

Abstract
Collagen fibrils form extracellular networks that regulate cell functions and provide mechanical strength to tissues. Collagen fibrillogenesis is an entropy-driven process promoted by warming and reversed by cooling. Here, we investigate the influence of noncovalent interactions mediated by the collagen triple helix on fibril stability. We measure the kinetics of cold-induced disassembly of fibrils formed from purified collagen I using turbimetry, probe the fibril morphology by atomic force microscopy, and measure the network connectivity by confocal microscopy and rheometry. We demonstrate that collagen fibrils disassemble by subunit release from their sides as well as their ends, with complex kinetics involving an initial fast release followed by a slow release. Surprisingly, the fibrils are gradually stabilized over time, leading to thermal memory. This dynamic stabilization may reflect structural plasticity of the collagen fibrils arising from their complex structure. In addition, we propose that the polymeric nature of collagen monomers may lead to slow kinetics of subunit desorption from the fibril surface. Dynamic stabilization of fibrils may be relevant in the initial stages of collagen assembly during embryogenesis, fibrosis, and wound healing. Moreover, our results are relevant for tissue repair and drug delivery applications, where it is crucial to control fibril stability.
Introduction
Collagens are the most abundant structural proteins in the extracellular matrix of vertebrates. They form filamentous frameworks that provide mechanical strength to connective tissues such as tendon, skin, and bone (1). Moreover, collagen plays a key role in the regulation of important cell functions such as migration and differentiation (2). The main fibril-forming collagen in mammals is type I collagen, which consists of two identical α1(I) chains and a α2(I) chain intertwined into a rod-like triple helix. In vivo, collagen self-assembles into fibrils upon enzymatic conversion of soluble procollagen precursors into insoluble tropocollagen monomers (3). Tropocollagen has a triple helical region of ∼1000 amino acid residues flanked on both sides by short, nonhelical regions known as the N- and C-telopeptides (Fig. 1 A, see figure caption for definition of length scales). The monomers are very long (300 nm) and thin (1.5 nm) and behave in solution as flexible polymers with a persistence length of ∼10 nm (4,5) (Fig. 1 B). They self-assemble into supramolecular fibrils with a precise axial stagger between neighbors of 67 nm, known as the D-period (6) (Fig. 1, C and D). Because each molecule spans a nonintegral number of periods (4.46 D), each period contains an overlap and a gap region that cause a cross-striated appearance in electron micrographs (Fig. 1 E). The D-banded axial periodicity is essential to ensure a high tensile strength (7,8), to regulate cellular recognition and binding of extracellular matrix molecules (9), and to template bone mineralization (10). Accordingly, mutations in type I collagen that cause defects in assembly are associated with severe connective tissue disorders (11).
Figure 1.
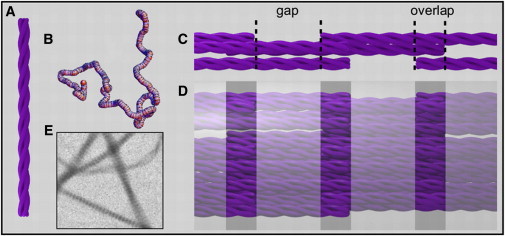
(color online) Schematic representation of the hierarchical self-assembly of type I collagen. (A) Collagen molecules consist of three polypeptide chains twisted into a rod-like triple helix (not shown to scale; in reality the length is 300 and the diameter 1.5 nm). (B) The monomers are flexible polymers with a persistence length of ∼10 nm. Shown is a typical conformation obtained by Monte Carlo simulations (courtesy of H. Amuasi). (C) Pentameric aggregate of collagen monomers in a D-staggered parallel arrangement. Note the straightened conformation of the collagen molecules. Each tropocollagen spans 4.46 D periods, giving rise to an overlap and a gap region within each period. (D) Fibrils are constructed from many pentamers. (E) Transmission electron micrograph (2 μm wide) of negatively stained collagen fibrils assembled at 37°C in a physiological buffer clearly shows D-banding with an axial periodicity of 67 nm.
In vitro assembly studies have provided key insights into the mechanism by which D-staggered collagen fibrils form. It was shown early on that purified collagen can form fibrils with the same axial periodicity as native fibrils without any accessory extracellular matrix components (12). Similar to native fibrils, the fibrils are tens of micrometers in length and ∼100 nm in diameter (13). Thus, all the required structural information is encoded in the primary sequence. The kinetics of fibril assembly has been studied extensively, usually by measuring the increase in solution turbidity that accompanies fibril formation. Turbidimetric growth curves of collagen exhibit an initial lag phase, followed by a growth phase during which the turbidity increases in a sigmoidal fashion (13–16). This behavior is characteristic of a cooperative nucleation and growth mechanism. However, uncertainty remains about the exact nature of the nucleus and the degree of cooperativity (17–21) and about the mechanism of growth (22–25). There is some evidence that five-stranded microfibrils are already formed during the turbidimetric lag phase, which subsequently grow linearly and laterally during the growth phase (16,18). Several studies indicate that collagen monomers undergo conformational changes during the lag phase (23,26–28). Indeed, tropocollagen has to convert from a fluctuating, coiled conformation in solution (Fig. 1 B) to a straightened conformation inside fibrils (Fig. 1 C).
Collagen assembly is driven by noncovalent interactions, which are reversible upon changes in temperature or solution conditions. Assembly is promoted by warming and reversed by cooling, and is thus an endothermic process, similar to the assembly of other biopolymers such as actin and microtubules (29,30) and opposite to typical synthetic supramolecular materials (31). Assembly is thus driven by an increase in entropy, resulting from the release of water molecules from the surface of tropocollagen helices (32). Opinions differ as to the relative contribution of different classes of noncovalent interactions to the thermodynamic driving force for assembly. Hydrophobic interactions are likely to play an important role. Atomistic simulations showed that water molecules are released from hydrophobic amino acid residues when the molecules pack into a fibril (33). Moreover, analysis of the amino acid sequence of collagen showed that hydrophobic residues occur in clusters and that D-staggered packing optimizes the alignment of hydrophobic side chains between apposing tropocollagens (34,35). Yet, there is also an alternative interpretation of the endothermic nature of collagen assembly, namely that water is released from polar residues. According to this interpretation, the dominant driving force is provided by hydration forces associated with water-mediated hydrogen bonding between polar residues (36–38). This interpretation is supported by observations that sugars and polyols strongly reduce the attraction between collagen helices and suppress fibrillogenesis (19,39,40). These compounds are expected to disrupt water-mediated as well as direct hydrogen bonding between polar residues. Recent simulations indeed indicate that clusters of hydrogen-bonded water exist in collagen fibrils, but the relative importance of hydration forces and hydrophobic forces could not be determined (33). The dependence of fibril assembly on solution pH and ionic strength suggests that electrostatic interactions also reinforce the stability and/or axial specificity of assembly (20,41,42). The D-staggered packing arrangement appears to optimize alignment between complementary charges on opposing helices (34). Some studies also implicate specific ions such as divalent phosphate in providing salt bridges and improving axial order (15,42,43). Finally, there is evidence that the telopeptides further enhance the specificity of molecular recognition (44). When the telopeptides are removed by pepsin or other proteases, nucleation is slowed down and the activation energy is increased (45–47). Computational models show that telopeptides dock to specific helix recognition sites and induce reciprocal conformational changes (48,49). It remains unknown whether this interaction is only catalytic in the initial stages of assembly or contributes also to the driving force.
