

Abstract
Signal transduction originates at the membrane, where the clustering of signaling proteins is a key step in transmitting a message. Membranes are difficult to study, and their influence on signaling is still only understood at the most rudimentary level. Recent advances in the biophysics of membranes, surveyed in this review, have highlighted a variety of phenomena that are likely to influence signaling activity, such as local composition heterogeneities and long-range mechanical effects. We discuss recent mechanistic insights into three signaling systems—Ras activation, Ephrin signaling and the control of actin nucleation—where the active role of membrane components is now appreciated and for which experimentation on the membrane is required for further understanding.
The influence of the membrane surface environment on the behavior of proteins that are localized to the vicinity of the membrane is still poorly understood. This contrasts starkly with our now quite sophisticated understanding of the structural and chemical aspects of the individual protein components1. This lack of knowledge is a natural consequence of the fact that membranes are difficult to study in vitro and that detailed quantitative information is difficult to obtain in vivo2. The relative inaccessibility of membranes to classical methods of study effectively cloaks their role in signaling biochemistry. We suggest that what is known about membranes in signal transduction is just a glimmer of a much larger story, with many key aspects of membrane function still hidden beneath the cloak.
Cell signaling relies on modular domains that generate protein-protein interactions at the membrane3,4. Equally critical are domains that recognize specific phospholipids and are thereby responsive to changes in membrane composition5, as best exemplified by the family of protein kinase C (PKC) isozymes6,7. The PKC isozymes transduce signals arising from the hydrolysis of phospholipids in the membrane and have a protein kinase domain linked to C1 domains and a C2 domain (Fig. 1). The C1 domains bind to diacylglycerol (DAG), whereas the C2 domain binds to negatively charged lipids, such as phosphatidylinositol-4,5,-bisphosphate (PIP2) and to Ca2+ ions6,7. The activation of PKC involves priming by phosphorylation, followed by the recruitment of PKC to the membrane by PIP2, Ca2+ and DAG7,8. One important principle that emerged from PKC studies is that the individual lipid-binding modules of PKC have insufficient affinity for their target lipids and so require that both PIP2 and DAG be present for effective activation of the enzyme. This requirement for multiple targeting signals is often observed in cell signaling and has been referred to as ‘coincidence detection’ (refs. 9, 10).
Figure 1.

Activation of PKC at the membrane. (a) Inactive PKC is associated with heat-shock protein 90 (HSP90) at the membrane. (b) The activation of cell-surface receptors results in the production of DAG in the membrane and an increase in cytoplasmic Ca2+. PKC is primed for activation by phosphorylation, which is carried out by two protein kinases, phosphoinositide-dependent kinase 1 (PDK1) and mammalian target of rapamycin complex (mTORc, not shown), which release it from HSP90 and the membrane. These two signals, along with PIP2 in the membrane, result in the recruitment of PKC to the membrane and its allosteric activation. Schematic diagrams are adapted from previous work2,6.
What we know about the mechanism of PKC raises questions that are difficult to address without in vitro reconstitution and quantitative analysis on membranes. For example, how does variation in the levels of PIP2 affect the activity of the enzyme, and how would proteins such as the PKC substrate MARCKS, which have been suggested to locally concentrate PIP2 (ref. 11), alter the dynamics of activation? To address such questions, we have to directly view the signaling proteins in action on the membrane. This has posed a formidable challenge in the past, but the good news is that new strategies to image, synthesize and control membranes (in vitro, in vivo and through computational modeling) are coming of age. We believe that this is a particularly exciting frontier of current research, where major breakthroughs can be expected in the near future.
To help spur the imagination, we review here a wide range of physical properties of membranes along with specific instances of their involvement in signal transduction. These range from relatively obvious effects, such as local concentration enhancement, to mechanisms for long-range cooperativity and emergent properties such as force sensing. More detailed discussion will focus on (i) activation of Ras by the nucleotide exchange factor Son of Sevenless (SOS), (ii) juxtacrine triggering of the ephrin type-A receptor 2 (EphA2) tyrosine kinase by its membrane-associated ephrin-A1 ligand and (iii) the stimulation of actin polymerization by proteins of the WASP/WAVE family. In each of these cases, various features of the membrane environment are intertwined with the signaling reactions themselves.
Membrane physical chemistry
At perhaps the most basic level, membranes provide physical surfaces. Adsorption of molecular reactants to the membrane surface, which is also fluid, generally increases their relative probability of an encounter and, correspondingly, their reaction kinetics. This is often referred to as a local concentration effect12. However, additional constraints affecting molecular mobility and orientation (entropic effects) can also slow reaction rates13. Localization to a membrane surface almost certainly changes intrinsic reaction rates, but there is no simple way to quantitatively predict this effect. Solution kinetic measurements for reactions that naturally occur on membranes should be treated as qualitative indicators only.
An excellent example of an emergent property resulting from protein binding to membrane surfaces is the self-organizing wave pattern of bacterial Min proteins14 (Fig. 2). These proteins are crucial for accurate cell division and undergo spatiotemporal oscillations in vivo. A simple mixture of MinD, an ATPase, and MinE, a protein that stimulates the ATPase activity of MinD, has recently been observed to spontaneously organize into propagating wave patterns on supported membrane surfaces in vitro in the presence of ATP. Although the emergent behavior is complex, the system can be quantitatively understood in terms of a reaction-diffusion model for membrane-surface interactions14. Importantly, the basic combination of properties that lead to this behavior is not so difficult to achieve, suggesting that dynamic spatial organization could be involved in a wide range of signaling processes. Indeed, similar wave patterns of actin polymerization can be observed along the membranes of eukaryotic cells15.
Figure 2.

Self-organizing spiral waves formed by Min proteins on the membrane surface. Left, only labeled MinE is shown (MinD, 1 mM; MinE, 1 mM); right, labeled MinD and MinE are shown (MinD, 1 mM; MinE, 1 mM). Figure is adapted from previous work14.
