M, Pawełczyk T. (2001) Expression level of adenosine kinase in rat tissues. Lack of phosphate effect on the enzyme activity. Acta Biochim Pol 48:745–754 [PubMed] [Google Scholar]
- Sakowicz M, Pawelczyk T. (2002) Insulin restores expression of adenosine kinase in streptozotocin-induced diabetes mellitus rats. Mol Cell Biochem 236:163–171 [DOI] [PubMed] [Google Scholar]
- Samuelson LC, Farber RA. (1985) Cytological localization of adenosine kinase, nucleoside phosphorylase-1, and esterase-10 genes on mouse chromosome 14. Somat Cell Mol Genet 11:157–165 [DOI] [PubMed] [Google Scholar]
- Sawynok J. (2007) Adenosine and ATP receptors. Handb Exp Pharmacol 177:309–328 [DOI] [PubMed] [Google Scholar]
- Sawynok J, Liu XJ. (2003) Adenosine in the spinal cord and periphery: release and regulation of pain. Prog Neurobiol 69:313–340 [DOI] [PubMed] [Google Scholar]
- Schiffer D, Cordera S, Cavalla P, Migheli A. (1996) Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci 139 (Suppl):27–33 [DOI] [PubMed] [Google Scholar]
- Schmitt LI, Sims RE, Dale N, Haydon PG. (2012) Wakefulness affects synaptic and network activity by increasing extracellular astrocyte-derived adenosine. J Neurosci 32:4417–4425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnebli HP, Hill DL, Bennett LL., Jr (1967) Purification and properties of adenosine kinase from human tumor cells of type H. Ep. No. 2. J Biol Chem 242:1997–2004 [PubMed] [Google Scholar]
- Schulte G, Fredholm BB. (2002) Signaling pathway from the human adenosine A(3) receptor expressed in Chinese hamster ovary cells to the extracellular signal-regulated kinase 1/2. Mol Pharmacol 62:1137–1146 [DOI] [PubMed] [Google Scholar]
- Schulte G, Fredholm BB. (2003) Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal 15:813–827 [DOI] [PubMed] [Google Scholar]
- Schumacher MA, Scott DM, Mathews II, Ealick SE, Roos DS, Ullman B, Brennan RG. (2000) Crystal structures of Toxoplasma gondii adenosine kinase reveal a novel catalytic mechanism and prodrug binding. J Mol Biol 298:875–893 [DOI] [PubMed] [Google Scholar]
- Sebastiao AM, Ribeiro JA. (2009a) Adenosine receptors and the central nervous system. Handb Exp Pharmacol 193:471–534 [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA. (2009b) Tuning and fine-tuning of synapses with adenosine. Curr Neuropharmacol 7:180–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P. (1987) Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse 1:133–152 [DOI] [PubMed] [Google Scholar]
- Sen B, Chakraborty A, Datta R, Bhattacharyya D, Datta AK. (2006) Reversal of ADP-mediated aggregation of adenosine kinase by cyclophilin leads to its reactivation. Biochemistry 45:263–271 [DOI] [PubMed] [Google Scholar]
- Sen B, Venugopal V, Chakraborty A, Datta R, Dolai S, Banerjee R, Datta AK. (2007) Amino acid residues of Leishmania donovani cyclophilin key to interaction with its adenosine kinase: biological implications. Biochemistry 46:7832–7843 [DOI] [PubMed] [Google Scholar]
