

Abstract
Purpose
Cerebral hypoxia is a major cause of neonatal seizures, and can lead to epilepsy. Pathological anatomical and physiological changes in the dentate gyrus have been associated with epileptogenesis in many experimental models, as this region is widely believed to gate the propagation of limbic seizures. However, the consequences of hypoxia-induced seizures for the immature dentate gyrus have not been extensively examined.
Methods
Seizures were induced by global hypoxia (5–7% O2 for 15 minutes) in rat pups on postnatal day 10. Whole-cell voltage-clamp recordings were used to examine A-type potassium currents (IA) in dentate granule cells in hippocampal slices obtained 1–17 days after hypoxia treatment.
Key Findings
Seizure-inducing hypoxia resulted in decreased maximum IA amplitude in dentate granule cells recorded within the first week but not at later times after hypoxia treatment. The decreased IA amplitude was not associated with changes in the voltage-dependence of activation or inactivation removal, or in sensitivity to inhibition by 4-aminopyridine (4-AP). However, consistent with the role of IA in shaping firing patterns, we observed in the hypoxia group a significantly decreased latency to first spike with depolarizing current injection from hyperpolarized potentials. These differences were not associated with changes in resting membrane potential or input resistance, and were eliminated by application of 10 mM 4-AP.
Significance
Given the role of IA to slow action potential firing, decreased IA could contribute to long-term hippocampal pathology after neonatal seizure-inducing hypoxia by increasing DG cell excitability during a critical window of activity-dependent hippocampal maturation.
Keywords: A-current, epilepsy, hippocampal slice, patch clamp
Introduction
Cerebral hypoxia is a major cause of neonatal seizures, and such seizures increase the risk of later epilepsy (Aicardi and Chevrie 1970, Hauser, et al. 1993, Volpe 1981). Experimentally, hypoxia-induced seizures in neonatal rat are associated with immediate and sub-acute dysregulation of a number of ion channels that may render the hippocampus hyper-excitable during this critical maturational period (Jensen, et al. 1991, Rakhade, et al. 2008, Sanchez, et al. 2005, Sanchez, et al. 2007, Sanchez, et al. 2001, Zhang, et al. 2006). To date, most of these channelopathies have been identified in hippocampal CA1 pyramidal neurons, as these represent the major output of the hippocampus.
The dentate gyrus is in a unique position for information transfer from the entorhinal cortex to the hippocampus (Andersen, et al. 1966), and can gate seizure propagation from the entorhinal cortex to the hippocampus proper (Collins, et al. 1988) (Dreier and Heinemann 1991, Heinemann, et al. 1990, Walther, et al. 1986). A wealth of data from experimental models has suggested that compromise of this gating role of the dentate gyrus is critical to, or at least permissive of, long-term epileptogenesis (Dudek and Sutula 2007). Thus, aberrant channel function that renders principal dentate gyrus neurons hyper-excitable could contribute critically to epileptogenesis, but such changes consequent to neonatal seizure-inducing hypoxia remain largely unexplored.
Potassium channels critically regulate neuronal excitability (Pongs 1999), and their dysfunction can result in epileptiform network activity in vitro (Gabriel, et al. 2004, Traub, et al. 2001) and in seizures and epilepsy in vivo (Binder, et al. 2006, Misonou, et al. 2004, Pena, et al. 2002, Pena and Tapia 2000). In particular, the “A-current” (IA) is a rapidly inactivating current that contributes to action potential repolarization and promotes single spike firing (i.e., inhibits burst firing) in many types of neuron and cardiac muscle (Castro, et al. 2001, Pongs 1999). IA is mediated by channels that are composed of molecular subunits from the Kv1 and Kv4 potassium channel families (Birnbaum, et al. 2004). In the dentate gyrus, Kv1.1, Kv1.2, and Kv1.4 α-subunits are mainly expressedin the middle third of the molecular layer (Rhodes, et al. 1997, Sheng, et al. 1994) and Kv4.1, 4.2 and Kv4.3 expressed in the granule cell layer (Serodio and Rudy 1998, Sheng, et al. 1994). A-type potassium channel function and regulation has been reported to be altered in animal models of status epilepticus, and may critically contribute to brain hyper-excitability and epileptogenesis (Bernard, et al. 2004, Ruschenschmidt, et al. 2006). Whether IA channel function is altered after perinatal seizure-inducing hypoxia and could contribute to consequent epileptogenesis has not been investigated.
In the current study, we asked if IA is pathologically altered in hippocampal DGCs by neonatal seizure-inducing hypoxia, and may serve to alter their intrinsic firing properties, potentially contributing to limbic network hyper-excitability.
Materials and Methods
Animals
Male Long-Evans rat pups (Charles River), aged postnatal day 10–17, were used for these experiments. Litters were housed with their dam on a 12-hour light/dark cycle. All procedures were in accordance with NIH guidelines on the ethical use of experimental animals.
