

Abstract
Aims
In children with acute lymphoblastic leukaemia (ALL) bone marrow activity can influence red blood cell (RBC) kinetics, the surrogate tissue for thiopurine methyltransferase (TPMT) measurements. The aim of this study was to investigate TPMT phenotype–genotype concordance in ALL, and the influence of TPMT on thiopurine metabolite formation.
Methods
We measured TPMT (activity, as units ml−1 packed RBCs and genotype) at diagnosis (n = 1150) and TPMT and thioguanine nucleotide (TGN) and methylmercaptopurine nucleotide (MeMPN) metabolites (pmol/8 × 108 RBCs) during chemotherapy (n = 1131) in children randomized to thioguanine or mercaptopurine on the United Kingdom trial ALL97.
Results
Median TPMT activity at diagnosis (8.5 units) was significantly lower than during chemotherapy (13.8 units, median difference 5.1 units, 95% confidence interval (CI) 4.8, 5.4, P < 0.0001). At diagnosis genotype–phenotype was discordant. During chemotherapy the overall concordance was 92%, but this fell to 55% in the intermediate activity cohort (45% had wild-type genotypes). For both thiopurines TGN concentrations differed by TPMT status. For mercaptopurine, median TGNs were higher in TPMT heterozygous genotype (754 pmol) than wild-type (360 pmol) patients (median difference 406 pmol, 95% CI 332, 478, P < 0.0001), whilst median MeMPNs, products of the TPMT reaction, were higher in wild-type (10 650 pmol) than heterozygous patients (3868 pmol) (P < 0.0001). In TPMT intermediate activity patients with a wild-type genotype, TGN (median 366 pmol) and MeMPN (median 8590 pmol) concentrations were similar to those in wild-type, high activity patients.
Conclusions
In childhood ALL, TPMT activity should not be used to predict heterozygosity particularly in blood samples obtained at disease diagnosis. Genotype is a better predictor of TGN accumulation during chemotherapy.
Keywords: childhood leukaemia, genotype–phenotype discordance, mercaptopurine, thioguanine, thioguanine nucleotides, thiopurine methyltransferase
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
In healthy children and adults thiopurine methyltransferase (TPMT) activities have a trimodal frequency distribution. In adult populations the concordance between TPMT genotype and phenotype is over 98%.
In childhood acute lymphoblastic leukaemia (ALL) the disease process and subsequent chemotherapy can influence TPMT activity measurements.
WHAT THIS STUDY ADDS
In childhood ALL, at disease diagnosis, the extent of the reduction in measured TPMT activity and the resulting TPMT genotype–phenotype discordance, previously reported in small patient cohorts with low numbers of variant alleles, has been confirmed in a large population of children with ALL. TPMT activity should not be used to predict heterozygosity.
During thiopurine chemotherapy, TPMT discordance is 45% in children with intermediate activity. TPMT genotype more accurately predicts mercaptopurine active metabolite accumulation and therefore should be used in preference to phenotyping for dosage recommendations.
Introduction
Mercaptopurine forms the core of long term maintenance chemotherapy in childhood acute lymphoblastic leukaemia (ALL) whilst, conventionally, thioguanine (INN, tioguanine) is used in ALL intensive treatment blocks and in the myeloid leukaemias 1, 2. The United Kingdom Medical Research Council (MRC) national trial ALL97 compared the efficiacy and toxicity of both thiopurines during the maintenance phases of chemotherapy.
Both drugs are directly inactivated by the polymorphic enzyme thiopurine methyltransferase (TPMT) 3 and both form cytotoxic thioguanine nucleotides (TGNs), metabolites linked to both treatment efficacy and toxicity 4, 5 However, mercaptopurine forms intermediate nucleotide metabolites prior to the TGNs (Figure 1). These mercaptopurine nucleotides are substrates for TPMT, and the resulting methylmercaptopurine nucleotides (MeMPNs) are formed at the expense of TGNs. Incorporation of drug derived TGN metabolites into DNA triggers cytotoxicity, a primary mode of action for thiopurine drugs 6, 7. The TGNs can also promote cytotoxicity by inhibiting DNA methylation 8 and induce apoptotic cell death by inhibition of intracellular signalling pathways 9–11.
Figure 1.
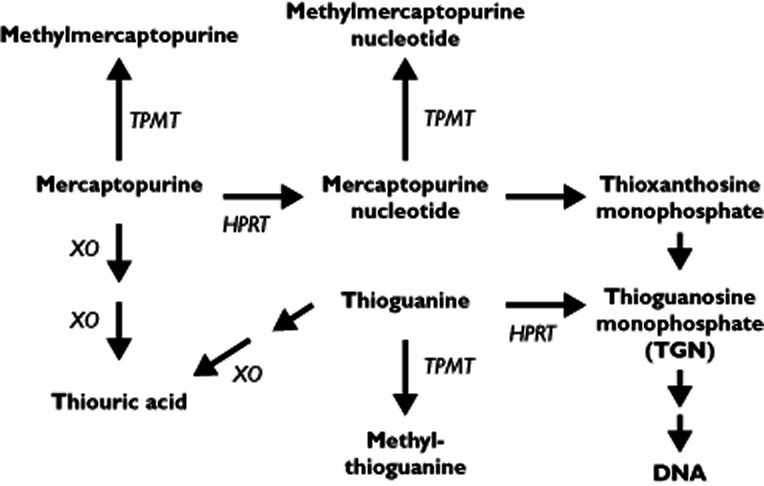
Thiopurine metabolism. Both mercaptopurine and thioguanine are methylated by thiopurine methyltransferase (TPMT), as is mercaptopurine nucleotide (thioinosine monophosphate). Nucleotide formation is catalyzed by hypoxanthine phosphoribosyltransferase (HPRT). Oxidation is catalyzed by xanthine oxidase (XO). Thioguanine requires deamination by guanase before catabolic oxidation. The thioguanine nucleotides (TGNs) are the mono-, di- and tri- phosphates of thioguanosine
Life-threatening bone marrow toxicity, due to excess production of TGN metabolites, occurs in the 0.3% of patients who are TPMT deficient 5, 12–14. Less severe myelosuppression can develop in TPMT heterozygotes, approximately 11% of patients, when taking standard doses of mercaptopurine 15, 16. Most protocols incorporate routine assessment of TPMT status at the start of thiopurine treatment by phenotype 17 or genotype 18. The phenotype concords with genotype in >98% of healthy adults 19. Screening for the TPMT*3 family of variant alleles (>92% of low activity alleles) and the TPMT*2 variant will detect >95% of TPMT deficient alleles 19, 20.
The clinical results of the ALL97 thiopurine randomization have been previously reported and showed no difference in efficacy between the two thiopurines. A significant reduction in central nervous system relapse with thioguanine was balanced by increased risks of death in remission and vascular liver disease 21. The latter toxicity was associated with TPMT heterozygosity 22. In this paper we report on the thiopurine metabolism studies within ALL97. The aim of these studies was to investigate the phenotype–genotype TPMT concordance in children with ALL and the influence of TPMT status on the inter- and intra-patient variability in thiopurine metabolism. Analyses of the association between thiopurine metabolism and clinical efficacy are on-going and will be reported separately.
Methods
Patients and chemotherapy
MRC ALL97 (registration number ISRCTN26727615) compared the efficacy and toxicity of dexamethasone and thioguanine (experimental arms) with prednisone and mercaptopurine (standard approach). Treatment centres in the UK and Ireland recruited patients aged 1 to 18 years with ALL diagnosed between January 1997 and June 2002. Treatment centres obtained local ethics committee approval and informed consent from patients and/or parents before entering children into the trial. The trial had an add-on pharmacogenetic and drug metabolism study to investigate inter- and intra-patient variability in response to oral thiopurines.
ALL97 underwent several modifications, details of which have been previously reported 21, 22. The steroid and thiopurine randomizations were retained and maintenance chemotherapy, upon which this study is based, remained the same. During maintenance patients received daily oral randomized thiopurine, weekly methotrexate and monthly intravenous vincristine followed by 5 days of randomized steroid. The thiopurine dose was titrated to toxicity from a standard protocol dose (thioguanine 40 mg m−2 and mercaptopurine 75 mg m−2, 100% protocol dose). The dosage titrations have been previously described 22.
