

Abstract
Modelling and simulation (M&S)-based approaches have been proposed to support paediatric drug development in order to design and analyze clinical studies efficiently. Development of anti-cancer drugs in the paediatric population is particularly challenging due to ethical and practical constraints. We aimed to review the application of M&S in the development of anti-cancer drugs in the paediatric population, and to identify where M&S-based approaches could provide additional support in paediatric drug development of anti-cancer drugs. A structured literature search on PubMed was performed. The majority of identified M&S-based studies aimed to use population PK modelling approaches to identify determinants of inter-individual variability, in order to optimize dosing regimens and to develop therapeutic drug monitoring strategies. Prospective applications of M&S approaches for PK-bridging studies have scarcely been reported for paediatric oncology. Based on recent developments of M&S in drug development there are several opportunities where M&S could support more informative bridging between children and adults, and increase efficiency of the design and analysis of paediatric clinical trials, which should ultimately lead to further optimization of drug treatment strategies in this population.
Keywords: anti-cancer drugs, chemotherapy, modelling, paediatric drug development, paediatric oncology, pharmacokinetics
Introduction
Drug treatment in the paediatric population is still frequently off-label, with dosing regimens commonly empirically derived from adult clinical trial data 1. However, changes in physiology during paediatric development may have substantial impact on drug pharmacokinetics (PK) and pharmacodynamics (PD) 2. A recent review of paediatric drug studies showed that for a substantial number of evaluated drugs, adjustments in dosing regimens and formulations were necessary 3. In paediatric oncology, the conduct of prospective drug development studies has been relatively limited as well, resulting in higher percentages of off-label use in this therapeutic area 4, 5. For instance, single agent phase I studies have only been conducted to a very limited extent 6. Study designs in paediatric oncology commonly evaluate safety and efficacy of anti-cancer agents by adding the treatment to existing regimens, which potentially leads to complex combination treatments that are difficult to evaluate. Practical limitations such as obtaining (additional) blood samples, and the low number of patients that are typically available for participation in clinical studies 7 further complicate the conduct of drug development studies. Thus, the need for informative and efficient clinical study designs in this particular patient population is of special relevance.
The need for improved pharmacotherapy in children and associated drug studies has also been recognized and encouraged by regulatory bodies through a number of programmes and guidelines 8, 9. The FDA has provided a paediatric study decision tree to help assessment of clinical studies that are necessary during paediatric drug development 10 (Figure 1). Based on this decision tree, three basic study types are distinguished: (i) PK bridging studies, (ii) exposure–response studies and (iii) studies evaluating safety and efficacy, to which we will refer to as a full drug development study. In addition to the studies in the FDA decision tree, studies aiming to perform post hoc drug-treatment optimization of toxicity or efficacy can also be distinguished. The current review is centered around these four types of studies.
Figure 1.
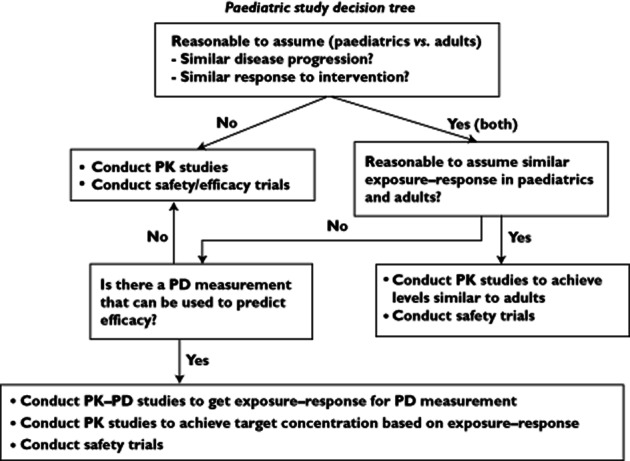
Paediatric decision tree to determine necessary paediatric clinical studies, as suggested by the FDAPK, pharmcokinetic; PD, pharmacodynamic
Paediatric PK bridging studies aim to determine dosing regimens that will lead to target exposures similar to adults. However, if differences in disease progression or the exposure–response relationship are expected, bridging cannot be based solely on adult target exposure levels. Differences in disease progression can be common in paediatric oncology, and are related to the differences in biology of paediatric malignancies, compared with adults (e.g. sarcoma vs. carcinoma) 11, which also lead to differences in response rate 12. In such cases, the FDA guidelines recommend either an exposure–response study, using surrogate biomarkers predictive of efficacy, or alternatively a full evaluation of safety and efficacy, e.g. a full drug development study.
Drug treatment optimization studies typically aim to identify either (i) patient characteristics predictive of (some of the) inter-individual variability in drug exposure, (ii) develop therapeutic drug monitoring and/or limited sampling strategies or (iii) investigate potential drug–drug interactions.
Modelling and simulation (M&S) in clinical pharmacology and drug development may be roughly defined as the area which involves development and application of mathematical and statistical models which describe PK and PD. Regulatory authorities have suggested that M&S based approaches may be used to support the development of dosing regimens in children 13–16. Several review articles have been published which discuss and demonstrate the potential value of M&S to support design and analysis of paediatric drug studies and associated decision making 17–20. An overview of the role of M&S in (paediatric) drug development is depicted in Figure 2.
Figure 2.
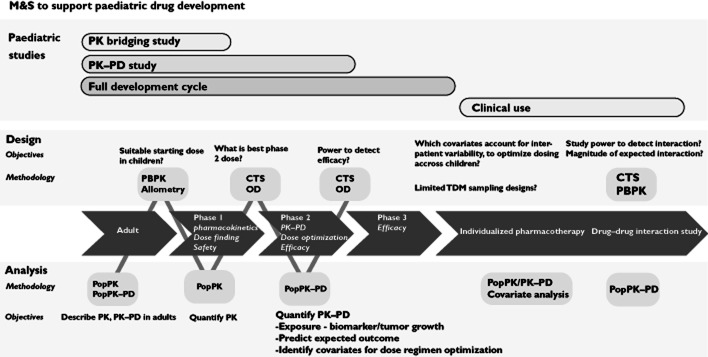
Schematic overview of the role of modelling and simulation (M&S) in paediatric drug development. CTS, clinical trial simulation; OD, optimal design; PK–PD, pharmacokinetic–pharmacodynamic modelling; PBPK, physiologically-based pharmacokinetic modelling
The overall objective of this review was to review systematically the literature for publications that applied M&S to support the development of anti-cancer drugs in the paediatric population. In this review, we will discuss identified analyses in the context of the earlier described four study types that are relevant to paediatric drug development: (i) PK bridging studies, (ii) exposure–response studies, (iii) full drug development studies and (iv) drug-treatment optimization studies. Additionally we aimed to identify and discuss M&S-based approaches that could potentially address some of the challenges associated with the design and analysis of paediatric drug development studies of anti-cancer drugs.
Methods
A systematic search on PubMed was performed in which search terms were chosen to identify publications where M&S were used to support development of anti-cancer drugs in the paediatric population. The search query consisted of five components addressing: (i) keywords for different possible types of M&S based analyses, (ii) keywords for different possible software packages, (iii) keywords indicating a paediatric study population and (iv) keywords indicating anticancer drugs were investigated. Additionally, with respect to this last component, we added a number of frequently used anti-cancer drugs also specifically.
The specific search query used was as follows: (‘population pharmacokinetic*’ OR ‘PK–PD model’ OR ‘pharmacokinetic model’ OR ‘pharmacodynamic model’ OR ‘NONMEM’ OR ‘PBPK’ OR ‘Physiologically-based model*’ OR ‘physiology-based model*’ OR ‘non-linear mixed effect’ OR ‘WinNonMix’ OR ‘*bugs’ OR ‘S-ADAPT’ OR ‘monolix’ OR ‘PK-Sim’ OR ‘Simcyp’) AND (‘paediatric'[TIAB] OR ‘paediatric’ OR ‘childhood'[TIAB] OR ‘paediatric'[TIAB] OR ‘children'[TIAB] OR ‘neonates’[TIAB] OR ‘toddlers’[TIAB] OR ‘adolescent'[TIAB]) AND (‘cancer'[TIAB] OR ‘oncology'[TIAB] OR ‘malignant*’[TIAB] OR ‘Leukemia’ OR ‘anticancer’ OR ‘anti-cancer’ OR ‘chemotherapy’ OR ‘neoplasms’ OR ‘*neoplastic*’ OR ‘etoposide'[TIAB] OR ‘methotrexate'[TIAB] OR ‘MTX’ OR OR ‘actinomycin D'[TIAB] OR ‘asparaginase'[TIAB] OR ‘busulphan'[TIAB] OR ‘carboplatin'[TIAB] OR ‘cyclophosphamide'[TIAB] OR ‘cytarabine'[TIAB] OR ‘irinotecan'[TIAB] OR ‘6-mercaptopurine'[TIAB] OR ‘clofarabine'[TIAB] OR ‘topotecan'[TIAB] OR ‘busulfan'[TIAB] OR ‘vincristine'[TIAB] OR ‘ifosfamide'[TIAB] OR ‘doxorubicin'[TIAB]) AND (‘0001/01/01'[PDAT]: ‘2012/10/01'[PDAT])
The search resulted in 117 hits on October 1 2012. After exclusion of review articles (n = 3), animal studies (n = 1), bio-analytical studies (n = 1), articles in a language other than English (n = 5), non-paediatric studies (n = 6), non-drug studies (n = 8), non-model based analyses (n = 4), non-oncology studies (n = 14) and other irrelevant articles (n = 2), 73 hits remained, which were included in this review.
Publications were subsequently categorized based on drug, study characteristics (number of patients, study type, indication and study objectives) and analysis characteristics (analysis type, software, estimation method, model evaluation method, model type, covariates identified). For purposes of clarity, indications were only referenced if more than 10 patients of a particular indication were included.
Results and discussion
An overview of the identified studies utilizing M&S-based approaches in paediatric oncology is depicted in Table 1. In Figure 3, the frequency of different drugs studied is depicted. Busulphan (n = 15, 20%) and methotrexate (n = 12, 16%) were the most frequently studied drugs. Also, multiple reports on etoposide (n = 4, 5%), topotecan (n = 4, 5%) and asparaginase (n = 4, 5%) were described, while an additional number of drugs was studied less frequently. The most frequently investigated indication was acute lymphoblastic leukemia (ALL), which is also the most common malignancy diagnosed in children. A large number of studies reported small numbers of mixed indications, or various indications requiring haematopoietic stem cell transplantation (Figure 4). Most analyses were reported to be associated with a single study (n = 37, 50%), while for 21% retrospective data collection was used. Combined (meta-) analysis of multiple studies was used for 13 studies. We also assessed the different model evaluation methods used (other than standard goodness of fit diagnostics), as depicted in Table 1.
Table 1.