Hypoxia treatment
Rat pups on postnatal day 10 were taken in pairs, weighed, their rectal temperatures recorded, and each was individually placed on a heating pad in a custom-made plexiglass chamber with an O2 sensor mounted inside and three gas inlets for N2 infusion. One pup (littermate control) of each pair was placed in a chamber that remained open to room air for the duration of hypoxia treatment of the second pup. For the treated pup, the chamber O2 concentration was lowered by N2 infusion to 7% within 30–40 seconds, maintained at 6–7% for an additional 4 minutes, lowered to 5–6% for 8 minutes, and then lowered by 1% per minute until the pup became apneic for 30 seconds, at which time the chamber lid was removed for exposure to room air. The total time of hypoxia exposure was 14–16 minutes. Using this approach, hypoxia-treated pups typically exhibited spontaneous convulsive seizures lasting 10–60 seconds beginning 2–4 minutes after hypoxia onset, which recurred throughout hypoxia exposure and continued for several minutes after return to room air. Each pair of rat pups was earmarked, maintenance of core temperature verified, and immediately returned to their dam prior to beginning treatment of the next pair.
Slice preparation and in vitro whole-cell recordings
Rat pups were killed by decapitation under isoflurane anesthesia. The brains were removed and immediately placed into ice-cold oxygenated artificial cerebrospinal fluid (ACSF). A transverse razor cut was made anterior to the cerebellum and perpendicular to the midbrain, and the cut end was glued to the stage of a vibratome (Leica) for slicing. 370 μm slices were cut in cold, continuously oxygenated ACSF, and then incubated for at least one hour in a custom-made holding chamber filled with continuously oxygenated ACSF at room temperature.
For recording, slices were transferred to a 0.5 ml submersion chamber (Warner Instruments). The chamber was continuously superfused with ACSF (1–2 ml/min by gravity flow) at room temperature. Patch pipettes were drawn on a Flaming-Brown micropipette puller (Model P-97, Sutter Instrument Co, US) to 1–2 μm tip diameter using borosilicate glass (inner diameter of 0.68 mm, outer diameter of 1.20 mm) (A-M systems, Carlsborg, WA) and the tips polished with Microforge (MF-830, Narishige, Japan). Filled electrodes had resistances between 3 and 7 MΩ. Whole-cell patch-clamp recordings were obtained from granule cells of the dentate gyrus under visual guidance using infrared differential interference contrast microscopy (Zeiss Axioskop FS2 w/Dage-MTI camera). Voltage-clamp recordings were obtained using a Multiclamp 700A amplifier and analog signals were digitized using a Digidata 1322A (Molecular Devices) for acquisition to a Windows-based computer. Analog data were low-pass filtered at 2 kHz and digitized at 10 kHz. Voltage-clamp protocols were generated and data acquired to computer using Clampex (PClamp, Molecular Devices). Input and series resistances were monitored intermittently throughout experiments by applying 5 or 10 mV hyperpolarizing voltage steps from a holding potential of −65 mV. Initial series resistances were estimated to be less than 20 megohms, and data were discarded if series resistance changed by more than 20%. For all experiments, one cell was recorded per slice, and no more than two slices per animal were used.
IA was recorded with 1 μM tetrodotoxin (TTX) and 10 mM tetraethylammonium (TEA) activated under voltage-clamp by first stepping the command potential from a holding potential (VH) of −40 mV to −100 mV for 150 ms to remove steady-state inactivation, followed by a series of test voltage command steps from −40 mV to + 30 mV in 10 mV increments. IA was isolated by inserting an inactivating 50 ms voltage step to −40 mV immediately before each test step, and subtracting the currents with this protocol from those using the activation protocol (see Figure 1). To study the voltage-dependence of IA inactivation, 150 ms conditioning voltage steps from −160 mV to −40 mV (in 20 mV increments) were applied prior to an activation step to +30 mV. For IA isolation, a 50 ms step to −40 mV was applied between the hyperpolarizing conditioning step and the activation step (see Figure 2), and IA was obtained by subtracting the currents in the second protocol from the currents in the first. Boltzmann fits to pooled data were generated using the equation I/Imax = 1 / (1 + exp [(V − V1/2 )/k]).
Figure 1. Decreased IA in DGCs after seizure-inducing hypoxia.
