

Abstract
Abacavir is a nucleoside analogue reverse transcriptase inhibitor (NRTI) indicated for the treatment of human immunodeficiency virus (HIV) infection as part of a multidrug, highly active antiretroviral therapy (HAART) regimen. Despite its efficacy in treating HIV, approximately 5% of individuals that receive abacavir develop an immune-mediated hypersensitivity reaction (HSR) that warrants immediate discontinuation of abacavir and switching to an alternative antiretroviral regimen. Abacavir HSR is associated with individuals that carry the *57:01 variant in the human leukocyte antigen B (HLA-B) gene. There is a large volume of evidence to show that those who carry HLA-B*57:01 are at significantly increased risk of developing HSR and should not receive abacavir. Using pharmacogenetic screening to ensure individuals who carry y HLA-B*57:01 do not receive abacavir can reduce the incidence of abacavir HSR and is now considered the standard of care before prescribing abacavir. Genetic testing for abacavir HSR is currently one of the best examples of integrating pharmacogenetic testing into clinical practice.
Keywords: abacavir, HIV, AIDS, genetic testing, HLA, drug hypersensitivity, pharmacogenetics
Introduction
Abacavir is a nucleoside analogue reverse transcriptase inhibitor (NRTI) first released in 1998 by GlaxoSmithKline for the treatment of HIV infection. It is available as a single agent (Ziagen; also available in generic formulations in the United States) as well as in the combination NRTI drugs Epzicom/Kivexa (with lamivudine) and Trizivir (with lamivudine and zidovudine). While abacavir is safe and effective for the treatment of HIV, approximately 5% of the population1 are at risk of developing potentially life-threatening hypersensitivity reactions to abacavir therapy2. While the presentation of symptoms predicted that abacavir HSR was immune-mediated, it took several years after its widespread use to discover genetic predictors of abacavir HSR. . A variant allele in the HLA-B gene, a critical part of the immune system, was shown to strongly correlate with the risk of developing abacavir HSR3. Following the publication of these findings, clinicians began to screen HIV patients for the presence of HLA-B*57:01 and selectively prescribe abacavir only to those that did not carry the risk allele. A number of clinical studies, most notably the landmark PREDICT-14 study, showed that pharmacogenetic testing for HLA-B*57:01 resulted in a significantly decreased incidence of abacavir HSR. Based on these findings, screening for HLA-B*57:01 is now considered routine clinical practice before prescribing abacavir. This review will summarize general background on abacavir and HLA-B*57:01, the clinical studies linking HLA-B genotype to abacavir HSR risk, the integration of pharmacogenetic testing into clinical practice, and the impact that genetic testing has had on abacavir HSR risk.
Signs and Symptoms of Abacavir Hypersensitivity
Per the black box warning on abacavir-containing products5, HSR is defined as a sign or symptom in two or more symptom groups (Table 1). These signs and symptoms are normally mild at onset, but will continue to increase in severity with continued abacavir exposure. True HSR occurs within the first days or weeks of abacavir exposure, with the median time to onset of symptoms around 11 days1. Symptoms of HSR that occur many months or years into abacavir exposure are unlikely to be true HSR and are more likely explained by infection or an adverse drug response to another medication. If HSR is suspected or cannot be ruled out, abacavir should be immediately withdrawn, an allergy to abacavir should be noted in the patient’s chart, and the patient should be switched to an alternative antiretroviral. Cessation of symptoms shortly after abacavir withdrawal should also provide confidence in the diagnosis of HSR. Abacavir re-challenge is contraindicated in a patient with suspected HSR due to the potential for severe and life-threatening reactions2, 6.
Table 1.
Grouping of HSR signs and symptoms.
| Signs and symptoms of abacavir HSR | |
|---|---|
| Group 1 | Fever |
| Group 2 | Rash |
| Group 3 (Gastrointestinal) | Nausea, vomiting, diarrhea, abdominal pain |
| Group 4 (Constitutional) | Malaise, fatigue, achiness |
| Group 5 (Respiratory) | Dyspnea, cough, pharyngitis |
Abacavir skin patch testing (SPT) has been performed in many research studies to confirm a clinical diagnosis of HSR. The method of testing is essentially the same as a standard skin patch test for allergens. Abacavir solution, typically in the range of 1%–10% in a petrolatum base, is applied to a patch placed on a patient’s skin, usually on the back7. Between 24 and 48 hours, the test results are then “read” by looking for an immune response at the sites of abacavir exposure, including redness, induration, itchiness, and blistering. The landmark PREDICT-1 study4 used abacavir SPT results to distinguish “immunologically confirmed” HSR from clinically diagnosed HSR. While PREDICT-1 and other studies8, 9 have shown exceptional correlation between positive SPT results and the presence of HLA-B*57:01, it is unclear what the false negative rate for this test truly is. Additionally, clinician error in applying the test or other patient factors, such as concomitant illnesses or medications, may contribute to false negative results. Because rechallenge with abacavir is contraindicated in individuals with clinically suspected HSR, it is unlikely that the accuracy of this test will ever be fully determined. As such, SPT is part of the research setting, but is not recommended for use in clinical practice to confirm hypersensitivity.
Abacavir Mechanism of Action
Like many other NRTIs, abacavir is a prodrug that requires intracellular phosphorylation10 to produce its active metabolite, carbovir triphosphate11 (Figure 1). Carbovir triphosphate is able to inhibit viral reverse transcriptase by competing with endogenous deoxyguanosine triphosphate at the active site. Upon incorporation into the newly synthesized viral DNA, carbovir triphosphate is able to induce chain termination due to its lack of the 3’ hydroxyl group necessary to form the critical phosphodiester bonds in the backbone of DNA, effectively halting the reverse transcription process.
Figure 1. Metabolism and activation of abacavir.

Abacavir is rapidly absorbed following oral dosing and is quickly taken up into cells via passive diffusion, where it is then phosphorylated by adenosine phosphotransferase to produce abacavir. A cytosolic deaminase converts abacavir monophosphate to carbovir monophosphate, which is then subject to processing by other intracellular kinases to produce the active metabolite, carbovir triphosphate.