Overview of identified M&S-based publications in pediatric oncology
| Drug | Study characteristics | Analysis characteristics | Reference | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nr. Pat (P/A)† | Age (years)‡ | Type§ | Indication¶ | Objectives†† | Type‡‡ | Software§§ | Evaluation¶¶ | Model††† | Covariates‡‡‡ | ||
| 6-MP | 19/0 | 10 (3–17) | Study | ALL | PK-Cov | PPK | NM (FOCE) | VPC, SPLIT | 1cmt M:1cmt | BSA, PGTPMT | Hawwa et al. 58 |
| Actinomycin D | 36/0 | 1.6–20.3 | Study | Oth | PK-Cov Contaminations | PPK | NM (FOCE) | VPC | 3cmt | BWT, AGE | Edwards 82 |
| Asparaginase | 168/0 | 5.8 (4–10.2) | Retro | ALL | PK-Cov | PPK | NM (FO) | 1cmt | BSA | Hempel et al. 83 | |
| Asparaginase | NA/0 | NA | NA | ALL | PdrK-Cov, SimDose | PPK | NM (SIM) | 1cmt | Avramis et al. 84 | ||
| Asparaginase | 118/0 | 5 (1–9) | Study | ALL | PK-Cov | PPK | NM (NS) | 1cmtNL | Avramis et al. 85 | ||
| Asparaginase | 32/0 | 4.5 (1–14) | Study | ALL | PK-Cov | PPK | NM (FOCE) | VPC, PPC | 2cmt | BSA, BWT | Borghorst et al. 86 |
| Busulfan | 24/0 | 6.3 (0.3–16.7) | Study | HSCT | PK-Cov, SimDose | PPK | NM (FOCE) | BS | 1cmt | BWT | Booth et al. 36 |
| Busulfan | 24/0 | 6 (0.5–16.7) | Retro | HSCT | PK-Cov, SimDose | PPK | NM (FOCE) | BS | 1cmt | BWT | Nguyen et al. 87 |
| Busulfan | 48/0 | 10.4 (0.4–18.1) | Retro | HSCT | PK-Cov | PPK | NM (FOCE) | 1cmt | BSA | Schiltmeyer et al. 88 | |
| Busulfan | 12/62 | 5.8 (1.3–12) | Retro | HSCT | PK-Cov, TDM | PPK | NM (FOCE) | 1cmt | BWT, ALT, CMED | Sandstrom et al. 88 | |
| Busulfan | 20/0 | 1.1 (0.1–7.8) | Study | HSCT | PK-Cov, LSS | PPK | NM (NS) | 1cmt | BWT, DIS | Hassan et al. 89 | |
| Busulfan | 94/0 | 9.2 (0.4–18.8) | Meta | HSCT | PK-Cov, SimDose | PPK | NM (FOCE) | VPC, SPLIT | 1cmt | BSA, BWT | Trame et al. 33 |
| Busulfan | 77/0 | 5 (0.2–23) | Meta | HSCT | PK-Cov | PPK | NM (FOCE) | VPC | 1cmt | BWT, PGGST | Zwaveling et al. 90 |
| Busulfan | 29/0 | 5.6 (0.1–18.3) | Study | HSCT | PK-Cov, SimDose | PPK | NM (FOCE) | VPC | 1cmt | Age, PGGST | Johnson et al. 64 |
| Busulfan | 103/0 | 1.5 (0.2–11.3) | Study | HSCT | PK-Cov | PPK | NM (FOCE) | BS | 1cmt | BWT, Age, CMED, FRM, DIS | Nakamura et al. 91 |
| Busulfan | 100/0 | 0.1 (5.3–18.1) | Study | HSCT | PK-Cov | PPK | USCP (NPAG) | 1cmt | BWT, DIS | Bertholle et al. 92 | |
| Busulfan | 6/0 | 6 (1.5–14) | Study | HSCT | PK-Cov, LSS | PPK | NM (FOCE) | 1cmt | Cremers et al. 60 | ||
| Busulfan | 48/0 | 2.5 (0.2–15) | Meta | HSCT | PK-Cov, SimDose | PPK | NM (NS) | VPC, SPLIT | 1cmt | BWT | Paci et al. 93 |
| Busulfan | 245/0* | (0.2–26) | Meta | HSCT | PK-Cov | PPK | NM (FOCE) | BS | 2cmt | BWT | Bartelink 94 |
| Busulfan | 158/0* | (0.1–26) | Meta | HSCT | PK-Cov | PPK | NM (FOCE) | External evaluation | 2cmt | BWT | Bartelink 95 |
| Busulfan | 63/0 | 3.0 (0.7–13) | Study | HSCT | PK-Cov | PPK | NM (FOCE) | 1cmt | BWT | Veal 96 | |
| Carboplatin | 13/56 | 31 (2–60) | Meta | NBL, Oth | PK-Cov, SimDose | PPK | NM (FOCE) | 2cmt | HT, BWT, Age, CLCR, DUR | Lindauer et al. 97 | |
| Carboplatin | 75/75 | 6.5 (1.2–17.5) | Retro | MED, NBL, Oth | PK-Cov | PPK | MPK (NS) | 2cmt | BWT, SCR, NEPH | Urien et al. 98 | |
| Carboplatin | 57/57 | 5 (0.2–18) | Retro | CNS, NBL, Oth | PK-Cov | PPK | NM (NS) | PPC | 2cmt | BWT, SCR, NEPH | Chatelut et al. 99 |
| Cisplatin | NA/0 | NA | NA | PK-Cov | PBPK | NS (NA) | NA | PBPK | Evans et al. 25 | ||
| Clofarabine | 40/0 | 12.3 (2–19) | Meta | ALL, AML | PK-Cov | PPK | NM (FOCE) | BS | 2cmt | BWT, WBC | Bonate et al. 55 |
| Cyclofosfamide | 15/0 | 13.3 (5.4–21) | Study | STS | PK-Cov | PPK | NM (FOCE) | 1cmt | BSA, Sex | Chinnaswamy et al. 100 | |
| Cyclofosfamide | 22/0 | 3.2 (1.3–9.4) | Study | NBL | PK-Cov | PPK | NM (FO) | VPC | 1cmtNL M:1cmt | GFR | McCune et al. 61 |
| Cytarabine | 52/0 | (0.2–19) | Meta | ALL, AML, Oth | PK-Cov | PPK | NM (FO) | 2cmt M:1cmt | BSA, Age | Periclou et al. 101 | |
| Daunorubicin | 24/0 | 15.4 (2.8–23.2) | Retro | AML, Oth | PK-Cov | PPK | NM (FOCE) | 1cmt | BWT | Hempel et al. 102 | |
| Daunorubicin | 33/0 | (0.1–18.8) | Meta | AML | PK-Cov | PPK | NM (FO) | 2cmt M:1cmt | Hempel et al. 103 | ||
| Doxorubicin | 22/0 | 15 (3.3–21.5) | Study | Oth | PK-Cov | PPK | NM (NS) | 3cmt M:1cmt | BSA | Thompson et al. 62 | |
| Erlotinib | 46/42 | (2–19) | Meta | CNS | PK-Cov, SkinTox | PPK | NM (FOCE) | VPC | 1cmt | BWT, PGCYP, PGABC | White et al. 74 |
| Etoposide | NA/0 | NA | NA | PK-Cov | PBPK | PKSIM (NA) | NA | PBPK | Kersting et al. 24 | ||
| Etoposide | 67/0 | 3.5 (0.3–16.7) | Study | MED, NBL, Oth | PK-Cov | PPK | MLX (SAEM) | VPC | 2cmt | BWT | Urien et al. 104 |
| Etoposide | 31/0 | 8 (0.8–23.7) | Retro | NBL, Oth | PK-Cov | PPK | Ppharm (NS) | VPC | 3cmt | BWT | Wurthwein et al. 105 |
| Etoposide | NA/0 | NA | NA | PK-Cov, SimDose | SIM | Excel (NA) | NA | NA | Wurthwein et al. 106 | ||
| Etoposide | 26/0 | 8.5 (2.0–19.0) | Study | SLT | PK-Cov | PPK | NM (FOCE) | VPC | 1cmt | Baheti 107 | |
| Ifosfamide | 32/0 | (1–18) | Study | Oth | PK-Cov | PPK | NM (FO) | 1cmt M:1cmt | Kerbusch et al. 108 | ||
| Imatinib | 41/0 | (6–24) | Meta | PHL, SLT | PK-Cov | PPK | NM (FOCE) | PPC | 1cmt M:2cmt | BWT | Menon et al. 109 |
| Irinotecan | 11/0 | 12 (3–17) | Study | SLT | PK-Cov | PPK | NM (FOCE) | 2cmt | BWT | Kimura et al. 63 | |
| Irinotecan | 82/0 | (1–21) | Study | SLT, Oth | PK-Cov | PPK | NM (FOCE) | PPC | 2cmt M:2cmt | BWT | Thompson et al. 110 |
| Isotretinoin | 29/0 | 3.2 (1.1–18.7) | Study | NBL | PK-Cov | PPK | NM (FOCE) | 1cmt | Veal et al. 111 | ||
| Melphalan | 59/0 | 0 (0.3–18) | Study | HSCT | PK-Cov, LSS | PPK | NM (FOCE) | SPLIT | 2cmt | BWT, GFR, CMED | Nath et al. 112 |
| Methotrexate | 64/0 | 5 (1.6–16.8) | Retro | ALL, Oth | PK-Cov | PPK | NM (FOCE) | VPC | 2cmt | BWT, PGCT | Faganel et al. 113 |
| Methotrexate | 340/0 | 5 (0.4–17.8) | Study | ALL | PK-Cov, Relapse | PPK | NM (NS) | 2cmt | BWT | Johnsson et al. 79 | |
| Methotrexate | 69/0 | 6.7 (1–15) | Meta | ALL | PK-Cov, EFS | PPK | NM (NS) | 2cmt | Martelli et al. 80 | ||
| Methotrexate | 131/0 | (1.3–17.1) | Meta | ALL | PK-Cov, GITox | PPK | NM (FOCE) | 2cmt | BWT, Age, DIS | Buitenkamp et al. 75 | |
| Methotrexate | 118/0 | 7.4 (1.3–13.6) | Study | ALL | PK-Cov | PPK | NM (FOCE) | SPLIT | 2cmt | BSA, Age | Zhang et al. 114 |
| Methotrexate | 24/0 | 14.8 (10.8–18.8) | Retro | OSC | PK-Cov | PPK | NM (FOCE) | VPC, SPLIT | 2cmt | BWT, Age | Colom et al. 115 |
| Methotrexate | 79/0 | 6.9 (2–16) | Study | ALL | PK-Cov, LSS | PPK | NM (FOCE) | SPLIT | 2cmt | BWT | Plard et al. 116 |
| Methotrexate | 49/0 | 7 (0.5–17) | Retro | ALL | PK-Cov | PPK | NM (FOCE) | SPLIT | 2cmt | BWT, Age | Aumente et al. 117 |
| Methotrexate | NA/0 | NA | NA | PK-Cov | PBPK | BM (NA) | NA | PBPK | Li et al. 68 | ||
| Methotrexate | 23/0 | 6 (0.8–15) | Retro | ALL | PK-Cov, LSS | PPK | Ppharm (EM) | SPLIT | 2cmt | BWT, Age | Odoul et al. 118 |
| Methotrexate | 194/0 | NS (NS-NS) | Study | ALL | PK-Cov | PPK | ADAPT (MCPEM) | 2cmt M:2cmt | DIS | Panetta et al. 67 | |
| Methotrexate | 37/0 | 14 (4–21) | Retro | OSC, Oth | PK-Cov, MCT, RT, VOM | PPK | USCP (IT2B/NPEM) | 2cmt | Aquerreta et al. 76 | ||
| MK-0752 | 23/0 | 8.1 (2.6–17.1) | Study | CNS | PK-Cov | PPK | NM (FOCE) | 1cmt | Fouladi et al. 119 | ||
| Benzylguanine | 25/0 | NS (NS-21) | Study | CNS | PK-Cov, TDM | IPK | MLAB (NS) | 1cmt M:1cmt | Neville et al. 120 | ||
| Paclitaxel | 30/0 | (2.3–22.8) | Study | SLT | PK-Cov, NEUTR | IPK | ADAPT (ITS) | SPLIT | 2cmtNL | Sonnichsen et al. 70 | |
| Phenylacetate | 27/0 | 10 (1.4–20) | Study | Oth | PK-Cov, TDM | IPK | MLAB (NS) | 1cmtNL M:1cmt | Thompson et al. 121 | ||
| Prednisolone | 23/0 | 5.4 (2.4–15.2) | Study | ALL | PK-Cov, TDM | PPK | WNM (FO) | 2cmt | BSA | Petersen et al. 122 | |
| Pyrazoloacridine | 22/0 | (1–25) | Study | Oth | PK-Cov, NEUTR, TDM | IPK | MLAB (NS) | 2cmt | Berg et al. 69 | ||
| Temozolomide | 39/0 | 7.1 (0.7–21.9) | Study | CNS | PK-Cov | PPK | ADAPT (NS) | 1cmt M:1cmt | BSA, Age | Panetta et al. 123 | |
| Temozolomide | NA/0 | NA | NA | PK-Cov, LSS | OD | ADAPT (Unknown) | NA | Kirstein et al. 124 | |||
| Thioguanine | 18/0 | 18 (4–25) | Study | Oth | PK-Cov | IPK | MLAB (NS) | 2cmtNL | Kitchen et al. 125 | ||
| Topotecan | 162/0 | 9.1 (0.1–22) | Meta | MED, NBL, SLT, Oth | PK-Cov | PPK | NM (NS) | BS | 2cmt | BSA, Age, GFR, CMED | Schaiquevich et al. 126 |
| Topotecan | 6/0 | 4.9 (3.2–8.4) | Study | MED | PK-Cov | PPK | ADAPT (MAP) | 2cmt | Freeman et al. 127 | ||
| Topotecan | 40/0 | 12 (3–20) | Retro | SLT | PK-Cov, NEUTR | IPK | ADAPT (NS) | 2cmt | Zamboni et al. 71 | ||
| Topotecan | 17/0 | 12 (1–16) | Meta | CNS | PK-Cov | IPK | ADAPT (MAP) | 3cmt | Baker et al. 128 | ||
| Vincristine | 26/0 | (2–16) | Study | SLT | PK-Cov | PPK | NM (NS) | VPC, BS | 2cmt | PGCYP, PGABC | Guilhaumou et al. 129 |
| Vincristine | 70/0 | (1–16) | Study | ALL | PK-Cov | IPK | ADAPT (MAP) | 2cmt | Groninger et al. 130 | ||
| Vincristine | 17/0 | 3.8 (1.3–12.4) | Study | ALL | PK-Cov | IPK | ADAPT (NS) | 2cmt | de Graaf et al. 131 | ||
| NA/0 | NA | NA | PK-Cov, TDM | PPK | NM (PRIOR) | 1cmt | Barrett et al. 81 | ||||
Not specified (NS), Not applicable (NA).
