

Abstract
Eukaryotic cells use numerous mechanisms to ensure that no segment of their DNA is inappropriately re-replicated, but the importance of this stringent control on genome stability has not been tested. Here we show that re-replication in Saccharomyces cerevisiae can strongly induce the initial step of gene amplification, increasing gene copy number from one to two or more. The resulting amplicons consist of large internal chromosomal segments that are bounded by Ty repetitive elements and are intrachromosomally arrayed at their endogenous locus in direct head-to-tail orientation. These re-replication–induced gene amplifications are mediated by nonallelic homologous recombination between the repetitive elements. We suggest that re-replication may be a contributor to gene copy number changes, which are important in fields such as cancer biology, evolution, and human genetics.
A central tenet of eukaryotic cell biology is that cells replicate their DNA only once every cell cycle. Although it has become an article of faith that this regulation is important because re-replication would threaten genome stability (1), that faith has never been experimentally tested. Preventing re-replication in eukaryotic cells requires blocking re-initiation at thousands of origins scattered throughout the genome, and eukaryotic cells use numerous overlapping mechanisms to do this (2, 3). By disrupting these mechanisms in a controlled manner, we are now able to examine re-replicated cells for possible genomic alterations.
The alterations we first looked for were heritable increases in gene copy number. Such increases are observed in the gene amplifications commonly associated with cancers (4, 5), the gene duplications important for molecular evolution (6), and the copy number variations prevalent in human genomes (7). Although re-replication was once a leading model for copy number increases, specifically during gene amplification (8, 9), the model has long been abandoned for lack of experimental support for it (10). Hence, re-replication is not seriously considered as a possible source of heritable copy number changes (4, 11, 12). Here, we demonstrate that re-replication can readily induce copy number changes, provoking reconsideration of re-replication as a potential source of such changes in cancer and evolution (8).
We have developed a system to detect and quantify early amplification events arising from a transient and limited pulse of re-replication at a defined genomic locus in the budding yeast Saccharomyces cerevisiae (Fig. 1A and figs. S1 and S2A). We took advantage of our ability to induce re-replication predominantly from a single origin (ARS317) by conditionally deregulating the replication initiation proteins Mcm2-7 and Cdc6 (MC2A genetic background) (13). We also adapted a copy number assay in which cells with a single copy of the ade3-2p allele turn pink, and cells with two or more copies turn red (14).
Fig. 1.

Re-replication greatly stimulates gene amplification. (A) Induction of re-replication from ARS317. Strains containing an ade3-2p copy number reporter cassette integrated at Chr IV567kb were arrested in G2/M phase and treated with galactose to trigger re-initiation of ARS317. aCGH analysis of re-replication (>2C) is shown for Chr IV. All other chromosomes maintained a 2C copy number. Top: YJL6974, a non–re-replicating strain with ARS317 in cassette. Middle: YJL6555, a re-replicating strain with no ARS317 in cassette. Bottom: YJL6558, a re-replicating strain with ARS317 in cassette. (B) ARS317 re-replication stimulates gene amplification. Left: Frequency of one-half to one-eighth red-sectored colonies (mean ± SEM, n = 2 to 7 induction replicates; table S1) after 0 or 3 hours of galactose induction of YJL6974, YJL6555, and YJL6558. Right: Amplification frequency estimated by multiplying sector frequency by the fraction of sectored colonies displaying reporter cassette amplification (tables S2 and S4). (C) Red sectors induced by re-replication display gene amplifications. Thirty-five red sectors derived from YJL6558 were analyzed by aCGH and classified on the basis of the copy number profile of Chr IV. A schematic of Chr IV (top) shows the positions of Ty elements (triangles), the centromere (circle), and the ade3-2p cassette (black bar).
ARS317 and ade3-2p were combined in a re-replicating reporter cassette (fig. S1A) that was integrated at either of two loci on chromosome IV (Chr IV567kb and Chr IV1089kb) in a haploid MC2A background from which the endogenous ARS317 was deleted. After arresting cells at the G2/M phase [time (t) = 0 hours], we transiently induced re-replication until half of the ARS317 origins re-initiated (t = 3 hours), then plated for isolated colonies at both time points. The mostly pink colonies were screened for heritable amplification of ade3-2p by looking for colonies with red sectors (fig. S1B). Colonies with one-half, one-quarter, or one-eighth red sectors were scored to focus on amplifications arising within three generations of the re-replication pulse (table S1).
