

Abstract
Neurodegenerative diseases such as Huntington disease, Parkinson’s disease, and Alzheimer’s disease are caused by the accumulation of aggregate prone proteins. Pathogenic proteins misfold, aggregate, and escape the cell’s normal degradative pathways. Protein aggregates subsequently lead to the toxic disruption of normal cellular processes leading, ultimately, to disease. Several lines of evidence suggest that reducing the burden of these toxic aggregates is therapeutic. One mechanism proposed to facilitate the degradation or clearance of these protein inclusions is macroautophagy. While autophagic treatment paradigms for neurodegeneration are still in the early stages of preclinical development, it is essential to identify and validate methods to measure the activation of autophagy in human patients. These methods will serve as important biomarkers necessary to test compound efficacy and monitor clinical improvement.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-013-0180-y) contains supplementary material, which is available to authorized users.
Keywords: Autophagy, Neurodegeneration, Biomarkers
Introduction
Autophagy or “self-eating” has been implicated in the pathogenesis and treatment of many degenerative disorders, most notably neurodegenerative diseases [1]. The fact that autophagy is disrupted in some neurodegenerative diseases further suggests that enhancing autophagy will be therapeutic in protein aggregate disorders [2]. One form of autophagy—macroautophagy (herein referred to as autophagy)—is an intracellular degradative process that sequesters and traffics regions of cytoplasm to the lysosome [3]. During times of nutrient deprivation or stress, cells degrade protein, liberating free amino acids. Autophagy is also necessary for the basal turnover of protein and organelles. Consistent with this, loss of autophagy in the central nervous system (CNS) leads to neuronal loss, ubiquitinated inclusions, and mitochondrial dysfunction [4, 5]. Whether enhancing autophagy will be therapeutic in neurodegenerative diseases associated with protein aggregation is unresolved.
The Therapeutic Potential of Autophagy
While dogma suggests that autophagy is the non-selective bulk degradation of proteins and organelles, recent studies demonstrate that targeted or selective autophagy of substrates can occur [6]. For example, damaged and depolarized mitochondria are selectively marked for autophagic engulfment via the E3 ubiquitin ligase, parkin [7]. Similarly, ubiquitinated protein aggregates are targeted to autophagosomes via ubiquitin adaptor proteins, such as HDAC6 and p62 [8, 9]. These cargo-selective autophagic targeting factors may serve as therapies in protein aggregate disorders.
In the case of the ubiquitin proteasome system (UPS), a protein is selectively ubiquitinated and then degraded via the proteasome [10]. The total level of ubiquitinated proteins or the catalytic activity of the proteasome serves as a reliable surrogate marker of UPS activity in human tissue [10, 11]. In the case of autophagy, a protein or organelle is sequestered into an autophagosome which then fuses with lysosomes where degradation occurs. The wide range of potential substrates and cellular contents degraded via autophagy poses a unique problem in assaying autophagic degradation in a cell or tissue. To circumvent this, studies have identified several proteins that are degraded selectively via autophagy, such as p62 and LC3 [12, 13]. In addition to being autophagic substrates, these proteins are integral components of the autophagic machinery. Therefore, in response to an autophagic stimulus, these substrates are both synthesized and degraded, making it difficult to reliably assess their levels in cells and tissue. The field of autophagy has emphasized that steady-state levels of any autophagic substrate are unreliable reporters and stress the necessity to evaluate autophagic processes using dynamic assays [14]. Thus, the true measure of the autophagic processes is the rate of degradation of cargo or a protein that is selectively engulfed by the autophagosome and subsequently degraded via the lysosome (Fig. 1). This is termed “autophagic flux” and is discussed more extensively later [14]. High throughput screening of compound libraries has identified hundreds of autophagy inducing candidates [15–18]. However, as these compounds move toward a therapeutic reality, it will be essential to confirm and identify their molecular targets and, more importantly, establish their abilities to truly increase degradation of substrates via enhanced autophagic flux in vitro.
Fig. 1.