Adult patients present but number of patients was not separately stated..
Children vs. Adults.
Age, median (range), years, for paediatric subjects. If only other measure then median or range were available, these were used.
Study types were retrospective (Retro), pooled meta-analysis of multiple studies or datasets (Meta) or data collected as part of clinical study (Study).
Indications: Acute lymphoblastic leukemia (ALL), Haematopoietic stem cell transplantation (HSCT), Neuroblastoma (NBL), Other (Oth), Medulloblastoma (MED), CNS malignancy (CNS), Acute myeloid leukemia (AML), Soft tissue sarcoma (STS), Philadelphia positive leukemia (PHL), Solid tumor (SLT), Osteosarcoma (OSC).
Study objectives: Characterize PK and/or covariates (PK-Cov), Limited sampling schedule (LSS), Therapeutic drug monitoring (TDM), Event-free survival (EFS), Gastrointestinal toxicitiy (GI-Tox), Mucosits (MCT), Renal toxicity (RT), Vomiting (VOM), Neutropenia (NEUTR).
Analysis types: Population pharmacokinetic/pharmacodynamic analysis (PPK), Individual level pharmacokinetic/pharmacodynamic (IPK) analysis, Physiologically-based pharmacokinetic model (PBPK), Optimal design (OD), Simulation study (SIM).
Software and estimation methods: nonmem (NM), Monolix (MLX), Berkeley Madonna (BM), USC-Pack (USCP), WinNonMix (WNM), First order conditional estimation (FOCE), First order estimation (FO), Simulation (SIM), Nonparametric estimation adaptive grid (NPAG), Stochastic approximation expectation maximization (SAEM), Expectation maximization (EM), Monte Carlo Parameteric Expectation Maximization Algorithm (MCPEM), Iterative two-stage Bayesian (IT2B), Non-parametric Expectation Maximization (NPEM,) Iterative two stage (ITS), Maximum a posteriori Bayesian estimation (MAP).
Model evaluation methods: Visual predictive check (VPC), External data or data-splitting procedure to evaluate model predictions (SPLIT), Posterior predictive check (PPC), Bootstrap analysis (BS).
Model type: For parent drug and metabolite (M). One (1), two (2) and three (3) compartmental (cmt) models, that may have a non-linear (NL) element.
Covariates identified in the analysis: Body surface area (BSA), Pharmacogenetic factors for SNP or enzyme activity xx (PGxx), Total body weight (BWT), Alanine transaminase (ALT), Concomitant or prior drug effect (CMED), Disease related factors (DIS), Height (HT), creatinine clerance (CLCR), Serum creatinine (SCR), Formulation (FRM).
Figure 3.
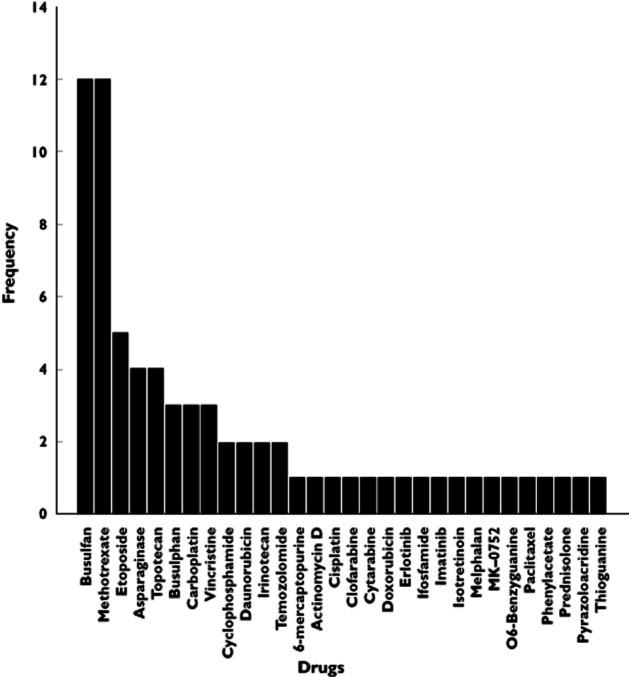
The frequency of anti-cancer drugs that were studied in the identified M&S-based analyses
Figure 4.
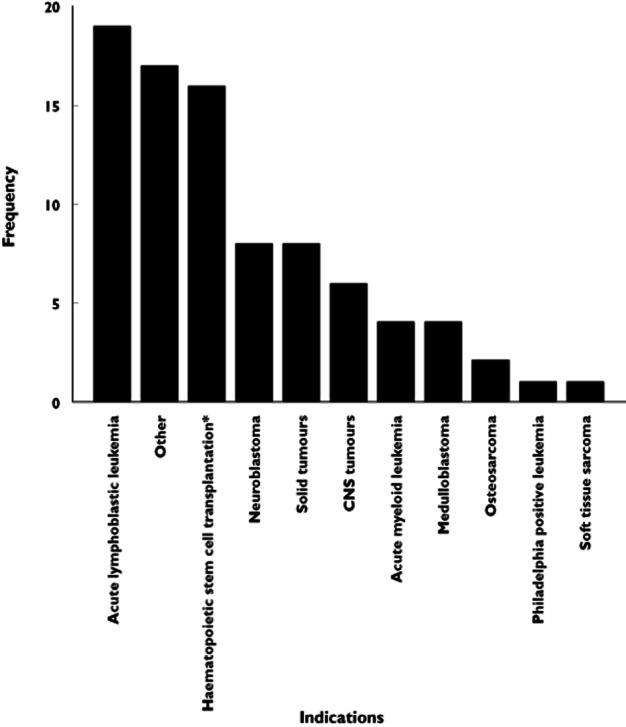
The frequency of different indications that were included in the identified M&S-based analyses. *Haematopoietic stem cell transplantation is no indication but a treatment for hematological malignancies
The identified articles are discussed below in the context of the four earlier mentioned types of paediatric studies: (i) pharmacokinetic bridging studies, (ii) exposure–response studies, (iii) full drug development studies and (iv) drug treatment optimization studies. For each of these studies, we discuss the applications of M&S in paediatric oncology drug development, and also address potential opportunities for additional application of M&S techniques.
Pharmacokinetic bridging studies
Three aspects of pharmacokinetic bridging studies are discussed: (i) determining starting dose in first-in-children PK studies, (ii) the procedure of dose regimen optimization during bridging and (iii) optimization of pharmacokinetic study designs.
Scaling approaches to determine starting dose in first-in-children PK studies
Prior to commencement of a trial, appropriate starting dose levels in children have to be determined. Historically, paediatric starting dose levels have been frequently set at 80% of the maximum tolerated dose in adult patients 21, scaled by body surface area BSA. Nonetheless, considering the narrow therapeutic window of anti-cancer agents, the use of scaling approaches that are supported by a scientific rational and prior knowledge is of special importance in paediatric oncology. Here, we discuss the two most commonly used scaling approaches, namely PBPK and allometry, although alternative approaches in which literature data were leveraged in a more empirical fashion have also been described 22.
Physiologically-based PK models
Physiologically-based PK (PBPK) models are multi-compartmental models that represent the major tissues, organs and drug effect pathways in an organism, that allow prediction of drug PK. These predictions are based on the physiological characteristics of tissues, and intrinsic physicochemical drug properties. A recent review has described the value of PBPK modelling in the paediatric population in more detail 23. In paediatric oncology, only three examples of PBPK modelling were identified.
Kersting et al. 24 described a PK bridging study where etoposide PK in children was predicted incorporating literature information of relevant drug metabolizing enzymes and age-dependent protein binding. The model predictions were compared with observed data in both children and adults and indicated adequate predictions across age. This example demonstrates how prior knowledge about physiology can be used to yield reasonable predictions of PK, and such approaches can thus potentially be applied for other drugs as well.
Evans et al. described a relatively simple PBPK model which predicted the concentration–time profiles of both free and total cisplatin in young children and adolescents 25. They showed how the model predictions adequately matched observed PK data in 14 patients. The described model structure was based on a model originally developed in dogs, and contained a limited number of compartments for plasma, skin, muscle, liver, gastrointestinal tract and kidneys. Subsequently, the model parameters were adjusted to reflect human paediatric physiology. Interesting in this approach was the adjustment of a model originally developed in animals, and how such an approach can also be used to make predictions on expected PK in the paediatric population.
Given the complexity of the different dynamical changes during paediatric development, PBPK methods are of special relevance in paediatric bridging studies for establishing a first-in-children dose. Rationally deriving a clinically relevant yet safe starting dose in paediatric oncology is complex, due to the small therapeutic windows of anti-cancer drugs, and frequently applied combination treatments. Here, PBPK methods can potentially be very useful because they allow incorporation of relevant physiological knowledge. Out of the scope of oncology, PBPK models have been successfully applied in the area of paediatric pharmacology for a number of examples 23, 26, 27. Application in paediatric oncology has thus far been limited, yet examples described can be considered promising.
Allometric scaling
Allometric scaling concerns the relationship between size and changes in physiology. It has been shown how size correlates with metabolic processes 28, based on a power relationship. The principles of allometry can be used for scaling clearance and volume between adults and children with respect to size and may therefore be a useful pharmacometric scaling tool in potentially determining rational starting doses when conducting a trial in children 29, 30. Although there is still debate 31 on the value of the allometric exponent on clearance, typically an estimate of 0.75 is used.
In the identified analyses in this review, allometric relationships with body weight were included very frequently (n = 33, 45%). In addition BSA was also frequently included as a covariate, and it has been shown that the relation between clearance and BSA is similar to allometric scaling using body weight (with an exponent of 0.75 on CL) 32. An illustrative example was provided by Trame et al. 33, who investigated different strategies for dosing regimen individualization for busulfan in paediatric oncology patients. They found that both BSA and allometric scaling-based dose regimens were adequate and similar for individualizing busulfan pharmacotherapy in children.
A recent comparison between allometric scaling and physiologically-based PK modelling for determining the first dose in children found that allometric scaling may not always be optimal in obtaining appropriate dose levels for children, especially in very young children 34, because this method does not take into account specific physiological changes relevant to drug exposure. In addition, an empirical comparison between allometry and PBPK in drug development showed that in general the performance was comparable, but that the magnitude of error in predicted exposure was much higher when using allometry 35, therefore in some cases leading to ineffective or toxic dose levels. These differences can most likely be attributed to the lack of specifically acknowledging developmental changes in metabolic capacity or organ function in the case of allometric scaling. Nonetheless, allometric scaling approaches do offer relatively simple ways to predict PK parameters in children to determine the paediatric starting dose, and can be incorporated in adult population PK models in a straightforward fashion.
Dose regimen optimization during bridging
Although a substantial number of identified M&S-based analyses aimed to characterize PK to optimize paediatric dosing regimens, however this was in most cases not in the context of an explicitly conducted paediatric bridging study. Rather, post hoc optimization of dose regimens of drugs already in use has been conducted.
One clear example where population PK M&S was used extensively in paediatric drug development was for i.v. busulfan. M&S-based analyses supported both paediatric labeing in the US 36 and Europe 37. The publication from Nguyen et al. 37 clearly described the application of M&S during the paediatric development. Population PK–PD M&S allowed characterization of inter-individual variability and understanding of associated patient related determinants of this variability. Subsequently, with a simulation analysis, optimal dosing regimens were derived across different age groups. The report by Nguyen et al. is illustrative because it clearly demonstrates the impact of M&S in the context of the paediatric clinical drug development process where adult human exposure needed to be bridged to paediatric patients, and to derive optimal paediatric dose regimens.