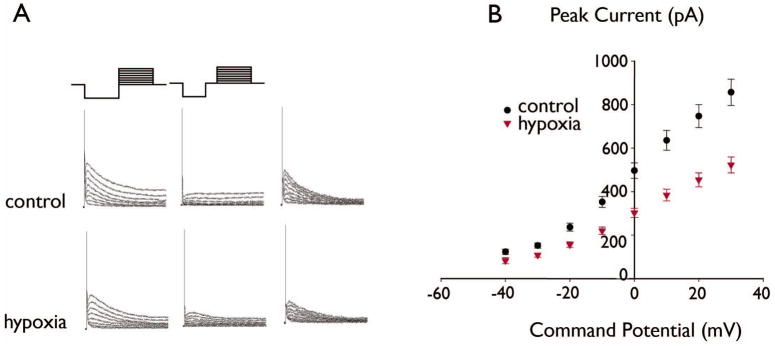
Figure 1 shows IA recorded in representative DGCs in slices from a control and hypoxia-treated rat. (A) Raw currents are shown in the left and middle panels, and subtracted currents are shown in the right panels (see Methods). For the cells shown, the peak subtracted amplitude activated at 30 mV was 963 pA for control compared to 608 pA for hypoxia. (B): Summary current-voltage relationships for IA in DGCs from the control and hypoxia-treated groups showed significantly decreased IA amplitudes in the hypoxia-treated group compared to controls (control, n=26; hypoxia, n=38, P<0.0001, ANOVA).
Figure 2. Voltage-dependence of IA was unchanged in DGCs after seizure-inducing hypoxia.

(A, B) Traces show the voltage-protocols and example subtracted currents used to measure the voltage-dependence of IA activation (A) and removal of inactivation (B). (C, D) Summary data and Boltzmann fits (see Methods) showed no differences between control and hypoxia-treated groups in the voltage-dependence of activation (C) or removal of steady-state inactivation (D).
For current-clamp recordings, RMP was measured without current injection, and steady hyperpolarizing current was injected to bring the membrane potential to −80 mV. Latencies to first spike in response to depolarizing current steps was measured using Clampfit as the time from the current step onset to the onset (foot) of the rising phase of the first action potential. Action potential amplitudes were measured as the voltage difference from the foot to the peak of the rising phase, and half-widths were measured as the spike width at the voltage halfway between the foot and peak of the first action potential.
Solutions
The ACSF consisted of (in mM): 126 NaCl, 3.3 KCl, 1.25 NaH2PO4, 1.3 MgSO4, 26 NaHCO3, 2 CaCl2 and 10 D-glucose, and was continuously bubbled with 95%O2/5%CO2. The internal patch solution consisted of (in mM): 145 K-gluconate,10 HEPES, 1 EGTA, 10 KCl, 0.1 CaCl2, 0.2 NaATP and 2 MgATP (pH adjusted to 7.3; 280–290 mOsm). Tetrodotoxin (TTX), 4-aminopyridine (4-AP), and tetraethylammonium (TEA) were dissolved in distilled water at high concentration, diluted to final concentration in ACSF, and applied by bath superfusion. All drugs were obtained from Sigma-Aldrich.
Data analysis and statistics
Data were analyzed off-line using Clampfit (Molecular Devices) on a Windows-based computer, and using AxoGraph 4.9 (Axon) and Igor Pro (Wavemetrics) on a Macintosh computer. Data are presented as mean ± SEM. Significant differences between control and experimental treatments were determined using two-way analysis of variance (ANOVA) for comparison of IA amplitudes, and Student’s t test for other measured parameters.
Results
Two types of dentate granule cells in the neonatal hippocampus
DGC properties were first characterized under current-clamp by examining voltage responses to injection of a family of 300-ms current pulses. According to the shape of the afterhyperpolarization following single action potentials (Dietrich, et al. 1999), two distinct response patterns were observed, termed Types I and II. Type I responses consisted of a delayed action potential followed by a distinctive large after-hyperpolarization (AHP), and were observed in the majority of cells in both the control (89/95, 93%) and hypoxia-treated (62/68, 91%) groups. Type II responses consisted of brief afterdepolarizations (ADPs) after single action potentials, and were observed in a small minority of neurons from both the control (6/95, 6.3%) and hypoxia-treated (6/68, 8.8%) groups. Given the low numbers of neurons encountered with Type II responses, these were excluded from further analyses. No significant differences were observed between control and hypoxia-treated groups in resting membrane potential (p=0.87), input resistance (P=0.37), or action potential amplitudes (P=0.79) (Table 1).
Table 1.
Classification of granule cells by AHP/ADP
AHP
|
ADP
|
|||
|---|---|---|---|---|
| Control (n=62) | Hypoxia (n=89) | Control (n=6) | Hypoxia (n=6) | |
| RMP (mV) | −70.3±0.48 | −69.4±0.41 | −71.6±1.62 | −71.3±1.11 |
| Rin (MΩ) | 1268.5±97.03 | 1217.9±108.02 | 412±51 | 227±84 |
| AP amplitude (mV) | 96±3.8 | 102.3±1.82 | 114.16±5.43 | 111.33±8.4 |
The top traces are current-clamp recordings of two types of dentate granule cells categorized according to their responses to 300 ms stepwise current injection (50 pA). Type I cells exhibited an afterhyperpolarization (AHP) after each action potential, whereas Type II cells showed an apparent afterdepolarization potential (ADP). There was no difference in resting membrane potential (RMP) (P=0.16), input resistance (IR) (P=0.99), or action potential (AP) amplitudes (P>0.05). (Values are means±SE; n is the number of cells.)