Despite several decades of intensive research on the process of collagen assembly, the molecular basis of the interactions responsible for collagen fibril stability (and mechanical strength) remains poorly understood. Here, we take an alternative approach to elucidate the role of noncovalent interactions mediated by the collagen triple helix in fibrillogenesis. Rather than studying the process of assembly, we focus on the kinetics of cold-induced fibril disassembly. As a model system, we use collagen whose telopeptides are removed by pepsin. This is necessary to prevent spontaneous formation of intermolecular covalent cross-links (17,46,50–52). We used several complementary methods to characterize the kinetics of cold-induced disassembly of collagen fibrils formed at body temperature (37°C): we used turbimetry to probe the fraction of collagen in fibril form, atomic force microscopy (AFM) to probe the fibril morphology, and confocal microscopy and rheometry to measure the network connectivity. We demonstrate that collagen fibrils disassemble by subunit release from their sides as well as their ends, with complex kinetics involving an initial fast release followed by a slow release. Despite the absence of telopeptides, the fibrils are gradually stabilized over time. This dynamic stabilization may reflect structural plasticity of the collagen fibrils arising from their complex structure. In addition, we propose that the polymeric nature of collagen monomers may lead to slow kinetics of subunit desorption from the fibril surface. Dynamic stabilization of fibrils may be relevant in the initial stages of collagen assembly in vivo (2).
Materials and Methods
Preparation of collagen gels
Bovine dermal collagen I (NUTRAGEN) was purchased as a 6.0 mg/ml solution in 0.01 M HCl from Advanced Biomatrix (San Diego, CA). This collagen is isolated from bovine hides by enzymatic digestion with pepsin, which removes portions from the N- and C-terminal telopeptides containing the lysine groups that are responsible for intermolecular cross-linking in vivo (53). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 4% gels was used to assess the purity and degree of intramolecular cross-linking. Gels were stained with Instant Blue Coomassie staining solution (Expedeon, Harston, UK). Samples with a final volume of 500 μL and collagen concentration of 1 mg/ml were prepared by diluting the stock solution with a buffer mixture, to give a final buffer composition of 10 mM phosphate and 160 mM NaCl at pH 7.2 (I = 0.17). This is a near-physiological buffer that promotes a native D-banding pattern. In the case of turbidity experiments, the buffer mixture was degassed in a vacuum before mixing with collagen, to prevent the formation of air bubbles. All preparation steps involving collagen were performed at 4°C. Unless otherwise noted, chemicals were purchased from Sigma Aldrich (St. Louis, MO). The extent of collagen polymerization at 37°C was tested by precipitating the fibrils by centrifugation for 10 min at 16,100 × g in a 5415R tabletop centrifuge (Eppendorf, Hamburg, Germany) at 37°C. The collagen concentration in the supernatant was measured using the sirius red assay (54). The supernatant (100 μl) was incubated for 30 min with 1 ml of 1 mg/ml Direct Red 80 in 1% picric acid. The solution was centrifuged for 10 min at 16,100 × g, the supernatant was removed, and the pellet was washed (to remove excess dye) with 750 μl of 0.01M HCl. The pellet was finally dissolved in 250 μl 0.5M NaOH. After 5 min, the solution absorbance at 550 nm was measured using a Nanodrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA) and calibrated against stock collagen in the same buffer.
Turbidity measurements
The kinetics of collagen polymerization at 37°C and subsequent depolymerization induced by cooling were probed by measuring the change in solution absorbance at a wavelength of 370 nm (A370) with a Lambda 35 dual-beam spectrophotometer (PerkinElmer, Waltham, MA) equipped with a PTP 6+6 Peltier temperature controller. Solutions of 1 mg/ml collagen were pipetted into a quartz cuvette (Quartz Suprasil precision cells 115B-S, Hellma Analytics, Mullheim, Germany) equilibrated to 37°C and absorbance readings were initiated within 15 s. The baseline value was taken as the first recorded turbidity value. The absorbance readings were converted into turbidity values, τ, by using the relation τ = (A370 ln (10))/L, where L is the optical path length (1.0 cm). It was previously shown by comparison with fibril precipitation experiments that the turbidity of collagen solutions is directly proportional to the amount of mass present in fibril form (13,15). Thus, the turbidity provides a reliable, and model-independent, measure of the relative degree of polymerization as a function of temperature. In some experiments we measured the wavelength dependence of the turbidity with 2 min time intervals; using light scattering theory modeling the fibers as rigid rods (55,56) it is possible to infer the fibril diameter and mass-length ratio (see the Supporting Material Note S1 and Fig. S1). Collagen fibrils were always first allowed to assemble for 2 h at 37°C, and the absorbance was recorded with 30 s intervals. Subsequently, fibril disassembly was induced by lowering the temperature to target values between 4 and 37°C. We used two different disassembly protocols: temperature step experiments and temperature ramp experiments. The actual time needed for the sample to reach a given target temperature in step experiments varied from 1 to 4 min, depending on the target temperature, as measured with a thermocouple. In temperature ramp experiments, the temperature was lowered at a constant rate between 0.2 and 129°C/h. The actual time dependence of the temperature was measured with a thermocouple and used in the analysis. The solution absorbance during disassembly was measured for 2 h at 30 s intervals in step experiments and at 15 min intervals during ramp experiments.
AFM and transmission electron microscopy (TEM)
To characterize fibril morphology and diameter, we imaged fibrils deposited on Formvar-coated glass coverslips using AFM (Dimension 3100 AFM, Veeco digital instruments, Plainview, NY). We were unable to image the fibrils in buffer, because the fibrils are very soft and easily pollute the AFM tip or dislodge from the surface. For this reason, we imaged the fibrils in air. For comparison, we also performed TEM at 80kV (Tecnai G2, FEI company, Hillsboro, OR) on samples that were prepared identically, but on Formvar-coated copper electron microscopy (EM)-grids. The glass substrates for AFM were cleaned with 70% ethanol and dried with a flow of N2, and then briefly dipped into a Formvar solution (1% in 1,2-dichloroethane) and slowly extracted to ensure a homogeneous layer. The Formvar was allowed to dry in air. The substrate was placed gently for 10 s on top of a collagen gel (polymerized in a humified petri dish) at the appropriate temperature. The substrate was washed 3 times with phosphate buffered saline (PBS) of pH 7.2 and 3 times with milliQ, water to prevent the formation of salt crystals. All solutions were of the appropriate temperature and excess liquid was blotted off with filter paper after each washing step. The substrate was dried at 37°C for at least 2 h. The fibrils were imaged in tapping mode using a TESPA doped silicon cantilever (Bruker AXS, Camarillo, CA) with a nominal tip radius of 8 nm and spring constant of 42 N/m. Images were flattened using the Nanoscope software (6.14R1, Veeco digital instruments), but otherwise unaltered. The fibril width and height were analyzed in ImageJ software (version 1.46a, National Institutes of Health, Bethesda, MD), averaging over 20 fibrils per experimental condition.
Confocal reflectance microscopy
The three-dimensional architecture of collagen gels in their native, hydrated state was examined by confocal microscopy with an Eclipse Ti inverted microscope (Nikon instruments Europe, Amstelveen, The Netherlands). We used the reflectance mode, to avoid the use of fluorescent labels, which can influence collagen (de)polymerization (57–59), using 457 nm light from an Ar laser (Melles Griot, Albuquerque, NM). Collagen gels were polymerized for 2 h at 37°C in glass chambers made from a microscope slide and coverslip separated by Parafilm spacers and sealed with vacuum grease. Gels were imaged either directly after polymerization or after subsequent cooling to 4°C. Image stacks were obtained at least 10 μm away from the glass surface by scanning through the z direction in steps of 0.5 μm over a range of 20 μm with a piezo-driven 100× oil immersion objective (Nikon). To optimize the signal/noise ratio, we used 2× line average. Maximum intensity z projections were made with ImageJ.
Cone-plate rheology
The linear viscoelastic properties of the collagen gels were measured using a stress-controlled MCR501 rheometer (Anton Paar, Graz, Austria) with a CP40-2 acrylic measuring cone with 40 mm diameter and 2° angle and a steel bottom plate heated to 37°C with a PTD200 Peltier system. The collagen solution was applied to the prewarmed rheometer, and allowed to polymerize in situ for 2 h before starting oscillatory shear measurements. The sample was in a closed hood to maintain a constant temperature and humidity. The frequency-dependent shear modulus G∗(ω) = σ(ω)/γ(ω) of the collagen gels was measured by applying an oscillatory strain γ(ω) decreasing logarithmically from 34 to 0.0628 rad/s, and recording the stress response, σ(ω). The strain amplitude was 0.5%, which is much smaller than the critical strain (∼5%) where nonlinearity sets in. G∗(ω) is a complex modulus with an in-phase (elastic) component, G′(ω), and an out-of-phase (viscous) component, G″(ω).