Macroautophagy encompasses multiple steps that include induction, nucleation of a pre-autophagic structure, expansion of the growing phagophore, sequestration, and cargo loading of cytoplasmic contents, vesicular trafficking, membrane fusion, and, finally, proteolytic digestion of autophagic contents. The entirety of this process is “autophagic flux”
Many studies have evaluated autophagy-enhancing compounds in vitro and then used them to enhance the clearance of pathologic protein aggregates (and, in some cases, improve behavioral phenotypes) in small animal models, for example rapamycin and rilmenidine in Huntington’s disease, lithium in amyotrophic lateral sclerosis, trehalose in frontotemporal dementia, and carbamazepine in α1-antitrypsin-associated liver disease [19–23]. These studies lend proof of concept to the notion that stimulating autophagy will be therapeutic. However, none of these studies have correlated the in vivo effect of a compound to enhance “autophagic flux” in a target tissue (brain, spinal cord, or liver), mobilize protein aggregates, and improve disease phenotype. Instead, they have, at best, demonstrated that a compound enhances autophagy in cell culture, and, when an animal model is treated with the compound, protein aggregate burden decreases and disease phenotype improves. Therefore, whether these compounds truly activate autophagy in vivo, in the target tissue, resulting in autophagy-dependent protein aggregate clearance and phenotypic improvement is not known. The identification of appropriate biomarkers that correlate with autophagic activation or inhibition is essential in order to validate any therapy purported to increase autophagy.
Monitoring Autophagic Degradation In Vivo
The goal of any autophagy-enhancing therapy in protein aggregate disease is to decrease protein aggregates within the target tissue. Several studies (mentioned above) using animal models have demonstrated that compounds and small molecules can decrease protein aggregates in the CNS [19–23]. This is an important biomarker for therapeutic efficacy (perhaps the most relevant marker as the goal of any autophagic therapy is to decrease protein aggregate burden), but its interpretation can be problematic. For example, the most commonly utilized autophagy-enhancing agent, rapamycin, is a mammalian target of rapamycin (mTOR) inhibitor [2]. mTOR integrates nutrient, energy, and growth signaling pathways to regulate cell growth, protein synthesis, and autophagy [24, 25]. When mTOR is activated by amino acids or exogenous growth factors, protein synthesis is activated and autophagy is diminished, whereas when mTOR is inhibited, as in treatment with rapamycin, protein synthesis is decreased and autophagy is activated. Therefore, a decrease in protein aggregate burden with mTOR inhibition could occur owing to a decrease in protein synthesis or enhanced autophagic clearance [26].
Protein aggregate burden can also be diminished via the activation of the proteasome independent of autophagic activity [27]. While the proteasome is likely ineffective at degrading large protein aggregates, it can degrade soluble aggregate prone species prior to aggregate formation. Similarly, protein aggregates can be decreased via the upregulation of protein chaperones that maintain aggregate prone proteins in states that are more amenable to proteasomal degradation [28]. Therefore, caution needs to be used when making an assumption that any autophagy-enhancing compound is truly working via an autophagic mechanism.
Autophagic Proteins
The quantitation of the levels and expression of select autophagic proteins may also serve as reliable biomarkers for autophagic activation. Under some conditions, such as starvation in skeletal muscle, there is a coordinated increase in the expression of multiple autophagic proteins [29, 30]. These include proteins that initiate autophagy, such as ATG5 and beclin, autophagosome machinery, and lysosomal components. However, the levels of these proteins can increase under conditions of cellular stress and even in response to the presence of protein aggregation [31]. Moreover, an increase in autophagic protein expression can be consistent with cell injury and death [32]. Finally, many autophagic proteins are integral components of the autophagosome and are synthesized and degraded during autophagic stimuli, making them difficult to assess (see the section Measuring “Autophagic Flux” In Vivo).
Autophagic Structures
Several studies have quantitated the number and size of autophagosomes utilizing immunohistochemical analysis or electron microscopy. In addition, the use of a green fluorescent protein-tagged LC3 protein either delivered to tissue or transgenically expressed can be used as a marker of autophagosomes [33]. However, an increase in autophagosomes does not always correlate with an increase in autophagic degradation. LC3 has been shown to incorporate into existing protein aggregates and even aggregate on its own [34]. Therefore, assuming that LC3 puncta are, indeed, autophagosomes may not be reliable.
