

Abstract
The soluble epoxide hydrolase (sEH) is a key enzyme in the metabolism of epoxy-fatty acids, signaling molecules involved in numerous biologies. Toward finding novel inhibitors of sEH, a library of known drugs was tested for inhibition of sEH. We found that fulvestrant, an anticancer agent, is a potent (KI = 26 nM) competitive inhibitor of sEH. From this observation we found that alkyl-sulfoxides represent a new kind of pharmacophore for the inhibition of sEH.
Keywords: soluble epoxide hydrolase, epoxy-eicosatrienoic acids, fulvestrant, competitive inhibition, X-ray structure
In mammals, epoxides of arachidonic acids (called epoxy-eicosatrienoic acids or EETs) and of other fatty acids are important lipid mediators that have key roles in the regulation of hypertension, inflammation, and other cardiovascular related diseases as well as in modulating both inflammatory and neuropathic pain.1 However, endogenous metabolism of these epoxy-fatty acids to their corresponding hydrated products by soluble epoxide hydrolase (sEH EC 3.3.2.10) generally reduces these biological activities.2–4 Both in vitro and in vivo studies have demonstrated that the anti-hypertensive and cardio protective effects mediated by the EETs are inversely dependent on the extent of sEH hydrolysis of the EETs.2–4 Thus, maintaining the in vivo concentration of EETs through sEH inhibition is a promising therapeutic pathway to treat cardiovascular and other diseases.2–4
Based on the catalytic mechanism and structure of sEH,3,5 1,3-disubstituted ureas, amides and carbamate were found to be potent and stable competitive inhibitors of sEH.6 Over the years, more potent urea-inhibitors that display better solubility and availability as well as significant biological activities in both in vitro and in vivo models were obtained.7 Interestingly, beside the effort of many groups and the screening of several libraries of small chemicals,8–12 to date only the Merck group was successful in replacing the urea or amide pharmacophore with a benzisoxazole, which inhibit the sEH in the nanomolar range.10 Recently, chalcone13 and benzoxazolone14 derivatives as well as salvionic acid A15 were reported to inhibit sEH; however, these compounds are at least a 1,000-fold less potent than the best sEH inhibitors available.7 These results underline the difficulty to discover novel pharmacophores that could potently inhibit the human sEH.
The urea function is not used often as pharmacophore for drugs, while amides are used more frequently. Interestingly, an anti-cancer agent (sorafenib), and an anti-bacterial agent (triclocarban) that are both containing ureas as central pharmacophore, have, beside their targeted action, potent sEH inhibitory activity leading to a reduction in pain and inflammation.16,17 In addition, we observed that co-inhibition of sEH with cyclooxygenase (COX), lipoxygenase or phosphodiesterase (PDE) has beneficial effects.4 The co-inhibition of sEH and COX-2 strongly reduces inflammation and pain, but also reduces dose and side effects of COX-2 inhibitors.18–20 Co-administration of PDE4 inhibitors enhances the action of sEH inhibitors and EETs in reducing allodynia and hyperalgesia of the enhanced pain state.21 In general, sEH inhibition appears to enhance the action and improve the safety profile of current pharmaceuticals.4 Thus, it is attractive to test the concept that active pharmaceutical ingredients with sEH inhibition will be more active and safer.
Toward testing this hypothesis, we, thus, report herein the screening of a non-targeted commercial chemical library of drugs: the Pharmakon 1600 (MicroSource Discovery Systems, Gaylordsville, CT) for sEH inhibition. This library is a unique collection of 1,600 known drugs (20 plates of 80 compounds at 10 mM in DMSO) from US and International Pharmacopeia, which have all reached clinical evaluation and not simply demonstrated biological activity experimentally. Most of the constituent drugs are still in the market.
Using recombinant purified human sEH and the fluorescent substrate PHOME ((3-phenyl-oxiranyl)-acetic acid cyano-(6-methoxy-naphthalen-2-yl)-methyl ester), we screened the library at a final concentration of 10 μM, following a previously described methodology.22 Overall, we obtained on average for the 20 plates S/B = 7.5 ± 0.4, S/N = 107 ± 25 and Z' = 0.90 ± 0.03 indicating that the assay performed very well. As shown on Fig. 1A, most compounds did not yield any significant inhibition (< 20%) of the human sEH, and only ten compounds gave a strong inhibition (> 80% inhibition). To confirm the potency of these compounds, fresh solutions in DMSO were prepared, and their inhibitory potency (IC50s) were determined using CMNPC (cyano(6-methoxy-naphthelen-2-yl)methyl trans-[(3-phenyl-oxiran-2-yl)methyl] carbonate)as substrate.23 As shown on Fig. 1B, only two compounds gave potent inhibition: fulvestrant (#15G10, IC50 = 6 ± 1 nM), and ebselen (#18H06, IC50 = 2,200 ± 300 nM). We recently reported that ebselen irreversibly inhibit sEH through binding to its N-terminal domain.24,25 For the other positive hit (fulvestrant), to our knowledge, this is the first report of inhibition of sEH by a compound that contains a sulfoxide as central pharmacophore. The observed potency of fulvestrant is of the same order as potent urea/amide containing inhibitors developed over the past decade.7
Figure 1.

A Primary screening results of the Pharmacon library. Percent of sEH inhibition for each compound tested at 10 μM. Compounds that gave more than 80% inhibition (dashed line) were selected for secondary screening. B. Secondary screening results. IC50s were measured using CMNPC as substrate.23 Results are mean ± standard deviation of at least three separate measurements.
