

Abstract
The toxicity of Gram-negative bacterial endotoxin (lipopolysaccharide, LPS) resides in its structurally highly conserved glycolipid component called lipid A. Our major goal has been to develop small-molecules that would sequester LPS by binding to the lipid A moiety, so that it could be useful for the prophylaxis or adjunctive therapy of Gram-negative sepsis. We had previously identified in rapid-throughput screens several guanylhydrazones as potent LPS binders. We were desirous of examining if the presence of the guanylhydrazone (rather than an amine) functionality would afford greater LPS sequestration potency. In evaluating a congeneric set of guanylhydrazone analogues, we find that C16 alkyl substitution is optimal in the N-alkylguanylhydrazone series; a homospermine analogue with the terminal amine N-alkylated with a C16 chain with the other terminus of the molecule bearing an unsubstituted guanylhydrazone moiety is marginally more active suggesting very slight, if any, steric effects. Neither C16 analogue is significantly more active than the N-C16-alkyl or N-C16-acyl compounds that we had characterized earlier, indicating that basicity of the phosphate-recognizing cationic group, is not a determinant of LPS sequestration activity.
Keywords: Endotoxin, Lipopolysaccharide, Sepsis, Shock, Lipopolyamines, Guanylhydrazones
Introduction
Endotoxins, or lipopolysaccharides (LPS), a structural component of the outer membrane of Gram-negative bacteria,1 play a pivotal role in septic shock, a syndrome of systemic toxicity which occurs frequently as a sequel to serious systemic Gram-negative infections.2 The activation by LPS of the innate immune response, mediated via toll-like receptor-4,3 leads to an uncontrolled production of numerous inflammatory mediators, including tumor necrosis factor-α (TNF-α), interleukin-1 β (IL-1β), and interleukin-6 (IL-6),4 precipitating a systemic inflammatory response. This culminates in the frequently fatal syndrome of multiple system organ failure.5 Despite continuing advances in antimicrobial chemotherapy, the incidence of sepsis has risen almost three-fold from 1979 through 2000,6 emphasizing an urgent, unmet need to develop therapeutic options specifically targeting the pathophysiology of sepsis.
The toxicity of LPS resides in its structurally highly conserved glycolipid component called lipid A,7 which is composed of a hydrophilic, bis-phosphorylated diglucosamine backbone, and a hydrophobic domain of 6 (E. coli) or 7 (Salmonella) acyl chains in amide and ester linkages (Fig. 1). Polymyxin B (PMB) is a membrane-active peptide antibiotic known to sequester LPS and abrogate its toxicity. The pronounced oto- and nephrotoxicity of PMB precludes its systemic use and has led to the development of an extracorporeal hemoperfusion cartridge based on PMB covalently immobilized on a polystyrene based fiber (Toraymyxin™, Toray Industries Inc., Tokyo).8;9 Approved for clinical use in Japan in late 2000, Toraymyxin provides a clinically validated proof-of-concept of the therapeutic potential of sequestering circulating LPS. A major goal in our laboratory has been to develop small-molecule analogues of PMB that would sequester LPS with a potency comparable to that of PMB and, importantly, be nontoxic and safe, so that it can be used parenterally for the prophylaxis or therapy of Gram-negative sepsis.
Fig. 1.

The structure of lipid A, the toxic core of enterobacterial Gram-negative lipopolysaccharide.
Our identification of the lipopolyamines as potential endotoxin-sequestering molecules was based on two simple heuristics that have been experimentally tested and validated10–15: (a) an optimal distance of ≈14 Å is necessary between protonatable functions in linear bis-cationic molecules for simultaneous ionic interactions with the anionic phosphates on lipid A (Fig. 1), and (b) additional, appropriately positioned hydrophobic group(s) are obligatory for the interaction to manifest in neutralization of endotoxicity. Leads obtained from high-throughput screening on focused libraries16–18 as well as from molecular modeling and in silico docking studies19 suggested that a long-chain alkyl, rather than an acyl group that we had previously characterized15 may allow for more favorable electrostatic interactions with the lipid A phosphate groups. Independent SAR leads obtained in iterative rounds of screening focused libraries10;17;20–24 have all converged on molecules bearing a homologated spermine (homospermine) scaffold. However, we had identified in such screens several guanylhydrazones as potent LPS binders.17 We were desirous of (i) determining if the SAR pertaining to the hydrophobic substituent derived for the acylpolyamines15 would also hold for molecules with similar homospermine scaffolds grafted with guanylhydrazone functionalities, (ii) if the presence of the guanylhydrazone functionality would afford greater LPS sequestration potency owing to reasons mentioned earlier, and (iii) if N-alkyl versus unsubstituted guanylhydrazone compounds would manifest in differential endotoxin neutralization activities due to possible steric effects. In order to examine these questions, we have synthesized a congeneric set of guanylhydrazone analogues. We find that, as for the acylhomospermine compounds, the C16 alkyl chain is optimal in the N-alkylguanylhydrazone series; a homospermine analogue with the terminal amine N-alkylated with a C16 chain with the other terminus of the molecule bearing an unsubstituted guanylhydrazone moiety is marginally more active suggesting very slight, if any, steric effects. Neither C16 analogue is significantly more active than the N-C16-alkyl23 or N-C16-acyl15 compounds that we had characterized earlier, indicating that a primary amine serves just as well as a guanylhydrazone in recognizing the lipid A phosphate groups.