- Shen HY, Li T, Boison D. (2010) A novel mouse model for sudden unexpected death in epilepsy (SUDEP): role of impaired adenosine clearance. Epilepsia 51:465–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Lusardi TA, Williams-Karnesky RL, Lan JQ, Poulsen DJ, Boison D. (2011) Adenosine kinase determines the degree of brain injury after ischemic stroke in mice. J Cereb Blood Flow Metab 31:1648–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Singer P, Lytle N, Wei CJ, Lan JQ, Williams-Karnesky RL, Chen JF, Yee BK, Boison D. (2012) Adenosine augmentation ameliorates psychotic and cognitive endophenotypes of schizophrenia. J Clin Invest 122:2567–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepel PN, Ramonet D, Stevens P, Geiger JD. (2005) Purine level regulation during energy depletion associated with graded excitatory stimulation in brain. Neurol Res 27:139–148 [DOI] [PubMed] [Google Scholar]
- Shneyvays V, Leshem D, Zinman T, Mamedova LK, Jacobson KA, Shainberg A. (2005) Role of adenosine A1 and A3 receptors in regulation of cardiomyocyte homeostasis after mitochondrial respiratory chain injury. Am J Physiol Heart Circ Physiol 288:H2792–H2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shneyvays V, Zinman T, Shainberg A. (2004) Analysis of calcium responses mediated by the A3 adenosine receptor in cultured newborn rat cardiac myocytes. Cell Calcium 36:387–396 [DOI] [PubMed] [Google Scholar]
- Siegmund B, Rieder F, Albrich S, Wolf K, Bidlingmaier C, Firestein GS, Boyle D, Lehr HA, Loher F, Hartmann G, et al. (2001) Adenosine kinase inhibitor GP515 improves experimental colitis in mice. J Pharmacol Exp Ther 296:99–105 [PubMed] [Google Scholar]
- Singer P, Feldon J, Yee BK. (2009) Are DBA/2 mice associated with schizophrenia-like endophenotypes? A behavioural contrast with C57BL/6 mice. Psychopharmacology (Berl) 206:677–698 [DOI] [PubMed] [Google Scholar]
- Singer P, McGarrity S, Shen H-Y, Boison D, Yee BK. (2012) Working memory and the homeostatic control of brain adenosine by adenosine kinase. Neuroscience 213:81–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Gupta RS. (2004) Genomic organization and linkage via a bidirectional promoter of the AP-3 (adaptor protein-3) mu3A and AK (adenosine kinase) genes: deletion mutants of AK in Chinese hamster cells extend into the AP-3 mu3A gene. Biochem J 378:519–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Hao W, Wu Z, Eigl B, Gupta RS. (1996) Cloning and characterization of cDNA for adenosine kinase from mammalian (Chinese hamster, mouse, human and rat) species. High frequency mutants of Chinese hamster ovary cells involve structural alterations in the gene. Eur J Biochem 241:564–571 [DOI] [PubMed] [Google Scholar]
- Singh B, Lin A, Wu ZC, Gupta RS. (2001) Gene structure for adenosine kinase in Chinese hamster and human: high-frequency mutants of CHO cells involve deletions of several introns and exons. DNA Cell Biol 20:53–65 [DOI] [PubMed] [Google Scholar]
- So EL. (2008) What is known about the mechanisms underlying SUDEP? Epilepsia 49 (Suppl 9):93–98 [DOI] [PubMed] [Google Scholar]
- Sommerschild HT, Kirkebøen KA. (2000) Adenosine and cardioprotection during ischaemia and reperfusion—an overview. Acta Anaesthesiol Scand 44:1038–1055 [DOI] [PubMed] [Google Scholar]
- Spácilová P, Naus P, Pohl R, Votruba I, Snásel J, Zábranská H, Pichová I, Ameral R, Birkus G, Cihlár T, et al. (2010) CycloSal-phosphate pronucleotides of cytostatic 6-(Het)aryl-7-deazapurine ribonucleosides: Synthesis, cytostatic activity, and inhibition of adenosine kinases. ChemMedChem 5:1386–1396 [DOI] [PubMed] [Google Scholar]