Decreased IA in dentate granule cells after seizure-inducing hypoxia
We observed decreased IA in DGCs recorded 1–5 days post-hypoxia without a change in voltage-dependence of activation. Mean IA amplitudes were significantly decreased in DGCs from the hypoxia-treated group compared to control (Figure 1; p < 0.0001, two-way ANOVA; n=26 cells control, 38 hypoxia). Boltzmann fits to the normalized currents showed no significant difference in the voltage-dependence of IA activation between the two groups (Figure 2C; p=0.85). For the control group, the mean voltage of half-activation (V-act1/2) was −4.6 ± 1.2 mV and average slope factor (K) was 13.7 ± 0.1 (n=6). For the hypoxia group, V-act1/2 = −7.4 ± 2.1 mV and K = −15.8 ± 0.7 (n=4).
The voltage-dependence of steady-state IA inactivation removal in DGCs was not significantly different between the two groups (Figure 2). In control DGCs, the mean voltage of half-inactivation removal (Vinact1/2) was −63.81 ± 3.31 mV and the mean slope factor was 16.32 ± 3.18 (n=7). In the hypoxia-treated group, mean Vinact1/2 was −57.97± 2.79 mV and the mean slope factor was 12.09 ± 2.65 (n=9). There was no significant difference in Vinact1/2 (P=0.12) or in slope factor (P=0.23) between the control and hypoxia-treated groups.
To examine the persistence of IA changes, we also examined IA in DG neurons in the second week post-hypoxia (P17-P21) when the sub-acute seizure activity from the initial insult would be expected to have ceased and in the third week post-hypoxia (P23-P27), when spontaneous seizures could be beginning (Rakhade, et al, 2011). In both groups, no difference was found between control (P17-P21, n=26; P23-P27, n=9) and hypoxia-treated groups (P17-P21, n=28; P23-P27, n=8) (data not shown). This indicated that the decrease in IA in DGCs persists for only up to approximately a week post-hypoxia.
Delayed but complete inhibition of IA in DGCs by 4-AP
Subtracted currents before and after 5 minutes of 10mM 4-AP application initially showed an apparent decrease in the 4-AP-induced IA inhibition after seizure-inducing hypoxia (Figure 3A). Pooled and normalized IA amplitudes confirmed that inhibition of IA was significantly greater in control DGCs compared to those from the hypoxia-treated group (Figure 3B, C). In control DGCs, 4-AP inhibited IA by 33.72 ± 2 % (n=11), and in DGCs from the hypoxia-treated group, 4-AP inhibited IA by 16 ± 4 % (n=9; p=0.03). However, continued application of 4-AP (at a constant fluid exchange rate of 2 ml/min) completely blocked IA in both control and hypoxia DGCs within 15 minutes (data not shown). Thus, although IA blockade by 4-AP was delayed in the hypoxia-treated group, complete inhibition was achieved in both groups with prolonged application. This raised the possibility of potential group differences in the binding kinetics of 4-AP, but importantly confirmed conditions sufficient to block IA in both groups for subsequent experiments.
Figure 3. 4-AP inhibition of IA in DGCs is slowed but intact after seizure-inducing hypoxia.

(A) Representative raw traces using the IA activation protocol with test step to +30 mV are shown before and after 5-minute application of 10 mM 4-AP. (B, C) Summary data illustrate greater apparent inhibition of IA by 4-AP after 5-minute application in the control group compared to the hypoxia-treated group. However, prolonged 4-AP application (>15 minutes) resulted in complete inhibition of IA in both groups (see Results).
Increased DGC membrane excitability after seizure-inducing hypoxia
IA inhibits burst firing by prolonging membrane hyperpolarization after single action potentials, thus delaying the onset of the next spike. To determine if decreased IA compromised this function in DGCs, we measured the latency to the first action potential relative to the onset of depolarizing current steps from either resting potential or −80 mV. DGCs in the hypoxia-treated group showed a shorter latency to the first action potential compared to controls when injected with depolarizing current from the more negative potential (Figure 4A). This latency was 19.8±4.69 ms for the control group (n=20), and was significantly decreased to 3.49±1.29 ms in hypoxia-treated group (n=9; P<0.01). Inhibition of IA by 15-minute application of 10 mM 4-AP significantly decreased the latency in the control group to 7.33 +/− 1.29 ms (P<0.01), but the decreased the latency in the hypoxia-treated group was not significant (P=0.15).
Figure 4. Decreased spike latency in DGCs upon rapid depolarization.