Results
Kinetics of collagen assembly and disassembly
Collagen self-assembly is entropy-driven and depends on noncovalent interactions (20,32). Fibrils can be assembled by heating a collagen solution from 4 to 37°C and disassembled by subsequent cooling. We use turbidimetry to analyze the kinetics of the disassembly process in response to a sudden stepwise lowering of the temperature, because the solution turbidity is a direct measure of the amount of mass present in fibril form (13,15). Fig. 2 a shows three typical assembly-disassembly experiments, with identical assembly conditions (1 mg/ml collagen in PBS, 2 h at 37°C) but differing in disassembly temperature (Tdis = 4, 17, 27°C). The change in turbidity during the assembly phase is highly reproducible, showing three distinct phases characteristic of a nucleation-and-growth process (14). There is an initial lag phase of ∼10 min with no turbidity change. Prior studies indicate that short, thin fibrils are already present at the end of the turbidimetric growth phase with ∼100 collagen molecules per unit length, but the structures are too small during the lag phase to give a measurable turbidity (16,22,60). This is confirmed by our own wavelength-dependent turbidity measurements (Note S1 and Fig. S2 a). The lag phase is followed by a growth phase with a sigmoidal increase in turbidity, during which the collagen fibrils grow in length and width. Wavelength-dependent turbidity data show that the mass-length ratio of the fibrils also increases sigmoidally during the growth phase (Fig. S2 a). Finally, all three turbidity curves reach a plateau phase where the turbidity reaches a constant level (2.57 ± 0.06), which varies by <4% among experiments. It actually takes 6 h for the turbidity to reach a constant value, and at 2 h, the turbidity has reached ∼93% of this maximum (consistent with precipitation experiments, showing that collagen assembly after 2 h is 87% complete). Nevertheless, for practical reasons we always assemble gels for only 2 h, and denote the fibrils at this point as mature, with a turbidity value τ37. Wavelength-dependent turbidity data indicate that the mature fibers have an average diameter of 61 nm (Fig. S2 b) with 1070 monomers per cross section (Note S1), consistent with prior reports for rat tail collagen (61).
Figure 2.
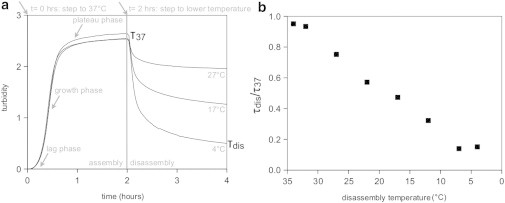
Turbidity measurement of the assembly and disassembly kinetics of a 1 mg/ml collagen solution. (a) Turbidity response for three collagen samples, each assembled for 2 h at 37°C and disassembled by cooling to different temperatures, Tdis. (b) Temperature dependence of τdis/τ37 (note the inverted temperature axis).
After 2 h, we reduce the temperature stepwise, which causes an immediate drop in the turbidity (Fig. 2 A). The slope of the turbidity response curve is relatively shallow in the first few minutes, because of the finite duration of the temperature step (1–4 minutes). The turbidity value τdis reached after 2 h of disassembly is strongly dependent on the disassembly temperature: the lower the temperature, the lower τdis, indicating that the fibrils release more mass. The kinetics of disassembly clearly does not follow a simple single exponential form. Empirically, the decay curves are well fit by a sum of two exponentials, corresponding to a fast decay with a characteristic decay time of ∼9 min, independent of disassembly temperature, followed by a slow decay with a decay time that increases from 200 min at 4°C to 2000 min at 27°C (Fig. S3). At 32°C, the turbidity is essentially constant at long times. As shown in Fig. S4, the turbidity during a disassembly experiment at 22°C is actually still not constant after 4 days.
To quantify the extent of disassembly, we take the ratio between the turbidity measured after disassembly and that measured for the mature fibrils, τdis/τ37, which is a measure of the degree of fibril stability. For practical reasons, we measure this ratio after 2 h of disassembly. Although the disassembly process is <80% complete at this time (Fig. S4), the rate of disassembly is already much smaller than directly after the temperature step, making this an acceptable choice. As shown in Fig. 2 B, τdis/τ37 decreases nearly linearly when the disassembly temperature is lowered from 32 to 7°C. The turbidity does not return to zero upon cooling, even to 4°C, which indicates that the collagen fibrils do not completely disassemble. To test whether collagen fibrils indeed remain in solution, we cycle samples cooled down to 22°C for 2 h back to 37°C. As shown in Fig. S5, disassembly is reversible, because heating to 37°C brings the turbidity back to its original value. Strikingly, there is no lag phase during the reassembly process, which strongly indicates that collagen fibrils indeed remain and can act as templates for fibril growth upon reheating. The wavelength dependence of the turbidity indicates that fibrils disassembled for 2 h at 4°C still contain 229 monomers per cross section, which is ∼20% of the mass/length ratio of mature fibrils (Fig. S2 a). The loss in mass from the sides (μdis/μ37) is comparable to the overall loss in fibril mass (τdis/τ37) at all disassembly temperatures (Fig. S6), suggesting that fibrils predominantly shed monomers from their sides.
The apparent stability of the fibrils at 4°C is surprising, because collagen solutions kept at 4°C and never exposed to 37°C do not develop any measurable turbidity (Fig. S7). This history dependence suggests that fibrils formed at 37°C and cooled to 4°C are kinetically stabilized. It should be noted that, although a collagen solution at 4°C does not produce aggregates detectable by the turbidity method, some aggregation does take place at 4°C. When a neutralized collagen solution is heated to 37°C after 17 h incubation at 4°C, the resulting turbidity curve has no lag phase (inset of Fig. S7). Apparently, there is a nonzero driving force for collagen to self-assemble at 4°C into structures that can act as nuclei for fibril growth. The rate of assembly after 17 h incubation at 4°C is slower than the rate of assembly of a freshly neutralized collagen solution (cf. Fig. 2 A), suggesting that the oligomeric structures formed at 4°C are different from the aggregates formed in the nucleation phase at 37°C. We conclude that there is a clear history dependence of the turbidity measured at 4°C, suggestive of kinetic stabilization.
To test whether the stability of collagen fibrils at low temperature is truly kinetic in origin, we examined whether collagen gels could be redissolved in 0.01 M HCl. Under these conditions, there is no driving force for collagen fibrillogenesis (62). We polymerize collagen at 1 mg/ml for 2 h and then dialyze against 0.01M HCl at 4°C. To test whether fibril disassembly is complete, we perform an assembly experiment at 37°C with the dialyzed solution. If the fibrils were fully dissolved, the assembly process should have a lag phase, similar to the first assembly step in Fig. S5. However, if parts of fibrils or other nuclei remain because they are being held together by covalent cross-links, there should be no lag phase. For the assembly experiment we use a concentration around 0.1 mg/ml, where the lag phase is longer and easier to recognize than at 1 mg/ml (13). We indeed find that the lag phase returns (Fig. S8), which implies that the collagen fibrils are in principle fully soluble. This indicates that there is some kinetic mechanism that stabilizes the fibrils in a near-physiological buffer at neutral pH. Moreover, this stabilization is apparently an intrinsic property of the collagen triple helix, because we use pepsin-treated collagen that lacks the telopeptides.
