

Abstract
We describe cell type–specific significance analysis of microarrays (cssam) for analyzing differential gene expression for each cell type in a biological sample from microarray data and relative cell-type frequencies. first, we validated cssam with predesigned mixtures and then applied it to whole-blood gene expression datasets from stable post-transplant kidney transplant recipients and those experiencing acute transplant rejection, which revealed hundreds of differentially expressed genes that were otherwise undetectable.
Traditional microarray analysis methods are oblivious to sample cell-type composition. They can neither distinguish between variations in gene expression resulting from an actual physiological change versus differences in cell-type frequency, nor identify the contributions of different cell types to the total measured gene expression. Therefore, their power to detect differentially expressed genes is strongly affected by the sample variation in cell-type frequencies1–3.
Ideally, one would perform between-group differential expression analysis for each of the cell types in a tissue. Experimental methods for isolating subsets of tissues, such as cell sorting or enrichment, are prohibitively expensive and may affect cell physiology and gene expression4,5. In theory, a statistics-based alternative is to quantify the relative abundance of each cell type in each sample, then deconvolve and compare cell type–specific average expression profiles for groups of mixed tissue samples (Fig. 1). Cell-type subset composition can be measured using labeled antibodies to cell-surface markers and flow cytometry, quantified by histology analyses6 or even estimated from the gene expression data by deconvolution from cell type–specific probes7–10. Though previous attempts at gene expression deconvolution have assumed deconvolution to be linear6–8, the relationship between the gene expression in mixed samples and the actual gene expression of the constituting cell subsets is unclear. This prevents assessment of the accuracy of deconvolution-derived profiles, their widespread application and development of such statistics-based techniques.
Figure 1.

Overview of csSAM. Different cell types are denoted by circles, diamonds and hexagons. csSAM identifies cell type–specific differential expression, as shown by the arrows on the right.
We tested the relationship between measured gene expression in mixed samples and the expression of genes in the isolated pure subsets, in a situation in which all factors are known. We analyzed tissue samples from the brain, liver and lung of a single rat in isolation (referred to as `measured pure tissue') as well as in ten different mixture ratios (referred to as `measured mixtures'; Supplementary Table 1) using Affymetrix expression arrays (Online Methods). Such mixtures mimic the common scenario in which biological samples in a dataset are heterogeneous and vary in the relative frequency of the component subsets from one another.
Next, we reconstituted mixture sample expression profiles by multiplying the measured pure tissue expression profiles by the frequency of the tissue subset in a given mixture sample. Overall, experimentally measured mixture data had high correlation with the reconstituted mixture data (r > 0.95; Supplementary Fig. 1). Probes for which data deviated from the diagonal comprised only a small fraction of the probes up to a twofold expression change cutoff (Supplementary Fig. 2); these probes were more abundant in experimentally measured mixtures than in reconstituted samples, likely because of nonlinear biases in sample amplification and normalization procedures or probe cross-hybridization (Supplementary Note 1, Supplementary Fig. 3 and Supplementary Table 2).
The high correlation that we observed between the measured and reconstituted mixtures suggests that statistical deconvolution of tissue-specific expression profiles from complex tissue samples using linear regression should yield accurate expression estimates for most genes. To test this, we applied linear regression fitting to the measured mixture samples using the mixture ratios (Online Methods). For each tissue, a comparison of the estimated expression profile of each subset to the measured expression pattern in the pure tissue showed a high correlation (Fig. 2), indicating that we could accurately deconvolute subset-specific expression patterns for the majority of genes from whole-sample measurements.
Figure 2.
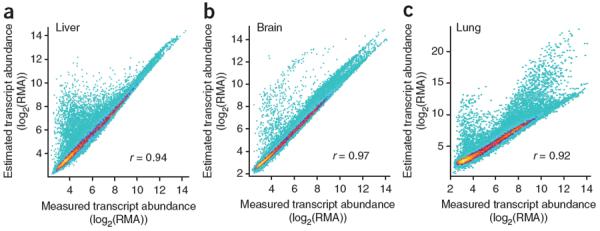
Statistical deconvolution of complex tissues yields accurate estimates of pure tissue-subset expression. (a–c) Density plots of estimated tissue-specific gene expression deconvoluted from mixed tissue samples plotted against measured gene expression from individual tissues. Color represents point density from a single probe (cyan) to 100 probes (yellow). RMA, robust multichip average.