Results and Discussion
Synthesis of the guanylhydrazone compounds
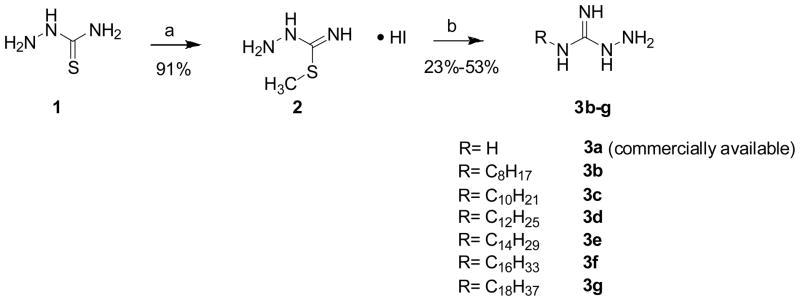
The syntheses of the 8 series involved a straightforward condensation of either commercially-available aminoguanidine 3a or N-alkylhydrazinecarboximidamides 3b–g with the N1,N2,N3,N4-tetraBoc-protected spermine-propanal precursor (6). N-Alkylhydrazinecarboximidamides were synthesized via S-methylation of hydrazinecarbothiamide, followed by displacement with the corresponding alkylamine (Scheme 1). N1,N2,N3-triBoc-protected spermine 4, synthesized as reported earlier15;22;23 was N-alkylated with 3-bromopropan-1-ol, and the secondary amine was Boc-protected in a one-pot procedure. The alcohol 5 was then oxidized to the corresponding aldehyde 6. The protected guanylhydrazone derivatives 7a–g were obtained in good yields via condensation of 6 with 3a (aminoguanidine) or 3b–g. The Boc groups were finally deprotected with TFA to afford the target compounds as orangish-yellow solids (Scheme 2). The synthesis of 14 (Scheme 3), bearing an N-alkyl substituent on one end and an unsubstituted guanylhydrazone on the other terminus was carried out starting from N1,N2,N3,N4-tetraBoc-protected spermine (9). N-Alkylation with n-C16H33I was carried out in the presence of NaH; the mono N-alkylated precursor 10 was isolated and subjected to N-alkylation on the other terminus with 3-bromopropoxy-tert-butyldimethylsilane. Sequential deprotection of the TBDMS group, oxidation of the resultant alcohol to the aldehyde, condensation with aminoguandine and finally, deprotection with TFA yielded 14 in 12% overall yield.
Scheme 1.
Reagents: a MeI, EtOH, 60°C, 30min.
b. R-NH2, EtOH, reflux, 1.5h.
Scheme 2.

Reagents: a (i) 3-bromopropan-1-ol, K2CO3, DMF, 60°C, 24h; (ii) Boc2O (excess), MeOH, r.t., 12h; b. PCC, DCM, r.t., 4.5h; c. aminoguanidine (3a) or 3b–3g, CH3COOH (cat.), EtOH, 85°C, 2h. d. CF3COOH, r.t., 30 min. The pentatrifluoroacetate salt form is assumed.
Scheme 3.

Reagents: a C16H33I, NaH, DMF, 0°C-r.t., 20h; b. 3-bromopropoxy-tert-butyl-dimethylsilane (excess), NaH, DMF, 0°C-r.t., 20h; c. (i) TBAF(1M in THF), 2h; (ii) PCC, DCM, r.t., 4.5h; d. 3a (aminoguanidine), CH3COOH (cat.), EtOH, 85°C, 2h; e. CF3COOH, r.t., 30 min. The pentatrifluoroacetate salt form is assumed.
LPS sequestration activities of the guanylhydrazone compounds
We had, as mentioned earlier, identified in rapid-throughput screens in focused libraries, several guanylhydrazones as potent LPS binders.17;25 It is to be noted that guanidines and, by extension, guanylhydrazones, are known to interact with phosphate groups via bidendate H-bonds by virtue of their planar geometry and multiple H-bond donor N-atoms (the ‘arginine fork’ principle26), which is not possible in the case of simple primary amines that constitute the cationic termini of the compounds that we have hitherto characterized.10;15;22;23;27 Since virtually all of our screening results point to superior LPS recognition by homospermine scaffolds, we wished to ‘graft’ guanylhydrazone functionalities on such a scaffold. It was of interest to examine if substantial differences in activities would be apparent in compounds with an N-alkylated guanylhydrazone functional group on one end and a free primary amine on the other or, conversely, an unsubstituted guanylhydrazone on one terminus, and an N-alkylated secondary amine on the other.
In the NF-κB reporter gene assay performed in TLR4-expressing HEK cells, we found a distinct enhancement in LPS-neutralizing potency with increasing alkyl chain lengths in the 8 series of compounds, with the optimal chain length being hexadecyl (8f); this SAR is virtually identical to that observed with the acylhomospermine compounds that we had examined earlier,15 which suggests that a C16 hydrocarbon chain is optimal for maximal interactions with the polyacyl domain of lipid A.28 The activity of compound 14, also with a hexadecyl substituent on the terminal amine rather than on the guanylhydrazone, was no different than that of 8f within the limits of statistical significance (Fig. 2). This would appear to indicate that long-chain substituents on the cationic termini do not adversely affect Coulombic interactions with the lipid A phosphates. Given the minor differences in potency between 8f and 14, we elected to characterize 8f in greater detail because of a higher toxicity for 14 in in vitro cellular assays (data not shown). As expected, 8f inhibits, in a dose-dependent manner, LPS-induced proinflammatory cytokine production in whole human blood ex vivo (Fig. 3); the IC50 values in human blood are considerably higher than in the NF-κB assay owing to the fact that these compounds are bound to albumin strongly (manuscript submitted) and albumin concentrations abound in the milieu of whole blood. Compound 8f also affords dose-dependent protection against lethal endotoxemic challenge in a murine model (Table 1), as we have observed with acylhomospermines.15
Fig. 2.

Relationship between alkyl chain length and potency of inhibition of LPS-induced NF-κB nuclear transactivation as measured in HEK cells expressing Tlr4 and an NF-κB-secreted alkaline phosphatase reporter gene construct, and of the induction of nitric oxide (NO) in murine macrophage J774.A1 cells. Compound 14 is depicted in open symbols; its alkyl chain length is also C16, but is denoted with an asterisk to differentiate it from 8f.
Fig. 3.

Inhibition of LPS-induced secretion of cytokines in human blood ex vivo by 8f as determined by multiplexed cytokine bead array assays. A representative experiment is depicted.
Table 1.
Dose-dependent protection by 8f in CF-1 mice challenged with a supralethal dose of 200 ng/mouse (LD100 = 100 ng) in cohorts of five animals. Lethality was recorded at 24 h post-challenge. Ratios denote dead/total.
| 8f Dose (μg/mouse) | Lethality in CF-1 Mice (alive/total) |
|---|---|
| 200 | 0/5* |
| 100 | 1/5* |
| 50 | 2/5 |
| 0 | 5/5 |
Asterixes indicate statistically significant values, p<0.05.