Rate dependence of fibril disassembly
The experiments described previously show the response of the system to an abrupt change of temperature. This is a nonequilibrium experiment, because the temperature is changed much faster than the equilibration time of fibril disassembly. To obtain more insight into the fibrils’ equilibration behavior, we perform a series of experiments where the temperature is lowered from 37 to 22°C at a constant rate. Because the temperature jump experiments show a fast disassembly process with a decay time of 9 min and a slow process with a time constant of several days, we choose a range of cooling rates from 0.2°C/h (which takes over 3 days) to 128.6°C/h (which takes 7 min). Fig. 3 shows a typical turbidity response curve, which encompasses three stages, as indicated by the dotted line showing the temperature: 1), assembly for 2 h at 37°C, 2), temperature ramp down to 22°C with a constant rate, and 3), a tail stage at a constant temperature of 22°C. In the assembly stage, fibrils are formed, producing a sigmoidal increase in turbidity. Next, the temperature is ramped down toward 22°C at a rate of 5°C/h. This does not lead to an immediate change in τ/τ37, likely because the fibrils are not yet fully assembled after 2 h, and continue to grow as long as the temperature is sufficiently close to 37°C. After a short delay, though, τ/τ37 drops at a constant rate until the target temperature is reached. Strikingly, τ/τ37 continues to decrease at 22°C, at a rate that gradually slows with time. We call this stage the tail stage.
Figure 3.
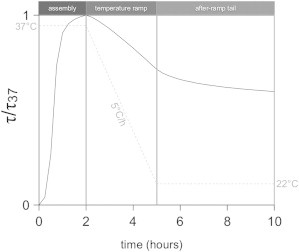
Normalized turbidity response (τ/τ37) of a 1 mg/ml collagen solution during a temperature ramp experiment. There are three stages, as indicated by the dotted line showing the temperature: 1), an assembly stage for 2 h at 37°C, 2), a temperature ramp stage in which the temperature is lowered to 22°C at a constant rate, and 3), a tail stage, during which the temperature is maintained at 22°C.
Remarkably, the fibrils’ response during the temperature ramp stage is independent of the cooling rate within experimental error (Fig. 4). Note that, because the sampling rate is limited to one measurement every 15 min, the faster ramps have only a few data points. Nevertheless, these data clearly show that τ/τ37 is a function of the temperature alone, and not of the rate of change of the temperature. This is true for a broad range of ramp rates, with the fastest rate being over 600 times faster than the slowest one. This result suggests that a certain fraction of collagen monomers on the surface of the fibrils is quickly exchangeable. However, this is clearly not true for all monomers, because after the ramp stage, the fibrils continue to disassemble at a slow rate. As shown in Fig. 5 A, the extent of disassembly at 22°C depends on the cooling rate of the preceding ramp stage. The higher the cooling rate during the ramp, the larger the decrease of τ/τ37. The disassembly curves have a complex functional form, which we did not attempt to fit. The initial rate of disassembly, measured as the initial slope of τ/τ37, increases approximately linearly with the cooling rate (Fig. 5 B). This thermal memory effect is strongly suggestive of a slow, time-dependent stabilization of the collagen fibrils: when the fibrils are cooled at a slower rate, there is more time for fibril stabilization. To test this hypothesis, we assemble collagen fibrils at 37°C for various lengths of time and measure the turbidity change in response to abrupt cooling to 22°C. In accordance with our hypothesis, we observe that the apparent stabilization (expressed in terms of τdis/τ37) increases from τdis/τ37 = 0.24 to 0.75 when the incubation time at 37°C is increased from 1 to 18 h (Fig. S9).
Figure 4.
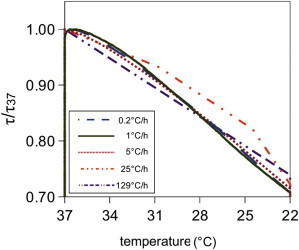
(color online) Normalized turbidity response (τdis/τ37) of five different collagen networks during the temperature ramp stage of temperature ramp experiments. In each case, the temperature is lowered from 37°C to 22°C, but at different rates, as indicated in the legend.
Figure 5.
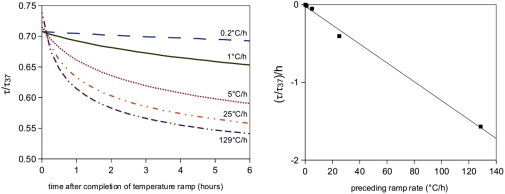
(Left) Normalized turbidity response (τdis/τ37) of five different collagen networks during the tail stage of temperature ramp experiments. For a larger preceding ramp rate, there is more disassembly during the tail stage. (Right) The initial slope of each tail response curve is a linear function of the preceding ramp rate.
Disassembly causes fibril thinning but leaves the fibril structure intact
Turbidimetry provides an ensemble measure of the degree of fibril disassembly, but gives no information on the changes in fibril morphology or network structure upon cooling. It is conceivable that disassembly might change the packing structure of the fibrils. To directly visualize effects of disassembly on fibril morphology, we use AFM to image collagen fibrils directly after assembly (2 h at 37°C) and after disassembly at either 22 or 4°C for 2 h. As shown in Fig. 6, mature collagen fibrils are long and straight and have an average apparent width of 221 ± 28 nm and height of 30 ± 9.6 nm. This flattening was also observed elsewhere (63), and is most likely a consequence of adsorption to the substrate and (partial) loss of hydration water upon drying in air (64). The measured fibril width and height are therefore not directly representative of the diameter of fibrils in their native, hydrated state. However, we can still use AFM to measure relative differences among different temperature conditions. In most of the fibrils the distinctive 67 nm D-periodic banding is visible, as exemplified by the axial height profile shown underneath the image.
Figure 6.

Atomic force micrographs of collagen fibrils assembled at 1 mg/ml, (a) directly after assembly or after subsequent 2 h disassembly at (b) 22°C or (c) 4°C. Representative height profiles are measured along a fibril in each image (white lines in the insets), showing that the 67 nm D-periodicity is present under all three conditions. Insets (0.78 × 0.78 μm2) show an enlarged view of the measured fibrils.
Disassembly at 22 or 4°C for 2 h clearly changes the fibril dimensions. The average apparent width decreases to 175 ± 25 nm at 22°C and 146 ± 34 nm at 4°C, whereas the average height decreases to 22 nm in both cases (± 9 nm at 22°C and ± 4 nm at 4°C). These changes indicate that the fibrils lose mass from their sides, consistent with the reduction of the fibril mass-length ratio measured by wavelength-dependent turbidimetry (Fig. S6). However, the major morphological features are not visibly affected by disassembly. The fibrils still appear long and straight, and retain their D-banding, indicating that the native intermolecular stagger is still intact. The fibrils still appear as flattened cylinders, structurally the same as mature fibrils, which suggests that no large groups of monomers break off during the disassembly process. We conclude that disassembly occurs all along the fibril by dissociation of monomers or small aggregates from the outer surface of the fibril, with the remaining fibril core retaining its structure and organization.
A striking feature of the AFM data is the material in the background. For the mature fibrils there is a small amount of material, consisting of amorphous aggregates. For the partially disassembled fibrils, the background material is more abundant, especially for the fibrils disassembled at 4°C. For fibrils disassembled at 4°C the background material looks fibrillar. However, it is uncertain whether the background material is also present in solution. We cannot rule out that it may result from the AFM sample preparation. The Formvar-coated glass substrate has a high affinity for collagen, and may therefore cause monomers or oligomers to aggregate on the surface (65). Similar aggregates appear in TEM images of fibrils on Formvar-coated electron microscopy (EM) grids (Fig. S10).
Disassembly minimally affects network connectivity
The AFM images demonstrate that the turbidity decrease upon cooling is, at least partly, caused by monomer loss from the sides. However, they do not reveal any effects on fibril length because the fibrils are many micrometers long (22). To test whether fibrils also lose mass from their ends, we probe the change in network connectivity at 4°C with two complementary methods. We use confocal reflectance microscopy (CRM) to visualize the networks in their native, hydrated state and reveal their connectivity, homogeneity, and mesh size (57). Moreover, we use cone-plate rheology to measure the elastic modulus of the gels, which is sensitive to network connectivity (66).