Abacavir Metabolism & Elimination
Unlike protease inhibitors and non-nucleoside reverse transcriptase inhibitors (NNTRIs), abacavir is not a significant substrate, inhibitor, or inducer of any members of the cytochrome P450 (CYP) family. This makes it an attractive choice for patients receiving other CYP substrates to avoid potential drug-drug interactions. Once absorbed, abacavir is extensively metabolized, with less than 2% of an oral dose being excreted into the urine as parent drug12. One pathway for metabolism of abacavir is through UDP-glucuronosyltransferases (UGTs) to produce the 5’-glucuronide (Figure 1). Although there have been a number of studies of the pharmacogenetics of glucuronidation of the protease inhibitor (PI) atazanavir, similar studies have not been done for abacavir. Therefore, it is unclear at the present time which UGT isoforms metabolize abacavir and whether genetic variation in these genes affects abacavir metabolism. Abacavir is also metabolized by alcohol dehydrogenase to produce an aldehyde intermediate that is then converted to a 5’-carboxylate derivative. Previous data have shown that ingestion of moderate amounts of ethanol can increase the area under the curve (AUC) and half-life of a single 600 mg dose of abacavir13, with a shifting of abacavir metabolism toward the glucuronidation pathway.
Approximately 85% of a given oral dose of abacavir is eliminated renally, with the majority of it as a roughly equal mix of the carboxylate and glucuronide metabolites and limited amounts as other minor metabolites and unmetabolized abacavir12. The remainder of the dose is eliminated fecally as a mixture of unmetabolized abacavir and the carboxylate metabolite. In pharmacokinetic studies performed in individuals with renal impairment (creatinine clearance < 60 mL/min) or on hemodialysis that had received abacavir for at least 2 months14, there were no observed changes in pharmacokinetic parameters. This may make abacavir an attractive choice in patients with impaired renal function15.
Function of HLA-B
HLA-B is one of many genes contained in the HLA locus on chromosome 6, the human equivalent of the major histocompatibility complex (MHC) found in virtually all vertebrates. HLA-B is a member of the major MHC class I genes, along with HLA-A and HLA-C. MHC class I genes encode proteins that heterodimerize with beta 2 microglobulin to form a functional complex that binds intracellular peptides in the endoplasmic reticulum16. After peptide loading, these complexes traffic to the cell surface and “present” the peptide to circulating T cells. Normally, the peptides presented are the normal degradation products of intracellular proteins and do not produce a response in T cells because they are recognized as “self.” However, sometimes the peptide presented is recognized as “non-self,” either because it originated from an intracellular pathogen, such as a virus or bacteria, or because its amino acid sequence is otherwise abnormal. This indicates that something has gone wrong inside the cell, such as genetic mutations and dysregulation that might lead to cancer. In such cases, the T cell will then activate and proceed to destroy the cell.
Variation in HLA Genes
The ability of an HLA molecule to successfully load and present a given peptide is directly related to that molecule’s amino acid sequence, particularly within the peptide binding groove. Due to the diverse and ever-evolving genomes of human pathogens, there is constant selective pressure for the appearance of new HLA alleles that better protect against infections caused by these pathogens. Additionally, because of codominant expression of HLA genes, it is evolutionarily advantageous for a given individual to have two different alleles of each HLA gene, effectively providing a significantly larger peptide binding repertoire than an individual who was homozygous at all HLA alleles. These effects have resulted in HLA genes that are some of the most polymorphic in the entire human genome17, but have also resulted in considerable differences in HLA allele distribution between populations.
While the evolution of HLA alleles is driven in response to environmental exposure to new pathogens and helps strengthen human immunity, this process is not without its drawbacks. The high variability in HLA alleles makes it exceedingly difficult to find suitable donors for organ and bone marrow transplants18. HLA mismatches between donor and recipient currently require lifelong use of immunosuppressants to prevent acute rejection of the organ, ultimately placing the recipient at increased risk of infection and cancer. Other than identical twins, it is highly unlikely to find a suitable organ donor that is a perfect match across all HLA loci. Certain HLA alleles have also been associated with an increased risk of developing a number of autoimmune conditions, such as ankylosing spondilitis19 and rheumatoid arthritis20. Despite decades of research, it is still unclear how these alleles contribute to an increased risk of autoimmune disease. It is also unclear how such alleles that predispose individuals to these debilitating conditions could reach reasonable frequencies in the population. It may be that at one time they provided an advantage against a particular pathogen, leaving behind the risk of autoimmunity as an unfortunate side effect.
Particular HLA alleles are also associated with an increased risk of rare, serious drug toxicities, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), collectively referred to as severe cutaneous adverse reactions (SCAR)21. Associations have been shown with HLA-B*15:02 and risk of carbamazepine-induced SJS in East Asian populations22, as well as HLA-B*58:01 for allopurinol-induced SCAR in European populations21. In addition to abacavir hypersensitivity, HLA-B*57:01 has also been associated with flucloxacillin-induced hepatotoxicity23, although the absolute risk is substantially less than abacavir hypersensitivity. Furthermore, while these HLA alleles are associated with increased risk for adverse events, they are by no means a guarantee that a reaction will occur, suggesting that other genetic and environmental factors also play a role in the development of these acute drug toxicities. In the case of flucloxacillin, only one in 500 to 1,000 drug-exposed patients carrying HLA-B*57:01 will actually develop hepatotoxicity23.
HLA-B*57:01 Allele Frequency in Worldwide Populations
The presence of HLA-B*57:01 can be detected in many worldwide populations and is highly variable. Table 2 summarizes the allele frequency ranges for many ethnic groups. Caucasian populations generally possess HLA-B*57:01 in the range of 6%–10%. African populations, however, have only a 1%–2% carrier rate and, as expected, have a lower incidence of abacavir HSR. East Asian populations typically have a 1%–3% carrier rate, though some populations have shown higher allele frequencies25, 26. However, HLA-B*57:01 is only present at a very low rate in Korean populations27 and is virtually absent in Japanese populations28. Strikingly, many Indian populations have very high HLA-B*57:01 allele frequency, with some in excess of a 15% carrier rate29.
Table 2.
Allele frequency of HLA-B*57:01 in various population groups24.
| Population Group | HLA-B*57:01 carrier frequency range |
|---|---|
| European | 1.4% – 10.2% |
| South American | 1.1% – 3.1% |
| African | 0.0% – 3.2% |
| Middle Eastern | 0.5% – 6.0% |
| Mexican | 0.0% – 4.0% |
| Asian | 0.0% – 6.7% |
| Southwest Asian (Indian) | 3.8% – 19.6% |
Potential Mechanisms of Abacavir Hypersensitivity
A number of different mechanisms have been proposed for mediating abacavir HSR. One proposed mechanism hypothesizes that abacavir itself may covalently bind to a peptide that is a normal ligand for HLA-B*57:01, effectively making abacavir a hapten against which the immune system begins to mount a defense30. Abacavir’s metabolism via alcohol dehydrogenase produces an aldehyde intermediate that then goes on to become abacavir carboxylate (Figure 1). Reactive aldehydes produced from endogenous compounds, such as the byproducts of lipid peroxidation, can form adducts with cellular proteins, producing cellular damage hypothesized to contribute to a wide variety of phenotypes, including neurodegeneration31 and cardiovascular disease32.