Accurate deconvolution of cell type–specific expression profiles enables the development and application of statistical techniques aimed at maximizing the information obtainable from a heterogeneous tissue gene expression assay. To estimate the specificity and sensitivity of statistical deconvolution to detect differentially expressed genes, we compared deconvoluted and measured differences in gene expression between tissues. Akin to fold change, all probes whose estimated abundance difference was greater than a set threshold were predicted to be differentially expressed. We compared these to a `gold standard' set of differentially expressed probes between tissues identified from the pure tissue sample measurements (Online Methods). Receiver operating characteristic (ROC) curve analysis showed the detection of differentially expressed genes by statistical deconvolution to be both highly specific and sensitive with an area under the curve of 0.85 and greater (Supplementary Fig. 4).
In real-life settings, differences are often assayed between groups of samples, each containing many cell types, and no `gold standard' gene list exists to tell true difference from noise. To test the utility of our method to address an important clinical problem in a complex tissue, we applied cell type–specific significance analysis of microarrays (csSAM) to human whole-blood gene expression array data from 24 kidney transplant recipients. Of these, 15 were experiencing acute rejection of the kidney, whereas 9 were stable after transplant. Blood cells represent a particularly complex tissue type, with over a dozen distinct cell types that can vary in frequency up to 10–20-fold between healthy individuals. In this case, data on white blood cell subsets from Coulter counter measurements was available for all individuals analyzed (Supplementary Table 3), distinguishing five major cell types: lymphocytes, monocytes, neutrophils, eosinophils and basophils.
We observed high variation in relative cell-type frequency between individuals but detected no significant differences in cell-type frequencies between the two groups (P ≥ 0.24 for all cell types). Whole-blood differential expression analysis using a previously published method, significance analysis of microarrays (SAM)11, revealed no differentially expressed genes between the two groups at a relatively permissive false discovery rate (FDR) of 0.3 and reduction in the number of multiple hypothesis tests (Fig. 3a and Supplementary Fig. 5).
Figure 3.

csSAM reveals cell type–specific differential expression undetectable at heterogeneous tissue level. (a–f) Differential expression analysis in whole blood (a) and the indicated cell types (b–f) between samples from individuals with acute rejection and stable post-transplant course.
Next, for each of the two groups of individuals, we deconvoluted the cell type–specific gene expression profile by linear regression analysis for each of the quantified cell types in each group of individuals. Each such cell type–specific expression profile represents the average for that cell type in that group of individuals. We used these deconvolved cell type–specific expression profiles to perform cell type–specific differential expression analysis (Online Methods). For each gene, in each cell type, we calculated the contrast in its deconvoluted expression between groups of individuals. We repeated the deconvolution and cell-type contrast procedure with permuted group-label data. To analyze differences in a gene's expression between two deconvolved cell types, we calculated FDR as the ratio of genes whose contrast exceeds a given threshold in the real dataset compared with the average number of genes exceeding the same threshold in the permuted dataset (Online Methods).
Though we detected no differentially expressed genes between the two groups in whole-blood analyses, sample heterogeneity may have masked biological differences. Applying the csSAM procedure to the kidney transplant dataset for each of the five quantified cell types, we identified 318 differentially expressed genes in monocytes at an FDR of 0.15 (Fig. 3b). We identified no genes as differentially expressed even at an FDR of 0.3 in any of the other cell types (Fig. 3c–e). However, repeated analysis by considering the one-tailed tests of up- and downregulated genes separately, identified differentially expressed genes between lymphocytes and neutrophils of these two groups of individuals as well as 137 genes upregulated in monocytes in samples from individuals experiencing acute kidney rejection at an FDR of 0.05 (Supplementary Fig. 6).
In conclusion, here we described the csSAM algorithm, which addresses the extensive loss of biological signal in microarray datasets when analyzing complex tissue samples that vary in cellular composition. What are the limitations of this methodology? First, probe saturation and cross-hybridization may result in inaccuracies of cell-specific expression profiles, though these do not seem to have a large effect on the accuracy of downstream differential expression analysis. Similarly, for those genes whose cellular expression changes in response to changes in the cell subset composition of their microenvironment, deconvolved cell type–specific expression profile may be inaccurate. Alternative, more sophisticated models to linear regression may be developed to address this problem. Unlike traditional methodologies, csSAM accuracy benefits from variation between samples. Though additional experiments would be needed to identify csSAM's lower detection boundaries, accurate estimates of rare cell types may be aided by sample enrichment or inclusion of highly variable samples, which will yield cell-type frequency–dependent changes in transcript amounts. The key advantage of csSAM is that it localizes the identified differential expression to a particular cellular context, which allows clear hypothesis formulation for follow-up experiments. Though the principal test case here involves blood cells, our methodology is readily usable with microarray analysis of any heterogeneous tissue and can be applied to other types of molecular measurements as well.