More complex membrane surface effects arise in juxtacrine signaling, in which both ligand and receptor reside in apposed membranes16,17. In two recent studies, the binding kinetics between T-cell receptors (TCRs) and their peptide major histocompatibility complex (pMHC) ligands was measured in situ by single-molecule microscopy and Förster resonance energy transfer (FRET)18 and by direct mechanical assays19. Both in situ measurements revealed dissociation kinetics much faster than corresponding solution measurements. Disruption of actin polymers reversed this effect (as measured by the FRET assay). In general, actin interactions with membranes have an important role in determining the overall membrane mechanical stiffness. The above-mentioned results illustrate a localized mechanical effect of the membrane and cytoskeleton on protein binding kinetics. Importantly, antigen recognition by T cells is thought to depend largely on the details of TCR-pMHC binding kinetics.
It is clear that the membrane is not a homogeneous fluid. It is a nanometer-scale emulsion of lipids, cholesterol and proteins in intimate association with the cortical cytoskeleton20. The concept of the membrane raft, a phase-separated domain or composition fluctuation, has been a topic of much debate in the context of cell-membrane organization. The debate has been exacerbated by attempts to oversimplify the complex but reasonably well-understood phenomena of miscibility and demixing in liquids. A comprehensive review clarifying some of these issues has recently been published21.
Cell membrane lipids and cholesterol show miscibility phase transitions and even critical-point phenomena at physiological temperatures. This has been observed in giant unilamellar vesicles (GUVs) formed from synthetic or purified components22–24 as well as giant plasma membrane vesicles (GPMVs) budded directly from living cells without dissolution or reconstitution25,26. The mismatch of interaction energies between unsaturated lipids, saturated lipids and cholesterol in a bilayer-membrane configuration is responsible for these phenomena. One must recognize that interaction energies exist just the same in the membranes of living cells; whether the system is actually phase-separated depends sensitively on the details of the environment. The most important issue is that the system is somewhat near a miscibility phase transition, one way or the other. As such, composition fluctuations of the lipid/cholesterol component of cell membranes can occur with minimal free-energy costs (for an analysis of these effects in multicomponent membrane systems, see ref. 27). This is the fundamental reason why critical point fluctuations become macroscopic, leading to well-known phenomena such as critical opalescence—when water turns white due to large density fluctuations near its critical point (for a general introduction to critical-point behavior as it relates to biomembranes, see ref. 28).
In the cell membrane, the consequences of lipid/cholesterol near immiscibility are manifold. For one, this amplifies the response of the membrane to any sort of perturbation (for example, electrostatic, mechanical or interactions with proteins). Such effects have been experimentally observed for proteins binding to membrane surfaces29–31, including actin polymerization32 and for electric field–induced membrane reorganization33. More generally, the way the membrane solvates other components, such as membrane proteins, and even mediates their interactions with each other will be affected by these intrinsic properties. Specific consequences of this latter point are less well understood, but computational modeling methods suitable to examine such problems are emerging34–36.
In one intriguing example, a recent computational study of the A2A G protein–coupled receptor in membrane environments reveals a structural instability of helix II in cholesterol-poor membranes. Cholesterol interaction apparently stabilizes this helix and has been proposed as a possible explanation for the observation that the A2A receptor couples to G protein only in the presence of cholesterol37. Interactions of membrane lipids with ion channels are also of substantial interest38,39 and could be influenced by membrane demixing effects39,40.
The phase separation and composition fluctuations mentioned above represent spatial organizations that exist in the liquid state, but these are not static structures. Effective diffusion coefficients of lipids in live cell membranes are typically in the 0.1–1 μm2 s−1 range, about two orders of magnitude slower than three-dimensional diffusion in the cytoplasm41. Care is warranted when comparing two- and three-dimensional diffusion, as the two processes are quite different. First, the oft-cited Stokes-Einstein relation, which predicts an inverse scaling of the diffusion coefficient with particle radius, is the result of calculating hydrodynamic drag in three dimensions. It has no two-dimensional analog due to the fact that there are no solutions to the slow viscous flow equations in two dimensions (the Stokes paradox)42. Rates of two-dimensional diffusion in membranes result from the integration of a large number of environmental factors and can vary greatly. Diffusion is not a molecular property—it is a property of the entire system. Correspondingly, molecular mobility in membranes is not a reliable indicator of molecular structure (for example, degree of clustering). Single-particle tracking and mobility measurements have been successfully used to characterize overall membrane organization41,43.
Whereas membranes are fluid in two dimensions, they are elastic in the third dimension. Bending of membranes creates a mechanism for long-range lateral force transmittance, even though the membrane itself is liquid. Consequences of this include the exotic stripe and hexagonal patterns of domains that form in phase-separated membranes23,36,44–46 as well as protein-protein interactions and possibly regulation39,47,48. An important corollary of these observations is that forcibly bending membranes necessarily imposes differential forces on structures in the membrane. Experimental studies of this effect using curvature-patterned substrates to impose defined bending modulations on membranes indicate that coupling to membrane phase-separated domains is strong enough to occur under physiological geometries49. Indeed, distinctive effects of modest curvature modulation of live T-cell immunological synapses can be observed.
Membrane bending effects manifest within the T-cell immunological synapse, and all juxtacrine signaling interfaces for that matter, on the molecular scale as well. The simple fact that intermembrane receptor–ligand complexes have differing lengths (from ~10 to 50 nm or more) leads to interesting binding cooperativity that could be called allostery at a distance. Once one receptor–ligand complex has formed, the two membranes are pinned at that particular spacing in the vicinity of the complex. This both favors more binding of similar-size complexes nearby and disfavors interactions among complexes of different sizes. This effect can lead to pattern formation and a type of phase-separation phenomena of receptor spatial organization within intercellular junctions16,50,51.