Inducing re-replication for 3 hours from the ade3-2p-ARS317 cassette integrated at Chr IV567kb (strain YJL6558) caused a 42-fold increase in red sectors to 3.3% of all colonies (Fig. 1B). Suppressing this re-replication by removing deregulated Cdc6 from the MC2A background (YJL6974) or ARS317 from the cassette (YJL6555) resulted, respectively, in only 4- and 12-fold increases in red sectors (Fig. 1B), most of which had not amplified the ade3-2p cassette. Re-replication also induced colony sectoring by 38-fold when the ade3-2p-ARS317 cassette was relocated to Chr IV1089kb (fig. S2B and table S1).
Array comparative genomic hybridization (aCGH) on 35 red sectors derived from YJL6558 (table S2) confirmed that most [31 of 35 (31/35)] had at least two copies of an internal chromosomal segment encompassing the ade3-2p-ARS317 cassette. Amplicons ranged in size from 135 to 470 kb (Fig. 1C) with boundaries mapping within a few kilobases of Ty elements or, rarely, long terminal repeats (LTRs) oriented in direct repeat. Similar aCGH results were obtained for red sectors arising from re-replication of the ade3-2p cassette integrated at Chr IV1089kb (fig. S2C and table S3). In contrast, few amplifications were observed among the less frequent red sectors isolated from the control strains YJL6974 (3/32) (table S4) and YJL6555 (1/6).
Using the aCGH data to convert sectoring to amplification frequency (10), we estimated relative amplification frequencies of 1:8:230 for YJL6974:YJL6555:YJL6558, respectively, and an absolute frequency of 3 × 10−2 for YJL6558 (Fig. 1B and table S2). This frequency roughly translates into an order of magnitude rate of 10−2 per generation (10), which is significantly higher than spontaneous rates of segmental duplications (10−7 to 10−6 per generation) or higher-order amplifications (10−10) reported for budding yeast (15, 16).
Re-replication generates slowed or stalled forks and DNA damage (17–20), perturbations that have been implicated in genomic alterations (21). Nonetheless, neither disruption of DNA replication by means of hydroxyurea or temperature-sensitive replication mutations (fig. S3, A and B) nor treatment of cells with the DNA-damaging agent phleomycin (fig. S3, C to E, and table S5) induced substantial ade3-2p amplification comparable to re-replication. Thus, re-replication appears to be particularly effective at inducing gene amplification (10), and we refer to these events as re-replication–induced gene amplification (RRIGA).
To investigate the mechanism of RRIGA, we determined the position, orientation, and boundaries of 20 segmental amplifications arising from re-replication of the ade3-2p cassette integrated at Chr IV567. All 20 were located to Chr IV, because this chromosome increased in size by an amount consistent with the length and copy number of the additional amplicon(s) (Fig. 2A and table S1). No other chromosomes increased in size, and probing for the ADE3 sequences on the ade3-2p cassette confirmed that the amplicons resided only on Chr IV (Fig. 2A).
Fig. 2.

Structure of gene amplifications induced by re-replication. (A) Amplicons remain on the endogenous chromosome. Pulsed-field gel electrophoresis–separated chromosomes from 24 sectors analyzed in Fig. 1C (table S2) were visualized by ethidium bromide staining (left), then probed for ADE3, which detects both ade3 on Chr VII and ade3-2p in the reporter cassette (right). YJL6558 (the parental strain), YJL7100, YJL7106, YJL7113, and YJL7118 did not contain amplifications. The Chr IV doublet pattern for YJL7098 and YJL7099 is consistent with the partial loss of amplification from the population. (B) Amplicons are tandemly arrayed in loco in direct head-to-tail orientation. Schematics of an unamplified amplicon and three possible orientations for amplicons tandemly duplicated in loco are shown. Predicted PCR junction fragments (10) are shown for five sets of primers that flank amplicon boundaries (+, PCR product expected; −, no PCR product expected). Representative PCR products are shown for parental strain YJL6558 and 19 of the 20 amplified strains displayed in (A).
We then tested the hypothesis that the amplicons were tandemly arrayed at their endogenous locus. The three possible arrangements for such tandem duplications (head to tail, head to head, or tail to tail) each generate a particular set of junctions and boundaries that are distinguishable by polymerase chain reaction (PCR) (Fig. 2B). Moreover, we could confirm the presence of Ty or LTR elements at these boundaries by using primers that flank these elements. Of the 20 amplifications examined, PCR products from 19 of them established that RRIGA amplicons are indeed tandemly arrayed at their endogenous locus in head-to-tail orientation and are bounded by Ty or LTR elements in direct repeat (Fig. 2B) (10).
Such structures could arise from nonallelic homologous recombination (NAHR). To examine this possibility, we sequenced the PCR products for interamplicon junctions from four independent amplifications spanning kilobases 515 to 650 on Chr IV. The sequences revealed hybrid Ty elements generated by precise crossovers between Ty2-1 at 515 kb and Ty1-1 at 650 kb (Fig. 3A and fig. S4). In addition, RRIGA was greatly reduced by deletion of RAD52, which is essential for homologous recombination (Fig. 3, B and C, and tables S1 and S6), but was not affected by deletion of DNL4, which is required for nonhomologous end-joining (NHEJ). Thus, RRIGA in budding yeast is mediated by NAHR between repetitive elements flanking a re-replicated chromosomal segment.