It was of interest to examine if 8f with its considerably higher pKa than an amine, planar geometry and multiple H-bond donor atoms, would be more potent than the N-acyl15- and N-alkyl-23 homospermine leads that are currently in preclinical development. Although much to our disappointment, we found no discernible differences in potency between these compounds in a battery of in vitro as well as animal experiments (data not shown), these results are, nonetheless, instructive in that they indicate that basicity of the terminal cationic functionality, per se, is not a determinant of LPS-sequestering activity, and that no increments in activity are to be gained by incorporating strongly basic functional groups. Indeed, our preliminary acute toxicity studies in rodents indicate that 8f is considerably more toxic than the aforementioned homospermine leads. These data, collectively, have led us to consider the exploration of analogues with multiple electron-donating groups placed adjacent to the amines on the scaffold; such compounds are expected to be considerably less basic, and will the subject of a future publication.
Experimental Section
Chemistry
All of the solvents and reagents used were obtained commercially and used as such unless noted otherwise. Moisture or air-sensitive reactions were conducted under argon atmosphere in oven-dried (120°C) glass apparatus. Solvents were removed under reduced pressure using standard rotary evaporators. Flash column chromatography was carried out using silica gel 635 (60–100 mesh), while thin-layer chromatography was carried out on silica gel CCM pre-coated aluminum sheets. All yields reported refer to isolated material judged to be homogenous by TLC, NMR spectroscopy and mass spectrometry.
General Procedure for the Synthesis of Compounds 3b–g29
A solution of hydrazinecarbothioamide (1, 20.0 g, 219.4 mmol) in anhydrous ethanol (200 mL) was brought to 60 °C and methyliodide was added. After stirring for 30 min, the reaction suspension was filtered and white solid was yielded as compound 2. The white solid was washed with ether (46.9 g, 91%). A solution of compound 2 (1.0 equiv.) and the corresponding amine (1.0 equiv.) in ethanol was heated under reflux for 1.5 h. The solution was cooled and diluted with ether, giving a gummy pink precipitate. The crude product was extracted with hot ethanol and concentrated under reduced pressure and the residue was recrystallized from CH3NO2 to yield compound 3b–g (23–53%). Representative data for the series are given below. 3b: 1H NMR (400 MHz, MeOD) δ 0.86–0.97 (m, 3H), 1.26–1.45 (m, 10H), 1.55–1.66 (br m, 2H), 3.17–3.25 (m, 2H); 13C NMR (126 MHz, MeOD) δ 14.43, 23.71, 27.72, 30.34, 32.96, 42.96, 42.18, 159.68; MS (ESI) calculated for C9H22N4 m/z 186.18 found 187.22 (MH+). 3g: 1H NMR (400 MHz, MeOD) δ 0.80 (s, J = 7.0 Hz, 3H), 1.04–1.36 (m, 30H), 1.44–1.61 (m, 2H), 2.76–2.85 (m, 1H), 3.03–3.14 (m, 1H), 4.51 (br s, 1H); 13C NMR (126 MHz, MeOD) δ 14.45, 23.75, 27.46, 27.72, 28.63, 30.13, 30.39, 30.52, 30.79, 33.09, 40.79, 42.17, 152.67; MS (ESI) calculated for C19H42N4 m/z 326.34, found 327.36 (MH+).
Synthesis of 5
To a solution of 4 (930 mg, 1.85 mmol) in anhydrous DMF (8 mL) was added K2CO3 (768 mg, 5.55 mmol), followed by 3-bromopropanol (194 μL, 2.22 mmol). The reaction mixture was stirred at 60 °C for 18 h. The reaction mixture was diluted with dichloromethane and the resulting solution was successively washed with water (2X), brine (1X) and dried over Na2SO4. After removal of solvent under vacuum, the residue was purified by flash column chromatography (Hexane:EtOAc = 7:3) to afford the title compound 5 as a viscous oil (514 mg, 42%). 1H NMR (400 MHz, CDCl3) δ 1.44 (br s, 36H), 1.57–1.77 (m, 8H), 3.09–3.40 (m, 11H), 3.55–3.79 (m, 7H), 4.89 (br s, 3H); 13C NMR (126 MHz, CDCl3) δ 27.34, 27.42, 31.81, 35.86, 58.16, 78.60, 156.20; MS (ESI) calculated for C33H64N4O9 m/z 660.46, found 683.49 (M+Na+).
Synthesis of 6
To a solution of 5 (500 mg, 0.76 mmol) in anhydrous DCM (20 mL) was added pyridinium chlorochromate (245 mg, 1.14 mmol) and the solution was stirred at ambient temperature for 4.5 h. After removal of solvent under high vacuum, the residue was purified by flash column chromatography (Hexane: EtOAc = 3:1) to afford the title compound 6 as a viscous oil (349 mg, 70%). 1H NMR (400 MHz, CDCl3) δ 1.40–1.55 (m, 36H), 1.61–1.82 (m, 5H), 2.66–2.77 (m, 2H), 3.07–3.32 (m, 11H), 3.48–3.56 (m, 2H), 9.81 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 24.55, 25.00, 27.26, 27.39, 27.42, 27.46, 27.87, 36.28, 40.05, 42.29, 42.64, 43.12, 43.61, 44.15, 44.76, 45.51, 45,80, 78.37, 78.93, 154.42, 155.09, 199.95; MS (ESI) calculated for C33H62N4O9 m/z 658.45, found 681.37 (M+Na+).
General Procedure for the Synthesis of Compounds 7a–g
To a solution of 6 (100 mg, 0.15 mmol) in anhydrous ethanol (5 mL) was added compound 3a–g (1.3 eq) followed by catalytic amount of acetic acid. The solution was refluxed at 85 °C for 2 h. After removal of solvent under high vacuum, the residue was purified by flash column chromatography (DCM: MeOH = 9:1) to afford 7a–g as a yellow viscous oil (70% – 100%).