There is arguably no aspect of membrane structure more pertinent to signal transduction than the fact that many receptors and signaling molecules form clusters. These have sometimes been equated with the lipid/cholesterol-mediated membrane rafts mentioned earlier. It is becoming increasingly clear that actin also plays a major role in cluster assembly as do protein-protein interactions between membrane proteins themselves. Whereas the balance of forces that lead to cluster formation and content determination remain a major open frontier in membrane-signaling research, the existence of such clusters is not in doubt. Many observations (some direct) report signaling cluster formation and sometimes also actin associatation, along with a specific connection to signaling function52–59. The most important aspect of signaling-cluster formation in membranes, from our present perspective, is that the assembly process itself becomes a highly capable mechanism of regulating signaling events, as illustrated in the specific examples that follow.
Activation of Ras by SOS at the membrane
The textbook model for how the small GTP-binding protein Ras is activated involves the recruitment of SOS from the cytoplasm to activated receptors at the membrane, where it can interact with Ras60,61 (Fig. 3a). This model is based on the idea that, if one protein, such as Ras, is localized to the membrane, then simply recruiting another protein, such as SOS, to the membrane can increase the interaction between the two proteins substantially12,62.
Figure 3.
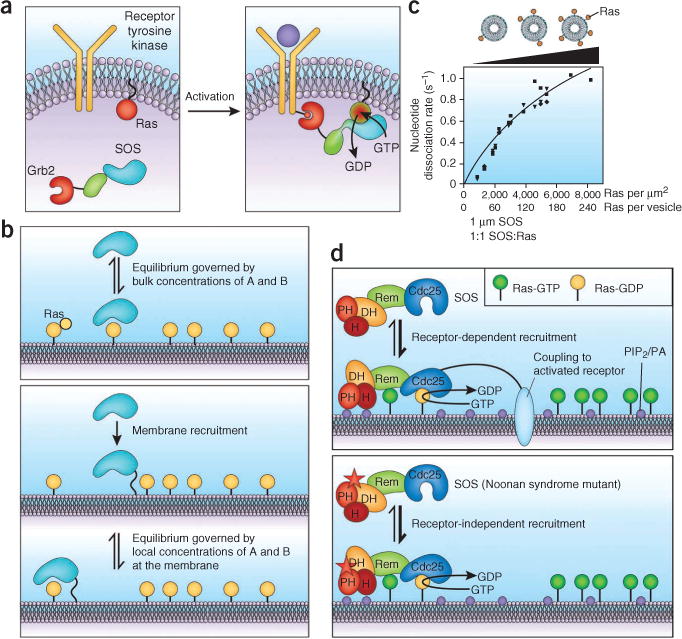
The activation of Ras by SOS. (a) In the textbook model for Ras activation, activated growth factor receptors recruit SOS to the membrane, where it finds and activates Ras. (b) Membranes enhance the binding affinity between two proteins only if both are localized to the same membrane. (c) The presence of the allosteric binding sites for Ras on SOS greatly increases the activity of the SOScat domain by recruiting SOS to the membrane. The specific activity of the SOScat domain is dependent on the Ras surface density. (d) The regulatory domains of SOS block the allosteric Ras binding site and prevent uncontrolled Ras activation by SOS. Activation involves the coordinated action of phospholipids at the membrane as well as receptor recruitment of SOS. The regulatory domains are destabilized by Noonan syndrome mutations, leading to constitutive activation. PA, phosphatidic acid; H, histone domain. Rem and Cdc25 are the catalytic modules of SOS. Panels c and d are adapted from previous work67.
Colocalization at the membrane can increase the local concentration of molecules by a factor of ~1,000 relative to the situation where they are free to move in the cytoplasm, thereby driving complex formation63,64. This increase in apparent affinity occurs only if both molecules in an interacting pair are localized to the membrane. If only one of the two proteins are membrane anchored, then dissociation of the complex will lead to the rapid diffusion away from the membrane of the cytosolic partner before it can be captured by other membrane-bound proteins (Fig. 3b). For this reason, one of the hallmarks of signal-transduction systems is the provision of multiple membrane attachment points, some of which are switchable, allowing the conversion of a weak interaction into a strong one upon activation of the signal.
A completely unexpected finding showed that the textbook model for the activation of Ras by SOS is far too simplistic. This was the discovery that SOS has a second binding site for Ras that is specific for nucleotide-bound Ras, with Ras-GTP binding to this site with higher affinity than does Ras-GDP65. Ras turns out to be an allosteric activator of SOS—the catalytic module of SOS (SOScat) is intrinsically inactive, and the binding of Ras to the distal site causes the active site to open. This sets up a positive feedback loop between Ras and SOS, because the initial weak activation of SOS by Ras-GDP is replaced by much stronger activation by Ras-GTP65,66.
The presence of an allosteric Ras binding site in SOScat means that SOS can be anchored to membranes directly by Ras. The importance of this effect is shown by experiments in which Ras is tethered to lipid bilayers, either in vesicles or on supported membranes67. Membrane tethering of Ras has a dramatic effect, increasing the specific activity of SOScat by up to 500-fold compared to reactions in which SOS and Ras are both in solution. As a result, the activity of SOS depends on the surface density of Ras molecules rather than the bulk concentration (Fig. 3c).
The uncontrolled activation of SOS by Ras is prevented by regulatory domains that are located N-terminal to the catalytic module. A Dbl homology–pleckstrin homology (DH-PH) module blocks the allosteric Ras binding site, locking SOS in the inactive conformation 68. A domain with histone folds, located before the DH-PH unit, further stabilizes the inactive conformation67,69,70. The DH-PH unit can be released by the interaction of the pleckstrin homology domain with PIP2 or phosphatidic acid in the membrane, but the N-terminal histone domain provides resistance to activation67,71. The histone domain itself has affinity for membranes and can bind to acidic phospholipids, such as PIP2 or phosphatidic acid70,72.