In the case of a Huntington’s disease model, it was shown that autophagosomes were, formed and could be enhanced when autophagy was stimulated [31]. However, these autophagosomes failed to contain autophagic cargo, in particular, huntingtin-positive protein aggregates resulting in a reduction in global autophagic degradation [31]. This is a hugely problematic observation. It suggests that autophagic flux (turnover of autophagosomes) could occur and even be increased yet not engulf pathologic aggregates. This finding clearly emphasizes the need to utilize multiple biomarkers when considering an autophagic treatment for degenerative disease.
An increase in autophagosomes can also correlate with a decrease in their degradation [35]. This can make it difficult to assess whether an increase in steady state autophagosomes is due to enhanced autophagosome biogenesis of functional and degradative structures or a constipation of non-degradative autophagosomes.
Autophagic Pathways
Some autophagy-enhancing compounds have clearly identified pharmacologic targets that can be measured to test efficacy. One example is rapamycin, which activates autophagy by inhibiting mTOR [24]. Therefore, measures of mTOR activity or the phosphorylation of its downstream targets can be useful biomarkers. However, as the number of compounds that have putative autophagy enhancing effects grows, the mechanism of action may be less clear or due to off-target effects. In the case of rilmenidine, currently in clinical trials for Huntington’s disease, an obvious surrogate marker of drug efficacy is less clear. Rilmenidine is a centrally-acting antihypertensive that acts on α2-adrenoceptors and imidazoline I1 receptors [20]. Measuring activation of these receptors or monitoring patient blood pressure may be helpful to evaluate rilmendine efficacy with regard to cardiovascular effects, but is unlikely to be relevant to its proposed autophagy-enhancing function.
Identifying a clear pharmacologic target that can serve as surrogate biomarker is an important concept in drug development in which a chemical compound platform may need to be diversified in order to identify compounds with improved therapeutic efficacy. Two examples of compounds that have demonstrated autophagy promoting effects without clear pharmacologic targets are trehalose and spermidine [2]. In the case of these types of autophagic compounds, one would need to measure autophagic function to confirm efficacy.
Autophagy-Specific Cargo
Proteins can be degraded via two principal proteolytic pathways—the UPS and autophagy. Most proteins, depending upon their state (soluble, misfolded, or aggregated), can be degraded via both pathways. This can make the interpretation that a substrate is truly degraded via enhanced autophagy difficult. Several autophagy-specific/selective substrates have been proposed, most notably the autophagosome marker LC3II and p62/sequestosome. LC3 is converted to LC3II upon autophagic stimulation and is then conjugated to the growing phagophore membrane via phosphotidylethanolamine. Upon fusion with the lysosome, the autophagosome and LC3II are both degraded. p62 is a member of a growing class of autophagic adaptor proteins that bind ubiquitinated cargo and LC3, facilitating the degradation of select cargo [36]. In performing this function, p62 is degraded along with its associated cargo within the autophagosome. However, just as other substrates can be degraded via the UPS or autophagy, p62 may also be degraded within the autophagosome or via the proteasome [37].
Several studies have suggested that the selection of autophagic cargo is dictated by the type of ubiquitin chain that tags the degradation destined protein [38]. For example, when the UPS is inhibited with agents such as epoxomicin, there is selective accumulation of K48-linked ubiquitin chains [39]. In contrast, when lysosomal degradation is blocked in cell culture, there is an enrichment of K63 linked ubiquitin chains [39]. These data suggest that measuring the levels of ubiquitinated proteins and, perhaps, the types of ubiquitin chains can be surrogate biomarkers for autophagic activity.