The population PK–PD models extensively used in the analysis by Nguyen, but also in most other analyses identified, involved compartmental PK models that employed non-linear mixed effect (NLME) modelling, in which different levels of variability can be estimated, and which allow analysis of sparsely sampled datasets 38. Because of these advantages, most of the identified M&S analyses (n = 59, 80%) used a NLME approach to analyze the data and to characterize inter-individual variability. The majority of identified analyses used nonmem (n = 50, 68%) or ADAPT (n = 9, 10%) for data analysis, while the remaining analyses (n = 15, 21%) used a broad range of less frequently used software packages.
Informative sampling designs
Recently, Foo et al. 39 suggested an adaptive optimal design methodology for paediatric PK bridging studies which allowed adjustment of optimal study designs during execution of the trial, adaptively assessing when sufficient paediatric patients were included in a clinical trial. Although this example was only described in the context of a simulation study, it is potentially promising because it aims to optimize the number of paediatric patients in a clinical study combined with the often limited number of patients available with paediatric malignancies.
The applied method in the aforementioned example, optimal design, is a statistical methodology that aims to optimize a study design with respect to a design criterion 40. To define an optimal design, a prior model is necessary. Most commonly, optimal designs have been applied to optimize parameter estimation precision, in order to identify optimal sparse sampling designs that allow adequate estimation precision. Optimal design methods may, however, also be used to optimize other design parameters such as number of subjects, groups, dose levels or even study power 41. Because of the practical and ethical limitations of PK studies in the paediatric oncology population, optimal design approaches can be especially relevant, in order to allow informative studies with minimal sampling and/or subjects.
A challenge to the application of optimal design (or any other design optimization method), is that prior knowledge (e.g. prior model) may sometimes be limited, which complicates application of such design optimization methods. If no prior model is available, it could potentially be considered to use a PBPK model to support the optimization 27. In addition, especially in paediatric oncology, the number of practical restrictions in terms of study design can be substantial, thereby restricting the design space to optimize a clinical study design.
In our review, however, we did not identify any studies that applied optimal design to derive optimal sparse sampling schedules, or any other design optimization, although the benefit of such methods has been demonstrated in adults 39, 42–45. Given the limited number of patients frequently available and potential other limitations in paediatric oncology drug development, design of an informative clinical study design is important and optimal design or approaches such as clinical trial simulation can be relevant tools to optimize clinical study designs where feasible.
Exposure–response studies
Development of biomarkers for treatment response in paediatric oncology is an active field, with many recent and potentially promising developments 46–49. In adult oncology, exposure–response models have been developed 50, 51. However in this review focusing on paediatric oncology, exposure–response models for biomarkers (potentially) predictive for efficacy were not identified.
Nonetheless, development of biomarkers as surrogate measures of efficacy is of special importance for the evaluation of anti-cancer drugs in children. Although phase II studies frequently use outcome-based measures such as progression free survival (PFS), other, longitudinal continuous (bio-) markers could potentially be more informative and sensitive measures in early phase clinical trials. For instance Bruno et al. 52 suggested the use of change in tumour size as a more informative endpoint of phase II trials. Additionally, when analyzing biomarkers in a model-based framework, measures of outcome (e.g. PFS, overall survival (OS)) may be linked to biomarkers of disease progression. In adult oncology for instance, Claret et al. 53 developed PK–PD-outcome models that quantify the relationship between drug exposure, tumour growth inhibition and outcome (PFS/OS). Thus, recent developments in adult oncology indicate potentially promising results of exposure–response modelling of anti-cancer drugs, but unfortunately no applications have been reported yet in the development of paediatric anti-cancer drugs.
Full drug development studies
One example of a paediatric anti-cancer drug in which M&S has supported a full drug development study, was the development of clofarabine 54, 55, which was first approved by the FDA for acute lymphoblastic leukemia (ALL) in paediatric patients instead of an adult indication 56. Bonate et al. described a population PK analysis of clofarabine and intracellular clofarabine triphosphates, based on available data from multiple clinical studies in paediatric patients, also identifying predictors of inter-individual variability 55. In this study, it was shown that both the white blood cell count and body weight were clinically important predictors for the expected drug exposure. This analysis was later also extended with adult data and the metabolite 6-ketoclofarabine 54.
Paediatric drug development studies generally suffer from increased risks for dropout, but also large (age-related) variability in patient characteristics, which may in turn affect the outcome of a clinical trial. In addition, for full drug development studies in which PK, safety and efficacy are characterized, substantial numbers of patients and time are required. Therefore, specifically, the conduct of such studies in the area of paediatric oncology is highly challenging, and unexpected events or other sources of variability may impact on the outcome of a clinical trial. Specifically for such trials, clinical trial simulation (CTS) is a M&S methodology which may be considered for a priori evaluation of the likelihood of a trial meeting its objectives, because it allows evaluation of the impact of variability introduced by a range of unexpected events during trial execution (subject dropout, missed samples, lack of compliance) 57. In CTS, a clinical trial with the expected exposure and/or response profiles of individual patients can be simulated, using developed exposure–response models. Subsequently, random events such as missing data can be applied to the dataset, and subsequently the obtained simulated trial data can be analyzed using the planned statistical analysis procedures. In the context of paediatric oncology however, CTS approaches have not been reported.
Drug treatment optimization studies
Even if a drug is already clinically used, paediatric dosing regimens of anti-cancer agents can potentially be optimized further with respect to either pharmacokinetics (e.g. target exposures), or with respect to toxicity profiles or based on ultimate outcome (e.g. efficacy measures). In such treatment optimization studies, it is often important to understand and quantify variability between patients, which is why population PK–PD methods are frequently applied.
Optimization of pharmacokinetics
Most of the studies (n = 59 80%) which were identified in this review (Table 1) descriptively analyzed PK in paediatric oncology patients using population PK modelling approaches, with the ultimate aim to identify patient covariates predictive for inter-individual variability in PK parameters. The analyses identified were generally built using either therapeutic drug monitoring (TDM) data or data obtained from phase I studies.
The frequency of inclusion of various patient characteristics as covariates is illustrated in Figure 5. In seven studies (9.5%), adult data were also co-analyzed, which could be considered a useful approach to support characterization of body size and maturation effects on PK parameters. In some studies 58–64, covariates predictive of inter-individual variability in PK parameters were investigated while the sample size was relatively small. However, accurate identification of covariate effects requires a representative distribution of the covariate of interest. Thus, conclusions related to influential covariates derived from such studies should be considered carefully 65.
Figure 5.
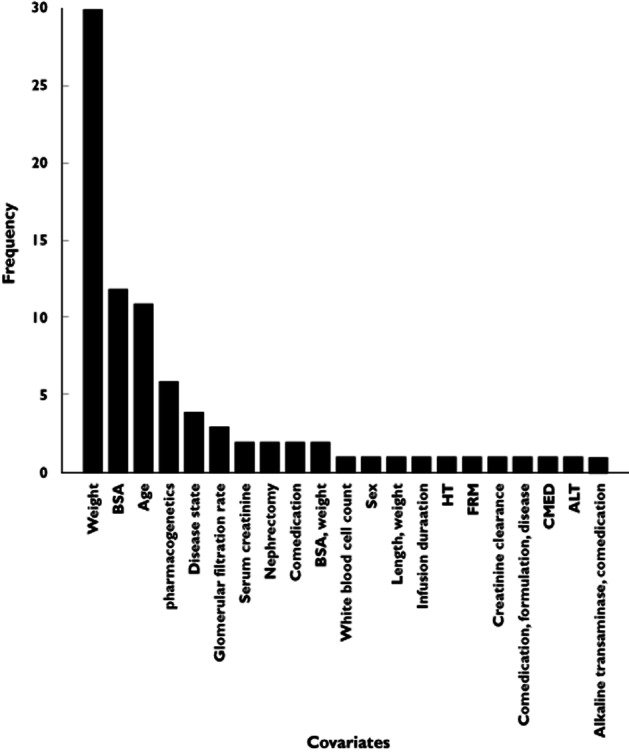
The frequency covariates that were included in the identified M&S-based analyses. ALT, alanine transaminase; BSA, body surface area; CMED, comedication; FRM, formulation; HT, height
Eleven studies developed population PK models to develop or optimize TDM or limited sampling (LS) strategies. In this context, M&S can be used, for instance, to perform simulations studies to evaluate the success rate of different TDM or LS strategies. Additionally, optimal design techniques may be used to derive the most informative sampling times for obtaining information about the expected drug exposure. Specifically for TDM approaches, inter-occasion variability (IOV) is an important component of variability to consider, because it may inflate residual variability estimates and cause bias in parameter estimates 66. When IOV is large, TDM strategies may be less useful. Yet, a substantial number of the identified analyses did not report evaluation of IOV (70%), although in some cases this may be related to availability of only single occasion data.
Recently, integration of physiological components for dose optimization strategies in the context of paediatric oncology was demonstrated by Panetta et al. who described an analysis in which the intracellular PK of methotrexate metabolites in leukemia cells was characterized, and related to ALL cell lineages. Also a folate-pathway model was included in this framework to derive optimal treatment schedules 67. Overall this analysis demonstrated how insight can be obtained into intracellular pharmacology of drugs and the relationship with treatment efficacy.
PBPK methods can also be used to investigate the impact on PK for specific conditions. Li et al. used PBPK modeling to investigate specifically the effect of malignant effusions in paediatric oncology patients on the disposition of methotrexate 68. Thus even when a general paediatric dose regimen has already been established, additional co-morbidities may arise that will further affect PK and of which the effect may not be comparable between adults and children. Since PBPK methods aim to represent human physiology, these models can be adapted to include representations of any relevant factors such as the investigated effect of malignant effusion in this example.
Optimization for toxicity and outcome measures
A limited number of studies was performed specifically describing models for toxicity or outcome measures (n = 7). Haematological toxicity is a commonly occurring dose-limiting toxicity for many anti-cancer drugs. Exposure–response models describing the time course and variability of blood cell counts may therefore be useful to optimize treatment with respect to the incidence of severe haematological toxicity. In the field of paediatric oncology, Berg et al. 69 first described the relationship between decrease in absolute neutrophil count and exposure to pyrazolacridine in children and young adults using an empirical post hoc sigmoid Emax model, which allowed assessment of limiting exposure levels in these patients. Later, Sonnichsen et al. 70 described the actual time course of the neutrophil count. However in this analysis, the authors did not consider the impact of exposure on the shape (e.g. magnitude of decrease) of the neutrophil count time course, which limits the use of such a model in investigating alternative dose regimens. Finally Zamboni et al. 71 also described the time course of neutropenia induced by topotecan using a model with a number of transit compartments. This analysis also incorporated the relationship between drug exposure and the response in neutrophil count decline, allowing the model to be used for investigation of alternative dose regimens. In adult oncology, Friberg et al. 72 developed a model similar to the analysis described by Zamboni et al. also incorporating transit compartments accounting for the maturation process of neutrophils in the bone marrow. The model by Friberg et al. was, however, developed in the context of population PK–PD modelling, allowing improved quantification of variability and analysis of more sparse data, which is frequently the case for clinical data of haematological toxicity. The model by Friberg et al. (Figure 6) is now considered to be well established as it has been applied to a range of anti-cancer agents. Very recently, an adapted version of the model by Friberg et al. has also been implemented for paediatric oncology patients 73 for topotecan. Potentially, (paediatric) maturational effects of haematopoesis could also be incorporated in this model, but it has not yet been investigated if such effects are present and clinically relevant.
Figure 6.