7a: yield 80%; 1H NMR (400 MHz, CDCl3) δ 1.35–1.52 (m, 36H), 1.56–1.91 (m, 5H), 2.39–2.61 (m, 2H), 2.97–3.33 (m, 11H), 3.34–3.62 (m, 3H), 7.32–7.63 (m, 3H); 13C NMR (126 MHz, CDCl3) δ 15.27, 25.52, 26.03, 27.08 27.63, 27.98, 28.05,28.29, 28.46, 28.50, 37.38, 37.71, 43.07, 43.76, 44.21, 44.72, 46.52, 46.81, 53.45, 65.86, 78.98, 79.18. 79.50, 79.79, 80.00 80.24, 155.50, 155.86, 156.10, 156.23; MS (ESI) calculated for C34H66N8O8: m/z 714.50, found 715.48 (MH+).
7b: yield 100%; 1H NMR (400 MHz, CDCl3) δ 0.80–0.90 (m, 3H), 1.16–1.54 (m, 52H), 1.55–1.82 (m, 6H), 2.49 (br s, 2H), 2.96–3.56 (m, 16H), 6.69–7.21 (br m, 1H), 7.63 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 14.1, 22.61, 25.55, 26.66, 28.45, 28.71, 31.75, 37.41, 37.70, 42.25, 44.25, 44.94, 46.40, 46.57, 53.50, 79.04, 79.24, 79.47,79.55, 79.95, 80.14, 154.38, 154.46, 155.53,156.12,156.26; MS (ESI) calculated for C42H82N8O8 m/z 826.63, found 827.66 (MH+).
7c: yield 73%; 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.8 Hz, 3H), 1.14–1.56 (m, 56H), 1.55–1.80 (m, 6H), 2.49 (br s, 2H), 2.99–3.53 (m, 16H), 6.69–7.20 (br m, 1H), 7.63 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 13.10, 14.25, 21.64, 24.57, 27.43, 30.84, 36.40, 36.42, 36.69, 41.17. 43.26, 43.89, 45.46, 45.83, 52.45, 64.84, 78.52, 78.83, 78.85, 78.92, 78.96, 154.51, 154.57, 155.13, 155.21, 155.25; MS (ESI) calculated for C44H86N8O8 m/z 854.66, found 855.70 (MH+).
7d: yield 80%; 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.8 Hz, 3H), 1.20–1.54 (m, 60H), 1.59–1.81 (m, 6H), 2.50 (br s, 2H), 3.03–3.53 (m, 16H), 6.70–7.22 (br m, 2H), 7.66 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 14.13, 15.27, 22.68, 26.69, 28.51, 28.69, 29.34, 29.59, 29.64, 37.42, 37.53, 37.69, 42. 23, 43.79, 44.26, 44.69, 44.83, 44.91, 44.96, 45.00, 65.86, 79.02, 79.06, 79.37, 79.54, 79.89, 80.00, 80.11, 80.15, 155.27, 155.31, 155.50, 156..06, 156.11, 156.24; MS (ESI) calculated for C46H90N8O8 m/z 882.69, found 883.72 (MH+).
7e: yield 75%; 1H NMR (400 MHz, CDCl3) δ 0.85 (t, J = 6.8 Hz, 3H), 1.14–1.56 (m, 64H), 1.55–1.79 (m, 6H), 2.49 (br s, 2H), 2.97–3.55 (m, 16H), 6.67–7.19 (br m, 2H), 7.61 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 14.13, 22.68, 26.68, 29.19, 29.69, 31.91, 37.42, 37.70, 42.29, 42.70, 42.73, 43.78, 44.24, 44.70, 44.94, 44.97, 45.02, 46.40, 46.85, 79.02, 79.37, 79.53, 79.83, 79.90, 80.07 80.13, 153.92, 153.98, 154.53, 155.51, 156.06, 156.12, 156.25; MS (ESI) calculated for C48H94N8O8 m/z 910.72, found 911.75 (MH+).
7f: yield 71%; 1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 6.8 Hz, 3H), 1.15–1.58 (m, 68H), 1.58–1.81 (m, 6H), 2.50 (br s, 2H), 2.97–3.58 (m, 16H), 6.67–7.19 (br m, 2H), 7.66 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 13.11, 21.66, 24.54, 25.68, 27.43, 28.18, 28.68, 30.90, 36.42, 36.69, 41.20, 43.22, 43.90, 44.50, 45.53, 52.46, 78.03, 78.36, 78.53, 78.83, 79.12, 147.00, 148.09, 154.52, 155.10, 155.25; MS (ESI) calculated for C50H98N8O8 m/z 938.75, found 939.78 (MH+).
7g: yield 70%; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 6.6 Hz, 3H), 1.17–1.57 (m, 70H), 1.59–1.78 (br s, 8H), 2.49 (s, 1H), 2.97–3.54 (m, 18H), 6.72–7.23 (br m, 2H), 7.61 (br s, 1H); 13C NMR (126 MHz, CDCl3) δ 8.59, 13.96, 14.07, 14.12, 23.32, 25.65, 30.83, 32.19, 38.94, 40.61, 40.82, 42.33, 45.96, 62.23, 62.59, 62.76, 64.20, 64.98, 68.85, 74.42, 126.97, 131.75, 153.62, 169.20; MS (ESI) calculated for C52H102N8O8 m/z 966.78, found 967.77 (MH+).
General Procedure for the Synthesis of Compounds 8a–g
The resulting 7a–g was dissolved in excess dry trifluoroacetic acid and stirred at room temperature for 30 min. Trifluoroacetic acid was removed by purging nitrogen and the residue was thoroughly washed with diethyl ether to obtain the title compounds as yellow flaky solid 8a–g (100%).
8a: 1H NMR (500 MHz, MeOD) δ 1.17–1.36 (m, 2H), 1.78–1.90 (br s, 4H), 2.06–2.23 (m, 4H), 2.75–2.84 (m, 2H), 3.05 (m, 12H), 3.40 (t, J = 7.1 Hz, 2H), 7.54 (t, J = 4.1 Hz, 1H); 13C NMR (126 MHz, MeOD) δ 36.38, 43.55, 44.39, 44.42, 44.59, 46.80, 115.43, 117.75, 147.12, 155.70; MS (ESI) calculated for C14H34N8 m/z 314.29, found 158.17 (MH++H+ [doubly-charged species]), 315.33 (MH+).