The trial showed a survival benefit with dexamethasone 23 but an excess of thioguanine-related veno-occlusive disease 21, 22. At closure it was recommended that children randomized to thioguanine, who had not finished maintenance chemotherapy, should be transferred to the mercaptopurine arm.
Thiopurine studies
TPMT phenotype and genotype were measured in a diagnostic lithium heparin blood sample (5 ml). During thiopurine maintenance chemotherapy, TPMT and thiopurine metabolite concentrations were measured in blood samples taken immediately before a monthly vincristine injection. Samples were requested after at least 7 days at the standard protocol dose of thiopurine, or the maximum tolerated dose, but not within 2 months of a red cell transfusion. The first sample was requested at the earliest convenient point when the criteria above were met. A second sample was requested to be taken at the start of year 2 and a third sample at the end of year 2. If the patient's treatment had been reduced or interrupted at any time point, the sample was taken on recovery of cell counts during the next cycle of thiopurine treatment as per the criteria stated above.
Thiopurine assays
Thiopurine metabolite concentrations and TPMT activities were measured by high performance liquid chromatography 22, 24, 25. Metabolite assays are stated in the text in pmol which represent pmol/8 × 108 red blood cells (RBCs) and TPMT activities are stated in units which represent units ml−1 packed RBCs 24. The lower limit of detection and quantitation for TGN metabolites were 6 pmol and 30 pmol (coefficient of variation, CV, 8.2%), respectively and for MeMPNs 15 pmol and 60 pmol (CV 9.8%), respectively. The interassay coefficients of variation, for 25 assays over a 12 month period, for the thiopurine quality control samples at 300, 3000 and 12 000 pmol/8 × 108 RBCs were 4.6%, 3.8% and 5%, respectively with an accuracy of 1.3%, 1.6% and 3.3%. RBC TPMT activity assay 24 was modified to measure directly the methylmercaptopurine reaction product 22 with no interference from methylmercaptopurine nucleotide or nucleoside drug metabolites. The lower limit of detection and quantitation was 6 pmol (0.75 units TPMT, CV 9%) with a control patient quality control CV of 7.8%.
Routinely, blood samples were genotyped for TPMT*3A, TPMT*3B and TPMT*3C, by amplification of exons 7 and 10 as previously described 22. TPMT*3A is an exon 7 and 10 double mutant. Aliquots (10 μl) of exon 7 (460G > A) and exon 10 (719A > G) PCR product were digested with the restriction enzymes Mwo1 for 4 h at 60°C and Acc1 for 4 h at 37°C, respectively, and analyzed on a 4% polyacrylamide gel. Exon 7 (TPMT*3B) gave wild-type fragments of 226 bp and 100 bp. The variant allele, 326 bp, was not digested. Exon 10 (TPMT*3C) gave variant allele fragments of 268 bp and 169 bp. The wild-type, 437 bp, was not digested. TPMT*2 was detected by sequencing exon 5 of the TPMT gene 20. To identify novel sequence variations, the TPMT open reading frame from exon 3 to exon 10 was sequenced using intron based primers 20 on an ABI 3730 capillary sequencer, using dye-primer technology (Applied Biosystems, Warrington, UK).
Stability studies
To investigate the stability of TGN and MeMPN metabolite concentrations in the samples forwarded from UK treatment centres, blood samples arriving in the laboratory were left at room temperature (22°C) over several days. Aliquots of whole blood were taken at day 1 (24 h post-sampling), day 3, day 6 and day 9, and washed resuspended RBCs prepared for metabolite assays. The then established TGN assay used washed RBCs 25. In addition, if the sample volume permitted, an aliquot of whole blood was removed at each time interval. From both the washed RBC and whole blood samples a 200 μl aliquot was removed for RBC counting prior to freezing at −30°C. Stability studies on TPMT have been previously reported 26.
Statistical analysis
The Anderson-Darling test was used to examine the fit of observations to a normal distribution. Differences between groups were compared by the Chi-square statistic, the Kruskal–Wallis one way analysis of variance, Wilcoxon matched pairs test or the Mann–Whitney test. Metabolite and TPMT activity values are stated as median and range. The median differences (and the 95% confidence interval, CI) were calculated from the point estimates of all the median differences. Correlations were assessed using the Spearman-rank correlation coefficient (rs).
Results
Blood samples
At least one sample was obtained from 1565 children (81% of children in the trial) entered into ALL97 (n = 803) and ALL97/99 (n = 762). Not all samples, as requested in the study protocol, were provided for each child. In addition, due to the small sample volume forwarded from some children, TPMT phenotype, genotype and drug metabolites could not be measured in every sample. Table 1 summarizes the samples available for analysis.
Table 1.
Blood samples available for thiopurine analysis
| Total | TG | MP | |
|---|---|---|---|
| Individual children | 1565 | ||
| Individual TPMT genotypes | 1320 (84%) | ||
| Individual children at disease diagnosis | |||
| TPMT activity measured | 1150 | ||
| TPMT genotypes | 950 (83%) | ||
| Individual children during chemotherapy | |||
| TPMT activity measured | 1131 | 428 | 703 |
| TPMT activity and genotype | 1117 (99%) | 423 | 694 |
| TPMT activity and metabolites | 1114 (98%) | 424 | 690 |
| TPMT activity, genotype and metabolites | 1100 (97%) | 419 | 681 |
| TPMT activities measured at diagnosis and during chemotherapy | 755 (67%) | ||
| TPMT genotypes available in the above cohort | 741 (66%) | ||
| Children with multiple samples at the protocol directed time intervals | 378 (33%) | 134 | 244 |
MP, mercaptopurine cohort; TG, thioguanine cohort; TPMT, thiopurine methyltransferase. TPMT activity was used in the analysis if it was measured in blood samples processed within 5 days of sampling. Metabolite values were used in analysis if they were measured in whole blood samples up to 6 days old or, for washed red cells, up to 3 days old. For chemotherapy samples the total number is split between those taking TG or taking MP. Two TPMT deficient children are included in this table, one randomized to TG and one to MP.
A number of children switched from thioguanine to mercaptopurine but only had samples whilst taking mercaptopurine. Samples were taken from 754 children when taking mercaptopurine (667 randomized to mercaptopurine, 87 after switching to mercaptopurine from thioguanine) and 457 when taking thioguanine (456 randomized to thioguanine, one given thioguanine but randomized to mercaptopurine). The first blood sample that met the study criteria was used as the representative sample in the subsequent analysis of thiopurine metabolism. The representative samples for mercaptopurine were taken at a median of 12.7 months after the start of induction therapy (range 1.6–39.6 months). For thioguanine samples the representative sample was at a median of 6.2 months (range 1.9–32.9 months).
Stability studies
TGN and MeMPN metabolites were measured in washed RBCs from 53 children (25 mercaptopurine, 28 thioguanine). Matched comparisons showed no loss of metabolites between the day 1 and day 3 samples. By day 6, thioguanine derived TGNs had decreased significantly compared with day 1 (day 1 median 1892 pmol, day 6 median 1054 pmol; median difference 502 pmol, 95% CI 283, 737, P = 0.002). Similarly, by day 6, mercaptopurine derived TGNs had fallen by 35% compared with day 1 (day 1 median 360 pmol, day 6 median 234 pmol; median difference 153 pmol, 95% CI 90, 251, P < 0.001), and mercaptopurine derived MeMPNs had fallen by 20% (day 1 median 11 084 pmol, day 6 median 8886 pmol, median difference 1704 pmol, 95% CI 1194, 4615, P < 0.001).