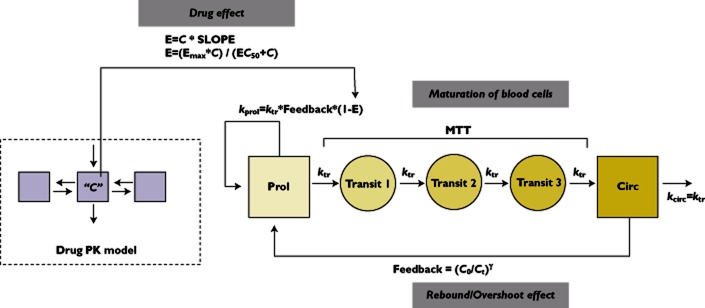
Semi-physiological model for haematological toxicity developed by Friberg et al. for adult patients. C = Drug concentration, E = Effect, Emax = Maximum effect, EC50 = Half-maximum effect concentration, SLOPE = Drug effect, ktr = Transition rate constant, MTT = Mean transition time
Model-based analyses have also been reported for other types of toxicities in paediatric oncology. Usually, these were implemented as post hoc logistic regression analyses linking exposure to the probability of toxicity, in contrast to the more integrated models as described for haematological toxicity. For instance White-Koning et al. reported on a exposure–response model for erlotinib-induced skin toxicity in adult and paediatric patients 74. In this PK–PD analysis, it was demonstrated that the higher recommended dose in children compared with adults for erlotinib is mainly due to pharmacokinetic rather than pharmacodynamic differences. Another PK–PD model in paediatric cancer patients was reported that described the relationship between methotrexate exposure and the probability of gastrointestinal toxicity in patients with and without Down's syndrome 75. Patients with Down's syndrome have decreased tolerance to methotrexate, but this analysis allowed exclusion of the impact of pharmacokinetic differences between patients with and without this syndrome. Finally, Aquerreta et al. 76 developed a combined PK–PD model quantifying the probability for developing renal toxicity, mucositis and vomiting for paediatric oncology patients treated with methotrexate. The model could be used to optimize rationally high dose methotrexate treatment, and it also confirmed upper thresholds of methotrexate plasma concentrations that should be avoided in paediatric patients.
With the increasing survival of paediatric oncology patients, the long term cardiac toxicity of anthracyclines is receiving substantial interest. Recently in adults an exposure–response study for cardiotoxicity was described and utilized to optimize treatment 77, 78. A similar approach could also be of relevance in the paediatric oncology population.
In summary, a number of illustrative examples have been published demonstrating how PK–PD models for toxicity can be developed for paediatric oncology patients, to help in the understanding of potential mechanisms or factors that may play a role in the development of various toxicities. Nonetheless, the applications are limited compared with the much larger number of exposure–toxicity analyses that have been described for adults. The ultimate application of such models by optimizing dosing regimens for toxicity have however not been conducted. One possible exception is the study by Panetta et al. 73, who described a population PK–PD model that incorporated a tumour growth inhibition model based on paediatric xenograft data, together with a (clinical) model for topotecan-induced neutropenia, in order to investigate optimal treatment regimens taking into account both efficacy and toxicity in paediatric patients with neuroblastoma. This analysis demonstrated how useful computational approaches can be in evaluating potential dose regimens and for leveraging of preclinical data. Nonetheless, this example has not yet been verified or supported by a clinical study or any other clinical observations.
Besides toxicity, post hoc logistic regression analyses have also been reported for measures of outcome. Jönson et al. 79 reported a model-based analysis accounting for the probability of relapse after methotrexate. It was found that dosing regimens based on body weight for methotrexate may give more predictable PK but could potentially also improve outcome measures in these patients. Furthermore, Martelli et al. considered inclusion of event-free survival in their PK analysis 80, but here no clear relationship could be identified.
Finally, an example of the link of model-based analysis and routine patient care was recently provided by Barrett et al. 81, who described the integration of hospital database systems with a Bayesian model-based framework for determination of optimal dose adjustment strategies in individual patients. They showed how the management of paediatric drug treatment can be greatly enhanced by the use of this system, especially for drugs with narrow therapeutic windows that may easily lead to suboptimal treatment or toxicities.
Conclusion
We reviewed the application of M&S-based analysis in paediatric oncology in the context of the four types of clinical studies that can typically be performed: PK bridging studies, exposure–response analyses, full drug development studies and drug treatment optimization studies. M&S-based approaches have been used successfully in other areas of paediatric drug development and in adult-oncology, but the application of M&S to support paediatric drug development proactively has been very limited.
Overall, most studies identified were descriptive PK studies that aimed to characterize PK in paediatric patients, and to identify potential predictors of variability in PK parameters, in order to optimize further dose regimens of already clinically used drugs or to optimize TDM strategies of such drugs. Although these analyses have been useful to further optimize drug treatment, formal analyses related to bridging of exposure were much more limited. The clinical development of busulphan and clofarabine are illustrative examples for the overall role M&S approaches can play in a clinical drug development process.
The use of PBPK modelling for first-in-children dose selection is promising, but only a limited number of examples have currently been published, most likely because this is a relatively new development in the field of quantitative clinical pharmacology. Although in many cases, conventional empirical dose selection approaches 21 may still be considered, scaling methods such as PBPK could be useful to provide scientific support for the selected starting dose-level.
With respect to exposure–response studies, no reports in paediatric oncology have been described. Nonetheless, exposure–response analyses could still be considered promising when biomarkers in paediatric patients are further developed, but this task is complex also due to the intrinsic differences in disease biology between children and adults.
Given the low incidence of paediatric malignancies and ethical and practical constraints in this particularly sensitive group of patients, efficient design and analysis of clinical studies is crucial, and M&S approaches can potentially support and streamline the paediatric drug development process of anti-cancer drugs, since they allow integration of (prior) knowledge, efficient analysis of sparse or heterogeneous data, and can be used to support decision making, thereby stressing the relevance of these methods to be used more in the field of paediatric oncology drug development
Competing Interests
All authors have completed the Unified Competing Interest form at httpwww.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
References
- 1.Mathis L, Rodriguez W. Drug therapy in pediatrics: a developing field. Dermatol Ther. 2009;22:257–261. doi: 10.1111/j.1529-8019.2009.01239.x. [DOI] [PubMed] [Google Scholar]
- 2.Rose K, Della PO. Development of paediatric medicines: concepts and principles. Handb Exp Pharmacol. 2011;205:111–124. doi: 10.1007/978-3-642-20195-0_5. [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez W, Selen A, Avant D, Chaurasia C, Crescenzi T, Gieser G, Di Giacinto J, Huang S-M, Lee P, Mathis L, Murphy D, Murphy S, Roberts R, Sachs HC, Suarez S, Tandon V, Uppoor RS. Improving pediatric dosing through pediatric initiatives: what we have learned. Pediatrics. 2008;121:530–539. doi: 10.1542/peds.2007-1529. [DOI] [PubMed] [Google Scholar]
- 4.van den Berg H, Tak N. Licensing and labelling of drugs in a paediatric oncology ward. Br J Clin Pharmacol. 2011;72:474–481. doi: 10.1111/j.1365-2125.2011.03977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van den Berg H, van den Anker JN, Beijnen JH. Cytostatic drugs in infants: a review on pharmacokinetic data in infants. Cancer Treat Rev. 2012;38:3–26. doi: 10.1016/j.ctrv.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Boklan J. Little patients, losing patience: pediatric cancer drug development. Mol Cancer Ther. 2006;5:1905–1908. doi: 10.1158/1535-7163.MCT-06-0179. [DOI] [PubMed] [Google Scholar]
- 7.Aleksa K, Koren G. Ethical issues in including pediatric cancer patients in drug development trials. Paediatr Drugs. 2002;4:257–265. doi: 10.2165/00128072-200204040-00005. [DOI] [PubMed] [Google Scholar]
- 8. Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for paediatric use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004. E. 2006. Available at http://eur-lex.europa.eu/LexUriServ/ (last accessed 12 October 2010)
- 9. FDAAA. Title IV: Pediatric Research Equity Act of 2007 (PREA) and Title V: Best Pharmaceuticals for Children Act of 2007 (BPCA). 2007. September 27, 2007 (Public Law No. 110-85)
- 10. Guidance for industry exposure-response relationships – study design, data analysis, and regulatory applications. U.S. Department of Health and Human Services Food and Drug Administration. 2003. Available at httpwww.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072109.pdf (last accessed 10 October 2012)
- 11.Pizzo P, Poplack D. Chapter 1. Epidemiology of childhood cancer. In: Pine JW Jr, editor. Principles and Practice of Pediatric Oncology. 6th edn. Philadelphia, PA: Lippincott Williams & Wilkins; 2011. [Google Scholar]
- 12.Simone J, Lyons J. Superior cancer survival in children compared to adults: a superior system of cancer care? 1998. Available at http://www.simoneconsulting.com/PDF/children_survival.pdf (last accessed 4 October 2012)
- 13.Manolis E, Pons G. Proposals for model-based paediatric medicinal development within the current European Union regulatory framework. Br J Clin Pharmacol. 2009;68:493–501. doi: 10.1111/j.1365-2125.2009.03484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van den Broek MPH, Huitema ADR, van Hasselt JGC, Groenendaal F, Toet MC, Egberts TCG, de Vries LS, Rademaker CMA. Lidocaine (lignocaine) dosing regimen based upon a population pharmacokinetic model for preterm and term neonates with seizures. Clin Pharmacokinet. 2011;50:461–469. doi: 10.2165/11589160-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 15.van den Broek MPH, Groenendaal F, Toet M, van Straaten H, van Hasselt J, Huitema A, de Vries L, Egberts A, Rademaker C. Pharmacokinetics and clinical efficacy of phenobarbital in asphyxiated newborns treated with hypothermia: a thermopharmacological approach. Clin Pharmacokinet. 2012;51:671–679. doi: 10.1007/s40262-012-0004-y. [DOI] [PubMed] [Google Scholar]