8b: 1H NMR (400 MHz, MeOD) δ 0.85–0.97 (m, 3H), 1.27–1.48 (m, 10H), 1.76–1.94 (m, 4H), 2.03–2.28 (m, 4H), 2.74–2.85 (m, 2H), 3.06–3.31 (m, 15H), 3.36–3.43 (m, 15H), 7.31–7.82 (m, 1H); 13C NMR (126 MHz, MeOD) δ 13.01, 22.29, 22.70, 22.81, 23.97, 26.28, 28.53, 31.54, 36.41, 41.14, 43.71, 46.81, 115.55, 117.86, 161.59, 161.86; MS (ESI) calculated for C22H50N8 m/z 426.42, found 214.22 (MH++H+), 427.42 (MH+).
8c: 1H NMR (400 MHz, MeOD) δ 0.83–1.00 (m, 3H), 1.23–1.47 (m, 14H), 1.59–1.70 (br m, 2H), 1.75–1.93 (m, 4H), 2.05–2.25 (m, 4H), 2.76–2.83 (m, 2H), 3.05–3.31 (m, 14H), 3.35–3.43 (m, 4H), 7.37–7.67 (m, 1H); 13C NMR (126 MHz, MeOD) δ 13.03, 22.32, 22.70, 22.81, 23.98, 26.28, 31.64, 36.40, 41.14,. 43.71, 44.40, 44.43, 44.58, 146.44, 146.49, 146.54, 154.36; MS (ESI) calculated for C24H54N8 m/z 454.45, found 228.23 (MH++H+), 455.45 (MH+).
8d: 1H NMR (400 MHz, MeOD) δ 0.92 (t, J = 6.8 Hz, 3H), 1.24–1.46 (m, 18H), 1.58–1.70 (m, 2H), 1.77–1.90 (br m, 4H), 2.05–2.25 (m, 4H), 2.74–2.84 (m, 2H), 3.05–3.31 (m, 14H), 3.40 (t, J = 7.1 Hz, 2H), 7.40–7.67 (m, 1H); 13C NMR (126 MHz, MeOD) δ 13.03, 22.33, 22.70, 22.81, 23.97, 26.29, 28.40, 28.57, 29.07, 29.25, 29.34, 31.67, 36.40, 41.13, 43.71, 44.40, 44.43, 44.59, 115.43, 115.46, 117.47, 146.54, 146.57, 146.62, 154.37; MS (ESI) calculated for C26H58N8 m/z 482.48, found 242.25 (MH++H+), 483.49 (MH+).
8e: 1H NMR (400 MHz, MeOD) δ 0.92 (t, J = 6.8 Hz, 3H), 1.25–1.42 (m, 22H), 1.58–1.70 (m, 2H), 1.78–1.89 (br m, 4H), 2.04–2.25 (m, 4H), 2.73–2.84 (m, 2H), 3.04–3.30 (m, 14H), 3.40 (t, J = 7.1 Hz, 2H), 7.46–7.64 (m, 1H); 13C NMR (126 MHz, MeOD) δ 13.03, 22.33, 22.70, 22.81, 23.97, 26.29, 28.96, 31.67, 36.41, 41.15,. 43.71, 44.40, 44.43, 44.59, 115.44, 117.74, 146.59, 146.60, 154.37; MS (ESI) calculated for C28H62N8 m/z 510.51, found 256.31 (MH++H+), 511.60 (MH+).
8f: 1H NMR (400 MHz, MeOD) δ 0.92 (t, J = 6.8 Hz, 3H), 1.26–1.46 (m, 26H), 1.58–1.69 (m, 2H), 1.79–1.91 (br m, 4H), 2.05–2.24 (m, 4H), 2.75–2.84 (m, 2H), 3.05–3.31 (m, 13H), 3.40 (t, J = 7.1 Hz, 2H), 7.51–7.56 (m, 1H); 13C NMR (126 MHz, MeOD) δ 13.05, 22.33, 22.70, 22.81, 23.96, 26.29, 28.57, 31.67, 36.41, 41.15,. 43.72, 44.41, 44.44, 44.60, 115.47, 117.81, 146.55, 146.59, 146.63, 154.36; MS (ESI) calculated for C30H66N8 m/z 538.54, found 270.28 (MH++H+), 539.56 (MH+).
8g: 1H NMR (400 MHz, MeOD) δ 0.91 (t, J = 6.7 Hz, 3H), 1.21–1.47 (m, 29H), 1.58–1.69 (m, 2H), 1.77–1.88 (br m, 4H), 2.05–2.24 (m, 4H), 2.73–2.86 (m, 2H), 3.04–3.30 (m, 14H), 3.40 (t, J = 7.0 Hz, 2H), 7.51 (br s, 1H); 13C NMR (126 MHz, MeOD) δ 113.47, 22.75, 23.12, 23.23, 24.38, 26.71, 28.85, 32.09, 36.83, 41.57, 44.14, 44.84, 44.86, 45.02, 147.03, 147.09, 154.78; MS (ESI) calculated for C32H70N8 m/z 566.574, found 284.31 (MH++H+), 567.58 (MH+).
Synthesis of 10
Compound 9 (2.4 g, 3.98 mmol) was added to the suspension of NaH (30 equiv.) in DMF at 0 °C. After the reaction suspension was stirred at 0 °C for 30 min, 1-iodohexadecane (1.68 g, 1.0 equiv.) which was dissolved in 5 mL DMF was added to the reaction. The mixture was then stirred at room temperature for 24 h. The reaction solution was quenched with 1 M HCl and extracted with DCM (3X). The extracted DCM layers were washed with brine (1x) and dried over Na2SO4. After removal of the solvent under reduced pressure, the desired compound 10 was isolated at 16% EtOAc in hexane by flash column chromatography (35%). 1H NMR (500 MHz, CDCl3) δ 0.88 (t, J = 7.0 Hz, 3H), 1.25 (s, 27H), 1.37–1.54 (m, 40H), 1.61–1.78 (m, 4H), 3.01–3.33 (br m, 14H); 13C NMR (126 MHz, CDCl3) δ 14.13, 22.69, 25.55, 25.89, 25.95, 26.87, 28.45, 29.36, 29.70, 31.93, 37.29, 37.66 43.68, 44.18, 44.60, 44.66, 44.90, 46.52, 46.63, 46.80, 47.10, 79.12, 79.27, 155.48, 155.95, 156.11; MS (ESI) calculated for C46H90N4O8 m/z 826.68, found 849.71 (M+Na+).