ONLINE METHODS
Microarray analysis of rat brain, liver and lung
We mixed the cRNA derived from rat brain, liver and lung biospecimens from a single rat in 13 different proportions, three of which were from each of the tissues in isolate (100% lung, 100% brain and 100% liver). The 10 other mixtures included RNA from each of the three tissues at varying proportions. Each of the samples was analyzed in triplicate (Supplementary Table 1). Snap-frozen samples of rat liver, brain and lung were kept frozen while cutting them into pieces. cDNA synthesis and labeling was done with a starting amount of 1 μg, using the Affymetrix labeling kit, following the manufacturer's instructions. Each sample was hybridized to rat-specific RAE230_2 whole-genome expression arrays (Affymetrix), and the resulting cell files were processed by RMA normalization and used for deconvolution. The abundance of liver, brain and lung tissue in each mixture and their variation across mixtures paralleled those of the neutrophils, lymphocytes and monocytes, respectively, in our renal transplant dataset. Tissues were obtained from untreated animals that were handled according to the Swiss Animal Welfare Law (Tierschutzgesetz, 2005, 2008).
Human renal transplant dataset
Whole-blood gene expression measurements for 24 pediatric renal transplant recipients were analyzed on human-specific HGU133V2.0 (+) whole-genome expression arrays (Affymetrix). Informed consent was obtained from all of the subjects enrolled in this study, and the study protocols were approved by the ethics committee of Stanford University's School of Medicine. Of the 24 samples, 15 were from individuals showing acute rejection of the transplant and 9 were from individuals with stable post-transplant course. White blood cells were analyzed by using Coulter counter to obtain the percentages of monocytes, lymphocytes, eosinophils, basophils and neutrophils for each sample (Supplementary Table 3). Normalization of data from individuals with stable post-transplant course and acute rejection was preformed together by RMA and the output was used directly for SAM and csSAM.
Statistical deconvolution of cell type–specific expression profiles
Assume expression values Xij for sample i = 1, 2, … n and genes j = 1, 2, … p, and measured cell-type proportions W = wik for samples i = 1, 2, … n and cell types k = 1, 2, … K. Our model for a single group of samples is
where hkj is the gene expression for cell-type k and gene j, and eij is a random error. Letting X and W be matrices with entries Xij and Wij respectively, we fit this model by a standard least-squares regression of each column of X on W, to yield the coefficients in the corresponding column of H. As normalized microarray data do not directly correspond to transcript abundance, the issue of normalization and scale to use requires additional investigation and is likely to depend on transcript quantification technology. In the case of a single-channel array (for example, Affymetrix), we set any coefficients estimated as negative to zero. We interpreted the estimated hkj as the average gene expression for cell-type k in the group of samples.
For the two-group model with groups yi = 1 and 2, we assume for groups 1 and 2
respectively. We estimate h1kj and h2kj separately from the group 1 and 2 samples, respectively.
False discovery analysis in the rat experiment
Let Tj be the T-statistic for the true difference between brain and liver expression, for gene j. Define gene j to be truly higher in brain if Tj > 2. We considered the list of all such genes as the gold standard for upregulated brain genes. Let hj1, hj2 be the estimated expression for brain and liver from deconvolution, then we declare gene j substantially higher in brain if hj2 − hj1 > c, similarly for upregulated liver genes Tj < −2 and hj2 − hj1 < − c. We calculate the receiver operating characteristic (ROC) curves by varying threshold c and comparing the genes whose difference in estimated expression profiles was above the threshold to those comprising the gold standard.
csSAM tests for two-class differences
We considered five tests of differences between two classes: (i) whole (mixed) tissue differences, (ii) differences in cell subset composition, (iii) an adjustment test where the data is adjusted and a one-degree-of-freedom test is used for comparing the two groups, (iv) individual tests for each cell-type and (v) an omnibus test for differences across all cell types.
For the first test, we used SAM9 to test for differences between two classes, ignoring differences in cell-type composition. For the second test, for each cell type, we performed a t-test between the two groups to identify substantial differences in composition. Tests 3–5 are new. The third test, data adjustment, has an interesting statistical feature. Let be the average composition, that is, let the average of the rows of W and be the residuals from the fit. We form the adjusted data for each array,
in which is or and pnk is a constant defined in Supplementary Note 2. We have K different potential tests, one for each cell type. We define a single test by averaging these K tests with weights proportional to the cell-type average frequencies. We then compute the usual T-statistic Tj from the adjusted data and use it to test for differential expression. Thus, the adjusted data is well calibrated in the sense that the T-test based on this data is exactly equivalent to the usual statistical test for the corresponding contrast. This equivalence holds for any contrast vector , not just the average composition. In this sense, it is appropriate to treat the adjusted data as real data (see Supplementary Note 2 for a proof and Supplementary Fig. 7 for application of this test on our clinical dataset).