Whole blood samples were available from 20 children (15 mercaptopurine, 5 thioguanine), there was no significant difference between metabolite values measured at day 1, 3 or 6. The day 6 analysis showed a non-significant decline of 1.1% for thioguanine derived TGNs and 0.1% for mercaptopurine derived TGNs and 9.3% for MeMPN. Ten children, taking mercaptopurine, had a blood sample available at day 9. TGNs had decreased by 7.2% (day 1 median 335 pmol, day 9 median 311 pmol; median difference 30 pmol, 95% CI −6.5, 60, P = 0.154) and MeMPNs by 30% (day 1 median 12 289 pmol, day 9 median 8723 pmol; median difference 2060, 95% CI 699, 4874, P = 0.053).
Drug metabolites were stable for at least 6 days when measured in whole blood. If the blood sample volume received was low (<2 ml), metabolites were measured in washed RBCs (a preparation also used for the production of TPMT lysates), and samples processed within 3 days of sampling were used in data analysis.
TPMT phenotype
TPMT activities were measured in 1131 children during maintenance chemotherapy. There was no significant difference between the range of TPMT activities measured in children taking thioguanine (0 to 23 units, median 13.8, n = 428) compared with children taking mercaptopurine (0 to 26 units, median 13.8, n = 703). There was no gender difference in TPMT activities. TPMT activities measured in 1150 children at diagnosis (range 0 to 38 units, median 8.5) were significantly lower than those during chemotherapy (median difference 5.1 units, 95% CI 4.8, 5.4, P < 0.0001). TPMT activities were available for 755 children both at diagnosis (median 8.5 units, range 0 to 38) and on thiopurine (median 13.8 units, range 0–26.3). The disparity in TPMT activities remained when restricting the analysis to this matched cohort (median difference 5.3 units, 95% CI 5.0, 5.6, P < 0.0001).
Blood samples were received from four children who were TPMT deficient. Two of these children received randomized thiopurine on the trial protocol and were included in this analysis. Their TPMT activities at diagnosis and during chemotherapy were <0.75 units.
TPMT genotype
TPMT genotype was available for 1320 children, 1149 were white Caucasian and 171 belonged to other ethnic groups (71 Asian, 44 mixed race, 19 Black, 6 Oriental and 31 unknown or non-Caucasian). One hundred and twenty-three children had variant alleles (100 TPMT*1/*3A, 17 TPMT1/*3C, four TPMT*1/*2, one compound heterozygote TPMT*2/*3A and one homozygous TPMT*3A/*3A). The TPMT*3B allele was not detected. The TPMT*3 family allele frequency was 5.2% in white Caucasians (4.5% *3A, 0.7% *3C) and 1.75% in other ethnic groups (0.58% *3A, 1.17% *3C). TPMT*2 was only detected in white Caucasians (allele frequency 0.22%).
Concordance between genotype and phenotypic activity
TPMT genotypes were available for 1117 of the 1131 children with TPMT activities measured during chemotherapy. The break point between the ‘intermediate’ and ‘high’ TPMT activity ranges was examined at 9.5, 10.5 and 11.5 units, values based on the nadir of the TPMT frequency distributions in healthy control adults and children 3, 15 and in children taking chemotherapy 15. At 9.5 units the sensitivity for the detection of the variant allele was 78% (specificity 97%), at 10.5 units the sensitivity was 92% (specificity 92%) and at 11.5 units the sensitivity was 94% (specificity 85%). A 10.5 unit break point was used for data analysis. The overall concordance was 92%, but the specificity of 92% results in 8% of wild-type alleles (79 alleles) in the intermediate activity cohort and a concordance of 55% in this group (i.e. 45% of intermediate activity patients had a wild-type genotype). The concordance in the high activity group (above 10.5 units) was 99% (Figure 2). It should be noted that the intermediate activity cohort contained 16.5% of children rather than the 11% predicted by the TPMT polymorphism 3, 15.
Figure 2.
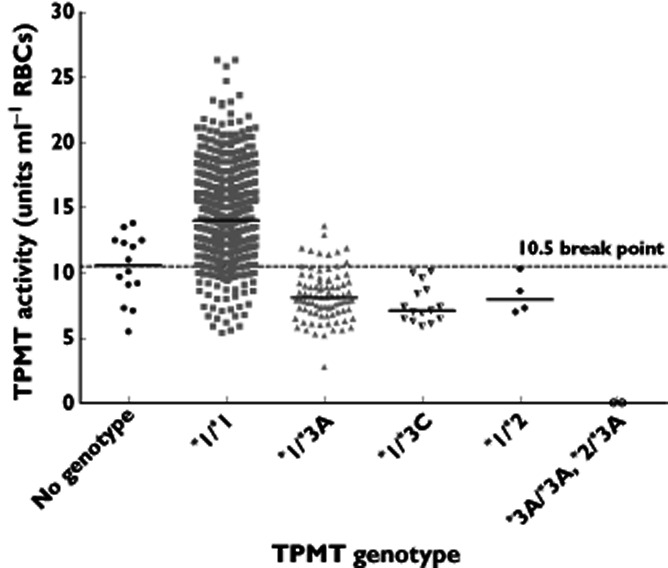
TPMT activities for 1131 children during thiopurine maintenance chemotherapy (median 13.8 units, range 0–26). Thiopurine genotype was available for 1117 children (1009 wild-type, 87 TPMT*1/*3A, 15 TPMT*1/*3C, four TPMT*1/*2 and two TPMT deficient children TPMT*3A/*3A, TPMT*2/*3A). At the 10.5 unit break point (dotted line) the sensitivity for the TPMT*3 variant allele was 92% (specificity for wild-type 93%). Solid line = median TPMT activities
TPMT genotypes were available for 950 of the 1150 children with TPMT activities measured at disease diagnosis. The distribution of TPMT activities was unimodal, with a negative skew, with overlapping activities for heterozygous and wild-type children. Of the 92 variant alleles, 81 were below the median TPMT activity of 8.5 units. At this cut-off the sensitivity for detecting the heterozygous genotype was 88%, but the specificity for wild-type (TPMT*1/*1) was only 53%. Other than for the detection of TPMT deficiency, TPMT activities at diagnosis per se had a poor predictive value with respect to heterozygosity and functional activity during chemotherapy. This is illustrated for the matched group of 755 children in whom 741 genotypes were available (Figure 3).
Figure 3.
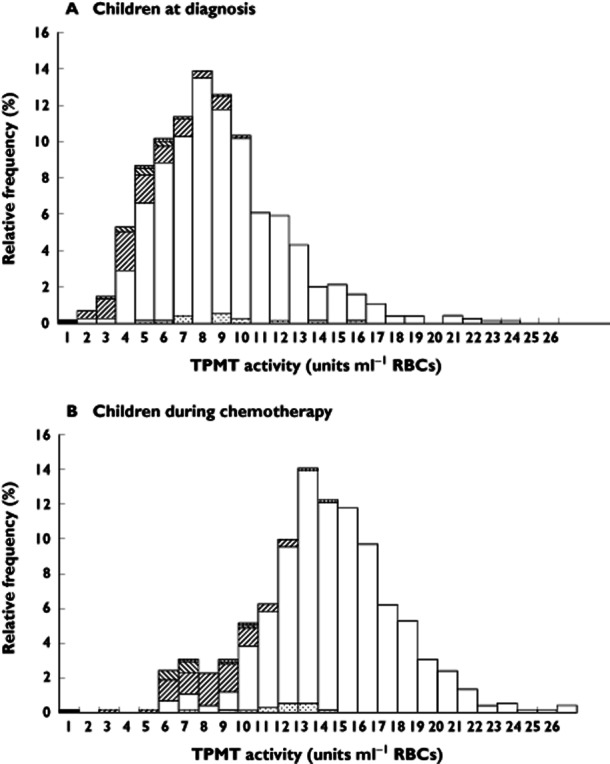
TPMT frequency distribution histograms for 755 children at disease diagnosis (median 8.5 units, range 0–38) and for those same children during thiopurine maintenance chemotherapy (median 13.8 units, range 0–26). The histograms are plotted to 26 units, histogram (A) omits one child who had an at diagnosis TPMT activity of 38 units. Thiopurine genotypes were available for 741 of the 755 children. ▪, TPMT*3A/*3A;  , TPMT*1/*2;
, TPMT*1/*2;
 , TPMT*1/*3C;
, TPMT*1/*3C;  , TPMT*1/*3A; □, TPMT*1/*1;
, TPMT*1/*3A; □, TPMT*1/*1;
 , no genotype
, no genotype
Sequencing non-concordant samples
Sequencing of the TPMT open reading frame in children with TPMT activities <12 units without TPMT*2 or TPMT*3 variant alleles (n = 186; 79 children with a wild-type genotype and ≤10.5 units TPMT i.e. intermediate activity) revealed five children with sequence variations: TPMT*1/*9 (7.4 units, mercaptopurine derived TGNs 502 pmol at 67 mg m−2 mercaptopurine), TPMT*1/*21 (6.8 units, thioguanine derived TGNs 4060 pmol at 41 mg m−2 thioguanine) and three novel variants TPMT*1/*32 (exon 5, 340G > A, Glu114Lys, rs115106679; activity 9.7 units, mercaptopurine derived TGNs 694 pmol at 75 mg m−2 mercaptopurine), TPMT*1/*33 (exon 7, 487C > T, Arg163Cys, rs112339338; activity 8.5 units, mercaptopurine derived TGNs 634 pmol at 54 mg m−2 mercaptopurine), TPMT*1/*34 (exon 5, 244C > T, Arg82Trp, rs111901354; activity 8.0 units, thioguanine derived TGNs 3006 pmol at 44 mg m−2 thioguanine).