The activation of SOS is therefore likely to be due to the combined effects of the binding of SOS to activated receptors and by the action of phospholipids such as PIP2 and phosphatidic acid on the pleckstrin homology and histone domains of SOS, releasing the blockage of the allosteric site and enabling Ras to engage the allosteric site (Fig. 3d). One intriguing possibility is that the activation of SOS by growth factor receptors might simply be due to allosteric release of an as-yet-undefined inhibitory interaction involving the Grb2 binding site rather than recruitment to the membrane. This possibility is suggested by the fact that deletion of the Grb2 binding site in SOS activates SOS and causes cell transformation73,74, and it is possible that the anchorage of SOS to the membrane actually occurs through interactions with Ras and phospholipids rather than the receptor.
The experiments with Ras localized to membranes were key to understanding the effects of SOS mutations that are found in patients with Noonan syndrome, a developmental disease in which the Ras-mitogen-activated protein (MAP) kinase pathway is activated inappropriately75. SOS constructs with Noonan syndrome mutations are clearly activated in cell-based assays76,77, but similar constructs do not show detectable activation when the interaction between Ras and SOS is studied in vitro with both Ras and SOS in solution. In contrast, when Ras is tethered to lipid bilayers, the Noonan syndrome mutations lead to a robust activation of SOS67. Several of the Noonan syndrome mutations in SOS cause a release of the autoinhibitory interactions, enabling the pleckstrin homology domain to bind to PIP2. For example, mutation of Arg552 to glycine disrupts the internal engagement of the histone domain to the rest of the regulatory domains69, allowing PIP2 to activate SOS67.
The responsiveness of SOS to the surface density of Ras may be a feature that is correlated with the dynamic partitioning of activated Ras into small and densely packed nanoclusters on the cell surface56,78. The density of Ras within these nanoclusters is comparable to densities of membrane-bound Ras that lead to greatly enhanced SOS activation in the in vitro studies (see Fig. 3c). The interaction of Ras with the surface of the lipid bilayer is altered by the exchange of GDP by GTP, which might provide one mechanism for segregation79. The high density of Ras-GTP within these clusters might provide anchorage for SOS at the membrane even after withdrawal of the signal from the receptor, which is consistent with the ability of the allosteric Ras binding site in SOS to sustain Ras activation. The Ras effector Raf kinase is preferentially activated in these nanoclusters80, which might also be a consequence of the high density of Ras within them. The catalytic activity of Raf is switched on by dimerization, which would be promoted by the binding of Raf to clusters of Ras at high densities81.
The allosteric site on SOS is essential for the sustained activation of the MAP kinase pathway downstream of Ras66. The positive feedback loop between Ras and SOS has also been shown to underlie a ‘digital’ response to T-cell receptor activation, in which the MAP kinase pathway is either on or off82. By making SOS dependent on Ras for activity, the cell is able to ensure that only strong signals lead to sustained Ras activation and that, once activated, SOS continues to signal even upon receptor inactivation. This property of the Ras-SOS system has been implicated in the ability of thymocytes to distinguish between positively and negatively selecting antigenic peptides83.
EphA2 receptor tyrosine kinase triggering by ephrin-A1
The ephrins are a family of cell-surface proteins that are linked to the membrane either by a glycosylphosphatidylinositol (GPI) anchor (class A) or by a single transmembrane segment (class B)84. Ephrins are activating ligands for the Eph receptors. The Eph receptors are tyrosine kinases and are also divided into two classes, depending on the ephrins with which they interact. EphA2 signaling is important in development and shows functional alterations in cancer85. Ephrin-A1, the natural ligand for EphA2, is a GPI-linked protein, and the two interact in a juxtacrine configuration. Thus, numerous spatial and mechanical aspects of both cell membranes have a direct impact on the receptor-ligand interaction.
This system has recently been reconstituted between live EphA2-expressing cells and supported membranes displaying ephrin-A1 (ref 86). Ligand binding, receptor clustering and phosphorylation are observed. Additionally, EphA2 receptor clusters become associated with actin and are actively transported within the intercellular junction while still engaged with ephrin-A1 on the apposed membrane.
The use of supported membranes enabled application of the spatial mutation technique87,88. Physical barriers to lateral mobility are prefabricated onto the underlying substrate by electron-beam lithography or other patterning methods89. Supported membrane lipids and associated proteins diffuse freely within the membrane but cannot cross the grid patterns of barriers90. Local clustering and formation of signaling complexes is allowed, but long-range transport is guided by the barriers. EphA2 receptors and other associated molecules in the live cell become subject to these mobility constraints through their interaction with ephrin-A1 shown on the supported membrane (Fig. 4a).
Figure 4.
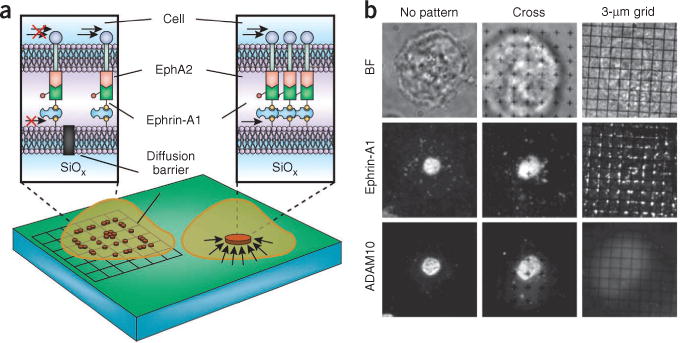
EphA2 spatial mutation experiment. (a) Schematic of a hybrid live cell–supported membrane junction between a live human epithelial cell expressing EphA2 and a patterned supported membrane expressing ephrin-A1. Physical barriers on the substrate block the lateral mobility of supported membrane molecules and associated molecules in the living cell. (b) Spatial mutation of EphA2– ephrin-A1 complexes alters recruitment of A disintegrin and metallopeptidase 10 (ADAM10). BF, bright field microscopy. Figure is adapted from previous work86.