Fig. 3.
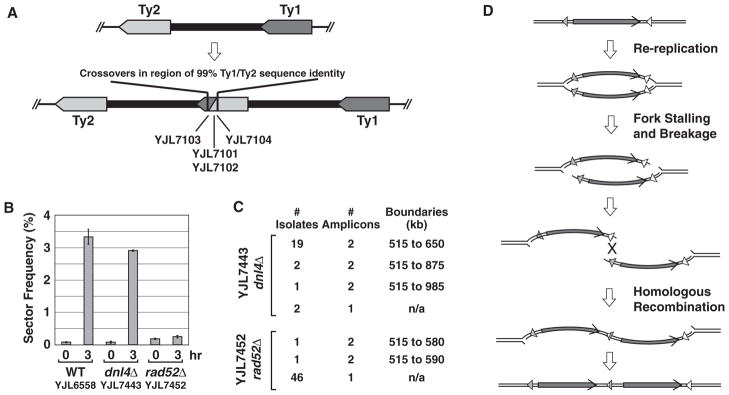
Role of nonallelic homologous recombination in RRIGA. (A) Schematic of hybrid recombinant Ty elements (fig. S4) found at the interamplicon junction of four isolates with segmental amplifications on Chr IV from 515 to 650 kb. (B) Re-replication–induced sectoring is dependent on HR and not NHEJ. YJL7443 (dnl4Δ) and YJL7452 (rad52Δ) are otherwise isogenic with re-replicating strain YJL6558. Re-replication–induced sectoring frequency (mean ±SEM, n = 2 to 7 induction replicates; table S1) was analyzed as described for Fig. 1B. (C) Re-replication–induced gene amplification is dependent on HR and not NHEJ. A representative aCGH analysis of Chr IV for 24 red sectors derived from YJL7443 (dnl4Δ) and 48 derived from YJL7452 (rad52Δ) is shown (table S6). (D) How re-replication might stimulate NAHR. Arrowheads, nonallelic or hybrid recombinant repetitive element; arrows, amplified segments.
How re-replication might stimulate NAHR with such high efficiency is illustrated in Fig. 3D. First, re-replication increases the copy number of a chromosome segment. Second, re-replication forks, which display compromised progression, may stall, collapse, and break with high frequency. Third, in contrast to stalled replication forks in S phase, isolated re-replication forks are unlikely to be rescued by converging forks from neighboring origins. Fourth, the re-replication bubble structure can facilitate recombinational repair between re-replicated segments in a variety of ways, such as by pairing double-stranded breaks that might occur at both forks in trans.
In short, re-replication appears capable of promoting several key events in an optimal temporal order and spatial context to stimulate gene amplifications. The critical events occur before repair pathways are recruited, leaving room for alternative ways to resolve broken re-replication bubbles (10). Hence, although RRIGA is preferentially mediated by NAHR in budding yeast, additional repair pathways, such as NHEJ, might be used by other species. In fact, repair of broken re-replication bubbles may well stimulate other genomic alterations besides RRIGA.
Our results demonstrate that loss of eukaryotic DNA replication control can indeed induce genome instability, and in the case of RRIGA does so with extraordinary efficiency. Such efficiency suggests that even lower levels of re-replication, below the sensitivity of current assays (<5 to 10%), may cause substantial induction of RRIGA (10). Thus, although some consider the multiple mechanisms used to control replication as redundant, because disrupting them individually does not lead to detectable re-replication (2), we view each as essential for the stringent control needed to safeguard genome stability (2, 3, 13).
Establishing that re-replication can induce copy number changes raises the question of whether re-replication actually does induce such changes in cancer and evolution (10). Several recent observations hint that RRIGA might indeed play a role in oncogenesis or tumor progression. For example, segmental duplications found in two tumor genomes bear striking similarity to RRIGA structures observed in budding yeast: Oncogenes are duplicated in a head-to-tail arrangement in loco, with repetitive Alu elements at the outer amplicon boundaries and a hybrid recombinant Alu element at the interamplicon junction (22, 23). Additionally, replication initiation proteins are overexpressed in a number of human cancers (1, 24), and modest overexpression of the replication proteins Cdt1 and Cdc6 can potentiate oncogenesis in mouse cells (25–27). We thus hope that our study provokes further investigation into the possible role of RRIGA in cancer and evolution.