- Spychala J, Datta NS, Takabayashi K, Datta M, Fox IH, Gribbin T, Mitchell BS. (1996) Cloning of human adenosine kinase cDNA: sequence similarity to microbial ribokinases and fructokinases. Proc Natl Acad Sci USA 93:1232–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spychala J, Mitchell BS. (2002) Cyclosporin A and FK506 decrease adenosine kinase activity and adenosine uptake in T-lymphocytes. J Lab Clin Med 140:84–91 [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, et al. (2003) Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 362:1028–1037 [DOI] [PubMed] [Google Scholar]
- Stone TW, Ceruti S, Abbracchio MP. (2009) Adenosine receptors and neurological disease: neuroprotection and neurodegeneration. Handb Exp Pharmacol 193:535–587 [DOI] [PubMed] [Google Scholar]
- Studer FE, Fedele DE, Marowsky A, Schwerdel C, Wernli K, Vogt K, Fritschy JM, Boison D. (2006) Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience 142:125–137 [DOI] [PubMed] [Google Scholar]
- Sullivan WJ, Jr, Chiang CW, Wilson CM, Naguib FN, el Kouni MH, Donald RG, Roos DS. (1999) Insertional tagging of at least two loci associated with resistance to adenine arabinoside in Toxoplasma gondii, and cloning of the adenosine kinase locus. Mol Biochem Parasitol 103:1–14 [DOI] [PubMed] [Google Scholar]
- Suzuki R, Stanfa LC, Kowaluk EA, Williams M, Jarvis MF, Dickenson AH. (2001) The effect of ABT-702, a novel adenosine kinase inhibitor, on the responses of spinal neurones following carrageenan inflammation and peripheral nerve injury. Br J Pharmacol 132:1615–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. (2008) Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology (Berl) 199:331–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatlisumak T, Takano K, Carano RA, Miller LP, Foster AC, Fisher M. (1998) Delayed treatment with an adenosine kinase inhibitor, GP683, attenuates infarct size in rats with temporary middle cerebral artery occlusion. Stroke 29:1952–1958 [DOI] [PubMed] [Google Scholar]
- Tawfik HE, Schnermann J, Oldenburg PJ, Mustafa SJ. (2005) Role of A1 adenosine receptors in regulation of vascular tone. Am J Physiol Heart Circ Physiol 288:H1411–H1416 [DOI] [PubMed] [Google Scholar]
- Tennyson CN, Klamut HJ, Worton RG. (1995) The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat Genet 9:184–190 [DOI] [PubMed] [Google Scholar]
- Tesch AM, MacDonald MH, Kollias-Baker C, Benton HP. (2002) Effects of an adenosine kinase inhibitor and an adenosine deaminase inhibitor on accumulation of extracellular adenosine by equine articular chondrocytes. Am J Vet Res 63:1512–1519 [DOI] [PubMed] [Google Scholar]
- Theofilas P, Brar S, Stewart K-A, Shen H-Y, Sandau US, Poulsen DJ, Boison D. (2011) Adenosine kinase as a target for therapeutic antisense strategies in epilepsy. Epilepsia 52:589–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Capogna M, Scanziani M. (1993) Presynaptic inhibition in the hippocampus. Trends Neurosci 16:222–227 [DOI] [PubMed] [Google Scholar]
- Tracey WR, Magee W, Masamune H, Oleynek JJ, Hill RJ. (1998) Selective activation of adenosine A3 receptors with N6-(3-chlorobenzyl)-5′-N-methylcarboxamidoadenosine (CB-MECA) provides cardioprotection via KATP channel activation. Cardiovasc Res 40:138–145 [DOI] [PubMed] [Google Scholar]