The important observation from this study is that triggering the EphA2 receptor with ephrin-A1 produced different results depending on how ephrin-A1 transport was constrained. The EphA2-expressing cell gives the receptor a lateral tug via an acto-myosin contractility process, and the outcome of the signaling process apparently depends on whether the ephrin-A1 ligand in the apposed cell resists this applied force. Constraint patterns on the micron scale and below led to observable changes in cytoskeleton organization as well as the discrete switching off of A disintegrin and metalloprotease 10 (ADAM 10) recruitment (Fig. 4b). EphA2 phosphorylation itself was not much affected by the spatial mutation. Thus, somewhere between receptor phosphorylation and the more downstream events, there is a step in the signaling cascade that is sensitive to the large-scale spatial organization of the EphA2 receptors or possibly even the tensile forces acting on them. Elaborate cooperation between the cell membrane and cytoskeleton enables this signaling system to respond to differences in the mechanical aspects of the microenvironment.
Activation of actin nucleation by WASP and WAVE
The migration of cells across surfaces relies on the formation of filopodia and lamellipodia, which are dynamic protrusions on the leading edge of the cell that enable forward crawling movement. The assembly of highly branched actin networks is critical for the generation of these structures and for their continued extrusion, and the nucleation of actin is triggered by Ras-related GTP-binding proteins of the Rho family91,92. GTP-bound forms of cell-division control protein 42 (Cdc42) and Rac stimulate the Arp3/2 complex, which binds to the sides of actin filaments and promotes the nucleation of branching filaments93. This remodeling of actin structure and the ensuing change in cellular dynamics is one of the most profound consequences of the cellular response to input signals.
The coupling between GTP-loaded Cdc42 and Rac and the Arp3/2 complex is mediated by members of the Wiskott-Aldrich syndrome protein (WASP) family, which are actin nucleation–promoting factors94,95. The active elements of WASP proteins are VCA domains (also called WCA domains), which couple actin and the Arp3/2 complex, thereby promoting the growth of branched actin filaments. WASP and the related neuronal WASP protein (n-WASP) are normally autoinhibited because the VCA domain is covered by other elements of the WASP protein, and the binding of Cdc42 to WASP releases this inhibition96. The related Scar/WAVE proteins form heteropentameric assemblies, and their regulation is more complex.
The importance of the membrane in the activation of WASP was shown by studies that examined the role of a basic region within WASP that is located adjacent to the Cdc42-binding domain9. This polybasic region bound in a switch-like manner to vesicles containing PIP2 (Hill coefficient of ~20)97. It was shown recently that the Arp2/3 complex has two binding sites for VCA domains, resulting in a greatly enhanced affinity for dimerized forms of WASP and other VCA-containing proteins98. Thus, to the extent that higher densities of PIP2 result in colocalization and dimerization of WASP, potentiating cooperative binding to Arp3/2, this could have a profound effect on actin nucleation.
The WAVE protein differs from WASP in that it has no domain that interacts directly with its activator, Rho-GTP. In contrast to WASP, WAVE is intrinsically active, and when isolated from the WAVE complex, it is able to trigger the nucleation of branched actin filaments by Arp3/2 (ref. 99). Recent studies from three different labs have shown that the WAVE complex does not normally disassemble to release free WAVE and that the intact complex is intrinsically inhibited100–102.
One study100 showed that very high concentrations of the GTP-bound form of Rac activated their reconstituted form of the WAVE complex. This is consistent with an autoinhibitory mechanism in which the VCA domain is masked by other components of the WAVE complex. One of the components of the complex is a Rac effector protein, and presumably, activation of the WAVE complex involves displacement of the inhibitory interaction by the binding of Rac to the complex. The results of one of these studies102 are particularly interesting because they showed that the activation of the WAVE complex involves the coordinated action of at least three different events, which are the binding of the WAVE complex to Rac-GTP and to acidic phospholipids in the membrane, particularly PIP3, and the phosphorylation of the WAVE complex.
Rac-GTP does not by itself activate the WAVE complex when present at physiologically relevant concentrations102. Rac is normally prenylated, and the addition of prenylated and membrane-bound Rac also does not activate the WAVE complex. However, when vesicles containing 10% PIP3 were included in the assay along with prenylated Rac-GTP, essentially complete activation of the WAVE complex was obtained102. Although PIP3 is the most potent activator of the phospho-lipids tested, several other negatively charged phospholipids also activated the WAVE complex when included with Rac. Vesicles that contain only lipids with uncharged head groups, such as phosphatidylcholine or phosphatidylethanolamine, were unable to support the activation of the WAVE complex by membrane-bound Rac-GTP.
PIP3-containing vesicles cannot activate the WAVE complex in the absence of Rac, showing that a coordinated interaction between the WAVE complex and these two activators is required102. The activation was highly cooperative with respect to the concentration of the wave complex, with a Hill coefficient greater than 4.0, indicating that the activation process involves the formation of a higher-order assembly of WAVE complexes on the surface of the membrane (Fig. 5).
Figure 5.

Model for activation of the WAVE complex. The WAVE complex is intrinsically inactive. The first step in activation is phosphorylation, which primes the complex for interaction with the membrane. Binding to acidic phospholipids, particularly PIP3 and Rac-GTP, is required for activation. Figure is modified from previous work102.
Concluding remarks
Our understanding of signal transduction in biology has undergone an exponential expansion extending over the last several decades. As a result of this success, it is now becoming possible to tackle the problem of putting signaling mechanisms into context—and the first step of this inevitably involves cell membranes. The challenge is to develop quantitative mechanistic understandings of membrane effects on systems of signaling proteins in much the same way that structural biology has helped us to understand the chemistry of individual proteins based on their three-dimensional atomic structures. This will not happen with conventional tools of the trade. Fortunately, qualitatively new strategies are emerging on numerous fronts, and it is an exciting time to examine molecular mechanisms of signal transduction at the membrane.