- 16.Lo YL, van Hasselt JGC, Heng SC, Lim CT, Lee TC, Charles BG. Population pharmacokinetics of vancomycin in premature Malaysian neonates: identification of predictors for dosing determination. Antimicrob Agents Chemother. 2010;54:2626–2632. doi: 10.1128/AAC.01370-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Hasselt JGC, Andrew MA, Hebert MF, Tarning J, Vicini P, Mattison DR. The status of pharmacometrics in pregnancy: highlights from the 3rd American Conference on Pharmacometrics. Br J Clin Pharmacol. 2012;74:932–939. doi: 10.1111/j.1365-2125.2012.04280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellanti F, Della PO. Modelling and simulation as research tools in paediatric drug development. Eur J Clin Pharmacol. 2011;67(Suppl. 1):75–86. doi: 10.1007/s00228-010-0974-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meibohm B, Läer S, Panetta JC, Barrett JS. Population pharmacokinetic studies in pediatrics: issues in design and analysis. AAPS J. 2005;7:E475–487. doi: 10.1208/aapsj070248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cella M, Gorter de Vries F, Burger D, Danhof M, Della PO. A model-based approach to dose selection in early pediatric development. Clin Pharmacol Ther. 2010;87:294–302. doi: 10.1038/clpt.2009.234. [DOI] [PubMed] [Google Scholar]
- 21.Marsoni S, Ungerleider RS, Hurson SB, Simon RM, Hammershaimb LD. Tolerance to antineoplastic agents in children and adults. Cancer treatment reports. 1985;69:1263–1269. [PubMed] [Google Scholar]
- 22.van Hasselt JGC, Green B, Morrish GA. Leveraging physiological data from literature into a pharmacokinetic model to support informative clinical study design in pregnant women. Pharm Res. 2012;29:1609–1617. doi: 10.1007/s11095-012-0671-2. [DOI] [PubMed] [Google Scholar]
- 23.Barrett JS, Della CAO, Läer S, Meibohm B. Physiologically Based Pharmacokinetic (PBPK) modeling in children. Clin Pharmacol Ther. 2012;92:40–49. doi: 10.1038/clpt.2012.64. Available at http://www.ncbi.nlm.nih.gov/pubmed/22669290 (last accessed 20 July 2012) [DOI] [PubMed] [Google Scholar]
- 24.Kersting G, Willmann S, Würthwein G, Lippert J, Boos J, Hempel G. Physiologically based pharmacokinetic modelling of high- and low-dose etoposide: from adults to children. Cancer Chemother Pharmacol. 2011;69:397–405. doi: 10.1007/s00280-011-1706-9. [DOI] [PubMed] [Google Scholar]
- 25.Evans WE, Crom WR, Tsiatis A, Green AA, Hayes FA, Pratt CB. Pharmacokinetic modeling of cisplatin disposition in children and adolescents with cancer. Cancer Chemother Pharmacol. 1982;10:22–26. doi: 10.1007/BF00257231. [DOI] [PubMed] [Google Scholar]
- 26.Leong R, Vieira MLT, Zhao P, Mulugeta Y, Lee C, Huang S-M, Burckart GJ. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin Pharmacol Ther. 2012;91:926–931. doi: 10.1038/clpt.2012.19. Available at http://dx.doi.org/10.1038/clpt.2012.19 (last accessed 23 April 2012) [DOI] [PubMed] [Google Scholar]
- 27.Khalil F, Läer S. Physiologically based pharmacokinetic modeling: methodology, applications, and limitations with a focus on its role in pediatric drug development. J Biomed Biotechnol. 2011;2011:1–13. doi: 10.1155/2011/907461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleiber M. Body size and metabolism. Hilgardia. 1932;6:315–351. [Google Scholar]
- 29.Holford NHG. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30:329–332. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- 30.Anderson BJ, Allegaert K, Holford NHG. Population clinical pharmacology of children: modelling covariate effects. Eur J Pediatr. 2006;165:819–829. doi: 10.1007/s00431-006-0189-x. [DOI] [PubMed] [Google Scholar]
- 31.Mahmood I. Theoretical versus empirical allometry: facts behind theories and application to pharmacokinetics. J Pharm Sci. 2010;99:2927–2933. doi: 10.1002/jps.22073. Available at http://www.ncbi.nlm.nih.gov/pubmed/20127826 (last accessed 26 November 2012) [DOI] [PubMed] [Google Scholar]
- 32.McLeay SC, Morrish GA, Kirkpatrick CMJ, Green B. The relationship between drug clearance and body size: systematic review and meta-analysis of the literature published from 2000 to 2007. Clin Pharmacokinet. 2012;51:319–330. doi: 10.2165/11598930-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 33.Trame MN, Bergstrand M, Karlsson MO, Boos J, Hempel G. Population pharmacokinetics of busulfan in children: increased evidence for body surface area and allometric body weight dosing of busulfan in children. Clin Cancer Res. 2011;17:6867–6877. doi: 10.1158/1078-0432.CCR-11-0074. [DOI] [PubMed] [Google Scholar]
- 34.Strougo A, Eissing T, Yassen A, Willmann S, Danhof M, Freijer J. First dose in children: physiological insights into pharmacokinetic scaling approaches and their implications in paediatric drug development. J Pharmacokinet Pharmacodyn. 2012;39:195–203. doi: 10.1007/s10928-012-9241-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teitelbaum Z, Lave T, Freijer J, Cohen AF. Risk assessment in extrapolation of pharmacokinetics from preclinical data to humans. Clin Pharmacokinet. 2010;49:619–632. doi: 10.2165/11533760-000000000-00000. Available at http://www.ncbi.nlm.nih.gov/pubmed/20690784 (last accessed 29 August 2012) [DOI] [PubMed] [Google Scholar]
- 36.Booth BP, Rahman A, Dagher R, Griebel D, Lennon S, Fuller D, Sahajwalla C, Mehta M, Gobburu JVS. Population pharmacokinetic-based dosing of intravenous busulfan in pediatric patients. J Clin Pharmacol. 2007;47:101–111. doi: 10.1177/0091270006295789. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen L. Integration of modelling and simulation into the development of intravenous busulfan in paediatrics: an industrial experience. Fundam Clin Pharmacol. 2008;22:599–604. doi: 10.1111/j.1472-8206.2008.00651.x. [DOI] [PubMed] [Google Scholar]
- 38.Sheiner L, Wakefield J. Population modelling in drug development. Stat Methods Med Res. 1999;8:183–193. doi: 10.1177/096228029900800302. [DOI] [PubMed] [Google Scholar]
- 39.Foo LK, Duffull S. Adaptive optimal design for bridging studies with an application to population pharmacokinetic studies. Pharm Res. 2012;29:1530–1543. doi: 10.1007/s11095-011-0659-3. [DOI] [PubMed] [Google Scholar]
- 40.Duffull SB. Design of clinical pharmacology trials. Clin Exp Pharmacol Physiol. 2001;28:905–912. doi: 10.1046/j.1440-1681.2001.03546.x. [DOI] [PubMed] [Google Scholar]
- 41.Ueckert S, Nyberg J, Hooker A. Explicit optimization of clinical trials for statistical power. 2011. p. 2251. Abstracts of the Annual Meeting of the Population Approach Group in Europe.
- 42.Green B, Duffull SB. Prospective evaluation of a d-optimal designed population pharmacokinetic study. J Pharmacokinet Pharmacodyn. 2003;30:145–161. doi: 10.1023/a:1024467714170. [DOI] [PubMed] [Google Scholar]
- 43.Maloney A, Karlsson MO, Simonsson USH. Optimal adaptive design in clinical drug development: a simulation example. J Clin Pharmacol. 2007;47:1231–1243. doi: 10.1177/0091270007308033. [DOI] [PubMed] [Google Scholar]
- 44.Jamsen KM, Duffull SB, Tarning J, Lindegardh N, White NJ, Simpson JA. Optimal designs for population pharmacokinetic studies of oral artesunate in patients with uncomplicated falciparum malaria. Malar J. 2011;10:181. doi: 10.1186/1475-2875-10-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ogungbenro K, Matthews I, Looby M, Kaiser G, Graham G, Aarons L. Population pharmacokinetics and optimal design of paediatric studies for famciclovir. Br J Clin Pharmacol. 2009;68:546–560. doi: 10.1111/j.1365-2125.2009.03479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mizia-Malarz A, Sobol G, Janowska J, Wos H, Zahorska-Markiewicz B. Prognostic value of proangiogenic cytokines in children with lymphomas. Pediatr Blood Cancer. 2009;53:1195–1199. doi: 10.1002/pbc.22179. [DOI] [PubMed] [Google Scholar]
- 47.Gessi M, Bueren AO, Rutkowski S, Pietsch T. p53 expression predicts dismal outcome for medulloblastoma patients with metastatic disease. J Neurooncol. 2011;106:135–141. doi: 10.1007/s11060-011-0648-8. [DOI] [PubMed] [Google Scholar]
- 48.Sandoval JA, Malkas LH, Hickey RJ. Clinical significance of serum biomarkers in pediatric solid mediastinal and abdominal tumors. Int J Mol Sci. 2012;13:1126–1153. doi: 10.3390/ijms13011126. Available at http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3269742&tool=pmcentrez&rendertype=abstract (last accessed 2 October 2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Dongen JJ, Seriu T, Panzer-Grümayer ER, Biondi A, Pongers-Willemse MJ, Corral L, Stolz F, Schrappe M, Masera G, Kamps WA, Gadner H, van Wering ER, Ludwig WD, Basso G, De Bruijn MA, Cazzaniga G, Hettinger K, van der Does-van den Berg A, Hop WC, Riehm H, Bartram CR. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352:1731–1738. doi: 10.1016/S0140-6736(98)04058-6. Available at http://www.ncbi.nlm.nih.gov/pubmed/9848348 (last accessed 1 September 2012) [DOI] [PubMed] [Google Scholar]
- 50.Keizer RJ, Funahashi Y, Semba T, Wanders J, Beijnen JH, Schellens JHM, Huitema ADR. Evaluation of α2-integrin expression as a biomarker for tumor growth inhibition for the investigational integrin inhibitor e7820 in preclinical and clinical studies. AAPS J. 2011;13:230–239. doi: 10.1208/s12248-011-9260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lindauer A, Di Gion P, Kanefendt F, Tomalik-Scharte D, Kinzig M, Rodamer M, Dodos F, Sörgel F, Fuhr U, Jaehde U. Pharmacokinetic/pharmacodynamic modeling of biomarker response to sunitinib in healthy volunteers. Clin Pharmacol Ther. 2010;87:601–608. doi: 10.1038/clpt.2010.20. Available at http://www.ncbi.nlm.nih.gov/pubmed/20376000 (last accessed 15 July 2012) [DOI] [PubMed] [Google Scholar]
- 52.Bruno R, Claret L. On the use of change in tumor size to predict survival in clinical oncology studies: toward a new paradigm to design and evaluate phase II studies. Clin Pharmacol Ther. 2009;86:136–138. doi: 10.1038/clpt.2009.97. [DOI] [PubMed] [Google Scholar]
- 53.Claret L, Girard P, Hoff PM, van Cutsem E, Zuideveld KP, Jorga K, Fagerberg J, Bruno R. Model-based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J Clin Oncol. 2009;27:4103–4108. doi: 10.1200/JCO.2008.21.0807. [DOI] [PubMed] [Google Scholar]
- 54.Bonate PL, Cunningham CC, Gaynon P, Jeha S, Kadota R, Lam GN, Razzouk B, Rytting M, Steinherz P, Weitman S. Population pharmacokinetics of clofarabine and its metabolite 6-ketoclofarabine in adult and pediatric patients with cancer. Cancer Chemother Pharmacol. 2011;67:875–890. doi: 10.1007/s00280-010-1376-z. [DOI] [PubMed] [Google Scholar]
- 55.Bonate PL, Craig A, Gaynon P, Gandhi V, Jeha S, Kadota R, Lam GN, Plunkett W, Razzouk B, Rytting M, Steinherz P, Weitman S. Population pharmacokinetics of clofarabine, a second-generation nucleoside analog, in pediatric patients with acute leukemia. J Clin Pharmacol. 2004;44:1309–1322. doi: 10.1177/0091270004269236. [DOI] [PubMed] [Google Scholar]
- 56.Pession A, Masetti R, Kleinschmidt K, Martoni A. Use of clofarabine for acute childhood leukemia. Biologics. 2010;4:111–118. doi: 10.2147/btt.s10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holford N, Ma SC, Ploeger BA. Clinical trial simulation: a review. Clin Pharmacol Ther. 2010;88:166–182. doi: 10.1038/clpt.2010.114. [DOI] [PubMed] [Google Scholar]
- 58.Hawwa AF, Collier PS, Millership JS, McCarthy A, Dempsey S, Cairns C, McElnay JC. Population pharmacokinetic and pharmacogenetic analysis of 6-mercaptopurine in paediatric patients with acute lymphoblastic leukaemia. Br J Clin Pharmacol. 2008;66:826–837. doi: 10.1111/j.1365-2125.2008.03281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sandström M, Karlsson MO, Ljungman P, Hassan Z, Jonsson EN, Nilsson C, Ringden O, Oberg G, Bekassy A, Hassan M. Population pharmacokinetic analysis resulting in a tool for dose individualization of busulphan in bone marrow transplantation recipients. Bone Marrow Transplant. 2001;28:657–664. doi: 10.1038/sj.bmt.1703229. [DOI] [PubMed] [Google Scholar]