Abacavir’s aldehyde metabolite forms adducts to the valine residues of cellular proteins33, lending some credibility to the hypothesis that one or more haptenated peptides may be the mediator of abacavir HSR. While this may occur in vivo, it remains unclear whether there are specific proteins with which abacavir will adduct,, allowing for a potential connection between these proteins and the peptide binding repertoire of HLA-B*57:01 versus other alleles.
Another proposed mechanism suggests that abacavir or one of its metabolites may interact with the HLA-B*57:01 protein, possibly in or near the peptide binding groove, to alter its binding repertoire. With its repertoire altered, HLA-B*57:01 would then begin to present novel self-peptides to immune cells, leading to the immune response. Recently published data show that abacavir treatment can indeed alter the peptide binding repertoire of a soluble form of HLA-B*57:01 expressed in a lymphoblastoid cell line34. While abacavir treatment resulted in loading of novel self-peptides, the C-terminal amino acids of these peptides were not consistent with peptides that are normally loaded onto HLA-B*57:01 in the absence of abacavir. However, when these peptides were synthesized and tested for binding to purified HLA-B*57:01 protein, it was found that these peptides bind even in the absence of abacavir and that addition of abacavir had no detectable effect on the binding affinity. This suggests that even though these peptides can bind to isolated and purified HLA-B*57:01, they are prevented from doing so under normal intracellular conditions. Of note, , flucloxacillin treatment had no effect on the binding repertoire of HLA-B*57:01 in this experimental system. Given that some of the novel self-peptides loaded onto HLA-B*57:01 in the presence of abacavir are from proteins expressed in the liver and might provide a possible link to the increase in hepatotoxicity risk of flucloxacillin, it would appear that abacavir and flucloxacillin may interact with HLA-B*57:01 through distinct mechanisms.
Another recent report35 also shows that abacavir can modify the peptides presented by HLA-B*57:01. While the peptides used in this experimental system come from peptide libraries, the results show a clear shift in the C-terminal residues of bound peptides after abacavir is added. Under normal circumstances, HLA-B*57:01 prefers large hydrophobic residues such as tryptophan and phenylalanine in the C-terminus of bound peptides, but this shifts to valine, alanine, and isoleucine when cotreated with abacavir. Additionally, the group was able to successfully crystallize HLA-B*57:01 in the presence of a bound peptide and abacavir. As previously hypothesized, abacavir binds directly in the peptide binding groove of HLA-B*57:01 and disrupts where the C terminus of endogenous peptide ligands would typically bind (Figure 2). Metabolites of abacavir were not tested, but may also have some effect on peptide binding. However, it is clear from the results of these experiments that abacavir is able to bind to HLA-B*57:01 directly and alter its peptide binding repertoire, lending credibility to the hypothesis that novel self-peptide presentation is responsible for or plays a significant part in abacavir HSR.
Figure 2. Binding of abacavir to HLA-B*57:01.
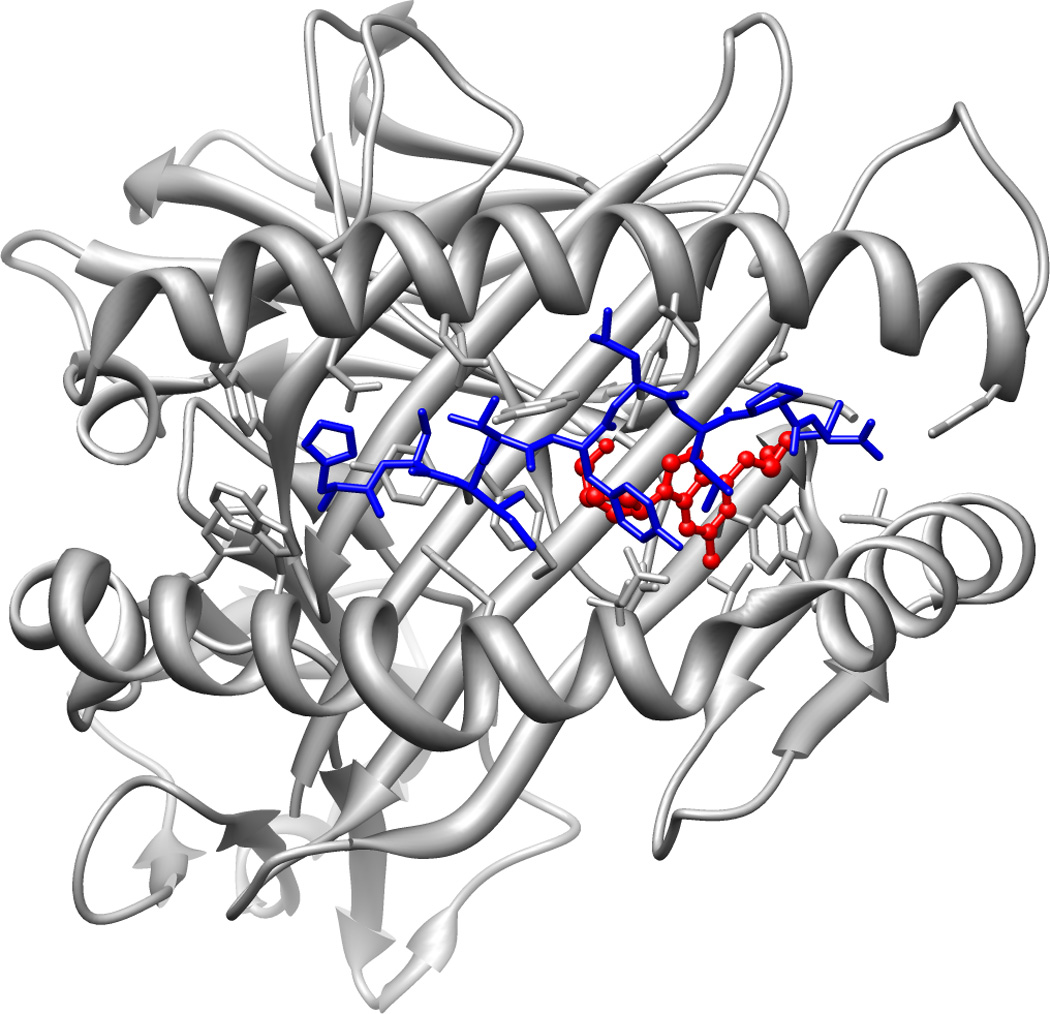
Abacavir (shown in red) binds within the F pocket of HLA-B*57:01 where the C-terminal residues of bound peptides would normally bind, altering its binding repertoire. Presentation of alternative peptides (shown in blue) to the immune system is hypothesized to trigger abacavir HSR. This image was rendered from PDB file 3UPR.