Acknowledgments
We thank the members of our laboratories and our collaborators for many stimulating discussions and for sharing their insights into the topics described here.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Bhattacharyya RP, Remenyi A, Yeh BJ, Lim WA. Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu Rev Biochem. 2006;75:655–680. doi: 10.1146/annurev.biochem.75.103004.142710. [DOI] [PubMed] [Google Scholar]
- 2.Engelman DM. Membranes are more mosaic than fluid. Nature. 2005;438:578–580. doi: 10.1038/nature04394. [DOI] [PubMed] [Google Scholar]
- 3.Scott JD, Pawson T. Cell signaling in space and time: where proteins come together and when they’re apart. Science. 2009;326:1220–1224. doi: 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21:140–146. doi: 10.1016/j.ceb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- 6.Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab. 2010;298:E395–E402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newton AC. Lipid activation of protein kinases. J Lipid Res. 2009;50(Suppl):S266–S271. doi: 10.1194/jlr.R800064-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosse C, et al. PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol. 2010;11:103–112. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- 9.Prehoda KE, Scott JA, Mullins RD, Lim WA. Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science. 2000;290:801–806. doi: 10.1126/science.290.5492.801. [DOI] [PubMed] [Google Scholar]
- 10.McLaughlin S, Wang J, Gambhir A, Murray D. PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 11.Gambhir A, et al. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys J. 2004;86:2188–2207. doi: 10.1016/S0006-3495(04)74278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kholodenko BN, Hoek JB, Westerhoff HV. Why cytoplasmic signalling proteins should be recruited to cell membranes. Trends Cell Biol. 2000;10:173–178. doi: 10.1016/s0962-8924(00)01741-4. [DOI] [PubMed] [Google Scholar]
- 13.Jung H, Robison AD, Cremer PS. Multivalent ligand-receptor binding on supported lipid bilayers. J Struct Biol. 2009;168:90–94. doi: 10.1016/j.jsb.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loose M, Fischer-Friedrich E, Ries J, Kruse K, Schwille P. Spatial regulators for bacterial cell division self-organize into surface waves in vitro. Science. 2008;320:789–792. doi: 10.1126/science.1154413. [DOI] [PubMed] [Google Scholar]
- 15.Weiner OD, Marganski WA, Wu LF, Altschuler SJ, Kirschner MW. An actin-based wave generator organizes cell motility. PLoS Biol. 2007;5:e221. doi: 10.1371/journal.pbio.0050221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qi SY, Groves JT, Chakraborty AK. Synaptic pattern formation during cellular recognition. Proc Natl Acad Sci USA. 2001;98:6548–6553. doi: 10.1073/pnas.111536798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manz BN, Groves JT. Spatial organization and signal transduction at intercellular junctions. Nat Rev Mol Cell Biol. 2010;11:342–352. doi: 10.1038/nrm2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huppa JB, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang J, et al. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464:932–936. doi: 10.1038/nature08944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayor S, Rao M. Rafts: scale-dependent, active lipid organization at the cell surface. Traffic. 2004;5:231–240. doi: 10.1111/j.1600-0854.2004.00172.x. [DOI] [PubMed] [Google Scholar]
- 21.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 22.Veatch SL, Keller SL. Organization in lipid membranes containing cholesterol. Phys Rev Lett. 2002;89:268101. doi: 10.1103/PhysRevLett.89.268101. [DOI] [PubMed] [Google Scholar]
- 23.Baumgart T, Hess ST, Webb WW. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 2003;425:821–824. doi: 10.1038/nature02013. [DOI] [PubMed] [Google Scholar]
- 24.Kaizuka Y, Groves JT. Structure and dynamics of supported intermembrane junctions. Biophys J. 2004;86:905–912. doi: 10.1016/S0006-3495(04)74166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baumgart T, et al. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc Natl Acad Sci USA. 2007;104:3165–3170. doi: 10.1073/pnas.0611357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veatch SL, et al. Critical fluctuations in plasma membrane vesicles. ACS Chem Biol. 2008;3:287–293. doi: 10.1021/cb800012x. [DOI] [PubMed] [Google Scholar]
- 27.Groves JT, Boxer SG, McConnell HM. Electric field effects in multicomponent fluid lipid membranes. J Phys Chem B. 2000;104:119–124. [Google Scholar]
- 28.Honerkamp-Smith AR, Veatch SL, Keller SL. An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes. Biochim Biophys Acta. 2009;1788:53–63. doi: 10.1016/j.bbamem.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamazaki V, Sirenko O, Schafer RJ, Groves JT. Lipid mobility and molecular binding in fluid lipid membranes. J Am Chem Soc. 2005;127:2826–2827. doi: 10.1021/ja042430l. [DOI] [PubMed] [Google Scholar]
- 30.Forstner MB, Yee CK, Parikh AN, Groves JT. Lipid lateral mobility and membrane phase structure modulation by protein binding. J Am Chem Soc. 2006;128:15221–15227. doi: 10.1021/ja064093h. [DOI] [PubMed] [Google Scholar]
- 31.Hammond AT, et al. Crosslinking a lipid raft component triggers liquid ordered-liquid disordered phase separation in model plasma membranes. Proc Natl Acad Sci USA. 2005;102:6320–6325. doi: 10.1073/pnas.0405654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu AP, Fletcher DA. Actin polymerization serves as a membrane domain switch in model lipid bilayers. Biophys J. 2006;91:4064–4070. doi: 10.1529/biophysj.106.090852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Groves JT, Boxer SG, McConnell HM. Electric field-induced critical demixing in lipid bilayer membranes. Proc Natl Acad Sci USA. 1998;95:935–938. doi: 10.1073/pnas.95.3.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Meyer F, Smit B. Effect of cholesterol on the structure of a phospholipid bilayer. Proc Natl Acad Sci USA. 2009;106:3654–3658. doi: 10.1073/pnas.0809959106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ayton GS, Voth GA. Systematic multiscale simulation of membrane protein systems. Curr Opin Struct Biol. 2009;19:138–144. doi: 10.1016/j.sbi.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reynwar BJ, et al. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature. 2007;447:461–464. doi: 10.1038/nature05840. [DOI] [PubMed] [Google Scholar]