For the fourth test, cell type–specific differential expression, we use the contrast as the test statistic and median-center its distribution. For the omnibus test, we compute the quantity
in which is the estimated standard error of the corresponding difference (see Supplementary Fig. 8 for application of this test on our clinical dataset).
Full R source code for csSAM and demonstrations are available in Supplementary Data. Updates will be available at http://buttelab.stanford.edu/doku.php?id=public:data.
Estimation of FDR for csSAM cell-specific tests
To estimate FDR, we fix X and W and permute y, the assignment of samples to groups, to yield y*. We then fit the two-group model to the data (X,W,y*). As for the cell-specific expression profiles of the original data, we median-center the contrast. In each case we estimate the FDR by V/R, where R is the number of genes exceeding a given threshold in the original data, and V is the average number of genes exceeding the same threshold in the permuted datasets. This yields an estimated FDR for genes for each individual cell-type comparisons as well as for the omnibus test. Use of a positive or negative threshold yields separate FDRs for upregulated or downregulated genes.
Supplementary Material
ACKNOWLEDGMENTS
We thank N. Hartmann, M. Letzkus and E.J. Oakeley for support with mixing experiments; A. Morgan, B. Narasimhan, R. Olshen, E. Eden, members of Stanford Molecular Profiling Colloquium and Biostats forum for enlightening discussions; O. Goldberger, M. Drukker, G. Nestorova, X. Ji and D. Hirschberg for generating mixture datasets; A. Skrenchuk and B. Oskotsky for computer support; and the Lucile Packard Foundation for Children's Health and the Hewlett Packard Foundation for support and computational resources. S.S.S.-O., M.M.D. and A.J.B. are supported by the US National Institutes of Health (NIH) (U19 AI057229). S.S.S.-O. was also supported by the Brennan family. R.T. is supported by the National Science Foundation (DMS-9971405) and NIH (contract N01-HV-28183). P.K. and M.M.S. are supported by NIH (U01 AI077821 and R21 AI075256). T.H. is supported by the National Science Foundation (DMS-0505676) and NIH (2R01 CA 72028-07).
Footnotes
Supplementary information is available on the Nature Methods website.
AUTHOR CONTRIBUTIONS
S.S.S.-O., R.T., D.L.B., F.S., M.M.D. and A.J.B. designed the experiments. S.S.S.-O., R.T., T.H. and P.K. developed the algorithms. M.M.S., F.S. and D.L.B. generated the data. S.S.S.-O., R.T., P.K., N.M.P. and D.L.B. analyzed the data. S.S.S.-O., R.T., P.K., M.M.D. and A.J.B. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturemethods/.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Whitney AR, et al. Proc. Natl. Acad. Sci. USA. 2003;100:1896–1901. doi: 10.1073/pnas.252784499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cobb JP, et al. Proc. Natl. Acad. Sci. USA. 2005;102:4801–4806. doi: 10.1073/pnas.0409768102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmer C, Diehn M, Alizadeh AA, Brown PO. BMC Genomics. 2006;7:115. doi: 10.1186/1471-2164-7-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feezor RJ, et al. Physiol. Genomics. 2004;19:247–254. doi: 10.1152/physiolgenomics.00020.2004. [DOI] [PubMed] [Google Scholar]
- 5.Debey S, et al. Pharmacogenomics J. 2004;4:193–207. doi: 10.1038/sj.tpj.6500240. [DOI] [PubMed] [Google Scholar]
- 6.Stuart RO, et al. Proc. Natl. Acad. Sci. USA. 2004;101:615–620. doi: 10.1073/pnas.2536479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lahdesmaki H, Shmulevich L, Dunmire V, Yli-Harja O, Zhang W. BMC Bioinformatics. 2005;6:54. doi: 10.1186/1471-2105-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang M, Master SR, Chodosh LA. BMC Bioinformatics. 2006;7:328. doi: 10.1186/1471-2105-7-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu P, Nakorchevskiy A, Marcotte EM. Proc. Natl. Acad. Sci. USA. 2003;100:10370–10375. doi: 10.1073/pnas.1832361100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abbas AR, Wolslegel K, Seshasayee D, Modrusan Z, Clark HF. PLoS One. 2009;4:e6098. doi: 10.1371/journal.pone.0006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tusher VG, Tibshirani R, Chu G. Proc. Natl. Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.