Measuring “Autophagic Flux” In Vivo
When evaluating a potential therapy or intervention that induces autophagy, it is essential to measure “autophagic flux” and not just induction of autophagy [40]. Autophagic flux is the turnover of a protein or organelle via autophagy. The autophagy pathway includes multiple steps, for example induction, sequestration of cytoplasmic contents, trafficking to and fusion with the lysosome, and, finally, lysosomal degradation (Fig. 1). Quantitation of the movement or “flux” of a protein substrate through these steps is the autophagic flux within the cell or tissue. Therefore, in order to accurately measure autophagic flux, it is essential to identify a substrate that is selectively degraded via autophagy. One candidate is the autophagosome protein LC3II. However, steady-state levels of LC3II protein are an unreliable measure of autophagic flux. An increase in LC3II protein levels can be consistent with enhanced LC3II conversion or a decrease in LC3II positive autophagosome degradation [14]. Moreover, autophagic flux can be elevated when steady state levels of LC3II appear unchanged. This is because in an intact autophagic system, LC3II is produced as rapidly as it is degraded. Therefore, any intervention that proposes to increase autophagy needs to be confirmed via an autophagic flux assay. Autophagic flux has traditionally been measured in cell culture by measuring LC3II protein levels with and without inhibitors of lysosomal fusion, such as bafilomycinA or vinblastine (Fig. 2). An increase in autophagic flux can only be determined when the LC3II levels are compared amongst conditions that include no treatment and treatment with an autophagy-inducing agent. In addition, both of these conditions need to be performed in the setting of co-treatment with an inhibitor of autophagosome degradation (i.e., lysosomal protease inhibitor or inhibitor of lysosome–autophagosome fusion) (Fig. 2). Some studies have performed these types of experiments in vivo. For example, we adapted the lysosomal fusion inhibitor model to skeletal muscle in vivo using the microtubule depolarizing agent colchicine [41]. We screened multiple lysomotropic and microtubule disrupting compounds for their ability to block LC3II degradation, and identified colchicine as a potent and safe inhibitor of autophagosome–lysosome fusion in mouse skeletal that increased basal levels of LC3II. When mice were starved for 24 hours or treated with rapamycin for 7 days, there was no change in LC3II levels in the skeletal muscle compared with untreated mice. However, when starved or rapamycin-treated mice were treated for 24 hours with colchicine there was an obvious increase in the levels of LC3II within the skeletal muscle as compared with control mice treated with colchicine alone, suggesting an increase in autophagic flux.
Fig. 2.

How to measure basal and induced autophagic flux. a An intact autophagic system produces and degrades LC3II/autophagosomes. b Blocking LC3II/ autophagosomes with compounds like BafA and colchicine reflect the production of LC3II in the cell or “flux.” c Interventions that enhance flux increase LC3II/ autophagosome production and degradation; therefore, on an immunoblot, LC3II levels may not change. d Blocking LC3II degradation in the setting of enhanced flux reveals the true increase in LC3II production. e Example of immunoblot and densitometric graph of autophagic flux assays. Condition A compared with B reflects basal flux, whereas comparing B to D reflects the amount of stimulated or enhanced autophagic flux
Using this type of in vivo autophagic flux assay, one could potentially screen multiple compounds with reported in vitro efficacy for their ability to enhance autophagic flux in vivo (Fig. 3). Similar in vivo assays have quantified autophagic flux in cardiac tissue with the lysomotrophic agent chloroquine and in the liver, heart, lung, kidney, and spleen utilizing the protease inhibitor leupeptin, but none have been able to evaluate autophagic flux in the CNS [42, 43].
Fig. 3.