- 37.Lyman E, et al. A role for a specific cholesterol interaction in stabilizing the Apo configuration of the human A(2A) adenosine receptor. Structure. 2009;17:1660–1668. doi: 10.1016/j.str.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt D, Jiang QX, MacKinnon R. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature. 2006;444:775–779. doi: 10.1038/nature05416. [DOI] [PubMed] [Google Scholar]
- 39.Phillips R, Ursell T, Wiggins P, Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature. 2009;459:379–385. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Groves JT, Boxer SG, McConnell HM. Lateral reorganization of fluid lipid membranes in response to the electric feld produced by a buried charge. J Phys Chem B. 2000;104:11409–11415. [Google Scholar]
- 41.Saxton MJ, Jacobson K. Single-particle tracking: applications to membrane dynamics. Annu Rev Biophys Biomol Struct. 1997;26:373–399. doi: 10.1146/annurev.biophys.26.1.373. [DOI] [PubMed] [Google Scholar]
- 42.Saffman PG, Delbruck M. Brownian motion in biological membranes. Proc Natl Acad Sci USA. 1975;72:3111–3113. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kusumi A, et al. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu Rev Biophys Biomol Struct. 2005;34:351–378. doi: 10.1146/annurev.biophys.34.040204.144637. [DOI] [PubMed] [Google Scholar]
- 44.Rozovsky S, Kaizuka Y, Groves JT. Formation and spatio-temporal evolution of periodic structures in lipid bilayers. J Am Chem Soc. 2005;127:36–37. doi: 10.1021/ja046300o. [DOI] [PubMed] [Google Scholar]
- 45.Groves JT. Bending mechanics and molecular organization in biological membranes. Annu Rev Phys Chem. 2007;58:697–717. doi: 10.1146/annurev.physchem.56.092503.141216. [DOI] [PubMed] [Google Scholar]
- 46.Ursell TS, Klug WS, Phillips R. Morphology and interaction between lipid domains. Proc Natl Acad Sci USA. 2009;106:13301–13306. doi: 10.1073/pnas.0903825106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mouritsen OG, Bloom M. Mattress model of lipid-protein interactions in membranes. Biophys J. 1984;46:141–153. doi: 10.1016/S0006-3495(84)84007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ursell T, Huang KC, Peterson E, Phillips R. Cooperative gating and spatial organization of membrane proteins through elastic interactions. PLOS Comput Biol. 2007;3:e81. doi: 10.1371/journal.pcbi.0030081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parthasarathy R, Yu CH, Groves JT. Curvature-modulated phase separation in lipid bilayer membranes. Langmuir. 2006;22:5095–5099. doi: 10.1021/la060390o. [DOI] [PubMed] [Google Scholar]
- 50.Shaw AS, Dustin ML. Making the T cell receptor go the distance: a topological view of T cell activation. Immunity. 1997;6:361–369. doi: 10.1016/s1074-7613(00)80279-4. [DOI] [PubMed] [Google Scholar]
- 51.Weikl TR, Groves JT, Lipowsky R. Pattern formation during adhesion of multicomponent membranes. Europhys Lett. 2002;59:916–922. [Google Scholar]
- 52.Campi G, Varma R, Dustin ML. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med. 2005;202:1031–1036. doi: 10.1084/jem.20051182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yokosuka T, et al. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
- 54.DeMond AL, Mossman KD, Starr T, Dustin ML, Groves JT. T cell receptor microcluster transport through molecular mazes reveals mechanism of translocation. Biophys J. 2008;94:3286–3292. doi: 10.1529/biophysj.107.119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lillemeier BF, et al. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat Immunol. 2010;11:90–96. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henis YI, Hancock JF, Prior IA. Ras acylation, compartmentalization and signaling nanoclusters. Mol Membr Biol (Review) 2009;26:80–92. doi: 10.1080/09687680802649582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sourjik V, Berg HC. Functional interactions between receptors in bacterial chemotaxis. Nature. 2004;428:437–441. doi: 10.1038/nature02406. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki K, Fujiwara TK, Sanematsu F, Iino R, Edidin M, Kusumi A. GPI-anchored receptor clusters transiently recruit Lyn and Gα for temporary cluster immobilization Lyn activation: single-molecule tracking study 1. J Cell Biol. 2007;177:717–730. doi: 10.1083/jcb.200609174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suzuki K, Fujiwara TK, Edidin M, Kusumi A. Dynamic recruitment of phospholipase Cγ at transiently immobilized GPI-anchored receptor clusters induces IP3-Ca2+ signaling: single-molecule tracking study 2. J Cell Biol. 2007;177:731–742. doi: 10.1083/jcb.200609175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schlessinger J, Bar-Sagi D. Activation of Ras and other signaling pathways by receptor tyrosine kinases. Cold Spring Harb Symp Quant Biol. 1994;59:173–179. doi: 10.1101/sqb.1994.059.01.021. [DOI] [PubMed] [Google Scholar]
- 61.Downward J. The GRB2/Sem-5 adaptor protein. FEBS Lett. 1994;338:113–117. doi: 10.1016/0014-5793(94)80346-3. [DOI] [PubMed] [Google Scholar]
- 62.Haugh JM, Lauffenburger DA. Physical modulation of intracellular signaling processes by locational regulation. Biophys J. 1997;72:2014–2031. doi: 10.1016/S0006-3495(97)78846-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuriyan J, Eisenberg D. The origin of protein interactions and allostery in colocalization. Nature. 2007;450:983–990. doi: 10.1038/nature06524. [DOI] [PubMed] [Google Scholar]
- 64.McLaughlin S, Aderem A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci. 1995;20:272–276. doi: 10.1016/s0968-0004(00)89042-8. [DOI] [PubMed] [Google Scholar]