(A) Raw voltage traces are shown to compare spikes evoked by stepwise current injection from rest (solid lines) and from −80 mV (dotted lines). In the control neuron (left), current injection from −80 mV resulted in increased latency to first spike compared to rest consistent with the removal of IA inactivation by membrane hyperpolarization and subsequent IA activation. In the neuron from the hypoxia-treated group (right), there was no significant increase in latency to first spike upon current injection from −80 mV compared to rest consistent with decreased IA activation. (B) Decreased IA was associated with increased action potential duration in DGCs after hypoxia. The bar graphs show summary action potential half-widths (left) and amplitudes (right) in each group with and without 4-AP. Action potential half-widths were significantly increased in the hypoxia-treated group compared to controls consistent with slowed repolarization consequent to decreased IA. However, 4-AP comparably increased action potential half-widths in both groups, suggesting that other 4-AP-sensitive currents that underlie action potential repolarization were unaffected by hypoxia treatment. Action potential amplitudes were not significantly different between groups. (C) Summary bar graphs show no differences in resting membrane potential between control and hypoxia-treated groups. 4-AP comparably depolarized RMP in both groups (P<0.001, paired t-test), and this effect also was not different between the control and hypoxia-treated groups. (D) Current-clamp recordings before and after 4-AP showed that the 4-AP-induced depolarization of the resting potential observed in both groups was sufficient to elicit spontaneous action potential firing in DGC neurons.
IA also is be strongly activated during action potentials, and promotes the rapid repolarization of membrane potential. Thus, decreased IA would also be expected to slow repolarization and increase the half-width of action potentials. Accordingly, we found that action potentials recorded from DGCs in the hypoxia-treated group were broader than controls. Mean action potential half-widths in the control group were 2.1±0.08 ms (n=12) compared to 2.7±0.35 ms in the hypoxia-treated group (n=15), P<0.05. Action potential half-widths were comparably increased after 15 minutes of 4-AP application in both groups (Fig. 4B), with an 75% increase (1.59 ± 0.4 ms) in the control group (P<0.05), and an 82% increase (2.23 ± 0.5 ms) in the hypoxia-treated group (P<0.001).
We also recorded resting membrane potential and spontaneous action potentials in DGCs under current-clamp before and after 4-AP to investigate the resting contribution of IA to intrinsic excitability. 10 mM 4-AP depolarized the RMP from −69.91±1.01 to −64.17±0.73 mV (P<0.05) in the control group, and from −66.2 ±1.25 mV to −61.07 ±1.88 mV (P<0.05) in the hypoxia-treated group (Figure 4C). Thus, the effect of 4-AP on RMP was not different between the control and hypoxia-treated groups, suggesting that other 4-AP-sensitive currents predominate in setting the RMP. Despite the small change of RMP in DGL neurons, action potentials were induced after 4-AP treatment. The membrane potential of DGL neuron was set to −70 mV with positive or negative current injection, close to the reported average resting membrane potential. 10 mM 4-AP depolarized DGL neurons both in control and hypoxia group and most of the cells recorded (10/11) induced trains of action potential as shown in Figure 4D.
Discussion
The key findings of this study were that IA was significantly decreased in the majority of DGCs recorded 1–5 days after seizure-inducing hypoxia, and that this was associated with more rapid spiking in response to depolarizing current injection from hyperpolarized membrane potentials. Thus, dysregulation of intrinsic firing properties secondary to altered IA could contribute to hyper-excitability of dentate granule cells sub-acutely after neonatal seizure-inducing hypoxia, at a time of maturation when activity-dependent anatomical and synaptic patterning is highly labile. This conceivably could have pro-epileptogenic consequences or promote cognitive delay, but such functional consequences remain to be addressed. Our findings only establish altered IA in DGCs as a potential contributing factor to limbic pathophysiology consequent to seizure-inducing neonatal hypoxia.
A-type K+ channels are crucial determinants of neuronal firing patterns, and could be particularly important in controlling seizures. Reducing the amplitude of A-type currents (IA) increases seizure susceptibility (Juhng, et al. 1999) and lack of A-type Kv4.2 potassium channels contributes the increased excitability and decreased seizure thresholds in MAM-exposed rats (Castro, et al. 2001). Decreased IA in hippocampal CA1 pyramidal neurons after kainate-induced status epilepticus has been observed to increase distal dendritic excitability by allowing increased back-propagation of action potentials to apical dendrites (Bernard, et al. 2004). Notably, after pilocarpine-induced status epilepticus, IA in DGCs did not exhibit similar changes to that observed in other hippocampal subregions, and in fact, appeared resistant to seizure-associated changes (Ruschenschmidt, et al. 2006). Nonetheless, these authors reported powerful regulation of IA recovery from inactivation in DGCs by the intracellular redox milieu, which could be profoundly altered by hypoxia or prolonged seizures. In the current study, we examined the voltage-dependence but not the time course of inactivation removal (and at room temperature), and thus, did not address this mode of IA regulation. However, our finding of decreased channel availability in the hypoxia-treated group suggests that oxidation-promoted speeding of recovery from IA inactivation did not occur under our recording conditions. Under physiological conditions, it is possible that redox changes could oppose the observed down-regulation of IA in DGCs, but this has yet to be explored, as well as other mechanisms of posttranslational regulation that could be triggered by hypoxia, seizures, or both at this early maturational stage.