- Tsuchiya A, Kanno T, Saito M, Miyoshi Y, Gotoh A, Nakano T, Nishizaki T. (2012) Intracellularly transported adenosine induces apoptosis in [corrected] MCF-7 human breast cancer cells by accumulating AMID in the nucleus. Cancer Lett 321:65–72 [DOI] [PubMed] [Google Scholar]
- Ugarkar BG, Castellino AJ, DaRe JM, Kopcho JJ, Wiesner JB, Schanzer JM, Erion MD. (2000a) Adenosine kinase inhibitors. 2. Synthesis, enzyme inhibition, and antiseizure activity of diaryltubercidin analogues. J Med Chem 43:2894–2905 [DOI] [PubMed] [Google Scholar]
- Ugarkar BG, Castellino AJ, DaRe JS, Ramirez-Weinhouse M, Kopcho JJ, Rosengren S, Erion MD. (2003) Adenosine kinase inhibitors. 3. Synthesis, SAR, and antiinflammatory activity of a series of l-lyxofuranosyl nucleosides. J Med Chem 46:4750–4760 [DOI] [PubMed] [Google Scholar]
- Ugarkar BG, DaRe JM, Kopcho JJ, Browne CE, 3rd, Schanzer JM, Wiesner JB, Erion MD. (2000b) Adenosine kinase inhibitors. 1. Synthesis, enzyme inhibition, and antiseizure activity of 5-iodotubercidin analogues. J Med Chem 43:2883–2893 [DOI] [PubMed] [Google Scholar]
- van Calker D, Müller M, Hamprecht B. (1979) Adenosine regulates via two different types of receptors, the accumulation of cyclic AMP in cultured brain cells. J Neurochem 33:999–1005 [DOI] [PubMed] [Google Scholar]
- Vanderpoorten A, Shaw AJ, Cox CJ. (2004) Evolution of multiple paralogous adenosine kinase genes in the moss genus Hygroamblystegium: phylogenetic implications. Mol Phylogenet Evol 31:505–516 [DOI] [PubMed] [Google Scholar]
- Vannoni D, Bernini A, Carlucci F, Civitelli S, Di Pietro MC, Leoncini R, Rosi F, Tabucchi A, Tanzini G, Marinello E. (2004a) Enzyme activities controlling adenosine levels in normal and neoplastic tissues. Med Oncol 21:187–195 [DOI] [PubMed] [Google Scholar]
- Vannoni D, Di Pietro MC, Rosi F, Bernini A, Leoncini R, Tabucchi A, Carlucci F, Floccari F, Santoro A, Tanzini G, et al. (2004b) Metabolism of adenosine in human colorectal tumour. Nucleosides Nucleotides Nucleic Acids 23:1455–1457 [DOI] [PubMed] [Google Scholar]
- Varani K, Massara A, Vincenzi F, Tosi A, Padovan M, Trotta F, Borea PA. (2009) Normalization of A2A and A3 adenosine receptor up-regulation in rheumatoid arthritis patients by treatment with anti-tumor necrosis factor alpha but not methotrexate. Arthritis Rheum 60:2880–2891 [DOI] [PubMed] [Google Scholar]
- Varani K, Padovan M, Govoni M, Vincenzi F, Trotta F, Borea PA. (2010) The role of adenosine receptors in rheumatoid arthritis. Autoimmun Rev 10:61–64 [DOI] [PubMed] [Google Scholar]
- Varani K, Padovan M, Vincenzi F, Targa M, Trotta F, Govoni M, Borea PA. (2011) A2A and A3 adenosine receptor expression in rheumatoid arthritis: upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res Ther 13:R197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, French J, Bartfai T, Baram TZ. (2011) The role of inflammation in epilepsy. Nat Rev Neurol 7:31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlajkovic SM, Guo CX, Dharmawardana N, Wong AC, Boison D, Housley GD, Thorne PR. (2010) Role of adenosine kinase in cochlear development and response to noise. J Neurosci Res 88:2598–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlajkovic SM, Guo CX, Telang R, Wong AC, Paramananthasivam V, Boison D, Housley GD, Thorne PR. (2011) Adenosine kinase inhibition in the cochlea delays the onset of age-related hearing loss. Exp Gerontol 46:905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlajkovic SM, Housley GD, Thorne PR. (2009) Adenosine and the auditory system. Curr Neuropharmacol 7:246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodnala M, Fijolek A, Rofougaran R, Mosimann M, Mäser P, Hofer A. (2008) Adenosine kinase mediates high affinity adenosine salvage in Trypanosoma brucei. J Biol Chem 283:5380–5388 [DOI] [PubMed] [Google Scholar]