Initial Studies Linking HLA-B*57:01 to Abacavir HSR
The first retrospective study linking HLA-B*57:01 to abacavir HSR risk was published in early 2002 and involved the first 200 patients exposed to abacavir in the Western Australian HIV Cohort Study3 (Table 3). The patients, who were primarily male and white, were HLA-typed across multiple HLA genes, including HLA-A, HLA-B, HLA-C, HLA-DR, and HLA-DQ. A single clinician, who was blinded to each of these patient’s s HLA genotype, reviewed their medical records to look for evidence of hypersensitivity per established diagnostic criteria. These included at least two HSR symptoms within the first 6 weeks of abacavir exposure. Additional criteria included resolution of symptoms within 72 hours after abacavir withdrawal and the absence of a likely alternative explanation for the patient’s symptoms.
Table 3.
Summary of pharmacogenetic studies and their key findings with regard to HLA-B*57:01 and abacavir HSR.
| Year Published | Authors | Study Type | Key findings |
|---|---|---|---|
| 2002 | Mallal et al3 | Retrospective |
|
| 2002 | Hetherington et al36 | Retrospective |
|
| 2004 | Hughes et al37 | Retrospective |
|
| 2004 | Martin et al30 | Retrospective |
|
| 2006 | Rauch et al38 | Prospective |
|
| 2008 | Mallal et al4 | Prospective |
|
| 2008 | Saag et al8 | Retrospective |
|
Among the cohort, 18 “definite” cases of abacavir HSR were identified, all of whom were white. As well, 167 abacavir-tolerant (control) individuals and an additional 15 individuals who experienced nonspecific symptoms were identified but did not meet the established criteria for HSR diagnosis. HLA-B*57:01 was present in 14 (78%) of the “definite” abacavir HSR cases versus only 4 (2.4%) in the abacavir-tolerant controls (odds ratio [OR]117; 95% confidence interval [CI]: 29–481; p < 0.0001). Additionally, the HLA-DR7 and HLA-DQ3 alleles were present in HSR cases at a significantly higher level than controls. Across the entire cohort, the haplotype of HLA-B*57:01, HLA-DR7, and HLA-DQ3 was found to have a positive predictive value (PPV) of 100% and negative predictive value (NPV) of 97%. These investigators then began performing genetic screening in all abacavir-eligible patients and did not prescribe abacavir to individuals that carry the HLA-B*57:01, HLA-DR7, HLA-DQ3 haplotype. However, because their cohort was mostly male and white, these findings required replication in females and other ethnicities.
Only weeks after these findings were published, GlaxoSmithKline released their own retrospective case-control study36 replicating the predictive value for HLA-B*57:01, but again noted the need to replicate these findings in females and other ethnic groups. In a multicenter, retrospective follow-up study37 published in 2004, GlaxoSmithKline investigators replicated their original findings in white males and extended the predictive value of HLA-B*57:01 to white females and Hispanics. The GlaxoSmithKline study showed no association between HLA-B*57:01 and HSR in black patients, the majority of whom were African American37. Among the cases, there were fewer black HLA-B*57:01 carriers than whites or Hispanics, which may simply be due to HLA-B*57:01 being less common in blacks compared with the other ethnic groups (Table 2). The low frequency of the risk allele and the possible misclassification of abacavir cases in this group could have limited the ability to detect a significant association between genotype and HSR.
The Western Australian HIV Cohort Study published their own follow-up study30 in 2004 consisting of a re-analysis of their original 200 patients3 and an additional 48 individuals who enrolled after prospective screening for HLA-B*57:01 was instituted. All patients were re-classified as cases or controls based upon updated diagnostic criteria, which included the results of SPT when available. The reclassification resulted in 18 HSR cases and 230 abacavir-tolerant controls. Again, HLA-B*57:01 was a significant predictor of abacavir HSR, being present in 17 (94.4%) of the cases and only 4 (1.7%) of the controls30. Being a carrier of HLA-B*57:01 was associated with an OR of 960 of developing HSR. Genotyping for the presence of HLA-B*57:01 had a PPV of 78.9% and an NPV of 99.4% for predicting incidence of HSR, supporting a role for the clinical utility in Caucasian populations.
In another follow-up study38, the authors from the Western Australia HIV Cohort Study focused on patients entering the study since 2002, all of whom were prospectively screened for HLA-B*57:01. The overall carrier rate of HLA-B*57:01 in this population of 260 treatment-naïve and treatment-experienced patients was 7.7%. Skin patch testing was performed in all cases of suspected HSR and in patients for who hypersensitivity could not be ruled out and who consented to testing. Among 151 individuals given abacavir during the course of the study, none of the 148 HLA-B*57:01- negative patients experienced HSR. Despite screening, three HLA-B*57:01 positive white patients received abacavir and all three experienced HSR confirmed by SPT. Two of these cases were due to abacavir being mistakenly prescribed before HLA typing results were reviewed. The third case was due to the patient willingly starting abacavir despite being a carrier of HLA-B*57:01. This patient was not a carrier of the HLA-DR7 and HLA-DQ3 alleles previously linked to HSR3, suggesting that carrying HLA-B*57:01 alone was sufficient to develop HSR. The HSR rate in this prospectively screened group of patients was 2.0%, representing a significant decrease from the 8.0% incidence of abacavir HSR prior to genetic screening38.
PREDICT-1
The results of PREDICT-1, the first randomized, double-blind, placebo-controlled trial for the evaluation of a pharmacogenetic test to reduce adverse events, were published in The New England Journal of Medicine in early 20084. The study enrolled 1,956 HIV- positive abacavir-naive individuals in 19 countries who were mostly white (84%) and male (72%). Participants were divided between the “standard of care” control group, which received no HLA-B*57:01 testing, and the prospective screening group, where all individuals were genetically tested and only HLA-B*57:01- negative individuals could be prescribed abacavir. All patients with clinically diagnosed HSR also had SPT performed within 6 to 10 weeks of the original reaction.