Sensitivity to 4-AP is characteristic of each IA subtype, but can vary depending on the molecular composition of the channels (Bekkers 2000, Korngreen and Sakmann 2000, Shibata, et al. 2000). The potassium channel blocker, 4-AP, significantly inhibited the IA-type channel recorded in DGCs in our study, further confirming that the transient potassium current is an IA-type current. Additionally, 4-AP eliminated the difference between groups in latency to spike onset upon rapid depolarizing current injection from a hyperpolarized potential, consistent with decreased IA as underlying the more rapid spiking in the hypoxia-treated group.
IA has been shown to contribute to the regulation of action potential firing rates as well as action potential duration (Honmou, et al. 1994, Kocsis, et al. 1982, Waddell and Lawson 1990). We observed that the duration of the first action potential was significantly increased by 4-AP, and consistently, AP duration was also prolonged in the hypoxia group compared to controls. Nonetheless, 4-AP had comparable effects on action potential duration and RMP in both the hypoxia and control groups, suggesting that additional 4-AP sensitive currents that contribute to these remained intact after hypoxia treatment.
Voltage-dependent potassium currents play important roles in regulating membrane excitability. IA has been implicated in determining the latency to first spike, and the threshold and repolarization of action potentials (Jagger and Housley 2002, Rudy 1988, Storm 1990). The first-spike latency in DGCs can be shaped by the expression, kinetic properties, and relative densities of IA. The presence of IA allows moderate hyperpolarizations (−80 mV) to evoke a well recognized increase in the delay to first spike. In the current study, the latency to the first spike was dramatically shortened in hypoxia DGCs compared to that of P10–15 control neurons. The shortened latency would certainly increase the probability of action potential generation and change the repetitive firing pattern of DGCs. These changes could have significant impact on the maturation of limbic circuits. Thus, the alterations in the excitability and firing pattern of DGCs in the neonatal brain could profoundly influence the activity of the whole hippocampus and vulnerability to epilepsy and other limbic pathologies.
Acknowledgments
This work was supported by NIH/NINDS R01 NS047385 (RMS); Epilepsy Foundation of America Postdoctoral Research Training Fellowship, National Natural Sciences Foundation of China No. 81100970 (BWP) and No. 81171127 (XHH); National Basic Research Program of China (No.2010CB529803 and No. 2011CDB465).
Footnotes
Ethical publication statement: We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Disclosure: None of the authors has any conflict of interest to disclose.
References
- Aicardi J, Chevrie JJ. Convulsive status epilepticus in infants and children. A study of 239 cases. Epilepsia. 1970;11:187–197. doi: 10.1111/j.1528-1157.1970.tb03880.x. [DOI] [PubMed] [Google Scholar]
- Andersen P, Holmqvist B, Voorhoeve PE. Entorhinal activation of dentate granule cells. Acta Physiol Scand. 1966;66:448–460. doi: 10.1111/j.1748-1716.1966.tb03223.x. [DOI] [PubMed] [Google Scholar]
- Bahremand A, Ziai P, Khodadad TK, Payandemehr B, Rahimian R, Ghasemi A, Ghasemi M, Hedayat T, Dehpour AR. Agmatine enhances the anticonvulsant effect of lithium chloride on pentylenetetrazole-induced seizures in mice: Involvement of L-arginine/nitric oxide pathway. Epilepsy Behav. 2010;18:186–192. doi: 10.1016/j.yebeh.2010.04.014. [DOI] [PubMed] [Google Scholar]
- Bekkers JM. Distribution and activation of voltage-gated potassium channels in cell-attached and outside-out patches from large layer 5 cortical pyramidal neurons of the rat. J Physiol. 2000;525(Pt 3):611–620. doi: 10.1111/j.1469-7793.2000.t01-2-00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53:631–636. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- Birnbaum SG, Varga AW, Yuan LL, Anderson AE, Sweatt JD, Schrader LA. Structure and function of Kv4-family transient potassium channels. Physiol Rev. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- Castro PA, Cooper EC, Lowenstein DH, Baraban SC. Hippocampal heterotopia lack functional Kv4.2 potassium channels in the methylazoxymethanol model of cortical malformations and epilepsy. J Neurosci. 