- Wang Y, Long MC, Ranganathan S, Escuyer V, Parker WB, Li R. (2005) Overexpression, purification and crystallographic analysis of a unique adenosine kinase from Mycobacterium tuberculosis. Acta Crystallogr Sect F Struct Biol Cryst Commun 61:553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman SJ, Berkow RL, Weetman RM, Baehner RL. (1985) 5-Azacytidine: acute central nervous system toxicity. Am J Pediatr Hematol Oncol 7:86–88 [PubMed] [Google Scholar]
- Whitley RJ, Ch’ien LT, Dolin R, Galasso GJ, Alford CA., Jr (1976) Adenine arabinoside therapy of herpes zoster in the immunosuppressed. NIAID collaborative antiviral study. N Engl J Med 294:1193–1199 [DOI] [PubMed] [Google Scholar]
- Wiesner JB, Ugarkar BG, Castellino AJ, Barankiewicz J, Dumas DP, Gruber HE, Foster AC, Erion MD. (1999) Adenosine kinase inhibitors as a novel approach to anticonvulsant therapy. J Pharmacol Exp Ther 289:1669–1677 [PubMed] [Google Scholar]
- Willems L, Headrick JP. (2005) Protecting murine hearts from ischaemia-reperfusion using selective inhibitors of adenosine metabolism. Clin Exp Pharmacol Physiol 32:179–183 [DOI] [PubMed] [Google Scholar]
- Williams-Karnesky RL, Stenzel-Poore MP. (2009) Adenosine and stroke: maximizing the therapeutic potential of adenosine as a prophylactic and acute neuroprotectant. Curr Neuropharmacol 7:217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CG, Martin RJ, Jaber M, Abu-Shaweesh J, Jafri A, Haxhiu MA, Zaidi S. (2004) Adenosine A2A receptors interact with GABAergic pathways to modulate respiration in neonatal piglets. Respir Physiol Neurobiol 141:201–211 [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. (1997) Presynaptic inhibition of elicited neurotransmitter release. Review Trends Neurosci 20:204–212 [DOI] [PubMed] [Google Scholar]
- Wu M, Sahbaie P, Zheng M, Lobato R, Boison D, Clark JD, Peltz G. (2013) Opiate-induced changes in brain adenosine levels and narcotic drug responses. Neuroscience 228:235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Fassett J, Hu X, Zhu G, Lu Z, Li Y, Schnermann J, Bache RJ, Chen Y. (2008) Ecto-5′-nucleotidase deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction. Hypertension 51:1557–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav V, Chu CK, Rais RH, Al Safarjalani ON, Guarcello V, Naguib FN, el Kouni MH. (2004) Synthesis, biological activity and molecular modeling of 6-benzylthioinosine analogues as subversive substrates of Toxoplasma gondii adenosine kinase. J Med Chem 47:1987–1996 [DOI] [PubMed] [Google Scholar]
- Yamada Y, Goto H, Ogasawara N. (1980) Purification and properties of adenosine kinase from rat brain. Biochim Biophys Acta 616:199–207 [DOI] [PubMed] [Google Scholar]
- Yamada Y, Goto H, Ogasawara N. (1981) Adenosine kinase from human liver. Biochim Biophys Acta 660:36–43 [DOI] [PubMed] [Google Scholar]