- 60.Cremers S, Schoemaker R, Bredius R, Den Hartigh J, Ball L, Twiss I, Vermeij P, Vossen J. Pharmacokinetics of intravenous busulfan in children prior to stem cell transplantation. Br J Clin Pharmacol. 2002;53:386–389. doi: 10.1046/j.1365-2125.2002.01555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCune JS, Salinger DH, Vicini P, Oglesby C, Blough DK, Park JR. Population pharmacokinetics of cyclophosphamide and metabolites in children with neuroblastoma: a report from the Children's Oncology Group. J Clin Pharmacol. 2009;49:88–102. doi: 10.1177/0091270008325928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thompson PA, Rosner GL, Matthay KK, Moore TB, Bomgaars LR, Ellis KJ, Renbarger J, Berg SL. Impact of body composition on pharmacokinetics of doxorubicin in children: a Glaser Pediatric Research Network study. Cancer Chemother Pharmacol. 2009;64:243–251. doi: 10.1007/s00280-008-0854-z. [DOI] [PubMed] [Google Scholar]
- 63.Kimura T, Kashiwase S, Makimoto A, Kumagai M, Taga T, Ishida Y, Ida K, Nagatoshi Y, Mugishima H, Kaneko M, Barrett JS. Pharmacokinetic and pharmacodynamic investigation of irinotecan hydrochloride in pediatric patients with recurrent or progressive solid tumors. Int J Clin Pharmacol Ther. 2010;48:327–334. doi: 10.5414/cpp48327. [DOI] [PubMed] [Google Scholar]
- 64.Johnson L, Orchard PJ, Baker KS, Brundage R, Cao Q, Wang X, Langer E, Farag-El Maasah S, Ross JA, Remmel R, Jacobson PA. Glutathione S-transferase A1 genetic variants reduce busulfan clearance in children undergoing hematopoietic cell transplantation. J Clin Pharmacol. 2008;48:1052–1062. doi: 10.1177/0091270008321940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ribbing J, Jonsson EN. Power, selection bias and predictive performance of the Population Pharmacokinetic Covariate Model. J Pharmacokinet Pharmacodyn. 2004;31:109–134. doi: 10.1023/b:jopa.0000034404.86036.72. [DOI] [PubMed] [Google Scholar]
- 66.Karlsson MO, Sheiner LB. The importance of modeling interoccasion variability in population pharmacokinetic analyses. J Pharmacokinet Pharmacodyn. 1993;21:735–750. doi: 10.1007/BF01113502. [DOI] [PubMed] [Google Scholar]
- 67.Panetta JC, Sparreboom A, Pui C-H, Relling MV, Evans WE. Modeling mechanisms of in vivo variability in methotrexate accumulation and folate pathway inhibition in acute lymphoblastic leukemia cells. PLoS Comput Biol. 2010;6:1–13. doi: 10.1371/journal.pcbi.1001019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li J, Gwilt P. The effect of malignant effusions on methotrexate disposition. Cancer Chemother Pharmacol. 2002;50:373–382. doi: 10.1007/s00280-002-0512-9. [DOI] [PubMed] [Google Scholar]
- 69.Berg SL, Blaney SM, Adamson PC, O'Brien M, Poplack DG, Arndt C, Blatt J, Balis FM. Phase I trial and pharmacokinetic study of pyrazoloacridine in children and young adults with refractory cancers. J Clin Oncol. 1998;16:181–186. doi: 10.1200/JCO.1998.16.1.181. [DOI] [PubMed] [Google Scholar]
- 70.Sonnichsen DS, Hurwitz CA, Pratt CB, Shuster JJ, Relling MV. Saturable pharmacokinetics and paclitaxel pharmacodynamics in children with solid tumors. J Clin Oncol. 1994;12:532–538. doi: 10.1200/JCO.1994.12.3.532. [DOI] [PubMed] [Google Scholar]
- 71.Zamboni WC, D'Argenio DZ, Stewart CF, MacVittie T, Delauter BJ, Farese AM, Potter DM, Kubat NM, Tubergen D, Egorin MJ. Pharmacodynamic model of topotecan-induced time course of neutropenia. Clin Cancer Res. 2001;7:2301–2308. [PubMed] [Google Scholar]
- 72.Friberg LE, Henningsson A, Maas H, Nguyen L, Karlsson MO. Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J Clin Oncol. 2002;20:4713–4721. doi: 10.1200/JCO.2002.02.140. [DOI] [PubMed] [Google Scholar]
- 73.Panetta JC, Schaiquevich P, Santana VM, Stewart CF. Using pharmacokinetic and pharmacodynamic modeling and simulation to evaluate importance of schedule in topotecan therapy for pediatric neuroblastoma. Clin Cancer Res. 2008;14:318–325. doi: 10.1158/1078-0432.CCR-07-1243. [DOI] [PubMed] [Google Scholar]
- 74.White-Koning M, Civade E, Geoerger B, Thomas F, Le Deley M-C, Hennebelle I, Delord J-P, Chatelut E, Vassal G. Population analysis of erlotinib in adults and children reveals pharmacokinetic characteristics as the main factor explaining tolerance particularities in children. Clin Cancer Res. 2011;17:4862–4871. doi: 10.1158/1078-0432.CCR-10-3278. [DOI] [PubMed] [Google Scholar]
- 75.Buitenkamp TD, Mathôt RAA, de Haas V, Pieters R, Zwaan CM. Methotrexate-induced side effects are not due to differences in pharmacokinetics in children with Down syndrome and acute lymphoblastic leukemia. Haematologica. 2010;95:1106–1113. doi: 10.3324/haematol.2009.019778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aquerreta I, Aldaz A, Giráldez J, Sierrasesúmaga L. Pharmacodynamics of high-dose methotrexate in pediatric patients. Ann Pharmacother. 2002;36:1344–1350. doi: 10.1345/aph.1A446. [DOI] [PubMed] [Google Scholar]
- 77.van Hasselt JGC, Schellens JHM, Mac Gillavry MR, Beijnen JH, Huitema ADR. Model-based evaluation and optimization of cardiac monitoring protocols for adjuvant treatment of breast cancer with trastuzumab. Pharm Res. 2012;29:3499–3511. doi: 10.1007/s11095-012-0845-y. [DOI] [PubMed] [Google Scholar]
- 78.van Hasselt JGC, Boekhout AH, Beijnen JH, Schellens JHM, Huitema ADR. Population pharmacokinetic-pharmacodynamic analysis of trastuzumab-associated cardiotoxicity. Clin Pharmacol Ther. 2011;90:126–132. doi: 10.1038/clpt.2011.74. [DOI] [PubMed] [Google Scholar]
- 79.Jönsson P, Skärby T, Heldrup J, Schrøder H, Höglund P. High dose methotrexate treatment in children with acute lymphoblastic leukaemia may be optimised by a weight-based dose calculation. Pediatr Blood Cancer. 2011;57:41–46. doi: 10.1002/pbc.22999. [DOI] [PubMed] [Google Scholar]
- 80.Martelli N, Mathieu O, Margueritte G, Bozonnat MC, Daurès J-P, Bressolle F, Hillaire-Buys D, Peyrière H. Methotrexate pharmacokinetics in childhood acute lymphoblastic leukaemia: a prognostic value? J Clin Pharm Ther. 2011;36:237–245. doi: 10.1111/j.1365-2710.2010.01179.x. [DOI] [PubMed] [Google Scholar]
- 81.Barrett JS, Mondick JT, Narayan M, Vijayakumar K, Vijayakumar S. Integration of modeling and simulation into hospital-based decision support systems guiding pediatric pharmacotherapy. BMC Med Inform Decis Mak. 2008;8:6. doi: 10.1186/1472-6947-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Edwards AYZ, Skolnik JM, Dombrowsky E, Patel D, Barrett JS. Modeling and simulation approaches to evaluate pharmacokinetic sampling contamination from central venous catheters in pediatric pharmacokinetic studies of actinomycin-D: a report from the children's oncology group. Cancer Chemother Pharmacol. 2012;70:83–94. doi: 10.1007/s00280-012-1878-y. Available at http://www.ncbi.nlm.nih.gov/pubmed/22623208 (last accessed 1 October 2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hempel G, Müller H-J, Lanvers-Kaminsky C, Würthwein G, Hoppe A, Boos J. A population pharmacokinetic model for pegylated-asparaginase in children. Br J Haematol. 2010;148:119–125. doi: 10.1111/j.1365-2141.2009.07923.x. [DOI] [PubMed] [Google Scholar]
- 84.Avramis VI, Spence SA. Clinical pharmacology of asparaginases in the United States: asparaginase population pharmacokinetic and pharmacodynamic (PK-PD) models (NONMEM) in adult and pediatric ALL patients. J Pediatr Hematol Oncol. 2007;29:239–247. doi: 10.1097/MPH.0b013e318047b79d. [DOI] [PubMed] [Google Scholar]
- 85.Avramis VI, Sencer S, Periclou AP, Sather H, Bostrom BC, Cohen LJ, Ettinger AG, Ettinger LJ, Franklin J, Gaynon PS, Hilden JM, Lange B, Majlessipour F, Mathew P, Needle M, Neglia J, Reaman G, Holcenberg JS, Stork L. A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a Children's Cancer Group study. Blood. 2002;99:1986–1994. doi: 10.1182/blood.v99.6.1986. [DOI] [PubMed] [Google Scholar]
- 86.Borghorst S, Pieters R, Kuehnel H-J, Boos J, Hempel G. Population pharmacokinetics of native Escherichia coli asparaginase. Pediatr Hematol Oncol. 2012;29:154–165. doi: 10.3109/08880018.2011.627978. [DOI] [PubMed] [Google Scholar]
- 87.Nguyen L, Fuller D, Lennon S, Leger F, Puozzo CIV. busulfan in pediatrics: a novel dosing to improve safety/efficacy for hematopoietic progenitor cell transplantation recipients. Bone Marrow Transplant. 2004;33:979–987. doi: 10.1038/sj.bmt.1704446. [DOI] [PubMed] [Google Scholar]
- 88.Schiltmeyer B, Klingebiel T, Schwab M, Mürdter TE, Ritter CA, Jenke A, Ehninger G, Gruhn B, Würthwein G, Boos J, Hempel G. Population pharmacokinetics of oral busulfan in children. Cancer Chemother Pharmacol. 2003;52:209–216. doi: 10.1007/s00280-003-0631-y. [DOI] [PubMed] [Google Scholar]
- 89.Hassan M, Fasth A, Gerritsen B, Haraldsson A, Syrůcková Z, van den Berg H, Sandström M, Karlsson M, Kumlien S, Vossen J. Busulphan kinetics and limited sampling model in children with leukemia and inherited disorders. Bone Marrow Transplant. 1996;18:843–850. [PubMed] [Google Scholar]
- 90.Zwaveling J, Press RR, Bredius RGM, van Derstraaten TRJHM, den Hartigh J, Bartelink IH, Boelens JJ, Guchelaar H-J. Glutathione S-transferase polymorphisms are not associated with population pharmacokinetic parameters of busulfan in pediatric patients. Ther Drug Monit. 2008;30:504–510. doi: 10.1097/FTD.0b013e3181817428. [DOI] [PubMed] [Google Scholar]
- 91.Nakamura H, Sato T, Okada K, Miura G, Ariyoshi N, Nakazawa K, Kitada M. Population pharmacokinetics of oral busulfan in young Japanese children before hematopoietic stem cell transplantation. Ther Drug Monit. 2008;30:75–83. doi: 10.1097/FTD.0b013e3181621cde. [DOI] [PubMed] [Google Scholar]
- 92.Bertholle-Bonnet V, Bleyzac N, Galambrun C, Mialou V, Bertrand Y, Souillet G, Aulagner G. Influence of underlying disease on busulfan disposition in pediatric bone marrow transplant recipients: a nonparametric population pharmacokinetic study. Ther Drug Monit. 2007;29:177–184. doi: 10.1097/FTD.0b013e318039b478. [DOI] [PubMed] [Google Scholar]
- 93.Paci A, Vassal G, Moshous D, Dalle J-H, Bleyzac N, Neven B, Galambrun C, Kemmel V, Abdi ZD, Broutin S, Pétain A, Nguyen L. Pharmacokinetic behavior and appraisal of intravenous busulfan dosing in infants and older children: the results of a population pharmacokinetic study from a large pediatric cohort undergoing hematopoietic stem-cell transplantation. Ther Drug Monit. 2012;34:198–208. doi: 10.1097/FTD.0b013e31824c2f60. [DOI] [PubMed] [Google Scholar]
- 94.Bartelink IH, Boelens JJ, Bredius RGM, Egberts ACG, Wang C, Bierings MB, Shaw PJ, Nath CE, Hempel G, Zwaveling J, Danhof M, Knibbe CAJ. Body weight-dependent pharmacokinetics of busulfan in paediatric haematopoietic stem cell transplantation patients: towards individualized dosing. Clin Pharmacokinet. 2012;51:331–345. doi: 10.2165/11598180-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 95.Bartelink IH, van Kesteren C, Boelens JJ, Egberts TCG, Bierings MB, Cuvelier GDE, Wynn RF, Slatter MA, Chiesa R, Danhof M, Knibbe CAJ. Predictive performance of a busulfan pharmacokinetic model in children and young adults. Ther Drug Monit. 2012;34:574–583. doi: 10.1097/FTD.0b013e31826051bb. [DOI] [PubMed] [Google Scholar]