TPMT status and thiopurine metabolism
Two TPMT deficient children were randomized for thiopurine, one to thioguanine and one to mercaptopurine. An initial thioguanine dose of 4 mg m−2 produced 3544 pmol TGNs, dose reduction to 2.5 mg m−2 daily produced 2252 pmol TGNs. The mercaptopurine child was treated on 7.5 mg m−2 on alternate days and TGNs were 1670 pmol. The mercaptopurine treated child lacked MeMPN metabolites.
For children with TPMT activity, thioguanine derived TGNs ranged from 36–6874 pmol, median 1940, at a median thioguanine dose of 40 mg m−2 (range 10–78) and mercaptopurine derived TGNs ranged from 0–2228 pmol, median 372, at a median mercaptopurine dose of 75 mg m−2 (range 6–125). Mercaptopurine derived MeMPNs ranged from 0–141 772 pmol, median 9590. There was no difference in drug dose or metabolite concentrations when analyzed by steroid group or gender. TGN concentrations negatively (thioguanine rs = −0.187, P < 0.001; mercaptopurine rs = −0.23, P < 0.001) correlated with TPMT activity. Mercaptopurine derived MeMPNs showed a positive correlation (rs = 0.104, P = 0.006) to TPMT.
Analysis of metabolites by genotype (n = 418 thioguanine, n = 680 mercaptopurine) indicated that, for both thiopurines, TGN concentrations were significantly higher in heterozygotes whilst mercaptopurine derived MeMPNs were significantly lower in the TPMT heterozygous cohort (Table 2).
Table 2.
Thiopurine methyltransferase genotype and metabolite formation
| Wild-type TPMT*1/*1 | Heterozygous TPMT *1/*3, *1/*2 | Median difference (95%CI) | |
|---|---|---|---|
| TG | |||
| Patients | 381 | 37 | |
| Dose (mg m−2) | 40 (10–78) | 40 (18–38) | −1.0 (−3, −0.1), P = 0.009 |
| TG-TGNs (pmol) | 1 904 (36–4 337) | 2 468 (174–6 730) | 478 (180, 772), P = 0.0026 |
| TPMT units | 14 (5.4–22.8) | 8.1 (5.2–12.9) | −5.8 (−6.5, −5), P < 0.0001 |
| MP | |||
| Patients | 613 | 67 | |
| Dose (mg m−2) | 75 (6–125) | 74 (14–113) | −1.0 (−3.0, 0.002), P = 0.09, NS |
| MP-TGNs (pmol) | 360 (0–1 216) | 754 (132–2 228) | 406 (332, 478), P < 0.0001 |
| MeMPNs (pmol) | 10 650 (0–141 772) | 3 868 (60–38 386) | −5524 (−7396, −3868), P < 0.0001 |
| TPMT (units) | 14 (5.6–26.3) | 7.9 (2.8–13.6) | −6.3 (−6.9, −5.7), P < 0.0001) |
MeMPNs, methylmercaptopurine nucleotides; MP, mercaptopurine cohort; TG, thioguanine cohort; TGNs, thioguanine nucleotides; TPMT, thiopurine methyltransferase. TGN and MeMPN units are pmol/8 × 108 red blood cells. TPMT units are units ml−1 packed red blood cells. The TPMT variant alleles quantified were TPMT *1/*2 (MP n = 3; TG n = 1), TPMT *1/*3A (MP n = 53; TG n = 34) and TPMT *1/*3C (MP n = 11; TG n = 4). Values are given as median (range), NS, not significant. The analysis excludes TPMT deficient children.
Within the intermediate activity cohort (TPMT ≤10.5 units), metabolites were compared between children with a wild-type and heterozygous genotype (Table 3). For children taking thioguanine there was no difference in TGN formation. However, mercaptopurine derived TGNs were significantly higher, and MeMPNs were significantly lower, in the heterozygous genotype children compared with the intermediate activity/wild-type genotype children. For both thiopurines, TPMT activities were lower in the heterozygous genotype children.
Table 3.
Metabolite accumulation within the thiopurine methyltransferase intermediate activity cohort; wild-type genotype and heterozygous genotype compared
| TPMT ≤10.5 units | Wild-type genotype | Heterozygous genotype | Median difference (95% CI) |
|---|---|---|---|
| TG patients | 35 | 33 | |
| TG-TGNs (pmol) | 2 360 (910–4 060) | 2 626 (174–6 730) | 246 (−214, 664), P = 0.29, NS |
| TPMT (units) | 9.9 (5.4–10.5) | 7.7 (5.2–10.5) | −1.7 (−2.4, −1.0), P < 0.0001 |
| MP patients | 44 | 63 | |
| TG-TGNs (pmol) | 366 (0–1 216) | 754 (132–2 228) | 369 (250, 502), P < 0.0001 |
| MeMPNs (pmol) | 8 590 (0–96 964) | 3 825 (60–38 386) | −4822 (−7278, −2518), P = 0.0001 |
| TPMT (units) | 9.7 (5.6–10.5) | 7.8 (2.8–10.5) | −1.4 (−2.0, −0.7), P < 0.0001 |
MeMPNs, methylmercaptopurine nucleotides; MP, mercaptopurine cohort; TG, thioguanine cohort; TGNs, thioguanine nucleotides; TPMT, thiopurine methyltransferase. TGN and MeMPN units are pmol/8 × 108 red blood cells. TPMT units are units ml−1 packed red blood cells. For the intermediate activity cohort (TG n = 68, MP n = 114) median TG derived TGNs = 2492 pmol, MP derived TGNs = 562 pmol, MeMPNs = 5542 pmol. TPMT genotypes were available for all 68 children on thioguanine but only for 107 of the 114 children on mercaptopurine. The analysis excludes TPMT deficient children. Values are given as median (range).
Comparisons between the heterozygote TPMT*1/*3A and TPMT*1/*3C children were not possible with thioguanine (32 TPMT*1/*3A, four TPMT*1/*3C), but within the mercaptopurine cohort, TPMT *1/*3A children (n = 53) had significantly higher TGN and MeMPN concentrations than TPMT *1/*3C children (n = 11) (Table 4), despite having similar drug dosages and TPMT activities.
Table 4.