- Yamada Y, Goto H, Ogasawara N. (1982) Differences of adenosine kinases from various mammalian tissues. Comp Biochem Physiol B 71:367–372 [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Burbiel JC, Maass A, Müller CE. (2003) Adenosine receptor agonists: from basic medicinal chemistry to clinical development. Expert Opin Emerg Drugs 8:537–576 [DOI] [PubMed] [Google Scholar]
- Yee BK, Singer P, Chen JF, Feldon J, Boison D. (2007) Transgenic overexpression of adenosine kinase in brain leads to multiple learning impairments and altered sensitivity to psychomimetic drugs. Eur J Neurosci 26:3237–3252 [DOI] [PubMed] [Google Scholar]
- Yellen G. (2008) Ketone bodies, glycolysis, and KATP channels in the mechanism of the ketogenic diet. Epilepsia 49 (Suppl 8):80–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogelzang NJ, Herndon JE, 2nd, Cirrincione C, Harmon DC, Antman KH, Corson JM, Suzuki Y, Citron ML, Green MR, Cancer and Leukemia Group B (1997) Dihydro-5-azacytidine in malignant mesothelioma. A phase II trial demonstrating activity accompanied by cardiac toxicity. Cancer 79:2237–2242 [DOI] [PubMed] [Google Scholar]
- Yu L, Gao YF, Li X, Qiao ZP, Shen JL. (2009) Double-stranded RNA specific to adenosine kinase and hypoxanthine-xanthine-guanine-phosphoribosyltransferase retards growth of Toxoplasma gondii. Parasitol Res 104:377–383 [DOI] [PubMed] [Google Scholar]
- Yu L, Gao YF, Qiao ZP, Li CL, Li X, Shen JL. (2008) Toxoplasma gondii: siRNA can mediate the suppression of adenosine kinase expression. Exp Parasitol 118:96–102 [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Heidari MR. (1993) Involvement of adenosine receptors in mouse thermoregulation. J Psychopharmacol 7:365–370 [DOI] [PubMed] [Google Scholar]
- Zhang G, Franklin PH, Murray TF. (1993) Manipulation of endogenous adenosine in the rat prepiriform cortex modulates seizure susceptibility. J Pharmacol Exp Ther 264:1415–1424 [PubMed] [Google Scholar]
- Zhang X, Shu L, Hosoi H, Murti KG, Houghton PJ. (2002) Predominant nuclear localization of mammalian target of rapamycin in normal and malignant cells in culture. J Biol Chem 277:28127–28134 [DOI] [PubMed] [Google Scholar]
- Zhang Y, El Kouni MH, Ealick SE. (2006) Structure of Toxoplasma gondii adenosine kinase in complex with an ATP analog at 1.1 angstroms resolution. Acta Crystallogr D Biol Crystallogr 62:140–145 [DOI] [PubMed] [Google Scholar]
- Zhang Y, El Kouni MH, Ealick SE. (2007) Substrate analogs induce an intermediate conformational change in Toxoplasma gondii adenosine kinase. Acta Crystallogr D Biol Crystallogr 63:126–134 [DOI] [PubMed] [Google Scholar]
- Zheng GZ, Lee C, Pratt JK, Perner RJ, Jiang MQ, Gomtsyan A, Matulenko MA, Mao Y, Koenig JR, Kim KH, et al. (2001) Pyridopyrimidine analogues as novel adenosine kinase inhibitors. Bioorg Med Chem Lett 11:2071–2074 [DOI] [PubMed] [Google Scholar]
- Zheng GZ, Mao Y, Lee CH, Pratt JK, Koenig JR, Perner RJ, Cowart MD, Gfesser GA, McGaraughty S, Chu KL, et al. (2003) Adenosine kinase inhibitors: polar 7-substitutent of pyridopyrimidine derivatives improving their locomotor selectivity. Bioorg Med Chem Lett 13:3041–3044 [DOI] [PubMed] [Google Scholar]
- Zhou QY, Li C, Olah ME, Johnson RA, Stiles GL, Civelli O. (1992) Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proc Natl Acad Sci USA 89:7432–7436 [DOI] [PMC free article] [PubMed] [Google Scholar]