- 96.Veal GJ, Nguyen L, Paci A, Riggi M, Amiel M, Valteau-Couanet D, Brock P, Ladenstein R, Vassal G. Busulfan pharmacokinetics following intravenous and oral dosing regimens in children receiving high-dose myeloablative chemotherapy for high-risk neuroblastoma as part of the HR-NBL-1/SIOPEN trial. Eur J Cancer. 2012;48:3063–3072. doi: 10.1016/j.ejca.2012.05.020. Available at http://www.ncbi.nlm.nih.gov/pubmed/22742881 (last accessed 1 October 2012) [DOI] [PubMed] [Google Scholar]
- 97.Lindauer A, Eickhoff C, Kloft C, Jaehde U. Population pharmacokinetics of high-dose carboplatin in children and adults. Ther Drug Monit. 2010;32:159–168. doi: 10.1097/FTD.0b013e3181cc6e21. [DOI] [PubMed] [Google Scholar]
- 98.Urien S, Doz F, Namouni F, Bastian G. Pharmacokinetic modelling of total and unbound plasma carboplatin–a population study in 75 children. Int J Clin Pharmacol Ther. 2002;40:270–278. doi: 10.5414/cpp40270. [DOI] [PubMed] [Google Scholar]
- 99.Chatelut E, Boddy AV, Peng B, Rubie H, Lavit M, Dezeuze A, Pearson AD, Roché H, Robert A, Newell DR, Canal P. Population pharmacokinetics of carboplatin in children. Clin Pharmacol Ther. 1996;59:436–443. doi: 10.1016/S0009-9236(96)90113-7. [DOI] [PubMed] [Google Scholar]
- 100.Chinnaswamy G, Errington J, Foot A, Boddy AV, Veal GJ, Cole M. Pharmacokinetics of cyclophosphamide and its metabolites in paediatric patients receiving high-dose myeloablative therapy. Eur J Cancer. 2011;47:1556–1563. doi: 10.1016/j.ejca.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 101.Periclou AP, Avramis VI. NONMEM population pharmacokinetic studies of cytosine arabinoside after high-dose and after loading bolus followed by continuous infusion of the drug in pediatric patients with leukemias. Cancer Chemother Pharmacol. 1996;39:42–50. doi: 10.1007/s002800050536. [DOI] [PubMed] [Google Scholar]
- 102.Hempel G, Reinhardt D, Creutzig U, Boos J. Population pharmacokinetics of liposomal daunorubicin in children. Br J Clin Pharmacol. 2003;56:370–377. doi: 10.1046/j.1365-2125.2003.01886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hempel G, Relling MV, de Rossi G, Stary J, de Lorenzo P, Valsecchi MG, Barisone E, Boos J, Pieters R. Pharmacokinetics of daunorubicin and daunorubicinol in infants with leukemia treated in the interfant 99 protocol. Pediatr Blood Cancer. 2010;54:355–360. doi: 10.1002/pbc.22266. [DOI] [PubMed] [Google Scholar]
- 104.Urien S, Doz F, Giraud C, Rey E, Gentet J-C, Chastagner P, Vassal G, Corradini N, Auvrignon A, Leblond P, Rubie H, Treluyer J-M. Developmental pharmacokinetics of etoposide in 67 children: lack of dexamethasone effect. Cancer Chemother Pharmacol. 2011;67:597–603. doi: 10.1007/s00280-010-1357-2. [DOI] [PubMed] [Google Scholar]
- 105.Würthwein G, Klingebiel T, Krümpelmann S, Metz M, Schwenker K, Kranz K, Lanvers C, Boos J. Population pharmacokinetics of high-dose etoposide in children receiving different conditioning regimens. Anticancer Drugs. 2002;13:101–110. doi: 10.1097/00001813-200201000-00012. [DOI] [PubMed] [Google Scholar]
- 106.Würthwein G, Boos J. Simulation tool for schedule-dependent etoposide exposure based on pharmacokinetic findings published in the literature. Anticancer Drugs. 2001;12:151–158. doi: 10.1097/00001813-200102000-00009. [DOI] [PubMed] [Google Scholar]
- 107.Baheti G, McGuire TR, Davda JP, Manouilov KK, Wall D, Gwilt PR, Gordon BB. Clinical pharmacology of etoposide in children undergoing autologous stem cell transplantation for various solid tumours. Xenobiotica. 2012 doi: 10.3109/00498254.2012.713530. DOI: 10.3109/00498254.2012.713530. [DOI] [PubMed] [Google Scholar]
- 108.Kerbusch T, de Kraker J, Mathjt RA, Beijnen JH. Population pharmacokinetics of ifosfamide and its dechloroethylated and hydroxylated metabolites in children with malignant disease: a sparse sampling approach. Clin Pharmacokinet. 2001;40:615–625. doi: 10.2165/00003088-200140080-00005. [DOI] [PubMed] [Google Scholar]
- 109.Menon-Andersen D, Mondick JT, Jayaraman B, Thompson PA, Blaney SM, Bernstein M, Bond M, Champagne M, Fossler MJ, Barrett JS. Population pharmacokinetics of imatinib mesylate and its metabolite in children and young adults. Cancer Chemother Pharmacol. 2009;63:229–238. doi: 10.1007/s00280-008-0730-x. [DOI] [PubMed] [Google Scholar]
- 110.Thompson PA, Gupta M, Rosner GL, Yu A, Barrett J, Bomgaars L, Bernstein ML, Blaney SM, Mondick J. Pharmacokinetics of irinotecan and its metabolites in pediatric cancer patients: a report from the children's oncology group. Cancer Chemother Pharmacol. 2008;62:1027–1037. doi: 10.1007/s00280-008-0692-z. [DOI] [PubMed] [Google Scholar]
- 111.Veal GJ, Cole M, Errington J, Pearson ADJ, Foot ABM, Whyman G, Boddy AV. Pharmacokinetics and metabolism of 13-cis-retinoic acid (isotretinoin) in children with high-risk neuroblastoma – a study of the United Kingdom Children's Cancer Study Group. Br J Cancer. 2007;96:424–431. doi: 10.1038/sj.bjc.6603554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nath CE, Shaw PJ, Montgomery K, Earl JW. Population pharmacokinetics of melphalan in paediatric blood or marrow transplant recipients. Br J Clin Pharmacol. 2007;64:151–164. doi: 10.1111/j.1365-2125.2007.02862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Faganel Kotnik B, Grabnar I, Bohanec Grabar P, Dolžan V, Jazbec J. Association of genetic polymorphism in the folate metabolic pathway with methotrexate pharmacokinetics and toxicity in childhood acute lymphoblastic leukaemia and malignant lymphoma. Eur J Clin Pharmacol. 2011;67:993–1006. doi: 10.1007/s00228-011-1046-z. [DOI] [PubMed] [Google Scholar]
- 114.Zhang C, Zhai S, Yang L, Wu H, Zhang J, Ke X. Population pharmacokinetic study of methotrexate in children with acute lymphoblastic leukemia. Int J Clin Pharmacol Ther. 2010;48:11–21. doi: 10.5414/cpp48011. [DOI] [PubMed] [Google Scholar]
- 115.Colom H, Farré R, Soy D, Peraire C, Cendros J-M, Pardo N, Torrent M, Domenech J, Mangues M-A. Population pharmacokinetics of high-dose methotrexate after intravenous administration in pediatric patients with osteosarcoma. Ther Drug Monit. 2009;31:76–85. doi: 10.1097/FTD.0b013e3181945624. [DOI] [PubMed] [Google Scholar]
- 116.Plard C, Piard C, Bressolle F, Fakhoury M, Zhang D, Yacouben K, Rieutord A, Jacqz-Aigrain E. A limited sampling strategy to estimate individual pharmacokinetic parameters of methotrexate in children with acute lymphoblastic leukemia. Cancer Chemother Pharmacol. 2007;60:609–620. doi: 10.1007/s00280-006-0394-3. [DOI] [PubMed] [Google Scholar]
- 117.Aumente D, Buelga DS, Lukas JC, Gomez P, Torres A, García MJ. Population pharmacokinetics of high-dose methotrexate in children with acute lymphoblastic leukaemia. Clin Pharmacokinet. 2006;45:1227–1238. doi: 10.2165/00003088-200645120-00007. [DOI] [PubMed] [Google Scholar]
- 118.Odoul F, Le Guellec C, Lamagnère JP, Breilh D, Saux MC, Paintaud G, Autret-Leca E. Prediction of methotrexate elimination after high dose infusion in children with acute lymphoblastic leukaemia using a population pharmacokinetic approach. Fundam Clin Pharmacol. 1999;13:595–604. doi: 10.1111/j.1472-8206.1999.tb00366.x. [DOI] [PubMed] [Google Scholar]
- 119.Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, Packer RJ, Goldman S, Gururangan S, Gajjar A, Demuth T, Kun LE, Boyett JM, Gilbertson RJ. Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol. 2011;29:3529–3534. doi: 10.1200/JCO.2011.35.7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Neville K, Blaney S, Bernstein M, Thompson P, Adams D, Aleksic A, Berg S. Pharmacokinetics of O(6)-benzylguanine in pediatric patients with central nervous system tumors: a pediatric oncology group study. Clin Cancer Res. 2004;10:5072–5075. doi: 10.1158/1078-0432.CCR-03-0123. [DOI] [PubMed] [Google Scholar]
- 121.Thompson P, Balis F, Serabe BM, Berg S, Adamson P, Klenke R, Aiken A, Packer R, Murry DJ, Jakacki R, Blaney SM. Pharmacokinetics of phenylacetate administered as a 30-min infusion in children with refractory cancer. Cancer Chemother Pharmacol. 2003;52:417–423. doi: 10.1007/s00280-003-0674-0. [DOI] [PubMed] [Google Scholar]
- 122.Petersen KB, Jusko WJ, Rasmussen M, Schmiegelow K. Population pharmacokinetics of prednisolone in children with acute lymphoblastic leukemia. Cancer Chemother Pharmacol. 2003;51:465–473. doi: 10.1007/s00280-003-0602-3. [DOI] [PubMed] [Google Scholar]
- 123.Panetta JC, Kirstein MN, Gajjar A, Nair G, Fouladi M, Heideman RL, Wilkinson M, Stewart CF. Population pharmacokinetics of temozolomide and metabolites in infants and children with primary central nervous system tumors. Cancer Chemother Pharmacol. 2003;52:435–441. doi: 10.1007/s00280-003-0670-4. [DOI] [PubMed] [Google Scholar]
- 124.Kirstein MN, Panetta JC, Gajjar A, Nair G, Iacono LC, Freeman BB, Stewart CF. Development of a pharmacokinetic limited sampling model for temozolomide and its active metabolite MTIC. Cancer Chemother Pharmacol. 2005;55:433–438. doi: 10.1007/s00280-004-0896-9. [DOI] [PubMed] [Google Scholar]
- 125.Kitchen BJ, Balis FM, Poplack DG, O'Brien M, Craig CE, Adamson PC. A pediatric phase I trial and pharmacokinetic study of thioguanine administered by continuous i.v. infusion. Clin Cancer Res. 1997;3:713–717. [PubMed] [Google Scholar]
- 126.Schaiquevich P, Panetta JC, Iacono LC, Freeman BB, Santana VM, Gajjar A, Stewart CF. Population pharmacokinetic analysis of topotecan in pediatric cancer patients. Clin Cancer Res. 2007;13(22 Pt 1):6703–6711. doi: 10.1158/1078-0432.CCR-07-1376. [DOI] [PubMed] [Google Scholar]
- 127.Freeman BB, Iacono LC, Panetta JC, Gajjar A, Stewart CF. Using plasma topotecan pharmacokinetics to estimate topotecan exposure in cerebrospinal fluid of children with medulloblastoma. Neuro-oncol. 2006;8:89–95. doi: 10.1215/15228517-2005-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Baker SD, Heideman RL, Crom WR, Kuttesch JF, Gajjar A, Stewart CF. Cerebrospinal fluid pharmacokinetics and penetration of continuous infusion topotecan in children with central nervous system tumors. Cancer Chemother Pharmacol. 1996;37:195–202. doi: 10.1007/BF00688317. [DOI] [PubMed] [Google Scholar]
- 129.Guilhaumou R, Simon N, Quaranta S, Verschuur A, Lacarelle B, Andre N, Solas C. Population pharmacokinetics and pharmacogenetics of vincristine in paediatric patients treated for solid tumour diseases. Cancer Chemother Pharmacol. 2011;68:1191–1198. doi: 10.1007/s00280-010-1541-4. [DOI] [PubMed] [Google Scholar]
- 130.Groninger E, Meeuwsen-de Boar T, Koopmans P, Uges D, Sluiter W, Veerman A, Kamps W, De Graaf S. Pharmacokinetics of vincristine monotherapy in childhood acute lymphoblastic leukemia. Pediatr Res. 2002;52:113–118. doi: 10.1203/00006450-200207000-00021. [DOI] [PubMed] [Google Scholar]
- 131.de Graaf SS, Bloemhof H, Vendrig DE, Uges DR. Vincristine disposition in children with acute lymphoblastic leukemia. Med Pediatr Oncol. 1995;24:235–240. doi: 10.1002/mpo.2950240405. [DOI] [PubMed] [Google Scholar]