Synthesis of 11
To a solution of tert-butyl dimethylsilylchloride (1.2 equiv.) and imidazole (1.3 equiv.) in anhydrous DMF was added 3-bromopropanol (1.0 equiv.) at 0°C.30 After 1 h, the reaction mixture was brought to room temperature and stirred for 8 h. Then the reaction solution was diluted with ether and the ethereal layer was washed with saturated NaHCO3 solution (1x) and 10% HCl and dried over Na2SO4. Compound 10 (670 mg, 0.81 mmol) was added to a suspension of NaH (40 equiv.) in DMF at 0 °C. After the reaction suspension was stirred at 0°C for 30 min, an excess of freshly prepared 3-bromopropoxy-tert-butyl-dimethylsilane (8.1 mmol) was added to the reaction and the solution stirred at room temperature for 20 h. The reaction solution was quenched with 1 M HCl and extracted with DCM (3x). The extracted DCM layers were washed with brine (1x) and dried over Na2SO4. After removal of the solvent under reduced pressure, the desired compound 10 (510 mg, 63%) was isolated at 13% EtOAc in hexane by flash column chromatography. 1H NMR (500 MHz, CDCl3) δ 0.01 (s, 6H), 0.82–0.89 (m, 12H), 1.14–1.32 (m, 28H), 1.41 (s, 40H), 1.64–1.78 (br m, 6H), 3.03–3.25 (br m, 16H), 3.52–3.64 (br m, 2H); 13C NMR (126 MHz, CDCl3) δ 14.13, 18.27, 22.69, 25.93, 26.88, 28.49, 29.37, 29.70, 31.93, 44.34, 44.62, 44.89, 46.65, 46.83, 47.08, 60.78, 79.20, 155.44; MS (ESI) calculated for C55H110N4O9Si m/z 998.80, found 1021.94 (M+Na+).
Synthesis of 12
To a solution of 11 (400 mg, 0.40 mmol) in anhydrous THF (20 mL) was added tetra-n-butyl-ammonium fluoride (1 M in THF, 174 μL, 0.17 mmol) and the solution was stirred at ambient temperature for 2 h. After removal of solvent under high vacuum, the residue was dissolved in 10 mL anhydrous DCM and pyridinium chlorochromate (130 mg, 0.60 mmol) was added. The reaction mixture was stirred at room temperature for 4.5 h. After removal of solvent under high vacuum, the residue was purified by flash column chromatography (Hexane: EtOAc = 3:1) to afford the title compound 12 as a viscous oil (260 mg, 74 %). 1H NMR (500 MHz, CDCl3) δ 0.88 (t, J = 6.9 Hz, 3H), 1.25 (br s, 27H), 1.45 (br s, 41H), 1.73 (br s, 5H), 2.70 (br s, 2H), 3.16 (br s, 14H), 3.51 (br s, 2H), 9.79 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 14.13, 22.69, 25.60, 25.95, 26.88, 27.52, 28.32, 29.37, 29.66, 30.95, 31.93, 41.09, 41.12, 43.32, 43.63, 44.61, 44.69, 45.80, 46.62, 46.78, 46.84, 46.86, 47.08, 79.11, 79.25, 79.34, 79.93, 155.47, 200.99; MS (ESI) calculated for C49H94N4O9 m/z 882.70, found 905.68 (M+Na+).
Synthesis of 13
To a solution of 12 (100 mg, 0.11 mmol) in anhydrous ethanol (5 mL) was added compound 3a (1.3 eq) followed by catalytic amount of acetic acid. The solution was refluxed at 85 °C for 2 h. After removal of solvent under high vacuum, the residue was purified by flash column chromatography (DCM: MeOH = 95:5) to afford the title compound 13 as yellow viscous oil (73%). 1H NMR (500 MHz, CDCl3) δ 0.88 (t, J = 6.9 Hz, 3H), 1.25 (br s, 27H), 1.45 (br s, 41H), 1.73 (br s, 4H), 2.43–2.56 (br m, 2H), 3.16 (br s, 14H), 3.05–3.32 (br m, 2H), 7.48 (br s, 3H); 13C NMR (126 MHz, CDCl3) δ 14.13, 22.69, 25.55, 25.59, 25.86, 25.97, 26.87, 27.51, 27.59, 28.47.28.50, 29.36, 29.42, 29.61, 29.66, 29.69, 30.95, 31.10, 31.92, 42.98, 43.08, 44.71, 46.61, 46.73, 47.12, 53.44, 79.29, 79.45, 79.97, 155.51, 155.96; MS (ESI) calculated for C50H98N8O8 m/z 938.75, found 939.75 (MH+).
Synthesis of 14
The resulting compound 13 was dissolved in excess dry trifluoroacetic acid and stirred at room temperature for 30 min. Excess solvent was removed by purging nitrogen and the residue was thoroughly washed with diethyl ether to obtain white solid 14 (100%). 1H NMR (500 MHz, DMSO-d6) δ 0.85 (t, J = 6.9 Hz, 3H), 1.18–1.34 (m, 24H), 1.51–1.69 (br m, 6H), 1.89–2.01 (br m, 4H), 2.62–2.69 (m, 2H), 2.83–3.06 (m, 13H), 3.19–3.27 (m, 2H), 7.49 (t, J = 4.0, 1H), 7.61–7.96 (br m, 3H), 8.64–8.95 (m, 6H), 12.01 (s, 1H); 13C NMR (126 MHz, DMSO) δ 13.92, 22.05, 22.32, 22.34, 22.64, 25.37, 25.83, 28.33, 29.00, 31.25, 42.87, 43.79, 43.86, 44.01, 46.10, 46.71, 115.80, 118.18, 147.44, 155.20; MS (ESI) calculated for C30H66N8 m/z 538.54, found 270.30 (MH++H+), 539.54 (MH+).