Fig. 7 shows two maximum intensity projections of 20 μm-thick sections of collagen gels, one taken of a gel formed at 37°C for 2 h (A) and the other taken after subsequent disassembly at 4°C for 2 h (B). At first sight, both images look indistinguishable: the networks are homogeneous and isotropic and have a similar fibril density. In contrast to the AFM images, no change in fibril diameter can be observed. Indeed, the fibrils have diameters below the diffraction limit, so a diameter reduction is only expected to show up in a CRM image when it is so extreme that the fibrils do not backscatter any light. In the mature gel at 37°C, there are no visible fibril ends and also time lapse imaging shows that the fibrils are motionless. Thus, we are unable to determine the lengths of the fibrils. In the gel that has been disassembled at 4°C we can still not recognize fibril ends in still images, but time lapse imaging reveals some dangling fibril ends as recognized by their transverse fluctuations. This effect is illustrated in Fig. 7 C, which shows five images with an undulating fibril end in the middle, recorded 4.75 s apart. The position and shape of the fibril in the first frame are marked with five white dots in each frame, which clearly reveals motion of the fibril end. The maximum intensity projection (panel all) shows that the fibril end undulates, whereas the other fibrils are motionless. Apparently, there is some endwise shrinkage of filaments. However, the loss in fibril mass (85% at 4°C) can clearly not be explained by endwise shrinkage alone, because then we should see only 15% of the original fibril density in the confocal images of disassembled gels. Indeed, the turbidity data show that the overall loss in fibril mass is close to the loss in mass-length ratio (Fig. S6): at 4°C, the overall mass loss relative to mature fibrils is 85%, whereas the loss from the sides is 79%.
Figure 7.
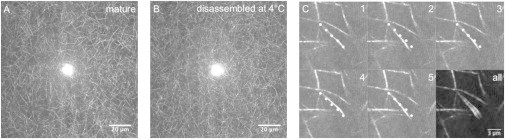
Maximum intensity projections of z-stacks (41 z planes spaced by 0.5 μm) of a 1 mg/ml collagen gel, (a) directly after 2 h of assembly at 37°C, and (b) after subsequent disassembly for 2 h at 4°C. The bright spot in the center of the images is an artifact of CRM caused by reflection. (c) Five frames of a time lapse movie (taken 4.75 seconds apart) of a 1 mg/ml collagen gel after partial disassembly at 4°C for 2 h. The dotted line indicates a fibril with a dangling end. The last image (all) shows a maximum intensity projection that clearly reveals fluctuations of the dangling fibril end.
To verify that disassembly indeed does not disrupt network connectivity, we also performed rheology on gels before and after disassembly. Fig. 8 shows that after assembly for 2 h at 37°C, the storage modulus, G′, is 30 Pa at 1 rad/s, consistent with prior reports (57,66). The gels behave as weak elastic solids, with a slight frequency dependence of G′ and a loss tangent G″/G′ of 0.25 (inset). After subsequent disassembly for 2 h at 4°C, the G′ is reduced ∼70-fold at an angular frequency of 10 rad/s. However, the sample is still a solid, with an elastic modulus that is only weakly dependent on frequency and a loss tangent that is still below 1 (0.08). Despite losing 85% of its mass, the fibril network remains space-spanning and elastic. The reduction of G′ may be caused by the increase in elastically inactive dangling ends observed by confocal microscopy. Prior rheology measurements of collagen networks have shown that G′ depends strongly on protein concentration, usually scaling as a power law in concentration with an exponent between 2 and 3 (61,66,67). In addition, the reduction of G′ may reflect the reduced fibril rigidity caused by the substantial reduction in fibril diameter. There is not yet a quantitative theoretical model that predicts the modulus of collagen networks. There is evidence that collagen networks deform in a highly nonaffine (nonuniform) manner (68). Nonaffinity is known to make networks much softer than they would be in the affine limit, but there is no quantitative (analytical) prediction of the extent of softening (69). When we reassemble the gel for 2 h at 37°C, G′ and G″ both return close to their original values. This is in accordance with the turbidity assay showing a return of the original solution turbidity (Fig. S2).
Figure 8.
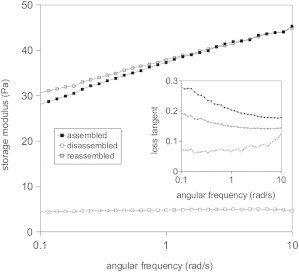
Network storage (elastic) modulus as a function of angular frequency, for a 1 mg/ml collagen gel subjected to a temperature cycle: fibrils were first assembled for 2 h at 37°C, then cooled down for 2 h at 4°C, and finally reassembled for 2 h at 37°C. The inset shows the corresponding loss tangents.
Discussion
Prior studies of collagen reversibility focused on covalent stabilization by the telopeptides, which is crucial for the tensile strength of collagen fibrils in vivo. Assembly of neutral-salt soluble or acid-solublized collagen, both of which possess intact telopeptides, is only partly reversible (52,70). Already during polymerization, allysine residues in the telopeptide regions condense with lysine or hydroxylysine residues in helix domains of adjacent molecules in the fibril, creating intermolecular cross-links. Upon aging of collagen gels at 37°C, assembly becomes progressively more irreversible (50,71). When cross-linking is prevented by using lathyritic collagen from animals fed with an inhibitor of lysyl oxidase, assembly is more reversible (72). Yet, there are a few prior turbidimetric reports hinting at a time-dependent increase in stability of fibrils formed from pepsin-collagen (73,74). The buffer composition was different from ours (pH 6.8, 300 mM phosphate), but the observations were comparable. In response to a sudden temperature drop, the turbidity decayed in a biphasic manner and gels aged at 37°C became progressively more insoluble. However, no imaging data were shown, so it is unclear what was the packing arrangement of monomers (which may not be D-staggered at 300 mM phosphate) or the relative contributions of endwise and sidewise disassembly. Similar observations were reported for lathyritic collagen, which also showed thermal memory effects despite the absence of cross-linking (72).
What is the mechanism of the time-dependent stabilization of fibrils? One viable mechanism is structural plasticity. This phenomenon was proposed to cause dynamic stabilization of two other supramolecular protein polymers, actin filaments and microtubules (75,76). Actin filaments, like collagen fibrils, display a biphasic disassembly response and a progressive stabilization upon aging. Based upon EM and x-ray scattering data, it was proposed that filaments can gradually change from a more disordered and unstable structural state into a more ordered and stable structural state. Recently, however, it was proposed that the reported stabilization of actin was due to photo-induced cross-linking induced by the intense illumination employed in the fluorescence microscopy assays used for monitoring disassembly (77). In our study, we robustly find time-dependent stabilization using techniques which do not employ illumination (EM, AFM, rheology). We propose that collagen fibrils may be relatively disordered directly after assembly, but acquire more order over time, by internal rearrangements. It is a formidable challenge, however, to directly demonstrate structural plasticity for collagen fibrils, given their enormously complex and semicrystalline three-dimensional architecture. While actin filaments and microtubules behave as linear polymers whose assembly dynamics are restricted to the ends, collagen fibrils have hundreds of monomers per cross section packed in an array whose lateral packing order remains poorly understood. Our EM and AFM images indicate that axial order is established immediately, probably because of the high degree of specificity of the noncovalent interactions that drive assembly. However, we have no information on the lateral packing order. X-ray diffraction measurements of native collagen fibrils show varying degrees of lateral disorder (6,78). Fibrils assembled in vitro were shown to have an even less well-ordered lateral packing (12). Synchrotron x-ray scattering measurements or EM microscopy of fibril cross sections may shed more light on time-dependent changes in lateral packing (79). Alternatively, antibodies might be developed that recognize specific structural states of collagen fibrils in analogy to actin and microtubules (80,81). A complementary approach may be computational modeling of collagen fibril assembly using coarse-graining approaches (82), possibly combined with atomistic simulations (5,33).
There are additional complexities in collagen assembly that may contribute to kinetic stabilization of the fibrils. Unlike actin and tubulin monomers, which are compact globular subunits, collagen monomers are long polymers with substantial flexibility (4,5). Both monomer additions to the surface of growing fibrils and monomer removal from disassembling fibrils require large conformational changes of the monomers, which may pose large kinetic barriers. There is indeed some evidence from hydrodynamic assays and vibrational spectroscopy of intramolecular conformational changes during fibril nucleation and growth (23,26–28). A tractable way to model these processes may be by using theories of polymer crystallization and desorption (83–85). Desorption of semiflexible polymers from a surface is known to be slow. Moreover, it is possible that collagen disassembly sets up a complex ecosystem of different supramolecular species that can exchange with the sides and ends of fibrils. For instance, oligomers may be released in addition to monomers and released monomers may potentially associate in solution.