Mercaptopurine metabolite accumulation compared within the heterozygous genotypes TPMT *1/*3A and TPMT*1/*3C
| Mercaptopurine | TPMT *1/*3A | TPMT *1/*3C | Median difference (95% CI) |
|---|---|---|---|
| Patients | 53 | 11 | |
| MP-TGNs (pmol) | 802 (132–2 228) | 492 (288–910) | 288 (23, 445), P = 0.031 |
| MeMPNs (pmol) | 4 542 (84–38 386) | 1 832 (60–10 746) | 2458 (129, 5605), P = 0.034 |
MeMPNs, methylmercaptopurine nucleotides; MP, mercaptopurine; TGNs, thioguanine nucleotides; TPMT, thiopurine methyltransferase. TGN and MeMPN units are pmol/8 × 108 red blood cells. Values are given as median (range).
Multiple assays
For the investigation of thiopurine metabolism over time blood samples were requested during year 1, at the start of year 2 and towards the end of year 2. Of the 766 children from whom multiple samples were received only 465 children (221 thioguanine, 244 mercaptopurine) provided samples at the requested time intervals. Of these, 87 children randomized to thioguanine switched to mercaptopurine during maintenance therapy and were excluded from the analysis.
Thioguanine
Multiple assays on thioguanine maintenance therapy were available for 134 children. Kruskal–Wallis analysis indicated that the thioguanine dose prescribed was lower at the end of therapy than at the start (P = 0.029) but there was no difference in TGN concentrations or TPMT activities. Matched analysis of the year 1 assay with the end of year 2 assay indicated a decrease of 2.5 mg m−2 in thioguanine dose over time, a similar decrease was observed in both genotype groups (Table 5A).
Table 5.
Thiopurine metabolism in the year 1 and in the end of year 2 assays. (A) Thioguanine analyzed by genotype, (B) Mercaptopurine analyzed by genotype
| Year 1 | End of year 2 | Wilcoxon difference (95% CI) | |
|---|---|---|---|
| (A) TG, wild-type TPMT, n = 114 | |||
| TG dose (mg m−2) | 40 (23–67) | 40 (8–114) | −2.5 (−5.0, −1.0), P = 0.005 |
| TG-TGNs (pmol) | 1 898 (36–5 366) | 1 952 (408–5 376) | 54 (−114, 204), P = 0.52, NS |
| TG, heterozygous TPMT, n = 18 | |||
| TG dose (mg m−2) | 38 (20–42) | 35 (8–60) | −3 (−8.5, 2.5), P = 0.23, NS |
| TG-TGNs (pmol) | 2 375 (174–6 730) | 2 026 (1 282–4 216) | −172 (−806, 899), P = 0.70, NS |
| (B) MP, wild-type TPMT, n = 225 | |||
| MP dose (mg m−2) | 75 (26–107) | 75 (23–116) | 0 (−1.5, 0.5), P = 0.59, NS |
| MP-TGNs (pmol) | 372 (18–994) | 324 (36–742) | −45 (−66, −24), P < 0.001 |
| MeMPNs (pmol) | 9 916 (0–55 952) | 14 747 (506–88 134) | 3877 (2286, 5541), P < 0.001 |
| TPMT (units) | 14.9 (10.6–26.3) | 13.9 (9.8–20.2) | −1.0 (−1.8, −0.5), P < 0.001 |
| MP, heterozygous TPMT, n = 18 | |||
| MP dose (mg m−2) | 73 (37–77) | 73 (34–78) | 0 (−1.0, 1.0), P = 0.92, NS |
| MP-TGNs (pmol) | 644 (360–1 928) | 802 (48–1 258) | 82 (−155, 311), P = 0.37, NS |
| MeMPNs (pmol) | 4 310 (247–41 084) | 4 886 (0–29 048) | 931 (−2858, 4317), P = 0.66, NS |
| TPMT (units) | 8.9 (6.0–13.6) | 7.4 (6.1–13.6) | 0.33 (−2.15, 3.4), P = 0.79, NS |
MeMPNs, methylmercaptopurine nucleotides; MP, mercaptopurine cohort; NS, not-significant; TG, thioguanine cohort; TGNs, thioguanine nucleotides; TPMT, thiopurine methyltransferase. TGN and MeMPN units are pmol/8 × 108 red blood cells. TPMT units are units ml−1 packed red blood cells. Table 5A Thioguanine n = 134, genotypes available for 132 children (17 TPMT*1/*3A, one TPMT*1/*3C). One wild-type had overt compliance problems and was prescribed thioguanine doses of >200% protocol by the end of year 2. If this child was removed from the analysis the maximum prescribed dose was 68 mg m−2 and the median dose difference at the end of treatment was −3.0 mg (−5.0 to −1.0), P = 0.002. Table 5B Mercaptopurine n = 244, genotypes available for 243 children (16 TPMT*1/*3A, one *1/*3C, one *1/*2). Values are presented as the median (range).
Mercaptopurine
Kruskal–Wallis analysis indicated that TGN concentrations were higher at the start of therapy (P < 0.001), whilst MeMPN concentrations were higher at the end (P < 0.001). TPMT activities were lower at the end of year 2 (P < 0.001), a difference (1.0 units TPMT) that was clinically not-significant. There was no significant difference in mercaptopurine dosage.
Matched analysis of the year 1 assay with the end of year 2 assay indicated a marginal decrease in TGN concentrations and TPMT activities over time but a profound increase in MeMPN concentrations. Splitting the data by genotype (225 wild-type TPMT, 18 heterozyotes) clearly showed that the increase in MeMPN production was in the wild-type cohort (Table 5B). Further analysis of the wild-type cohort indicated that MeMPNs increased throughout maintenance therapy. Comparing the year 1 assay vs. the start of year 2 assay gave median MeMPN concentrations of 9916 pmol vs. 11 084 pmol respectively, (median difference 2199 pmol, 95% CI 494, 4052, P < 0.001).
Compliance problems
A number of children were suspected of taking their thiopurine doses intermittently or not at all. Blood cell counts remained high despite long term thiopurines at the protocol standard, or higher, dose. Mercaptopurine derived TGNs and MeMPNs accumulate slowly and uniformly in a population of red cells over a period of days and weeks 27. Twenty children (2.7% of the 744 cohort) prescribed long term ≥100% mercaptopurine had metabolite concentrations persistently at or below the lower limit of detection. Thioguanine derived TGNs accumulate rapidly in the red cell after an oral dose. After a single 40 mg m−2 dose the inter-patient variation in TGNs concentrations at 6 h post-dose is 144 to 574 pmol 28, 29 and, after seven daily doses, 959 to 2361 pmol 28. Thus, TGNs <750 pmol whilst taking ≥100% thioguanine could be regarded as resulting from non-compliance. Twenty-eight children had thioguanine TGNs of <750 pmol (6.2% of the 450 children taking thioguanine). Twelve of these children (2.7%) had TGNs <500 pmol.
Discussion
We report, in children with ALL, that an intermediate TPMT phenotype, based on enzyme activity measurements, cannot be categorically defined either at disease diagnosis or during chemotherapy. At disease diagnosis TPMT activities are much reduced and are not reflective of on-therapy activities. This reduction has been previously reported 26, along with the resulting TPMT genotype–phenotype discordance, in small patient cohorts with low numbers of variant alleles 30, 31, and is now confirmed in a large population of children with ALL. The reduction in TPMT activities is well below the ranges recorded for healthy children 15, 30 and has been attributed to the disease process and the resulting anaemia of ALL with the associated measurement of a decayed TPMT enzyme 26. During chemotherapy TPMT activities increase to levels well above the range recorded for healthy children 15, 30, 32. These sizable leukaemia- and treatment-related changes in TPMT activities are not reported to occur in other clinical situations where thiopurine drugs are widely used 33. Any disease-dependent TPMT activity distribution fluctuation is clinically insignificant 34. For children with ALL, particularly at disease diagnosis, this is not so. TPMT reference ranges derived from healthy individuals, or patients on thiopurine immunosuppression, should not be used to derive presumed heterozygosity.