Nitric Oxide Assay
Nitric oxide production was measured as total nitrite in murine macrophage J774A.1 cells using the Griess assay31 as described previously.14 J774A.1 cells were plated at ~105/mL in a volume of 40 μL/well, in 384-well, flat-bottomed, cell culture treated microtiter plates and subsequently stimulated with 10 ng/mL lipopolysaccharide (LPS). Concurrent to LPS stimulation, serially diluted concentrations of test compounds were added to the cell medium and left to incubate overnight for 16 h. Polymyxin B was used as reference compound in each plate. Positive- (LPS stimulation only) and negative-controls (J774A.1 medium only) were included in each experiment. Nitrite concentrations were measured by adding 40 μl of supernatant to equal volumes of Griess reagents (50 μL/well; 0.1% NED solution in ddH2O and 1% sulfanilamide, 5% phosphoric acid solution in ddH2O) and incubating for 15 minutes at room temperature in the dark. Absorbance at 535 nm was measured using a Molecular Devices Spectramax M2 multifunction plate reader (Sunnyvale, CA). Nitrite concentrations were interpolated from standard curves obtained from serially diluted sodium nitrite standards.
Inhibition of LPS-induced NF-κB induction
The inhibition of induction of NF-κB, a key transcripitional activator of the innate immune system, was quantified using human embryonic kidney 293 cells cotransfected with TLR4 (LPS receptor), CD14 and MD2 (co-receptors), available from InvivoGen, Inc. (HEK-Blue™, San Diego, CA), as described elsewhere.32 Stable expression of secreted alkaline phosphatase (seAP) under control of NF-κB/AP-1 promoters is inducible by LPS, and extracellular seAP in the supernatant is proportional to NF-κB induction. HEK-4 cells were incubated at a density of ~105 cells/mL in a volume of 80 μL/well, in 384-well, flat-bottomed, cell culture-treated microtiter plates until confluency was achieved, and subsequently stimulated with 10 ng/mL lipopolysaccharide (LPS). Concurrent to LPS stimulation, serially diluted concentrations of test compounds were added to the cell medium using a rapid-throughput, automated protocol employing a Bio-Tek P2000 liquid handler as described above, and left to incubate overnight. Polymyxin B was used as reference compound in each plate. Positive- (LPS stimulation only) and negative-controls (HEK-detection medium only) were included in each experiment. seAP was assayed spectrophotometrically using an alkaline phosphatase-specific chromogen (present in HEK-detection medium as supplied by the vendor) at 620 nm.
Multiplexed cytokine assay ex vivo in human blood
100 μl aliquots of fresh whole blood, anticoagulated with EDTA, obtained by venipuncture from healthy human volunteers with informed consent and as per guidelines approved by the Human Subjects Experimentation Committee, was exposed to an equal volume of 20 ng/mL of E. coli 0111:B4 LPS, with graded concentrations of test compounds diluted in saline for 4 h in a 96-well microtiter plate.33;34 The effect of the compounds on modulating cytokine production was examined using a FACSArray multiplexed flow-cytometric bead array (CBA) system (Becton-Dickinson-Pharmingen, San Jose, CA) as described elsewhere.10;22;23
Mouse lethality experiments
Female, outbred, 9- to 11-week-old CF-1 mice (Charles River, Wilmington, MA) weighing 22–28 g were used as described elsewhere.10;14;22;23 The animals were sensitized to the lethal effects of LPS by D-galactosamine.35–37 The lethal dose causing 100% mortality (LD100) dose for the batch of LPS used (E. coli 0111:B4; procured from Sigma) was first determined by administering D-galactosamine (800 mg/kg) and LPS (0, 50, 100, 200 ng/mouse) as a single injection intraperitoneally (i.p.) in freshly prepared saline to cohorts of five animals in a volume of 0.2 mL. Mice received graded doses of compound diluted in saline, i.p. in one flank, immediately before a supralethal (200 ng) LPS challenge, which was administered as a separate i.p. injection into the other flank. Lethality was determined at 24 h post LPS challenge.
Supplementary Material
Acknowledgments
This work was supported from NIH grants 1R01 AI50107 and 1U01AI077947.
Footnotes
Supporting Information Paragraph: 1H and 13C NMR spectra of key intermediate compounds, elemental analyses of all key target compounds.