Dynamic stabilization of fibrils may be relevant in the initial stages of collagen assembly in vivo, particularly in growing tissues and during fibrosis and wound healing (2). It will be interesting to test whether other extracellular matrix compounds such as proteoglycans affect the kinetics of cold-induced disassembly of collagen (3). Our results may be of even more direct relevance in the context of biomedical applications. One important area is tissue optical clearing for biomedical optics and photomedicine (86). It is known that the turbidity of biological tissues can be reduced by immersion in chemical agents such as sugar alcohols and restored by immersion in physiological saline. It was proposed that these agents reversibly weaken noncovalent binding forces. Our results may help to explain the basis of this reversibility. A second application area where our results are relevant is in tissue regeneration and drug delivery (87). It is crucial to understand the molecular basis of collagen stability to control the kinetics of in vivo remodeling, resorption, and controlled drug release.
Conclusions
We demonstrate that collagen fibrils disassemble by subunit release from their sides as well as their ends, with complex kinetics involving an initial fast release followed by a slow release. Despite the absence of telopeptides, the fibrils are gradually stabilized over time. On a timescale of several days, the fibrils are stable even at temperatures as low as 4°C. Yet, when collagen is never heated and kept at 4°C, there is no formation of fibrils. This is clearly indicative of a kinetic stabilization. Thermal memory is also evident from temperature ramp experiments: when collagen solutions are cooled at a constant rate, the turbidity decreases in a rate-independent manner during cooling, but continues to decrease slowly at a rate that is correlated with the preceding cooling rate once the temperature has reached a constant value. Complementary experiments show that the stability of collagen fibrils formed at 37°C depends on age. We can rule out any irreversible changes such as denaturation, because the fibrils are perfectly soluble in acid and fibrils can be reverted to their original state by reheating to 37°C. We propose that the complex, hierarchical structure of collagen fibrils is responsible for the kinetic stabilization. The dynamic stabilization may reflect structural plasticity of the collagen fibrils arising from their complex structure or slow kinetics of subunit desorption from the fibril surface related to the polymeric nature of the collagen subunits. Dynamic stabilization of fibrils may be relevant in the initial stages of collagen assembly during tissue morphogenesis, fibrosis, and wound healing.
Acknowledgments
The authors thank C. Storm, H. Amuasi, B. Meijer, T. de Greef, P. van der Schoot, and B. Mulder for helpful discussions. This work is part of the Industrial Partnership Programme (IPP) Bio(-Related) Materials (BRM) of the Stichting voor Fundamenteel Onderzoek der Materie (FOM), which is financially supported by the Dutch Organization for Scientific Research (NWO). The IPP BRM is cofinanced by the Top Institute Food and Nutrition and the Dutch Polymer Institute.
Footnotes
Wim Pomp’s present address is Leiden Institute of Physics, Leiden University, Niels Bohrweg 2, 2333 CA Leiden, the Netherlands.
Supporting Material
References
- 1.Wess T.J. Collagen fibril form and function. Adv. Protein Chem. 2005;70:341–374. doi: 10.1016/S0065-3233(05)70010-3. [DOI] [PubMed] [Google Scholar]
- 2.Cox T.R., Erler J.T. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011;4:165–178. doi: 10.1242/dmm.004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kadler K.E., Hill A., Canty-Laird E.G. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008;20:495–501. doi: 10.1016/j.ceb.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun Y.L., Luo Z.P., An K.N. Direct quantification of the flexibility of type I collagen monomer. Biochem. Biophys. Res. Commun. 2002;295:382–386. doi: 10.1016/s0006-291x(02)00685-x. [DOI] [PubMed] [Google Scholar]
- 5.Buehler M.J., Wong S.Y. Entropic elasticity controls nanomechanics of single tropocollagen molecules. Biophys. J. 2007;93:37–43. doi: 10.1529/biophysj.106.102616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orgel J.P., Irving T.C., Wess T.J. Microfibrillar structure of type I collagen in situ. Proc. Natl. Acad. Sci. USA. 2006;103:9001–9005. doi: 10.1073/pnas.0502718103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gautieri A., Vesentini S., Buehler M.J. Hierarchical structure and nanomechanics of collagen microfibrils from the atomistic scale up. Nano Lett. 2011;11:757–766. doi: 10.1021/nl103943u. [DOI] [PubMed] [Google Scholar]
- 8.Yang L., van der Werf K.O., Bennink M.L. Micromechanical analysis of native and cross-linked collagen type I fibrils supports the existence of microfibrils. J. Mech. Behav. Biomed. Mater. 2012;6:148–158. doi: 10.1016/j.jmbbm.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Perumal S., Antipova O., Orgel J.P. Collagen fibril architecture, domain organization, and triple-helical conformation govern its proteolysis. Proc. Natl. Acad. Sci. USA. 2008;105:2824–2829. doi: 10.1073/pnas.0710588105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nudelman F., Pieterse K., Sommerdijk N.A. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat. Mater. 2010;9:1004–1009. doi: 10.1038/nmat2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prockop D.J., Kivirikko K.I. Collagens: molecular biology, diseases, and potentials for therapy. Annu. Rev. Biochem. 1995;64:403–434. doi: 10.1146/annurev.bi.64.070195.002155. [DOI] [PubMed] [Google Scholar]
- 12.Eikenberry E.F., Brodsky B. X-ray diffraction of reconstituted collagen fibers. J. Mol. Biol. 1980;144:397–404. doi: 10.1016/0022-2836(80)90098-4. [DOI] [PubMed] [Google Scholar]
- 13.Wood G.C., Keech M.K. The formation of fibrils from collagen solutions. 1. The effect of experimental conditions: kinetic and electron-microscope studies. Biochem. J. 1960;75:588–598. doi: 10.1042/bj0750588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cassel J., Mandelkern L., Roberts D. The kinetics of the heat precipitation of collagen. J. Am. Leather Chem. Assoc. 1962;57:556–575. [Google Scholar]
- 15.Williams B.R., Gelman R.A., Piez K.A. Collagen fibril formation. Optimal in vitro conditions and preliminary kinetic results. J. Biol. Chem. 1978;253:6578–6585. [PubMed] [Google Scholar]
- 16.Silver F.H., Birk D.E. Kinetic analysis of collagen fibrillogenesis: I. Use of turbidity—time data. Coll. Relat. Res. 1983;3:393–405. doi: 10.1016/s0174-173x(83)80020-x. [DOI] [PubMed] [Google Scholar]
- 17.Gelman R.A., Williams B.R., Piez K.A. Collagen fibril formation. Evidence for a multistep process. J. Biol. Chem. 1979;254:180–186. [PubMed] [Google Scholar]
- 18.Na G.C., Butz L.J., Carroll R.J. Mechanism of in vitro collagen fibril assembly. Kinetic and morphological studies. J. Biol. Chem. 1986;261:12290–12299. [PubMed] [Google Scholar]
- 19.Na G.C., Butz L.J., Carroll R.J. In vitro collagen fibril assembly in glycerol solution: evidence for a helical cooperative mechanism involving microfibrils. Biochemistry. 1986;25:958–966. doi: 10.1021/bi00353a003. [DOI] [PubMed] [Google Scholar]