The elevation of RBC TPMT activities during chemotherapy may reflect the influence of therapy on red cell kinetics and life-spans. Bone marrow suppression, and the subsequent activation of erythropoiesis, has been shown to elevate TPMT activity 35, young red cells having higher TPMT activities than old cells 26. Elevated TPMT activities are also observed in healthy neonates, (who have excess reticulocytes and nucleated red cells), compared with children 36 and in younger children compared with older children and adults 37. We could detect no influence of age on TPMT activities in our study, any age effect perhaps masked by the underlying chemotherapy. Despite these possible influences, the TPMT activity ranges measured during chemotherapy were similar for both thiopurines and, when measured under standard conditions, TPMT activities were reproducible over time. However, genotype–phenotype concordance in the intermediate activity range was poor with 45% of children having a wild-type genotype. Sequencing revealed only two rare and three novel TPMT alleles. This discordance in the intermediate activity range has previously been reported in small patient cohorts and healthy subjects 19, 31, 38. The first large scale TPMT genotype–phenotype concordance study in healthy blood donors (n = 1214) reported a clearly defined trimodal distribution and an overall concordance of >98% yet, after sequencing all discordant samples, reported 13 of 111 (11.7%) individuals with an intermediate activity wild-type in the open-reading frame 19. A systematic review 39 of gastroenterology patients prescribed thiopurines reported the near-perfect specificity of variant allele genotyping for identifying patients with intermediate TPMT enzyme activities but, in agreement with our study, the converse was not true. An intermediate activity range, defined by sensitivity and specificity analysis, contains a high proportion of false positives i.e. wild-type genotype.
Polymorphic tandem repeats in the TPMT promoter can modulate TPMT activity 38 but larger studies (n = 1211) have shown these effects to be quantitatively small 40. However, this may not be so for the influence of underlying chemotherapy 30. Methotrexate mediated inhibition of intracellular methylation could limit the supply of the methyl cofactor for the TPMT reaction. An association between the MTHFR 677C > T variant (reduced methylation, up to 10% of the population) and heterozygous TPMT phenotypes with wild-type genotypes has been reported 41.
The fact that genotype is a better indicator of constitutive TPMT function than phenotype, in children with ALL, is illustrated by mercaptopurine metabolite accumulation in the intermediate activity cohort. The wild-type genotype/intermediate activity children have similar TGN concentrations to children with TPMT high activity and wild-type genotype. If TPMT assessment is restricted to genotyping the TPMT*3 family and TPMT*2 alleles, the risks of misclassification of a TPMT deficient child have been reported to be 1 in 7416 patients for a result of TPMT*1/*3 or TPMT*1/*2 and 1 in 14 832 patients for a TPMT*1/*1 result 42. Measurement of TPMT activity in heterozygous genotype children at disease diagnosis would detect the former whilst the use of activity measurements alongside genotype, for all children, would be required for the latter.
TGN concentrations in the TPMT deficient children were within the same range as those previously reported 12–14, 16. TGN concentrations in the deficient child taking thioguanine were similar to those for mercaptopurine. For both thiopurines, heterozygous children accumulated significantly higher TGN concentrations compared with the wild-type cohort. This difference in TGN accumulation with respect to TPMT status has been reported for mercaptopurine 16, but not thioguanine, and is a reflection of increased drug methylation by higher TPMT activities. For mercaptopurine, TGNs were effective at much lower concentrations, some three to five-fold lower, than thioguanine derived TGNs. This effect has been attributed to the ability of MeMPNs to inhibit de novo purine synthesis 43. Thus the TGN competitive inhibitor is successful at lower concentrations. The difference in mercaptopurine metabolite accumulation between TPMT*1/*3A and TPMT*1/*3C children has not been previously reported and may be due to differences in the degradation half-lives of the mutant variant alleles 44, 45. Alternatively these observations may be due to problems with tablet taking or the small sample size of the latter cohort. In agreement with previous reports 46, the TPMT allele frequency differed between ethnic groups with white Caucasians having the highest frequency of low activity alleles and TPMT*3A predominating.
The analysis of thiopurine metabolism over time revealed potentially clinically significant findings. For thioguanine, the dose required to keep cell counts within the target range decreased, but the range recorded was wide and possible non-compliance with oral chemotherapy hinders the interpretation of these findings. Judged by very low, or absent, metabolite concentrations 2.7% of children were non-compliant, which may impact on treatment outcome. The use of drug metabolite ratios has estimated that there is strong evidence of non or partial compliance in 10% of ALL children 47, and electronic tablet counting has estimated that up to 17% of ALL children take less than 80% of their prescribed pills 48. The latter has been confirmed in a recent study and linked to a higher incidence of disease relapse 49.
For mercaptopurine, despite similar dosages and TGN concentrations, MeMPN concentrations increased as maintenance therapy progressed. There is some debate as to whether excess MeMPN accumulation is a cause, or a reflection, of hepatotoxic events 50, 51. MeMPN concentrations above 5000 pmol/8 × 108 RBCs, measured in the first year of therapy in children with ALL, have been associated with hepatotoxicity 52, MeMPNs concentrations approximately half the group median concentration reported in the present UK study. However, a degree of hepatotoxicity, as demonstrated by elevated liver function tests, is associated with a lower relapse risk 53. Whatever the mechanism underlying the increased MeMPN formation over time observed in the current study, the clinical significance warrants further investigation.
The data reported here will allow a more informed use of TPMT measurements in children and young adults with ALL. In this patient group genotype is superior to phenotype in the classification of TPMT heterozygosity. Previous observations with respect to TGN accumulation and TPMT status have been confirmed and extended to thioguanine, findings which may help guide the use of thiopurine drugs. The effective delivery of thiopurines is important in the successful treatment of ALL 15, 49. It is of some concern that we observed overt non-compliance with oral thiopurines in about 3% of children and this was a confounding factor in the interpretation of our data. These findings indicate a possible role for metabolite monitoring, or compliance surveillance, throughout maintenance.
Acknowledgments
The thiopurine studies within ALL97 were supported by Leukaemia and Lymphoma Research, London, UK. We wish to thank all the clinicians who entered patients into this trial and the participating children and their families. We wish to thank John Lilleyman for his contributions to thiopurine research within the UK ALL trials and for his helpful discussions, sound advice and encouragement.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available from request from the corresponding author) and declare support from Leukaemia and Lymphoma Research (LL, CSC, AV) and the Medical Research Council (RW, SMR) for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