Supplementary Material
Acknowledgments
We thank R. Bainton for the use of his microscope; A. Johnson for the use of his pulsed-field gel electrophoresis system and microscope; and A. Sil, D. Morgan, A. Johnson, D. Toczyski, E. Blackburn, P. O’Farrell, T. Tlsty, J. Haber, S. Elledge, and F. Winston for helpful discussions. This work was supported by grants to J.J.L. from the Sandler Program in Basic Sciences, the Stewart Trust Fund, UCSF Research Evaluation and Allocation Committee, UCSF Academic Senate, UC Cancer Research Coordinating Committee, and NIH (grant R01 GM59704). B.M.G. was supported by an NSF Predoctoral Fellowship and a Department of Defense Breast Cancer Predoctoral Fellowship (W81XWH-04-1-0409). K.J.F. was supported by an NSF Predoctoral Fellowship. Microarray data are deposited with the Gene Expression Omnibus (accession number GSE22018).
Footnotes
References and Notes
- 1.Blow JJ, Gillespie PJ. Nat Rev Cancer. 2008;8:799. doi: 10.1038/nrc2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arias EE, Walter JC. Genes Dev. 2007;21:497. doi: 10.1101/gad.1508907. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen VQ, Co C, Li JJ. Nature. 2001;411:1068. doi: 10.1038/35082600. [DOI] [PubMed] [Google Scholar]
- 4.Albertson DG. Trends Genet. 2006;22:447. doi: 10.1016/j.tig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 5.Lockwood WW, et al. Oncogene. 2008;27:4615. doi: 10.1038/onc.2008.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Innan H, Kondrashov F. Nat Rev Genet. 2010;11:97. doi: 10.1038/nrg2689. [DOI] [PubMed] [Google Scholar]
- 7.Zhang F, Gu W, Hurles ME, Lupski JR. Annu Rev Genomics Hum Genet. 2009;10:451. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schimke RT, Sherwood SW, Hill AB, Johnston RN. Proc Natl Acad Sci USA. 1986;83:2157. doi: 10.1073/pnas.83.7.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stark GR, Debatisse M, Giulotto E, Wahl GM. Cell. 1989;57:901. doi: 10.1016/0092-8674(89)90328-0. [DOI] [PubMed] [Google Scholar]
- 10.See supporting material on Science Online.
- 11.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Nat Rev Genet. 2009;10:551. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koszul R, Fischer G. C R Biol. 2009;332:254. doi: 10.1016/j.crvi.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 13.Green BM, Morreale RJ, Ozaydin B, Derisi JL, Li JJ. Mol Biol Cell. 2006;17:2401. doi: 10.1091/mbc.E05-11-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koshland D, Kent JC, Hartwell LH. Cell. 1985;40:393. doi: 10.1016/0092-8674(85)90153-9. [DOI] [PubMed] [Google Scholar]
- 15.Payen C, Koszul R, Dujon B, Fischer G, Haber JE. PLoS Genet. 2008;4:e1000175. doi: 10.1371/journal.pgen.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorsey MJ, Hoeh P, Paquin CE. Curr Genet. 1993;23:392. doi: 10.1007/BF00312624. [DOI] [PubMed] [Google Scholar]
- 17.Archambault V, Ikui AE, Drapkin BJ, Cross FR. Mol Cell Biol. 2005;25:6707. doi: 10.1128/MCB.25.15.6707-6721.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green BM, Li JJ. Mol Biol Cell. 2005;16:421. doi: 10.1091/mbc.E04-09-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melixetian M, et al. J Cell Biol. 2004;165:473. doi: 10.1083/jcb.200403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaziri C, et al. Mol Cell. 2003;11:997. doi: 10.1016/s1097-2765(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 21.Aguilera A, Gómez-González B. Nat Rev Genet. 2008;9:204. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 22.O’Neil J, et al. J Exp Med. 2007;204:3059. doi: 10.1084/jem.20071637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strout MP, Marcucci G, Bloomfield CD, Caligiuri MA. Proc Natl Acad Sci USA. 1998;95:2390. doi: 10.1073/pnas.95.5.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez MA, Tachibana KE, Laskey RA, Coleman N. Nat Rev Cancer. 2005;5:135. doi: 10.1038/nrc1548. [DOI] [PubMed] [Google Scholar]
- 25.Arentson E, et al. Oncogene. 2002;21:1150. doi: 10.1038/sj.onc.1205175. [DOI] [PubMed] [Google Scholar]
- 26.Liontos M, et al. Cancer Res. 2007;67:10899. doi: 10.1158/0008-5472.CAN-07-2837. [DOI] [PubMed] [Google Scholar]
- 27.Seo J, et al. Oncogene. 2005;24:8176. doi: 10.1038/sj.onc.1208881. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.