2001;21:6626–6634. doi: 10.1523/JNEUROSCI.21-17-06626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins WF, 3rd, Davis BM, Mendell LM. Modulation of EPSP amplitude during high frequency stimulation depends on the correlation between potentiation, depression and facilitation. Brain Res. 1988;442:161–165. doi: 10.1016/0006-8993(88)91445-x. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Milroy A, Jan YN, Jan LY, Lowenstein DH. Presynaptic localization of Kv1.4-containing A-type potassium channels near excitatory synapses in the hippocampus. J Neurosci. 1998;18:965–974. doi: 10.1523/JNEUROSCI.18-03-00965.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich D, Clusmann H, Kral T, Steinhauser C, Blumcke I, Heinemann U, Schramm J. Two electrophysiologically distinct types of granule cells in epileptic human hippocampus. Neuroscience. 1999;90:1197–1206. doi: 10.1016/s0306-4522(98)00574-0. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Heinemann U. Regional and time dependent variations of low Mg2+ induced epileptiform activity in rat temporal cortex slices. Exp Brain Res. 1991;87:581–596. doi: 10.1007/BF00227083. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Sutula TP. Epileptogenesis in the dentate gyrus: a critical perspective. Prog Brain Res. 2007;163:755–773. doi: 10.1016/S0079-6123(07)63041-6. [DOI] [PubMed] [Google Scholar]
- Gabriel S, Njunting M, Pomper JK, Merschhemke M, Sanabria ER, Eilers A, Kivi A, Zeller M, Meencke HJ, Cavalheiro EA, Heinemann U, Lehmann TN. Stimulus and potassium-induced epileptiform activity in the human dentate gyrus from patients with and without hippocampal sclerosis. J Neurosci. 2004;24:10416–10430. doi: 10.1523/JNEUROSCI.2074-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia. 1993;34:453–468. doi: 10.1111/j.1528-1157.1993.tb02586.x. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Clusmann H, Dreier J, Stabel J. Changes in synaptic transmission in the kindled hippocampus. Adv Exp Med Biol. 1990;268:445–450. doi: 10.1007/978-1-4684-5769-8_49. [DOI] [PubMed] [Google Scholar]
- Honmou O, Utzschneider DA, Rizzo MA, Bowe CM, Waxman SG, Kocsis JD. Delayed depolarization and slow sodium currents in cutaneous afferents. J Neurophysiol. 1994;71:1627–1637. doi: 10.1152/jn.1994.71.5.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagger DJ, Housley GD. A-type potassium currents dominate repolarisation of neonatal rat primary auditory neurones in situ. Neuroscience. 2002;109:169–182. doi: 10.1016/s0306-4522(01)00454-7. [DOI] [PubMed] [Google Scholar]
- Jensen FE, Applegate CD, Holtzman D, Belin TR, Burchfiel JL. Epileptogenic effect of hypoxia in the immature rodent brain. Ann Neurol. 1991;29:629–637. doi: 10.1002/ana.410290610. [DOI] [PubMed] [Google Scholar]
- Juhng KN, Kokate TG, Yamaguchi S, Kim BY, Rogowski RS, Blaustein MP, Rogawski MA. Induction of seizures by the potent K+ channel-blocking scorpion venom peptide toxins tityustoxin-K(alpha) and pandinustoxin-K(alpha) Epilepsy Res. 1999;34:177–186. doi: 10.1016/s0920-1211(98)00111-9. [DOI] [PubMed] [Google Scholar]
- Kocsis JD, Waxman SG, Hildebrand C, Ruiz JA. Regenerating mammalian nerve fibres: changes in action potential waveform and firing characteristics following blockage of potassium conductance. Proc R Soc Lond B Biol Sci. 1982;217:77–87. doi: 10.1098/rspb.1982.0095. [DOI] [PubMed] [Google Scholar]
- Korngreen A, Sakmann B. Voltage-gated K+ channels in layer 5 neocortical pyramidal neurones from young rats: subtypes and gradients. J Physiol. 2000;525(Pt 3):621–639. doi: 10.1111/j.1469-7793.2000.00621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A, Wooltorton JR, Watkins CS. Voltage-activated potassium channels in mammalian neurons and their block by novel pharmacological agents. Gen Pharmacol. 1998;30:13–24. doi: 10.1016/s0306-3623(97)00034-7. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Pena F, Bargas J, Tapia R. Paired pulse facilitation is turned into paired pulse depression in hippocampal slices after epilepsy induced by 4-aminopyridine in vivo. Neuropharmacology. 2002;42:807–812. doi: 10.1016/s0028-3908(02)00024-2. [DOI] [PubMed] [Google Scholar]
- Pena F, Tapia R. Seizures and neurodegeneration induced by 4-aminopyridine in rat hippocampus in vivo: role of glutamate- and GABA-mediated neurotransmission and of ion channels. Neuroscience. 2000;101:547–561. doi: 10.1016/s0306-4522(00)00400-0. [DOI] [PubMed] [Google Scholar]
- Pongs O. Voltage-gated potassium channels: from hyperexcitability to excitement. FEBS Lett. 1999;452:31–35. doi: 10.1016/s0014-5793(99)00535-9. [DOI] [PubMed] [Google Scholar]