References
- 1.Hann I, Vora A, Richards S, Hill F, Gibson B, Lilleyman J, Kinsey S, Mitchell C, Eden OB. Benefit of intensified treatment for all children with acute lymphoblastic leukaemia: results from MRC UKALL XI and MRC ALL97 randomised trials. Leukemia. 2000;14:356–363. doi: 10.1038/sj.leu.2401704. [DOI] [PubMed] [Google Scholar]
- 2.Hann IM, Stevens RF, Goldstone AH, Rees JKH, Wheatley K, Gray R, Burnett AK. Randomized comparison of DAT versus ADE as induction therapy in children and younger adults with acute myeloid leukaemia. Results of the Medical Research Council's 10th AML Trial (MRC AML 10) Blood. 1997;89:2311–2318. [PubMed] [Google Scholar]
- 3.Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32:651–662. [PMC free article] [PubMed] [Google Scholar]
- 4.Balis FM Holcenberg JS, Poplack DG, Ge J, Sather HN, Murphy RF, Ames MM, Waskerwitz MJ, Tubergen DG, Zimm S, Gilchrist GS, Bleyer WA. Pharmacokinetics and pharmacodynamics of oral methotrexate and mercaptopurine in children with lower risk acute lymphoblastic leukaemia: a joint Children's Cancer Group and Pediatric Oncology Branch study. Blood. 1998;92:3569–3577. [PubMed] [Google Scholar]
- 5.Lennard L, Van Loon JA, Weinshilboum RM. Pharmacogenetics of acute azathioprine toxicity: relationship to thiopurine methyltransferase genetic polymorphism. Clin Pharmacol Ther. 1989;46:149–154. doi: 10.1038/clpt.1989.119. [DOI] [PubMed] [Google Scholar]
- 6.Tidd DM, Paterson ARP. A biochemical mechanism for the delayed cytotoxic reactions of 6-mercaptopurine. Cancer Res. 1974;34:738–746. [PubMed] [Google Scholar]
- 7.Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull. 2006;79 and 80:153–170. doi: 10.1093/bmb/ldl020. [DOI] [PubMed] [Google Scholar]
- 8.Hogarth LA, Redfern CPF, Teodoridis JM, Hall AG, Anderson H, Case MC, Coulthard SA. The effect of thiopurine drugs on DNA methylation in relation to TPMT expression. Biochem Pharmacol. 2008;76:1024–1035. doi: 10.1016/j.bcp.2008.07.026. [DOI] [PubMed] [Google Scholar]
- 9.Tiede I, Fritz G, Strand S, Poppe D, Dvorsky R, Strand D, Lehr HA, Wirtz S, Becker C, Atreya R, Mudter J, Hildner K, Bartsch B, Holtman M, Blumberg R, Walczak H, Iven H, Galle PR, Ahmadian MR, Neurath MF. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest. 2003;111:1133–1145. doi: 10.1172/JCI16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poppe D, Tiede I, Fritz G, Becker C, Bartsch B, Wirtz S, Strand D, Tanaka S, Galle PR, Bustelo XR, Neurath MF. Azathioprine suppresses Ezrin-Radixin-Moesin-dependent T cell-APC conjugation through inhibition of Vav guanosine exchange activity on Rac proteins. J Immunol. 2006;176:640–651. doi: 10.4049/jimmunol.176.1.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bourgine J, Garat A, Allorge D, Crunelle-Thibaut A, Lo-Guidice JM, Colombel JF, Brolly F, Billaut-Laden I. Evidence for a functional genetic polymorphism of Rho-GTPase Rac1. Implication in azathioprine response? Pharmacogenet Genomics. 2011;21:313–324. doi: 10.1097/FPC.0b013e3283449200. [DOI] [PubMed] [Google Scholar]
- 12.Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase deficient child with acute lymphoblastic leukaemia. J Pediatr. 1991;119:985–989. doi: 10.1016/s0022-3476(05)83063-x. [DOI] [PubMed] [Google Scholar]
- 13.Lennard L, Lewis IJ, Michelagnoli M, Lilleyman JS. Thiopurine methyltransferase deficiency in childhood lymphoblastic leukaemia: 6-mercaptopurine dosage strategies. Med Pediatr Oncol. 1997;29:252–255. doi: 10.1002/(sici)1096-911x(199710)29:4<252::aid-mpo3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 14.McBride KL, Gilchrist GS, Smithson WA, Weinshilboum RM, Szumlanski CL. Severe 6-thioguanine-induced marrow aplasia in a child with acute lymphoblastic leukaemia and inherited thiopurine methyltransferase deficiency. J Pediatric Hematology. Oncology. 2000;22:441–445. doi: 10.1097/00043426-200009000-00011. [DOI] [PubMed] [Google Scholar]
- 15.Lennard L, Lilleyman JS, Van Loon JA. Weinshilboum RM Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336:225–229. doi: 10.1016/0140-6736(90)91745-v. [DOI] [PubMed] [Google Scholar]
- 16.Relling MV, Hancock ML, Rivera GK, Sandlund JT, Ribeiro RC, Krynetski EY, Pui C-H, Evans WE. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 1999;91:2001–2008. doi: 10.1093/jnci/91.23.2001. [DOI] [PubMed] [Google Scholar]
- 17.Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenetic test whose time has come. J Clin Pathol. 2010;63:288–295. doi: 10.1136/jcp.2009.069252. [DOI] [PubMed] [Google Scholar]
- 18.Relling MV, Gardner EE, Sandborn WJ, Pui C-H, Stein CM, Carrillo M, Evans WE, Klein TE. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther. 2011;89:387–391. doi: 10.1038/clpt.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaeffeler E, Fisher C, Brockmeier D, Wernet D, Moerike K, Eichelbaum M, Zanger UM, Schwab M. Comprehensive analysis of thiopurine S-methyltransferase phenotype-genotype correlation in a large population of German-caucasians and identification of novel TPMT variants. Pharmacogenetics. 2004;14:407–417. doi: 10.1097/01.fpc.0000114745.08559.db. [DOI] [PubMed] [Google Scholar]
- 20.Otterness D, Szumlanski C, Lennard L, Klemetsdal B, Aarbakke J, Park-Hah JO, Iven H, Schmeigelow K, Branum E, O'Brien J, Weinshilboum RM. Human thiopurine methyltransferase pharmacogenetics: gene sequence polymorphisms. Clin Pharmacol Ther. 1997;62:60–73. doi: 10.1016/S0009-9236(97)90152-1. [DOI] [PubMed] [Google Scholar]
- 21.Vora AJ, Mitchell CD, Lennard L, Eden TOB, Kinsey SE, Lilletman JS, Richards S. Toxicity and efficacy of thioguanine compared with mercaptopurine in childhood lymphoblastic leukaemia: results of the UK Medical Research Council Randomised Trial ALL97. Lancet. 2006;368:1339–1348. doi: 10.1016/S0140-6736(06)69558-5. [DOI] [PubMed] [Google Scholar]
- 22.Lennard L, Richards S, Cartwright CS, Mitchell C, Lilleyman JS, Vora AJ. The thiopurine methyltransferase genetic polymorphism is associated with thioguanine-related veno-occlusive disease of the liver in children with acute lymphoblastic leukaemia. Clin Pharmacol Ther. 2006;80:375–383. doi: 10.1016/j.clpt.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell CD, Richards SM, Lilleyman J, Vora A, Eden OB. Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomised trial. Br J Haematol. 2005;129:734–745. doi: 10.1111/j.1365-2141.2005.05509.x. [DOI] [PubMed] [Google Scholar]
- 24.Lennard L, Singleton HJ. High performance liquid chromatographic assay of human red blood cell thiopurine methyltransferase activity. J Chromatogr B. 1994;661:25–33. doi: 10.1016/0378-4347(94)00327-0. [DOI] [PubMed] [Google Scholar]
- 25.Lennard L, Singleton H. High performance liquid chromatographic assay of the methyl and nucleotide metabolites of 6-mercaptopurine: quantitation of red blood cell 6-thioguanine nucleotide, 6-thioinosinic acid and 6-methylmercaptopurine metabolites in a single sample. J Chromatogr. 1992;583:83–90. doi: 10.1016/0378-4347(92)80347-s. [DOI] [PubMed] [Google Scholar]