References
- 1.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zähringer U, Seydel U, Di Padova F, et al. FASEB J. 1994;8:217. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 2.Hurley JC. Drug Saf. 1995;12:183. doi: 10.2165/00002018-199512030-00004. [DOI] [PubMed] [Google Scholar]
- 3.Ulevitch RJ. Immunol Res. 2000;21:49. doi: 10.1385/IR:21:2-3:49. [DOI] [PubMed] [Google Scholar]
- 4.Dinarello CA. Curr Top Microbiol Immunol. 1996;216:133. doi: 10.1007/978-3-642-80186-0_7. [DOI] [PubMed] [Google Scholar]
- 5.Bone RC. Clin Chest Med. 1996;17:175. doi: 10.1016/s0272-5231(05)70307-5. [DOI] [PubMed] [Google Scholar]
- 6.Martin GS, Mannino DM, Eaton S, Moss M. N Engl J Med. 2003;348:1546. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 7.Holst O, Ulmer AJ, Brade H, Rietschel ET. On the chemistry and biology of bacterial endotoxic lipopolysaccharides. In: Masihi N, editor. Immunotherapy of infections. Marcel Dekker, Inc; New York, Basel, Hong Kong: 1994. pp. 281–308. [Google Scholar]
- 8.Vesentini S, Soncini M, Zaupa A, Silvestri V, Fiore GB, Redaelli A. Int J Artif Organs. 2006;29:239. doi: 10.1177/039139880602900210. [DOI] [PubMed] [Google Scholar]
- 9.Vincent JL, Laterre PF, Cohen J, Burchardi H, Bruining H, Lerma FA, Wittebole X, De Backer D, Brett S, Marzo D, Nakamura H, John S. Shock. 2005;23:400. doi: 10.1097/01.shk.0000159930.87737.8a. [DOI] [PubMed] [Google Scholar]
- 10.Burns MR, Wood SJ, Miller KA, Nguyen T, Cromer JR, David SA. Bioorg Med Chem. 2005;13:2523. doi: 10.1016/j.bmc.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 11.Burns MR, Jenkins SA, Wood SJ, Miller K, David SA. J Comb Chem. 2006;8:32. doi: 10.1021/cc0500755. [DOI] [PubMed] [Google Scholar]
- 12.David SA, Bechtel B, Annaiah C, Mathan VI, Balaram P. Biochim Biophys Acta. 1994;1212:167. doi: 10.1016/0005-2760(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 13.David SA, Mathan VI, Balaram P. J Endotoxin Res. 1995;2:325. [Google Scholar]
- 14.David SA, Silverstein R, Amura CR, Kielian T, Morrison DC. Antimicrob Agents Chemother. 1999;43:912. doi: 10.1128/aac.43.4.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller KA, Suresh Kumar EVK, Wood SJ, Cromer JR, Datta A, David SA. J Med Chem. 2005;48:2589. doi: 10.1021/jm049449j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burns MR, Jenkins SA, Wood SJ, Miller K, David SA. J Comb Chem. 2006;8:32. doi: 10.1021/cc0500755. [DOI] [PubMed] [Google Scholar]
- 17.Wood SJ, Miller KA, David SA. Comb Chem High Throughput Screen. 2004;7:733. doi: 10.2174/1386207043328229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wood SJ, Miller KA, Lushington GH, Burns MR, David SA. Comb Chem High Throughput Screen. 2006;9:27. doi: 10.2174/138620706775213903. [DOI] [PubMed] [Google Scholar]
- 19.Guo JX, Wood SJ, David SA, Lushington GH. Bioorg Med Chem Lett. 2006;16:714. doi: 10.1016/j.bmcl.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 20.Burns MR, Jenkins SA, Vermeulen NM, Balakrishna R, Nguyen TB, Kimbrell MR, David SA. Bioorg Med Chem Lett. 2006;16:6209. doi: 10.1016/j.bmcl.2006.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burns MR, Jenkins SA, Wood SJ, Miller K, David SA. J Comb Chem. 2006;8:32. doi: 10.1021/cc0500755. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen TB, Adisechan A, Suresh Kumar EVK, Balakrishna R, Kimbrell MR, Miller KA, Datta A, David SA. Bioorg Med Chem. 2007;15:5694. doi: 10.1016/j.bmc.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sil D, Shrestha A, Kimbrell MR, Nguyen TB, Adisechan AK, Balakrishna R, Abbo BG, Malladi S, Miller KA, Short S, Cromer JR, Arora S, Datta A, David SA. Antimicrob Agents Chemother. 2007;51:2811. doi: 10.1128/AAC.00200-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wood SJ, Miller KA, Lushington GH, Burns MR, David SA. Comb Chem High Throughput Screen. 2006;9:27. doi: 10.2174/138620706775213903. [DOI] [PubMed] [Google Scholar]
- 25.Khownium K, Wood SJ, Miller KA, Balakrishna R, Nguyen TB, Kimbrell MR, Georg GI, David SA. Bioorg Med Chem Lett. 2006;16:1305. doi: 10.1016/j.bmcl.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 26.Calnan BJ, Tidor B, Biancalana S, Hudson D, Frankel AD. Science. 1991;252:1167. doi: 10.1126/science.252.5009.1167. [DOI] [PubMed] [Google Scholar]
- 27.Burns MR, Jenkins SA, Kimbrell MR, Balakrishna R, Nguyen TB, Abbo BG, David SA. J Med Chem. 2007;50:877. doi: 10.1021/jm061198m. [DOI] [PubMed] [Google Scholar]
- 28.David SA. J Molec Recognition. 2001;14:370. doi: 10.1002/jmr.549. [DOI] [PubMed] [Google Scholar]
- 29.Buchheit KH, Gamse R, Giger R, Hoyer D, Klein F, Kloppner E, Pfannkuche HJ, Mattes H. J Med Chem. 1995;38:2331. doi: 10.1021/jm00013a010. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen C, Ruda GF, Schipani A, Kasinathan G, Leal I, Musso-Buendia A, Kaiser M, Brun R, Ruiz-Perez LM, Sahlberg BL, Johansson NG, Gonzalez-Pacanowska D, Gilbert IH. J Med Chem. 2006;49:4183. doi: 10.1021/jm060126s. [DOI] [PubMed] [Google Scholar]
- 31.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Anal Biochem. 1982;126:131. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 32.Khownium K, Wood SJ, Miller KA, Balakrishna R, Nguyen TB, Kimbrell MR, Georg GI, David SA. Bioorg Med Chem Lett. 2006;16:1305. doi: 10.1016/j.bmcl.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 33.Fennrich S, Fischer M, Hartung T, Lexa P, Montag-Lessing T, Sonntag HG, Weigandt M, Wendel A. Dev Biol Standards. 1999;101:131. [PubMed] [Google Scholar]
- 34.Remick DG, Newcomb DE, Friedland JS. Whole-blood assays for cytokine production. In: Evans TJ, editor. Septic shock. Methods and protocols. Humana Press; New Jersey: 2000. pp. 101–114. [DOI] [PubMed] [Google Scholar]
- 35.Cook EB, Stahl JL, Lowe L, Chen R, Morgan E, Wilson J, Varro R, Chan A, Graziano FM, Barney NP. J Immunol Methods. 2001;254:109. doi: 10.1016/s0022-1759(01)00407-0. [DOI] [PubMed] [Google Scholar]
- 36.Freudenberg MA, Galanos C. Infect Immun. 1991;59:2110. doi: 10.1128/iai.59.6.2110-2115.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tracey KJ, Cerami A. Annu Rev Cell Biol. 1993;9:317. doi: 10.1146/annurev.cb.09.110193.001533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.