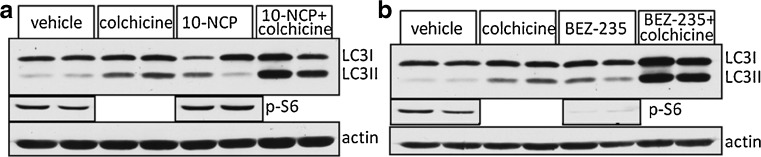
In vivo autophagic flux in skeletal muscle using mammalian target of rapamycin (mTOR)-independent (a) and mTOR-dependent (b) compounds. Mice are treated for 7 days with compound and then LC3 levels are measured in vehicle, 24-hour colchicine, compound or compound + colchicine on day 6. The levels of pS6 demonstrate that BEZ-235 inhibits mTOR, whereas 10-NCP does not
Measuring “Autophagic Flux” in Humans
How might one measure autophagic flux in human tissue? More specifically, how might one measure autophagic flux in an inaccessible tissue such as the brain of human patients? Recently, Bateman et al. [44–46] devised methodology to evaluate the synthesis and clearance of two proteins involved in Alzheimer’s disease—amyloid beta (Aβ) and apolipoprotein E (apoE). They infused human patients with a stable isotope-labeled amino acid (13C6-leucine) and then measured the incorporation of this tracer within the Aβ peptide or apoE protein that was sampled from the cerebrospinal fluid (CSF) using high resolution tandem mass spectrometry [46]. These studies were the first to document fractional synthesis and fractional clearance rates (FCR) for a CNS protein. It is conceivable that other pathologic aggregate prone proteins could be measured using similar strategies as some neurodegenerative proteins are detectable in the CSF, including tau, SOD-1 and TDP-43 [47–49]. As mentioned earlier, the mobilization of a pathologic protein aggregate or aggregate prone protein is one of the most relevant autophagic biomarkers for therapeutic efficacy. Therefore, methods that truly measure the FCR of the aggregate forming protein are very compelling and are becoming a valuable adjunctive tool for therapeutic trials [50]. The limitation, of course, is whether the protein is being degraded or cleared via an autophagic mechanism.
To circumvent that issue, one could envisage determining the FCR of an autophagy-specific/selective substrate, such as p62 or LC3II, in a similar manner. These proteins have not been reported to be present in the CSF space. However, in the case of an easily biopsied and tractable tissue, such as skeletal muscle, one could perform stable isotope labeling followed by high resolution tandem mass spectrometry looking at p62 or other autophagy-specific substrate from humans before and after an autophagic intervention. Interestingly, the FCR of skeletal muscle proteins has not been measured directly and only inferred from rates of fractional synthesis. The fractional synthesis rate of mixed proteins in skeletal muscle is ~0.04 %/h, which extrapolates to a FCR for mixed muscle protein of ~1 %/day [51]. Whether enhancing autophagy alters the FCR of total muscle protein or an autophagic substrate is not known.
Other Considerations Regarding Autophagy in Human Patients
Nearly all studies evaluating the rate and induction of autophagy in vivo have been performed in small animal models. For example, a detectable change in the degradation of autophagy proteins can be seen in the skeletal muscle of mice following 24 hours of nutrient deprivation [41]. Moreover, unlike a human, a mouse will lose ~20 % of its body weight when fasted for 24 hours [52]. Whether the autophagic capacity of a human that has evolutionarily adapted to not undergo prolonged periods of nutrient deprivation is similar to that of a rodent is unclear. It is conceivable that compounds or interventions that activate autophagy in rodents, with a high metabolic rate, may have no effect or an undetectable effect in humans.
Treatment paradigms for protein aggregate disorders will also need to be established. It is possible that continuous treatment with an autophagy-enhancing therapy will increase basal autophagic flux. Alternatively, intermittent dosing with an autophagy-stimulating compound may enhance the autophagic response without changing the overall basal rate of autophagic flux. Whether sustained enhancement of autophagic flux or intermittent stimulation of autophagy is more effective at decreasing protein aggregate burden is not known. Once potential compounds are established, concomitant biomarker development and usage may facilitate the answer to this question.
Will Autophagic Stimulation be Effective or Detrimental in Protein Aggregate Disease?
Supposing after autophagic biomarker development, a pharmacologic compound is identified that could potently initiate autophagy in the CNS. Will this intervention be effective or detrimental in neurodegeneration? As detailed earlier, the treatment of small animal models with autophagy-enhancing compounds has improved pathologic and behavioral phenotypes in some protein aggregate models [19–23]. However, in some models, activation of autophagy has worsened disease phenotype. For example, treatment of SOD1G93A-transgenic mice, which are a model of familial amyotrophic lateral sclerosis, with rapamycin reduced life span and hastened the onset of disease [53]. Similarly, the activation of autophagy with rapamycin abrogated muscle weakness and vacuolar pathology in an animal model of inclusion body myopathy, paget’s disease of the bone, and frontotemporal dementia due to mutations in valosin-containing protein [54]. These studies lend caution to the hope that enhancing autophagy will be beneficial in protein aggregate disorders.