- 20.Na G.C., Phillips L.J., Freire E.I. In vitro collagen fibril assembly: thermodynamic studies. Biochemistry. 1989;28:7153–7161. doi: 10.1021/bi00444a004. [DOI] [PubMed] [Google Scholar]
- 21.Suarez G., Oronsky A.L., Koch M.H. Synchrotron radiation x-ray scattering in the early stages of in vitro collagen fibril formation. Proc. Natl. Acad. Sci. USA. 1985;82:4693–4696. doi: 10.1073/pnas.82.14.4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bard J.B., Chapman J.A. Diameters of collagen fibrils grown in vitro. Nat. New Biol. 1973;246:83–84. doi: 10.1038/newbio246083a0. [DOI] [PubMed] [Google Scholar]
- 23.Gale M., Pollanen M.S., Goh M.C. Sequential assembly of collagen revealed by atomic force microscopy. Biophys. J. 1995;68:2124–2128. doi: 10.1016/S0006-3495(95)80393-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cisneros D.A., Hung C., Muller D.J. Observing growth steps of collagen self-assembly by time-lapse high-resolution atomic force microscopy. J. Struct. Biol. 2006;154:232–245. doi: 10.1016/j.jsb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Silver D., Miller J., Prockop D.J. Helical model of nucleation and propagation to account for the growth of type I collagen fibrils from symmetrical pointed tips: a special example of self-assembly of rod-like monomers. Proc. Natl. Acad. Sci. USA. 1992;89:9860–9864. doi: 10.1073/pnas.89.20.9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gelman R.A., Piez K.A. Collagen fibril formation in vitro. A quasielastic light-scattering study of early stages. J. Biol. Chem. 1980;255:8098–8102. [PubMed] [Google Scholar]
- 27.Bernengo J.C., Ronziere M.C., Veis A. A hydrodynamic study of collagen fibrillogenesis by electric birefringence and quasielastic light scattering. J. Biol. Chem. 1983;258:1001–1006. [PubMed] [Google Scholar]
- 28.George A., Veis A. FTIRS in H2O demonstrates that collagen monomers undergo a conformational transition prior to thermal self-assembly in vitro. Biochemistry. 1991;30:2372–2377. doi: 10.1021/bi00223a011. [DOI] [PubMed] [Google Scholar]
- 29.Kasai M. Thermodynamical aspect of G-F transformations of actin. Biochim. Biophys. Acta. 1969;180:399–409. doi: 10.1016/0005-2728(69)90124-8. [DOI] [PubMed] [Google Scholar]
- 30.Gaskin F., Cantor C.R., Shelanski M.L. Turbidimetric studies of the in vitro assembly and disassembly of porcine neurotubules. J. Mol. Biol. 1974;89:737–755. doi: 10.1016/0022-2836(74)90048-5. [DOI] [PubMed] [Google Scholar]
- 31.De Greef T.F., Smulders M.M., Meijer E.W. Supramolecular polymerization. Chem. Rev. 2009;109:5687–5754. doi: 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- 32.Kadler K., Hojima Y., Prockop D. Assembly of collagen fibrils de novo by cleavage of the type I pC-collagen with procollagen C-proteinase. The jounal of biological chemistry. J. Biol. Chem. 1987;260:15696–15701. [PubMed] [Google Scholar]
- 33.Streeter I., de Leeuw N.H. A molecular dynamics study of the interprotein interactions in collagen fibrils. Soft Matter. 2011;7:3373–3382. doi: 10.1039/C0SM01192D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hulmes D.J., Miller A., Woodhead-Galloway J. Analysis of the primary structure of collagen for the origins of molecular packing. J. Mol. Biol. 1973;79:137–148. doi: 10.1016/0022-2836(73)90275-1. [DOI] [PubMed] [Google Scholar]
- 35.Traub W. Molecular assembly in collagen. FEBS Lett. 1978;92:114–120. [Google Scholar]
- 36.Leikin S., Rau D.C., Parsegian V.A. Direct measurement of forces between self-assembled proteins: temperature-dependent exponential forces between collagen triple helices. Proc. Natl. Acad. Sci. USA. 1994;91:276–280. doi: 10.1073/pnas.91.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leikin S., Rau D.C., Parsegian V.A. Temperature-favoured assembly of collagen is driven by hydrophilic not hydrophobic interactions. Nat. Struct. Biol. 1995;2:205–210. doi: 10.1038/nsb0395-205. [DOI] [PubMed] [Google Scholar]
- 38.Kar K., Amin P., Brodsky B. Self-association of collagen triple helic peptides into higher order structures. J. Biol. Chem. 2006;281:33283–33290. doi: 10.1074/jbc.M605747200. [DOI] [PubMed] [Google Scholar]
- 39.Hayashi T., Nagai Y. Factors affecting the interactions of collagen molecules as observed by in vitro fibril formation. I. Effects of small molecules, especially saccharides. J. Biochem. 1972;72:749–758. doi: 10.1093/oxfordjournals.jbchem.a129953. [DOI] [PubMed] [Google Scholar]
- 40.Kuznetsova N., Chi S.L., Leikin S. Sugars and polyols inhibit fibrillogenesis of type I collagen by disrupting hydrogen-bonded water bridges between the helices. Biochemistry. 1998;37:11888–11895. doi: 10.1021/bi980089+. [DOI] [PubMed] [Google Scholar]
- 41.Harris J.R., Reiber A. Influence of saline and pH on collagen type I fibrillogenesis in vitro: fibril polymorphism and colloidal gold labelling. Micron. 2007;38:513–521. doi: 10.1016/j.micron.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 42.Li Y., Asadi A., Douglas E. pH effects on collagen fibrillogenesis in vitro: electrostatic interactions and phosphate binding. Mater. Sci. Eng. C. 2009;29:1643–1649. [Google Scholar]
- 43.Mertz E.L., Leikin S. Interactions of inorganic phosphate and sulfate anions with collagen. Biochemistry. 2004;43:14901–14912. doi: 10.1021/bi048788b. [DOI] [PubMed] [Google Scholar]
- 44.Prockop D.J., Fertala A. Inhibition of the self-assembly of collagen I into fibrils with synthetic peptides. Demonstration that assembly is driven by specific binding sites on the monomers. J. Biol. Chem. 1998;273:15598–15604. doi: 10.1074/jbc.273.25.15598. [DOI] [PubMed] [Google Scholar]
- 45.Snowden J.M., Swann D.A. The formation and thermal stability of in vitro assembled fibrils from acid-soluble and pepsin-treated collagens. Biochim. Biophys. Acta. 1979;580:372–381. doi: 10.1016/0005-2795(79)90149-1. [DOI] [PubMed] [Google Scholar]
- 46.Gelman R.A., Poppke D.C., Piez K.A. Collagen fibril formation in vitro. The role of the nonhelical terminal regions. J. Biol. Chem. 1979;254:11741–11745. [PubMed] [Google Scholar]
- 47.Helseth D.L., Jr., Veis A. Collagen self-assembly in vitro. Differentiating specific telopeptide-dependent interactions using selective enzyme modification and the addition of free amino telopeptide. J. Biol. Chem. 1981;256:7118–7128. [PubMed] [Google Scholar]
- 48.Malone J.P., Veis A. Heterotrimeric type I collagen C-telopeptideconformation as docked to its helix receptor. Biochemistry. 2004;43:15358–15366. doi: 10.1021/bi048304b. [DOI] [PubMed] [Google Scholar]
- 49.Malone J.P., George A., Veis A. Type I collagen N-telopeptides adopt an ordered structure when docked to their helix receptor during fibrillogenesis. Proteins. 2004;54:206–215. doi: 10.1002/prot.10526. [DOI] [PubMed] [Google Scholar]
- 50.Deshmukh K., Nimni M.E. Chemical changes associated with aging of collagen in vivo and in vitro. Biochem. J. 1969;112:397–405. doi: 10.1042/bj1120397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Na G.C. Monomer and oligomer of type I collagen: molecular properties and fibril assembly. Biochemistry. 1989;28:7161–7167. doi: 10.1021/bi00444a005. [DOI] [PubMed] [Google Scholar]
- 52.Brennan M., Davison P.F. Role of aldehydes in collagen fibrillogenesis in vitro. Biopolymers. 1980;19:1861–1873. doi: 10.1002/bip.1980.360191013. [DOI] [PubMed] [Google Scholar]