Overall, 3.4% of patients in the prospective screening group and 7.8% of those in the control group were clinically diagnosed with abacavir HSR, showing a significant reduction in the prospective screening group (OR 0.40; 95% CI: 0.25–0.62; p < 0.001). Furthermore, there were no cases of immunologically confirmed HSR in the prospective screening group compared with 23 (2.7%) in the control group (OR 0.03; 95% CI: 0.00–0.18; P<0.001). HLA-B*57:01 positivity had a sensitivity of 100% for predicting immunologically confirmed HSR, as well as a specificity of 96.9%, PPV of 47.9%, and NPV of 100%. Despite the strength of these findings, their applicability to the wider patient population was limited because the PREDICT-1 cohort was predominantly white and male.
In a multivariate analysis of covariates that might be associated with HSR, introduction of an NNRTI and concomitant use of a PI were both associated with clinically diagnosed HSR; these were the only factors associated with clinically diagnosed but not immunologically confirmed HSR. These results suggested that adverse drug reactions to other antiretrovirals were presumably being attributed to abacavir HSR. This highlights the need for clinical vigilance, as HLA-B*57:01 is not predictive for reactions that may present with similar symptoms.
SHAPE
Shortly after the publication of PREDICT-1, the results of the retrospective SHAPE study were published8. The purpose of this study was to evaluate the sensitivity and specificity of HLA-B*57:01 as a marker for SPT-confirmed abacavir HSR. Additionally, because of the lack of data on the predictive ability of HLA-B*57:01 in black individuals, the SHAPE study was specifically designed to validate HLA-B*57:01 screening in black and white cohorts that would be evaluated separately. A total of 130 white patients and 69 black patients identified by a previous clinical diagnosis of HSR underwent SPT. Among the two groups, 32.3% of the white patients and 7.2% of the black patients had a positive SPT and all were HLA-B*57:01 positive. In both white and black patients, presence of HLA-B*57:01 had a sensitivity of 100% for predicting immunologically confirmed HSR. Specificity was also very high in both populations-- 96% in white patients and 99% in black patients. This clinical trial validated the use of HLA-B*57:01 testing in blacks, although immunologically confirmed HSR is rare in this patient population overall.
Incorporation into Clinical Practice
Though the results of PREDICT-1 and SHAPE were formally published in early 2008, the findings had already been presented in the summer of 2007 at the International AIDS Society Conference. LabCorp, a US-based national reference laboratory, had been offering HLA-B*57:01 testing since early 2005 in response to the initial clinical studies and physician requests. Test volume was low, came from only a small number of physicians, and remained this way for several years. However, once the results of PREDICT-1 and SHAPE were presented, test volume increased 5 fold over a 6-month period39.
In late 2007, the recommendation for HLA-B*57:01 screening was added to the Department of Health and Human Services HIV Guidelines40 and test utilization continued to increase. In July 2008, the FDA issued an advisory to clinicians and updated the package inserts for all products containing abacavir, warning of the risk of HSR in HLA-B*57:01- positive individuals. The labeling change recommended screening all patients for HLA-B*57:01 prior to initiating abacavir therapy, regardless of a patient’s ethnicity. The label change also recommended screening all individuals of unknown HLA-B*57:01 status before reinitiation of abacavir, even if they had previously tolerated it without incident. Additionally, the FDA recommended that abacavir be immediately discontinued if HSR could not be ruled out, even when other diagnoses were possible. However, the momentum for increased testing had already been generated, making the FDA updates more of a confirmation of what clinicians were already practicing rather than a new recommendation.
Since that time, a number of HIV and pharmacogenetic guidelines have recommended the use of HLA-B*57:01 testing in all abacavir-naïve individuals24, 41–44, firmly establishing it as the standard of care before prescribing abacavir. More recent HIV clinical trials have demonstrated a decreased incidence of abacavir HSR following integration of baseline genetic screening. In the European ASSERT study45, all individuals were screened to confirm they were HLA-B*57:01-negative prior to being randomized to the abacavir/lamivudine or tenofovir/emtricitabine arms. In the abacavir treatment arm, only six individuals (3%) reported experiencing abacavir HSR, a significant decrease from historical averages prior to genetic screening1, 3. In the multicenter ARIES study46, only 4 of 515 HLA-B*57:01-negative patients had clinically suspected HSR. Moreover, when these patients later underwent SPT, all four results were negative.
Methods of Testing for HLA-B*57:01
There are many different ways to test for HLA-B*57:01. One such method is direct sequence-based typing, which amplifies the entire genomic region of HLA-B from both chromosomes using polymerase chain reaction (PCR). After amplification, the DNA fragments are then directly sequenced and checked against known HLA-B alleles. The results from the test are reported as the diplotype of both HLA-B alleles present (e.g. HLA-B*57:01 and HLA-B*07:02). Direct sequencing is considered the gold standard for HLA alleles, but it is also the most expensive, time consuming, and resource intensive. Because HLA-B*5701 is the only allele of interest in abacavir HSR, direct sequencing is not generally performed.
One common method of testing for HLA-B*57:01 uses allele-specific PCR47. In this method, oligonucleotide probes are specifically designed for HLA-B*57:01 and will not amplify any other HLA-B alleles48. Results are reported as either “positive” (i.e. the patient is a carrier of HLA-B*57:01) or “negative” (i.e. the patient does not carry HLA-B*57:01), making them easy to interpret. The results of samples assayed using allele- specific PCR maintain near perfect concordance with the results of sequence-based typing49 and are consistent between different testing sites.
Some labs also test for HLA-B*57:01 by assaying for a single nucleotide polymorphism (SNP) near the HLA-B gene. This SNP (rs2395029) is located in the HLA complex P5 (HCP5) gene approximately 100 kilo bases away from HLA-B. Testing for the presence of this SNP is less time and resource intensive than other methods, making it an attractive option for many labs. There is a strong linkage disequilibrium (LD) between rs2395029 and HLA-B*57:01, meaning that these variants are typically coinherited, in some Caucasian50, 51 and Hispanic52 populations. Therefore, testing for the presence of rs2395029 could be used as a surrogate marker for the presence of HLA-B*57:01. While complete LD has been observed in some populations, many others do not have complete LD between HLA-B*57:01 and rs239502953. When these variants are not inherited together, testing for rs2395029 could lead to an incorrect assumption about whether a patient carries HLA-B*57:01. This ultimately leads to decreased sensitivity and specificity and increased false positive and false negative rates for this test when compared to approaches that directly assess whether HLA-B*57:01 is present. While denying abacavir to someone that is truly HLA-B*57:01 negative is unlikely to be of serious clinical consequence, improperly prescribing abacavir to someone that carries HLA-B*57:01 places this patient at significant risk for HSR. Additionally, the linkage between rs2395029 and HLA-B*57:01 is not well studied in African and Asian populations, making this assay of questionable use in these groups. Despite these potential problems, some reference labs continue to use this test as a marker for HLA-B*57:01. Clinicians should be keenly aware of what methods are being used when they order HLA-B*57:01 testing and keep this in mind when examining cases of suspected HSR.