- 26.Lennard L, Chew TS, Lilleyman JS. Human thiopurine methyltransferase activity varies with red blood cell age. Br J Clin Pharmacol. 2001;52:539–546. doi: 10.1046/j.0306-5251.2001.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rostami-Hodjegan A, Lennard L, Lilleyman JS. The accumulation of mercaptopurine metabolites in age fractionated red blood cells. Br J Clin Pharmacol. 1995;40:217–222. doi: 10.1111/j.1365-2125.1995.tb05776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lennard L, Davies HA, Lilleyman JS. Is 6-thioguanine more appropriate than 6-mercaptopurine for children with acute lymphoblastic leukaemia? Br J Cancer. 1993;68:186–190. doi: 10.1038/bjc.1993.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lancaster DL, Patel N, Lennard L, Lilleyman JS. 6-Thioguanine in children with acute lymphoblastic leukaemia: influence of food on parent drug pharmacokinetics and 6-thioguanine numcleotide concentrations. Br J Clin Pharmacol. 2001;51:531–539. doi: 10.1046/j.0306-5251.2001.01391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brouwer C, De Abreu RA, Keizer-Garritsen JJ, Lambooy LHJ, Ament K, ter Reit PGJH, van Wering ER, Trijbels FJM, Veerman AJP, Hooherbrugge PM, Bokkerink JPM. Thiopurine methyltransferase in acute lymphoblastic leukaemia: biochemical and molecular biological aspects. Eur J Cancer. 2005;41:613–623. doi: 10.1016/j.ejca.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 31.Fakoury M, Andreu-Gallien J, Mahr A, Medard Y, Azougagh S, Vilmer E, Jacqz-Aigrain E. Should TPMT genotype and activity be used to monitor 6-mercaptopurine treatment in children with acute lymphoblastic leukaemia? J Clin Pharm Ther. 2007;32:633–639. doi: 10.1111/j.1365-2710.2007.00858.x. [DOI] [PubMed] [Google Scholar]
- 32.McLeod HL, Relling MV, Liu Q, Pui C-H, Evans WE. Polymorphic thiopurine methyltransferase in erythrocytes is indicative of activity in leukemic blasts from children with acute lymphoblastic leukemia. Blood. 1995;7:1897–1902. [PubMed] [Google Scholar]
- 33.Lindqvist M, Hindorf U, Almer S, Soderkvist P, Strom M, Hjortswang H, Peterson C. No induction of thiopurine methyltransferase during thiopurine treatment in inflammatory bowel disease. Nucleosides Nucleotides Nucleic Acids. 2006;25:1033–1037. doi: 10.1080/15257770600890814. [DOI] [PubMed] [Google Scholar]
- 34.Gisbert JP, Gomollon F, Cara C, Luna M, Gonzalez-Lama Y, Pajares JM, Mate J, Guijarro LG. Thiopurine methyltransferase activity in Spain: a study of 14,545 patients. Dig Dis Sci. 2007;52:1262–1269. doi: 10.1007/s10620-006-9119-z. [DOI] [PubMed] [Google Scholar]
- 35.de Boer NKH, van Bodegraven AA, de Graaf P, van der Hulst RWM, Zoetekouw L, van Kuilenburg ABP. Paradoxical elevated thiopurine S-methyltransferase activity after pancytopenia during azathioprine therapy: potential influence of red blood cell age. Ther Drug Monit. 2008;30:390–393. doi: 10.1097/FTD.0b013e31816c20b3. [DOI] [PubMed] [Google Scholar]
- 36.McLeod HL, Krynetski EY, Williams JA, Evans WE. Higher activity of polymorphic thiopurine S-methyltransferase in erythrocytes from neonates compared to adults. Pharmacogenetics. 1995;5:281–286. doi: 10.1097/00008571-199510000-00003. [DOI] [PubMed] [Google Scholar]
- 37.Serpe L, Calvo PL, Muntoni E, D'Antico S, Giaccone M, Avagnina A, Baldi M, Barbera B, Curti F, Pera A, Eandi M, Zara GP, Canaparo R. Thiopurine S-methyltransferase pharmacogenetics in a large-scale healthy Italian-Caucasian population: differences in enzyme activity. Pharmacogenomics. 2009;10:1753–1765. doi: 10.2217/pgs.09.103. [DOI] [PubMed] [Google Scholar]
- 38.Spire-Vayron de la Moureyre C, Debuysere H, Mastain B, Vinner E, Marez D, Lo Guidice J-M, Chevalier D, Brique S, Motte K, Colombel J-F, Turck D, Noel C, Flipo R-M, Pol A, Lhermitte M, Lafitte J-J, Libersa C, Broly F. Genotypic and phenotypic analysis of the polymorphic thiopurine S-methyltransferase gene (TPMT) in a European population. Br J Pharmacol. 1998;125:879–887. doi: 10.1038/sj.bjp.0702152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Booth RA, Ansari MT, Loit E, Tricco AC, Weeks L, Doucette S, Skidmore B, Sears M, Sy R, Karsh J. Assessment of thiopurine S-methyltransferase activity in patients prescribed thiopurines: a systematic review. Ann Intern Med. 2011;154:814–823. doi: 10.7326/0003-4819-154-12-201106210-00009. [DOI] [PubMed] [Google Scholar]
- 40.Yan L, Zhang S, Eiff B, Szumlanski CL, Powers M, O'Brien JF, Weinshilboum RM. Thiopurine methyltransferase polymorphic tandem repeat: genotype-phenotype correlation analysis. Clin Pharmacol Ther. 2000;68:210–219. doi: 10.1067/mcp.2000.108674. [DOI] [PubMed] [Google Scholar]
- 41.Arenas M, Simpson G, Lewis CM, Shobwale-Bakre E-M, Escuredo E, Fairbanks LD, Duley JA, Ansari A, Sanderson JD, Marinaki AM. Genetic variation in the MTHFR gene influences thiopurine methyltransferase activity. Clin Chem. 2005;51:2371–2374. doi: 10.1373/clinchem.2005.053157. [DOI] [PubMed] [Google Scholar]
- 42.Ford L, Kampanis P, Berg J. Thiopurine S-methyltransferase genotype-phenotype concordance: used as a quality assurance tool to help control the phenotype assay. Ann Clin Biochem. 2009;46:152–154. doi: 10.1258/acb.2008.008167. [DOI] [PubMed] [Google Scholar]
- 43.Nelson JA. Mechanisms of action of thiopurines used in the curative therapy for childhood leukaemias. Cancer Bull. 1992;44:470–473. [Google Scholar]
- 44.Salavaggione OE, Wang L, Wiepert M, Yee VC, Weinshilboum RM. Thiopurine S-methyltransferase pharmacogenetics: variant allele functional and comparative genomics. Pharmacogenet Genomics. 2005;15:801–815. doi: 10.1097/01.fpc.0000174788.69991.6b. [DOI] [PubMed] [Google Scholar]
- 45.Rutherford K, Daggett V. Four human thiopurine S-methyltansferase alleles severely affect protein structure and dynamics. J Mol Biol. 2008;379:803–814. doi: 10.1016/j.jmb.2008.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: insights, challenges and future directions. Oncogene. 2006;25:1629–1638. doi: 10.1038/sj.onc.1209372. [DOI] [PubMed] [Google Scholar]
- 47.Lennard L, Welch J, Lilleyman JS. Intracellular metabolites of 6-mercaptopurine in children with lymphoblastic leukaemia: a possible indicator of non-compliance. Br J Cancer. 1995;72:1004–1006. doi: 10.1038/bjc.1995.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lau RCW, Matsui D, Greenberg M, Koren G. Electronic measurement of compliance with mercaptopurine in pediatric pateints with acute lymphoblastic leukaemia. Med Pediatr Oncol. 1998;30:85–90. doi: 10.1002/(sici)1096-911x(199802)30:2<85::aid-mpo3>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 49.Bhatia S, Landier W, Shangguan M, Hageman L, Schaible AN, Carter AR, Hanby CL, Leisenring W, Yasui Y, Kornegay NM, Mascarenhas L, Ritchey AK, Casillas JN, Dickens DS, Meza J, Carroll WL, Relling MV, Wong FL. Nonadherence to oral mercaptopurine and risk of relapse in hispanic and non-hispanic white children with acute lymphoblastic leukemia: a report from the Children's Oncology Group. J Clin Oncol. 2012;30:2094–2101. doi: 10.1200/JCO.2011.38.9924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nygaard U, Toft N, Schmiegelow K. Methylated metabolites of 6-mercaptopurine are associated with hepatotoxicity. Clin Pharmacol Ther. 2004;75:274–281. doi: 10.1016/j.clpt.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 51.Gardiner S, Gearry RB, Burt MJ, Ding SJ, Barclay ML. Severe hepatoxicity with high 6-methylmercaptopurine nucleotide concentrations after thiopurine dose escalation due to low 6-thioguanine nucleotides. Eur J Gastroenterol Hepatol. 2008;20:1238–1242. doi: 10.1097/MEG.0b013e3282ffda37. [DOI] [PubMed] [Google Scholar]
- 52.de Beaumais TA, Fakhoury M, Medard Y, Azougagh S, Zhang D, Yakouben K, Jacqz-Aigrain E. Determinants of mercaptopurine toxicity in paediatric acute leukaemia maintenance therapy. Br J Clin Pharmacol. 2011;71:575–584. doi: 10.1111/j.1365-2125.2010.03867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmeigelow K, Pulczynska M. Prognostic significance of hepatotoxicity during maintenance chemotherapy for childhood acute lymphoblastic leukaemia. Br J Cancer. 1990;61:767–772. doi: 10.1038/bjc.1990.172. [DOI] [PMC free article] [PubMed] [Google Scholar]