- Rakhade SN, Zhou C, Aujla PK, Fishman R, Sucher NJ, Jensen FE. Early alterations of AMPA receptors mediate synaptic potentiation induced by neonatal seizures. J Neurosci. 2008;28:7979–7990. doi: 10.1523/JNEUROSCI.1734-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade SN, Klein PM, Huynh T, Hilario-Gomez C, Kosaras B, Rotenberg A, Jensen FE. Development of later life spontaneous seizures in a rodent model of hypoxia-induced neonatal seizures. Epilepsia. 2011;52(4):753–765. doi: 10.1111/j.1528-1167.2011.02992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes KJ, Strassle BW, Monaghan MM, Bekele-Arcuri Z, Matos MF, Trimmer JS. Association and colocalization of the Kvbeta1 and Kvbeta2 beta-subunits with Kv1 alpha-subunits in mammalian brain K+ channel complexes. J Neurosci. 1997;17:8246–8258. doi: 10.1523/JNEUROSCI.17-21-08246.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B. Diversity and ubiquity of K channels. Neuroscience. 1988;25:729–749. doi: 10.1016/0306-4522(88)90033-4. [DOI] [PubMed] [Google Scholar]
- Ruschenschmidt C, Chen J, Becker A, Riazanski V, Beck H. Functional properties and oxidative modulation of A-type K+ currents in hippocampal granule cells of control and chronically epileptic rats. Euro J Neurosci. 2006;23:675–685. doi: 10.1111/j.1460-9568.2006.04608.x. [DOI] [PubMed] [Google Scholar]
- Sanchez RM, Dai W, Levada RE, Lippman JJ, Jensen FE. AMPA/kainate receptor-mediated downregulation of GABAergic synaptic transmission by calcineurin after seizures in the developing rat brain. J Neurosci. 2005;25:3442–3451. doi: 10.1523/JNEUROSCI.0204-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez RM, Justice JA, Zhang K. Persistently decreased basal synaptic inhibition of hippocampal CA1 pyramidal neurons after neonatal hypoxia-induced seizures. Dev Neurosci. 2007;29:159–167. doi: 10.1159/000096220. [DOI] [PubMed] [Google Scholar]
- Sanchez RM, Koh S, Rio C, Wang C, Lamperti ED, Sharma D, Corfas G, Jensen FE. Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia-induced seizures. J Neurosci. 2001;21:8154–8163. doi: 10.1523/JNEUROSCI.21-20-08154.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serodio P, Rudy B. Differential expression of Kv4 K+ channel subunits mediating subthreshold transient K+ (A-type) currents in rat brain. J Neurophysiol. 1998;79:1081–1091. doi: 10.1152/jn.1998.79.2.1081. [DOI] [PubMed] [Google Scholar]
- Sheng M, Tsaur ML, Jan YN, Jan LY. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. 1992;9:271–284. doi: 10.1016/0896-6273(92)90166-b. [DOI] [PubMed] [Google Scholar]
- Sheng M, Tsaur ML, Jan YN, Jan LY. Contrasting subcellular localization of the Kv1.2 K+ channel subunit in different neurons of rat brain. J Neurosci. 1994;14:2408–2417. doi: 10.1523/JNEUROSCI.14-04-02408.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata R, Nakahira K, Shibasaki K, Wakazono Y, Imoto K, Ikenaka K. A-type K+ current mediated by the Kv4 channel regulates the generation of action potential in developing cerebellar granule cells. J Neurosci. 2000;20:4145–4155. doi: 10.1523/JNEUROSCI.20-11-04145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- Traub RD, Bibbig R, Piechotta A, Draguhn R, Schmitz D. Synaptic and nonsynaptic contributions to giant ipsps and ectopic spikes induced by 4-aminopyridine in the hippocampus in vitro. J Neurophysiol. 2001;85:1246–1256. doi: 10.1152/jn.2001.85.3.1246. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Neurology of the newborn. Major Probl Clin Pediatr. 1981;22:1–648. [PubMed] [Google Scholar]
- Waddell PJ, Lawson SN. Electrophysiological properties of subpopulations of rat dorsal root ganglion neurons in vitro. Neuroscience. 1990;36:811–822. doi: 10.1016/0306-4522(90)90024-x. [DOI] [PubMed] [Google Scholar]
- Walther H, Lambert JD, Jones RS, Heinemann U, Hamon B. Epileptiform activity in combined slices of the hippocampus, subiculum and entorhinal cortex during perfusion with low magnesium medium. Neurosci Lett. 1986;69:156–161. doi: 10.1016/0304-3940(86)90595-1. [DOI] [PubMed] [Google Scholar]
- Winkelman DL, Beck CL, Ypey DL, O’Leary ME. Inhibition of the A-type K+ channels of dorsal root ganglion neurons by the long-duration anesthetic butamben. J Pharmacol Exp Ther. 2005;314:1177–1186. doi: 10.1124/jpet.105.087759. [DOI] [PubMed] [Google Scholar]
- Zhang K, Peng BW, Sanchez RM. Decreased I-H in hippocampal area CA1 pyramidal neurons after perinatal seizure-inducing hypoxia. Epilepsia. 2006;47:1023–1028. doi: 10.1111/j.1528-1167.2006.00574.x. [DOI] [PubMed] [Google Scholar]