Conclusions
The story of abacavir HSR highlights a critical component of successful pharmacogenomic studies: a clearly defined phenotype. Because of the generalized nature of most HSR symptoms, the clinical presentation of HSR can be mistaken for the acute onset of a viral or bacterial infection or an adverse reaction to other medications. Prior to implementation of genetic screening, the clinical diagnosis of HSR likely included true cases of abacavir HSR along with a significant number of illnesses that manifested as similar symptoms4. Because rechallenge with abacavir is contraindicated, it was essentially impossible at the time to separate true abacavir HSR from a false-positive diagnosis. This has also resulted in significant differences in clinical diagnosis rates between studies1, 3, making comparisons between them difficult. After SPT began to identify true cases of HSR in a more agnostic manner, it was clear that many people clinically diagnosed with abacavir HSR were not, in fact, true cases30. Unsurprisingly, when cases and controls were reassigned based upon positive SPT results, HLA-B*57:01 became an even stronger predictor of HSR30.
The evolution of abacavir and HLA-B*57:01 from initial finding to standard of care may also serve as a useful guide for clinical implementation of other pharmacogenomic tests. Abacavir is a rarity in the world of pharmacogenomics because of our ability to successfully integrate genetic screening into normal clinical practice in just a matter of years. One of the longstanding challenges in pharmacogenomics has been the difficulty in translating all of the novel findings into guided clinical actions. Many pharmacogenetic tests have been clinically available for years, but are underused by clinicians54.
What has really been so special about abacavir? Part of its success may be that clinicians focusing on HIV treatment are a relatively small group. As such, it is comparatively easy to reach a significant portion of them at a scientific conference or with the latest journal article. This may have allowed information on abacavir pharmacogenetics to travel through the HIV community at a much quicker pace than pharmacogenetics would have been disseminated through much larger fields such as cardiovascular disease or diabetes. Additionally, HIV clinicians were already used to and familiar with genetic tests through interpreting viral resistance panels, so the barriers to adding another genetic test may have been lower than in other fields. While viral resistance panels deal directly with the genetics of the HIV virus and HLA-B*57:01 is a component of the entirely separate host genetics, the basic principle that specific genotypes guide particular therapeutic choices is the same. It may be possible to ease this barrier to implementation with the routine introduction of pharmacogenomics into medical and pharmacy school curricula. As well, the increasing number of drugs under development for individuals with a particular genotype will hopefully increase clinician understanding and appreciation for how genetics can influence and enhance therapeutic decisions.
Abacavir likely also benefited from the quick appearance of HSR, and which presented as a phenotype that was relatively easy to follow and manage. Many pharmacogenetic associations deal directly with drug pharmacokinetics or pharmacodynamics, requiring an extra stage of experimentation to determine what the optimal increased or decreased dosing of these drugs should be to match a particular genotype55, 56. If the gene in question has a significant number of isoforms or SNPs, this only adds to the complexity and difficulty. Dosing information is critical in the design of prospective clinical trials; unfortunately these trials could involve years of follow-up if the drug is used to prevent long-term outcomes and may require trial and error before optimal dosing is found. Abacavir is unusual in the speed at which genetic testing was implemented, perhaps because, a significant number of steps could be ignored that other pharmacogenetic tests have to address.
Consequently, it might be that abacavir and HLA-B*57:01 genotyping success lies in its simplicity. There was no complicated dosing to work out and the clinical trials did not require years of follow-up. The only choice available to us as clinicians regarding abacravir is to use the drug or not use the drug1, 2. While there is no doubt that the field of pharmacogenomics must push ahead to answer the tough questions, perhaps we should focus more on toxicities that can help us make decisions about whether to use a particular medication. Maybe then we can duplicate the lessons learned from abacavir and generate the next pharmacogenetic success story.
Acknowledgements
Supported by Predoctoral Training Grant GM-07175 from the National Institutes of Health.