Some studies have had less clear results. For example, even the same mechanism of action—enhanced autophagy—has proven to generate different effects in some animal models. SOD1G93A-transgenic mice, which had a worsened phenotype when treated with rapamycin [53], had improved strength and viability when treated with lithium chloride [21] . Both compounds increased the number of autophagic structures in spinal cord neurons of SOD1G93A-transgenic mice, yet had contrasting effects on disease pathogenesis [21, 53]. One could argue that the treatment paradigms were different, leading to the stark discrepancy. However, without knowing whether either of these compounds truly enhances autophagic flux in vivo, it is equally plausible that lithium and rapamycin do not have efficacy or lack efficacy in the case of rapamycin via an autophagic mechanism. Because of this type of uncertainty, further studies aimed at identifying compounds and biomarkers effective at enhancing autophagic flux are necessary.
Conclusion
Autophagic stimulation holds great promise in the treatment of protein aggregate diseases. By enhancing autophagy, protein aggregate burden will be diminished and cell death ameliorated. What therapies will be effective in the CNS and how to monitor autophagic degradation in vivo are current challenges toward making these treatments a reality.
Electronic supplementary material
(PDF 510 kb)
Acknowledgments
Dr Weihl is funded by the NIH (R01AG031867 and K02AG042095), as well as the Muscular Dystrophy Association (MDA218514).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
References
- 1.Banerjee R, Beal MF, Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci. 2010;33:541–549. doi: 10.1016/j.tins.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 4.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 5.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 6.Komatsu M, Ichimura Y. Selective autophagy regulates various cellular functions. Genes Cells. 2010;15:923–933. doi: 10.1111/j.1365-2443.2010.01433.x. [DOI] [PubMed] [Google Scholar]
- 7.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pandey UB, Nie Z, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 9.Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 10.Ying Z, Wang H, Wang G. The Ubiquitin proteasome system as a potential target for the treatment of neurodegenerative diseases. Curr Pharm Des 2012 Nov 2 [Epub ahead of print]. [DOI] [PubMed]
- 11.Yarasheski KE, et al. Whole-body and muscle protein metabolism are not affected by acute deviations from habitual protein intake in older men: the Hormonal Regulators of Muscle and Metabolism in Aging (HORMA) Study. Am J Clin Nutr. 2011;94:172–181. doi: 10.3945/ajcn.110.010959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 14.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L, Yu J, Pan H, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A. 2007;104:19023–19028. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farkas T, Hoyer-Hansen M, Jaattela M. Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux. Autophagy. 2009;5:1018–1025. doi: 10.4161/auto.5.7.9443. [DOI] [PubMed] [Google Scholar]
- 17.Tsvetkov AS, Miller J, Arrasate M, et al. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010;107:16982–16987. doi: 10.1073/pnas.1004498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams A, Sarkar S, Cuddon P, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 20.Rose C, Menzie FM, Renna M, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington's disease. Hum Mol Genet. 2010;19:2144–2153. doi: 10.1093/hmg/ddq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fornai F, Longone P, Cafaro L, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2008;105:2052–2057. doi: 10.1073/pnas.0708022105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hidvegi T, Ewing M, Hale P, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 23.Schaeffer V, Lavenir I, Ozcelik S, Tolnay M, Winker DT, Goedert M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain. 2012;135:2169–2177. doi: 10.1093/brain/aws143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.King MA, Hands S, Hafiz F, et al. Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol Pharmacol. 2008;73:1052–1063. doi: 10.1124/mol.107.043398. [DOI] [PubMed] [Google Scholar]