- 65.Margarit SM, et al. Structural evidence for feedback activation by Ras. GTP of the Ras-specifc nucleotide exchange factor SOS. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 66.Boykevisch S, et al. Regulation of ras signaling dynamics by Sos-mediated positive feedback. Curr Biol. 2006;16:2173–2179. doi: 10.1016/j.cub.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 67.Gureasko J, et al. Membrane-dependent signal integration by the Ras activator Son of sevenless. Nat Struct Mol Biol. 2008;15:452–461. doi: 10.1038/nsmb.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sondermann H, et al. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell. 2004;119:393–405. doi: 10.1016/j.cell.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 69.Sondermann H, Nagar B, Bar-Sagi D, Kuriyan J. Computational docking and solution x-ray scattering predict a membrane-interacting role for the histone domain of the Ras activator son of sevenless. Proc Natl Acad Sci USA. 2005;102:16632–16637. doi: 10.1073/pnas.0508315102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gureasko J, et al. Role of the histone domain in the autoinhibition and activation of the Ras activator Son of Sevenless. Proc Natl Acad Sci USA. 2010;107:3430–3435. doi: 10.1073/pnas.0913915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao C, Du G, Skowronek K, Frohman MA, Bar-Sagi D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat Cell Biol. 2007;9:706–712. doi: 10.1038/ncb1594. [DOI] [PubMed] [Google Scholar]
- 72.Yadav KK, Bar-Sagi D. Allosteric gating of Son of sevenless activity by the histone domain. Proc Natl Acad Sci USA. 2010;107:3436–3440. doi: 10.1073/pnas.0914315107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang W, et al. The Grb2 binding domain of mSos1 is not required for downstream signal transduction. Nat Genet. 1995;10:294–300. doi: 10.1038/ng0795-294. [DOI] [PubMed] [Google Scholar]
- 74.Corbalan-Garcia S, Margarit SM, Galron D, Yang SS, Bar-Sagi D. Regulation of Sos activity by intramolecular interactions. Mol Cell Biol. 1998;18:880–886. doi: 10.1128/mcb.18.2.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tartaglia M, Gelb BD. Noonan syndrome and related disorders: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2005;6:45–68. doi: 10.1146/annurev.genom.6.080604.162305. [DOI] [PubMed] [Google Scholar]
- 76.Roberts AE, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39:70–74. doi: 10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- 77.Tartaglia M, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39:75–79. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- 78.Plowman SJ, Muncke C, Parton RG, Hancock JF. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci USA. 2005;102:15500–15505. doi: 10.1073/pnas.0504114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abankwa D, et al. A novel switch region regulates H-ras membrane orientation and signal output. EMBO J. 2008;27:727–735. doi: 10.1038/emboj.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tian T, et al. Plasma membrane nanoswitches generate high-fdelity Ras signal transduction. Nat Cell Biol. 2007;9:905–914. doi: 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
- 81.Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009;461:542–545. doi: 10.1038/nature08314. [DOI] [PubMed] [Google Scholar]
- 82.Das J, et al. Digital signaling and hysteresis characterize ras activation in lymphoid cells. Cell. 2009;136:337–351. doi: 10.1016/j.cell.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Prasad A, et al. Origin of the sharp boundary that discriminates positive and negative selection of thymocytes. Proc Natl Acad Sci USA. 2009;106:528–533. doi: 10.1073/pnas.0805981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- 85.Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res. 2008;6:1795–1806. doi: 10.1158/1541-7786.MCR-08-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Salaita K, et al. Restriction of receptor movement alters cellular response: physical force sensing by EphA2. Science. 2010;327:1380–1385. doi: 10.1126/science.1181729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mossman KD, Campi G, Groves JT, Dustin ML. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 2005;310:1191–1193. doi: 10.1126/science.1119238. [DOI] [PubMed] [Google Scholar]
- 88.Groves JT. Spatial mutation of the T cell immunological synapse. Curr Opin Chem Biol. 2006;10:544–550. doi: 10.1016/j.cbpa.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 89.Castellana ET, Kataoka S, Albertorio F, Cremer PS. Direct writing of metal nanoparticle flms inside sealed microfluidic channels. Anal Chem. 2006;78:107–112. doi: 10.1021/ac051288j. [DOI] [PubMed] [Google Scholar]
- 90.Groves JT, Ulman N, Boxer SG. Micropatterning fluid lipid bilayers on solid supports. Science. 1997;275:651–653. doi: 10.1126/science.275.5300.651. [DOI] [PubMed] [Google Scholar]
- 91.Ridley AJ, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 92.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 93.Pollard TD. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu Rev Biophys Biomol Struct. 2007;36:451–477. doi: 10.1146/annurev.biophys.35.040405.101936. [DOI] [PubMed] [Google Scholar]
- 94.Stradal TE, et al. Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol. 2004;14:303–311. doi: 10.1016/j.tcb.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 95.Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8:37–48. doi: 10.1038/nrm2069. [DOI] [PubMed] [Google Scholar]
- 96.Leung DW, Rosen MK. The nucleotide switch in Cdc42 modulates coupling between the GTPase-binding and allosteric equilibria of Wiskott-Aldrich syndrome protein. Proc Natl Acad Sci USA. 2005;102:5685–5690. doi: 10.1073/pnas.0406472102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Papayannopoulos V, et al. A polybasic motif allows N-WASP to act as a sensor of PIP(2) density. Mol Cell. 2005;17:181–191. doi: 10.1016/j.molcel.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 98.Padrick SB, et al. Hierarchical regulation of WASP/WAVE proteins. Mol Cell. 2008;32:426–438. doi: 10.1016/j.molcel.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Machesky LM, et al. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci USA. 1999;96:3739–3744. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ismail AM, Padrick SB, Chen B, Umetani J, Rosen MK. The WAVE regulatory complex is inhibited. Nat Struct Mol Biol. 2009;16:561–563. doi: 10.1038/nsmb.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Derivery E, Lombard B, Loew D, Gautreau A. The Wave complex is intrinsically inactive. Cell Motil. Cytoskeleton. 2009;66:777–790. doi: 10.1002/cm.20342. [DOI] [PubMed] [Google Scholar]
- 102.Lebensohn AM, Kirschner MW. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. 2009;36:512–524. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]