References
- 1.Hetherington S, McGuirk S, Powell G, et al. Hypersensitivity reactions during therapy with the nucleoside reverse transcriptase inhibitor abacavir. Clin Ther. 2001;23:1603–1614. doi: 10.1016/s0149-2918(01)80132-6. [DOI] [PubMed] [Google Scholar]
- 2.Walensky RP, Goldberg JH, Daily JP. Anaphylaxis after rechallenge with abacavir. AIDS. 1999;13:999–1000. doi: 10.1097/00002030-199905280-00022. [DOI] [PubMed] [Google Scholar]
- 3.Mallal S, Nolan D, Witt C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359:727–732. doi: 10.1016/s0140-6736(02)07873-x. [DOI] [PubMed] [Google Scholar]
- 4.Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568–579. doi: 10.1056/NEJMoa0706135. [DOI] [PubMed] [Google Scholar]
- 5.Epzicom. Research Triangle Park, NC: ViiV Healthcare; 2012. May, [package insert]. [Google Scholar]
- 6.Escaut L, Liotier JY, Albengres E, Cheminot N, Vittecoq D. Abacavir rechallenge has to be avoided in case of hypersensitivity reaction. AIDS. 1999;13:1419. doi: 10.1097/00002030-199907300-00026. [DOI] [PubMed] [Google Scholar]
- 7.Shear NH, Milpied B, Bruynzeel DP, Phillips EJ. A review of drug patch testing and implications for HIV clinicians. AIDS. 2008;22:999–1007. doi: 10.1097/QAD.0b013e3282f7cb60. [DOI] [PubMed] [Google Scholar]
- 8.Saag M, Balu R, Phillips E, et al. High sensitivity of human leukocyte antigen-B*5701 as a marker for immunologically confirmed abacavir hypersensitivity in white and black patients. Clin Infect Dis. 2008;46:1111–1118. doi: 10.1086/529382. [DOI] [PubMed] [Google Scholar]
- 9.Giorgini S, Martinelli C, Tognetti L, et al. Use of patch testing for the diagnosis of abacavir-related hypersensitivity reaction in HIV patients. Dermatol Ther. 2011;24:591–594. doi: 10.1111/j.1529-8019.2012.01409.x. [DOI] [PubMed] [Google Scholar]
- 10.Yuen GJ, Weller S, Pakes GE. A review of the pharmacokinetics of abacavir. Clin Pharmacokinet. 2008;47:351–371. doi: 10.2165/00003088-200847060-00001. [DOI] [PubMed] [Google Scholar]
- 11.Faletto MB, Miller WH, Garvey EP, St Clair MH, Daluge SM, Good SS. Unique intracellular activation of the potent anti-human immunodeficiency virus agent 1592U89. Antimicrob Agents Chemother. 1997;41:1099–1107. doi: 10.1128/aac.41.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDowell JA, Chittick GE, Ravitch JR, Polk RE, Kerkering TM, Stein DS. Pharmacokinetics of [14C]abacavir, a human immunodeficiency virus type 1 (HIV-1) reverse transcriptase inhibitor, administered in a single oral dose to HIV-1-infected adults: a mass balance study. Antimicrob Agents Chemother. 1999;43:2855–2861. doi: 10.1128/aac.43.12.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDowell JA, Chittick GE, Stevens CP, Edwards KD, Stein DS. Pharmacokinetic interaction of abacavir (1592U89) and ethanol in human immunodeficiency virus-infected adults. Antimicrob Agents Chemother. 2000;44:1686–1690. doi: 10.1128/aac.44.6.1686-1690.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Izzedine H, Launay-Vacher V, Aymard G, Legrand M, Deray G. Pharmacokinetics of abacavir in HIV-1-infected patients with impaired renal function. Nephron. 2001;89:62–67. doi: 10.1159/000046045. [DOI] [PubMed] [Google Scholar]
- 15.Gupta SK, Eustace JA, Winston JA, et al. Guidelines for the management of chronic kidney disease in HIV-infected patients: recommendations of the HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis. 2005;40:1559–1585. doi: 10.1086/430257. [DOI] [PubMed] [Google Scholar]
- 16.Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA. Mechanisms of MHC class I-restricted antigen processing and cross-presentation. Immunol Rev. 2005;207:145–157. doi: 10.1111/j.0105-2896.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- 17.Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet. 2009;54:15–39. doi: 10.1038/jhg.2008.5. [DOI] [PubMed] [Google Scholar]
- 18.Hurley CK, Baxter Lowe LA, Logan B, et al. National Marrow Donor Program HLA-matching guidelines for unrelated marrow transplants. Biol Blood Marrow Transplant. 2003;9:610–615. doi: 10.1016/j.bbmt.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 19.Thomas GP, Brown MA. Genetics and genomics of ankylosing spondylitis. Immunol Rev. 2010;233:162–180. doi: 10.1111/j.0105-2896.2009.00852.x. [DOI] [PubMed] [Google Scholar]
- 20.Raychaudhuri S, Sandor C, Stahl EA, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44:291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A. 2005;102:4134–4139. doi: 10.1073/pnas.0409500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428:486. doi: 10.1038/428486a. [DOI] [PubMed] [Google Scholar]
- 23.Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41:816–819. doi: 10.1038/ng.379. [DOI] [PubMed] [Google Scholar]
- 24.Martin MA, Klein TE, Dong BJ, Pirmohamed M, Haas DW, Kroetz DL. Clinical pharmacogenetics implementation consortium guidelines for HLA-B genotype and abacavir dosing. Clin Pharmacol Ther. 2012;91:734–738. doi: 10.1038/clpt.2011.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng ML, Guo XJ, Zhang JY, et al. Study on the haplotypes of MICA and MICB microsatellite and HLA-B locus in the Guangzhou Han population. Tissue Antigens. 2004;64:281–285. doi: 10.1111/j.0001-2815.2004.00293.x. [DOI] [PubMed] [Google Scholar]
- 26.Hong W, Fu Y, Chen S, Wang F, Ren X, Xu A. Distributions of HLA class I alleles and haplotypes in Northern Han Chinese. Tissue Antigens. 2005;66:297–304. doi: 10.1111/j.1399-0039.2005.00474.x. [DOI] [PubMed] [Google Scholar]
- 27.Lee KW, Oh DH, Lee C, Yang SY. Allelic and haplotypic diversity of HLA-A, -B, -C, -DRB1, and -DQB1 genes in the Korean population. Tissue Antigens. 2005;65:437–447. doi: 10.1111/j.1399-0039.2005.00386.x. [DOI] [PubMed] [Google Scholar]
- 28.Inoue T, Ogawa A, Tokunaga K, et al. Diversity of HLA-B17 alleles and haplotypes in East Asians and a novel Cw6 allele (Cw*0604) associated with B*5701. Tissue Antigens. 1999;53:534–544. doi: 10.1034/j.1399-0039.1999.530603.x. [DOI] [PubMed] [Google Scholar]
- 29.Shankarkumar U, Sridharan B, Pitchappan RM. HLA diversity among Nadars, a primitive Dravidian caste of South India. Tissue Antigens. 2003;62:542–547. doi: 10.1046/j.1399-0039.2003.00118.x. [DOI] [PubMed] [Google Scholar]