- 27.Lee BH, Lee MJ, Park S, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rujano MA, Kampinga HH, Salomons FA. Modulation of polyglutamine inclusion formation by the Hsp70 chaperone machine. Exp Cell Res. 2007;313:3568–3578. doi: 10.1016/j.yexcr.2007.07.034. [DOI] [PubMed] [Google Scholar]
- 29.Zhao J, Brault JJ, Schild A, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 30.Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Martinez-Vicente M, Talloczy Z, Wong E, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butler D, Nixon RA, Bahr BA. Potential compensatory responses through autophagic/lysosomal pathways in neurodegenerative diseases. Autophagy. 2006;2:234–237. doi: 10.4161/auto.2729. [DOI] [PubMed] [Google Scholar]
- 33.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007;3:323–328. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- 35.Ju JS, Fuentealba RA, Miller SE, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009;187:875–888. doi: 10.1083/jcb.200908115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8:1986–1990. doi: 10.4161/cc.8.13.8892. [DOI] [PubMed] [Google Scholar]
- 37.Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- 38.Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ. 2012;20:21–30. doi: 10.1038/cdd.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dammer EB, Na CH, Xu P, et al. Polyubiquitin linkage profiles in three models of proteolytic stress suggest the etiology of Alzheimer disease. J Biol Chem. 2011;286:10457–10465. doi: 10.1074/jbc.M110.149633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ju JS, Varadhachary AS, Miller SE, Weihl CC. Quantitation of "autophagic flux" in mature skeletal muscle. Autophagy. 2010;6:929–935. doi: 10.4161/auto.6.7.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iwai-Kanai E, Yuan H, Huang C, et al. A method to measure cardiac autophagic flux in vivo. Autophagy. 2008;4:322–329. doi: 10.4161/auto.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haspel J, Shaik RS, Ifedigbo E, et al. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy. 2011;7:629–642. doi: 10.4161/auto.7.6.15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wildsmith KR, Basak JM, Patterson BW, et al. In vivo human apolipoprotein E isoform fractional turnover rates in the CNS. PLoS ONE 2012l7:e38013. [DOI] [PMC free article] [PubMed]
- 45.Bateman RJ, Munsell LY, Chen X, et al. Stable isotope labeling tandem mass spectrometry (SILT) to quantify protein production and clearance rates. J Am Soc Mass Spectrom. 2007;18:997–1006. doi: 10.1016/j.jasms.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12:856–861. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–349. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 48.Winer L, Srinivasan D, Chun S, et al. SOD1 in cerebral spinal fluid as a pharmacodynamic marker for antisense oligonucleotide therapy. Arch Neurol 2012 Nov 12 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 49.Kasai T, Tokuda T, Ishigami N, et al. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2009;117:55–62. doi: 10.1007/s00401-008-0456-1. [DOI] [PubMed] [Google Scholar]
- 50.Bateman RJ, Klunk WE. Measuring target effect of proposed disease-modifying therapies in Alzheimer's disease. Neurotherapeutics. 2008;5:381–390. doi: 10.1016/j.nurt.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balagopal P, Rooyackers OE, Adey DB, Ades PA, Nair KS. Effects of aging on in vivo synthesis of skeletal muscle myosin heavy-chain and sarcoplasmic protein in humans. Am J Physiol. 1997;273:E790–800. doi: 10.1152/ajpendo.1997.273.4.E790. [DOI] [PubMed] [Google Scholar]
- 52.O'Sullivan U, Gluckman PD, Breir BH, Woodall S, Siddiqui RA, McCutcheon SN. Insulin-like growth factor-1 (IGF-1) in mice reduces weight loss during starvation. Endocrinology. 1989;125:2793–2794. doi: 10.1210/endo-125-5-2793. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, et al. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7:412–425. doi: 10.4161/auto.7.4.14541. [DOI] [PubMed] [Google Scholar]
- 54.Ching JK, Elizabeth SV, Ju JS, Lusk C, Pittman SK, Weihl CC. mTOR dysfunction contributes to vacuolar pathology and weakness in valosin-containing protein associated inclusion body myopathy. Hum Mol Genet 2013 Jan 10 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 510 kb)