- 53.Kunii S., Morimoto K., Tonomura B. Actinidain-hydrolyzed type I collagen reveals a crucial amino acid sequence in fibril formation. J. Biol. Chem. 2010;285:17465–17470. doi: 10.1074/jbc.M110.110759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marotta M., Martino G. Sensitive spectrophotometric method for the quantitative estimation of collagen. Anal. Biochem. 1985;150:86–90. doi: 10.1016/0003-2697(85)90443-9. [DOI] [PubMed] [Google Scholar]
- 55.Carr M.E., Jr., Hermans J. Size and density of fibrin fibers from turbidity. Macromolecules. 1978;11:46–50. doi: 10.1021/ma60061a009. [DOI] [PubMed] [Google Scholar]
- 56.Yeromonahos C., Polack B., Caton F. Nanostructure of the fibrin clot. Biophys. J. 2010;99:2018–2027. doi: 10.1016/j.bpj.2010.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Y.-L., Kaufman L.J. Rheology and confocal reflectance microscopy as probes of mechanical properties and structure during collagen and collagen/hyaluronan self-assembly. Biophys. J. 2009;96:1566–1585. doi: 10.1016/j.bpj.2008.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Y.-L., Leone L.M., Kaufman L.J. Elastic moduli of collagen gels can be predicted from two-dimensional confocal microscopy. Biophys. J. 2009;97:2051–2060. doi: 10.1016/j.bpj.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brightman A.O., Rajwa B.P., Voytik-Harbin S.L. Time-lapse confocal reflection microscopy of collagen fibrillogenesis and extracellular matrix assembly in vitro. Biopolymers. 2000;54:222–234. doi: 10.1002/1097-0282(200009)54:3<222::AID-BIP80>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 60.Brokaw J., Doillon C., Silver F. Tubridimetry and morphological studies of type I collagen fiber assembly in vitro and the influence of fibronectin. Int. J. Biol. Macromol. 1985;7:135–140. [Google Scholar]
- 61.Piechocka I.K., van Oosten A.S., Koenderink G.H. Rheology of heterotypic collagen networks. Biomacromolecules. 2011;12:2797–2805. doi: 10.1021/bm200553x. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y., Silvent J., Giraud Gille M. Controlled collagen assembly to build dense tissue-like materials for tissue engineering. Soft Matter. 2011;7:11203–11210. [Google Scholar]
- 63.Strasser S., Zink A., Thalhammer S. Structural investigations on native collagen type I fibrils using AFM. Biochem. Biophys. Res. Commun. 2007;354:27–32. doi: 10.1016/j.bbrc.2006.12.114. [DOI] [PubMed] [Google Scholar]
- 64.Grant C.A., Brockwell D.J., Thomson N.H. Tuning the elastic modulus of hydrated collagen fibrils. Biophys. J. 2009;97:2985–2992. doi: 10.1016/j.bpj.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang F., Hörber H., Müller D.J. Assembly of collagen into microribbons: effects of pH and electrolytes. J. Struct. Biol. 2004;148:268–278. doi: 10.1016/j.jsb.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 66.Stein M., Vader D., Sander L. The micromechanics of three-dimensional collagen-I gels. Complexity. 2011;16:22–28. [Google Scholar]
- 67.Motte S., Kaufman L.J. Strain stiffening in collagen I networks. Biopolymers. 2013;99:35–46. doi: 10.1002/bip.22133. [DOI] [PubMed] [Google Scholar]
- 68.Lindström S.B., Vader D.A., Weitz D.A. Biopolymer network geometries: characterization, regeneration, and elastic properties. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2010;82:051905. doi: 10.1103/PhysRevE.82.051905. [DOI] [PubMed] [Google Scholar]
- 69.Broedersz C.P., Sheinman M., Mackintosh F.C. Filament-length-controlled elasticity in 3D fiber networks. Phys. Rev. Lett. 2012;108:078102. doi: 10.1103/PhysRevLett.108.078102. [DOI] [PubMed] [Google Scholar]
- 70.Ayad S., Wynn C.H. The effect of semicarbazide on the nature and stability of collagen fibrils. Biochem. J. 1970;118:61–65. doi: 10.1042/bj1180061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tanzer M.L. Intermolecular cross-links in reconstituted collagen fibrils. Evidence for the nature of the covalent bonds. J. Biol. Chem. 1968;243:4045–4054. [PubMed] [Google Scholar]
- 72.Payne K.J., King T.A., Holmes D.F. Collagen fibrillogenesis in vitro: an investigation of the thermal memory effect and of the early events occurring during fibril assembly using dynamic light scattering. Biopolymers. 1986;25:1185–1207. doi: 10.1002/bip.360250703. [DOI] [PubMed] [Google Scholar]
- 73.Hayashi T., Nagai Y. Time-dependent increase in stability of collagen fibrils formed in vitro. Effect of temperature. J. Biochem. 1974;75:651–654. doi: 10.1093/oxfordjournals.jbchem.a130433. [DOI] [PubMed] [Google Scholar]
- 74.Hayashi T. Time-dependent increase in the stability of collagen fibrils formed in vitro. I. Effects of pH and salt concentration on the dissolution of the fibrils. J. Biochem. 1978;84:245–249. doi: 10.1093/oxfordjournals.jbchem.a132124. [DOI] [PubMed] [Google Scholar]
- 75.Kueh H.Y., Brieher W.M., Mitchison T.J. Dynamic stabilization of actin filaments. Proc. Natl. Acad. Sci. USA. 2008;105:16531–16536. doi: 10.1073/pnas.0807394105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kueh H.Y., Mitchison T.J. Structural plasticity in actin and tubulin polymer dynamics. Science. 2009;325:960–963. doi: 10.1126/science.1168823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Niedermayer T., Jégou A., Lipowsky R. Intermittent depolymerization of actin filaments is caused by photo-induced dimerization of actin protomers. Proc. Natl. Acad. Sci. USA. 2012;109:10769–10774. doi: 10.1073/pnas.1121381109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hulmes D.J., Wess T.J., Fratzl P. Radial packing, order, and disorder in collagen fibrils. Biophys. J. 1995;68:1661–1670. doi: 10.1016/S0006-3495(95)80391-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hulmes D.J., Jesior J.C., Wolff C. Electron microscopy shows periodic structure in collagen fibril cross sections. Proc. Natl. Acad. Sci. USA. 1981;78:3567–3571. doi: 10.1073/pnas.78.6.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schoenenberger C.A., Buchmeier S., Jockusch B.M. Conformation-specific antibodies reveal distinct actin structures in the nucleus and the cytoplasm. J. Struct. Biol. 2005;152:157–168. doi: 10.1016/j.jsb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 81.Dimitrov A., Quesnoit M., Perez F. Detection of GTP-tubulin conformation in vivo reveals a role for GTP remnants in microtubule rescues. Science. 2008;322:1353–1356. doi: 10.1126/science.1165401. [DOI] [PubMed] [Google Scholar]
- 82.Amuasi H.E., Storm C. Off-lattice Monte Carlo simulation of supramolecular polymer architectures. Phys. Rev. Lett. 2010;105:248105. doi: 10.1103/PhysRevLett.105.248105. [DOI] [PubMed] [Google Scholar]
- 83.Maggs A., Huse D., Leibler S. Unbinding transitions of semi-flexible polymers. Europhys. Lett. 1989;8:615–620. [Google Scholar]
- 84.Benetatos P., Frey E. Depinning of semiflexible polymers. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2003;67:051108. doi: 10.1103/PhysRevE.67.051108. [DOI] [PubMed] [Google Scholar]
- 85.Kierfeld J. Force-induced desorption and unzipping of semiflexible polymers. Phys. Rev. Lett. 2006;97:058302. doi: 10.1103/PhysRevLett.97.058302. [DOI] [PubMed] [Google Scholar]
- 86.Hirshburg J.M., Ravikumar K.M., Yeh A.T. Molecular basis for optical clearing of collagenous tissues. J. Biomed. Opt. 2010;15:055002. doi: 10.1117/1.3484748. [DOI] [PubMed] [Google Scholar]
- 87.Pachence J.M. Collagen-based devices for soft tissue repair. J. Biomed. Mater. Res. 1996;33:35–40. doi: 10.1002/(SICI)1097-4636(199621)33:1<35::AID-JBM6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.