- 30.Martin AM, Nolan D, Gaudieri S, et al. Predisposition to abacavir hypersensitivity conferred by HLA-B*5701 and a haplotypic Hsp70-Hom variant. Proc Natl Acad Sci U S A. 2004;101:4180–4185. doi: 10.1073/pnas.0307067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Radak Z, Zhao Z, Goto S, Koltai E. Age-associated neurodegeneration and oxidative damage to lipids, proteins and DNA. Mol Aspects Med. 2011;32:305–315. doi: 10.1016/j.mam.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Chen CH, Sun L, Mochly-Rosen D. Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovasc Res. 2010;88:51–57. doi: 10.1093/cvr/cvq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Charneira C, Godinho AL, Oliveira MC, et al. Reactive aldehyde metabolites from the anti-HIV drug abacavir: amino acid adducts as possible factors in abacavir toxicity. Chem Res Toxicol. 2011;24:2129–2141. doi: 10.1021/tx200337b. [DOI] [PubMed] [Google Scholar]
- 34.Norcross MA, Luo S, Lu L, et al. Abacavir induces loading of novel self-peptides into HLA-B*57: 01: an autoimmune model for HLA-associated drug hypersensitivity. AIDS. 2012;26:F21–F29. doi: 10.1097/QAD.0b013e328355fe8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adam J, Eriksson KK, Schnyder B, Fontana S, Pichler WJ, Yerly D. Avidity determines T-cell reactivity in abacavir hypersensitivity. Eur J Immunol. 2012;42:1706–1716. doi: 10.1002/eji.201142159. [DOI] [PubMed] [Google Scholar]
- 36.Hetherington S, Hughes AR, Mosteller M, et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet. 2002;359:1121–1122. doi: 10.1016/S0140-6736(02)08158-8. [DOI] [PubMed] [Google Scholar]
- 37.Hughes AR, Mosteller M, Bansal AT, et al. Association of genetic variations in HLA-B region with hypersensitivity to abacavir in some, but not all, populations. Pharmacogenomics. 2004;5:203–211. doi: 10.1517/phgs.5.2.203.27481. [DOI] [PubMed] [Google Scholar]
- 38.Rauch A, Nolan D, Martin A, McKinnon E, Almeida C, Mallal S. Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study. Clin Infect Dis. 2006;43:99–102. doi: 10.1086/504874. [DOI] [PubMed] [Google Scholar]
- 39.Lai-Goldman M, Faruki H. Abacavir hypersensitivity: a model system for pharmacogenetic test adoption. Genet Med. 2008;10:874–878. doi: 10.1097/GIM.0b013e31818de71c. [DOI] [PubMed] [Google Scholar]
- 40.Department of Health and Human Services; Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Available at http://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf. [Google Scholar]
- 41.Gazzard BG, Anderson J, Babiker A, et al. British HIV Association guidelines for the treatment of HIV-1-infected adults with antiretroviral therapy 2008. HIV Med. 2008;9:563–608. doi: 10.1111/j.1468-1293.2008.00636.x. [DOI] [PubMed] [Google Scholar]
- 42.Aberg JA, Kaplan JE, Libman H, et al. Primary care guidelines for the management of persons infected with human immunodeficiency virus: 2009 update by the HIV medicine Association of the Infectious Diseases Society of America. Clin Infect Dis. 2009;49:651–681. doi: 10.1086/605292. [DOI] [PubMed] [Google Scholar]
- 43.Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF–UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics. 2011;12:113–124. doi: 10.2217/pgs.10.147. [DOI] [PubMed] [Google Scholar]
- 44.Swen JJ, Nijenhuis M, de Boer A, et al. Pharmacogenetics: From Bench to Byte- An Update of Guidelines. Clin Pharmacol Ther. 2011;89:662–673. doi: 10.1038/clpt.2011.34. [DOI] [PubMed] [Google Scholar]
- 45.Post FA, Moyle GJ, Stellbrink HJ, et al. Randomized comparison of renal effects, efficacy, and safety with once-daily abacavir/lamivudine versus tenofovir/emtricitabine, administered with efavirenz, in antiretroviral-naive, HIV-1-infected adults-48-week results from the ASSERT study. J Acquir Immune Defic Syndr. 2010;55:49–57. doi: 10.1097/QAI.0b013e3181dd911e. [DOI] [PubMed] [Google Scholar]
- 46.Squires KE, Young B, DeJesus E, et al. Safety and efficacy of a 36-week induction regimen of abacavir/lamivudine and ritonavir-boosted atazanavir in HIV-infected patients. HIV Clin Trials. 2010;11:69–79. doi: 10.1310/hct1102-69. [DOI] [PubMed] [Google Scholar]
- 47.Martin AM, Nolan D, Mallal S. HLA-B*5701 typing by sequence-specific amplification: validation and comparison with sequence-based typing. Tissue Antigens. 2005;65:571–574. doi: 10.1111/j.1399-0039.2005.00401.x. [DOI] [PubMed] [Google Scholar]
- 48.Hammond E, Mamotte C, Nolan D, Mallal S. HLA-B*5701 typing: evaluation of an allele-specific polymerase chain reaction melting assay. Tissue Antigens. 2007;70:58–61. doi: 10.1111/j.1399-0039.2007.00840.x. [DOI] [PubMed] [Google Scholar]
- 49.Hammond E, Almeida CA, Mamotte C, et al. External quality assessment of HLA-B*5701 reporting: an international multicentre survey. Antivir Ther. 2007;12:1027–1032. [PubMed] [Google Scholar]
- 50.Colombo S, Rauch A, Rotger M, et al. The HCP5 single-nucleotide polymorphism: a simple screening tool for prediction of hypersensitivity reaction to abacavir. J Infect Dis. 2008;198:864–867. doi: 10.1086/591184. [DOI] [PubMed] [Google Scholar]
- 51.Rodríguez-Nóvoa S, Cuenca L, Morello J, et al. Use of the HCP5 single nucleotide polymorphism to predict hypersensitivity reactions to abacavir: correlation with HLA-B*5701. J Antimicrob Chemother. 2010;65:1567–1569. doi: 10.1093/jac/dkq204. [DOI] [PubMed] [Google Scholar]
- 52.Sanchez-Giron F, Villegas-Torres B, Jaramillo-Villafuerte K, et al. Association of the genetic marker for abacavir hypersensitivity HLA-B*5701 with HCP5 rs2395029 in Mexican Mestizos. Pharmacogenomics. 2011;12:809–814. doi: 10.2217/pgs.11.31. [DOI] [PubMed] [Google Scholar]
- 53.Badulli C, Sestini R, Sbarsi I, et al. Tag SNPs of the ancestral haplotype 57.1 do not substitute HLA-B*57:01 typing for eligibility to abacavir treatment in the Italian population. Pharmacogenomics. 2012;13:247–249. doi: 10.2217/pgs.11.168. [DOI] [PubMed] [Google Scholar]
- 54.Ong FS, Deignan JL, Kuo JZ, et al. Clinical utility of pharmacogenetic biomarkers in cardiovascular therapeutics: a challenge for clinical implementation. Pharmacogenomics. 2012;13:465–475. doi: 10.2217/pgs.12.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grossman I. Routine pharmacogenetic testing in clinical practice: dream or reality? Pharmacogenomics. 2007;8:1449–1459. doi: 10.2217/14622416.8.10.1449. [DOI] [PubMed] [Google Scholar]
- 56.Eriksson N, Wadelius M. Prediction of warfarin dose: why, when and how? Pharmacogenomics. 2012;13:429–440. doi: 10.2217/pgs.11.184. [DOI] [PubMed] [Google Scholar]