To test the selectivity of fulvestrant toward sEH, we tested its ability to inhibit a series of related enzymes: sEH-phosphatase activity as well as human microsomal EH, human alkaline phosphatase and a series of esterases and amidases.26 Fulvestrant was found to only inhibit the sEH. We then tested if fulvestrant inhibition was reversible. After incubating 20 nM of human sEH with 10 μM of fulvestrant for 15 min, the enzyme was dialyzed to remove the small molecule. Following this procedure, we were able to recover more than 80% of the sEH activity, suggesting that fulvestrant inhibition of the sEH is reversible. To define the potency of fulvestrant as a sEH inhibitor, we determined its dissociation constant (KI; see Fig. 2) using a radioactivity-based assay.16 The fact that the KIapp values increase with the concentration of substrate (Fig. 2 insert) strongly suggests that fulvestrant is a competitive inhibitor of the sEH like the urea/amide inhibitors.3,6,7 Fulvestrant has a KI of 26 nM, which is approximately 10-fold higher than that of the very potent t-AUCB (KI = 1.5 nM), the compound used as positive control for the screening, but similar to the KI of other potent sEH inhibitors (7 < KI < 57 nM),16 especially of the one (TPAU) used in phase I and II clinical trials.27
Figure 2.
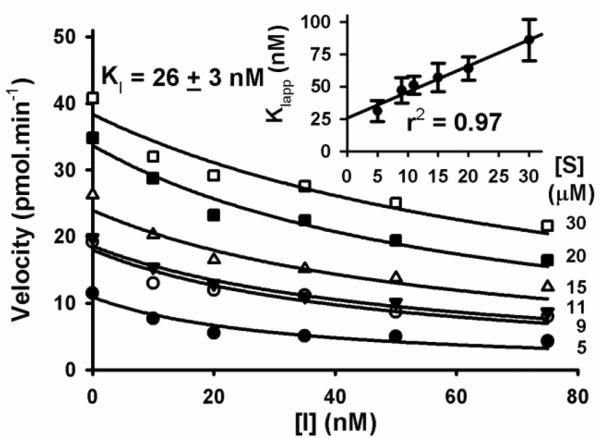
Determination of the KI of fulvestrant with the human sEH (1 nM) Using [3H]-tDPPO as substrate. For each substrate concentration (3.6 to 30.0 μM), the velocity is plotted as a function of sEH inhibitor concentration (0 to 75 nM), allowing the determination of an apparent inhibition constant (KIapp). KIapps are plotted as a function of the substrate concentration (insert). For [S] = 0, a KI value of 26 nM was found.
To confirm the binding site and mode of action of fulvestrant, we obtained crystals of the human sEH complexed with fulvestrant (see supplementary data for method and crystal details). The X-ray structure of the crystal (Fig. 3) showed that fulvestrant binds in the hydrolase catalytic pocket of sEH. Furthermore, the oxygen atom of the sulfoxide pharmacophore makes hydrogen bonds with two key catalytic residues: tyrosines 383 and 466 (Fig. 3), in a manner very similar to the way the carbonyl of urea inhibitors bind at the active site of sEH.3,5,7 Interestingly, interaction between the sulfur atom of fulvestrant and Asp335 is observed that is similar to the bond observed between the Nh groups of the urea and the catalytic Asp335 residue.3,5,7 Sulfoxides are highly polar groups with a strong positive charge on the sulfur atom,28 thus enhancing the feasibility of a strong bond between the sulfur atom and the enzyme catalytic Asp335. These observations confirm that fulvestrant is a competitive inhibitor of sEH. Close examination of the structure revealed that the fluorinated alkyl chain of fulvestrant occupies the smaller of the two lipophilic pockets in the sEH active site, while the steroid part of the molecule is directed towards the large deep pocket that opens toward solvent. Interestingly, while too far for establishing a hydrogen bond (5.37 Å) the hydroxyl group on the aromatic ring is pointing toward the amide nitrogen of Asn472. On the other end, the cyclopentyl ring of fulvestrant is at the entrance of the catalytic tunnel with the hydroxyl group on this ring facing outward toward the solvent. The rings are enveloped by numerous hydrophobic residues underlying the strong hydrophobicity of the pocket. Finally, the observation that the ring structure of fulvestrant can fit inside the catalytic cavity of sEH is quite unexpected and suggest that sEH can bind and probably hydrolyze, substrates that are larger than the ones for which it is reported to be active on.29
Figure 3.
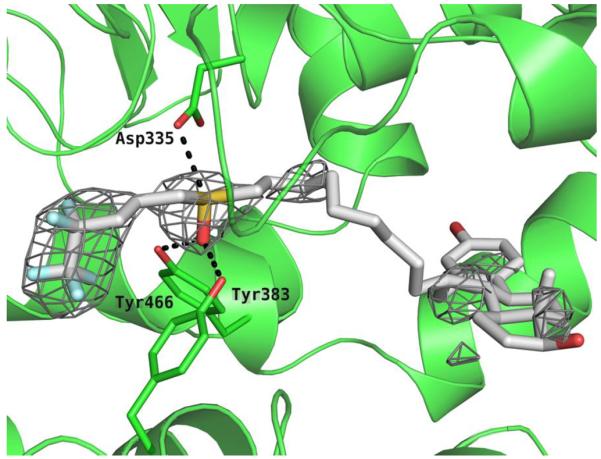
The active site of the hydrolase domain of human sEH complexed with fulvestrant. The Fo-Fc electron density map is contoured at 2.5σ. Non-bonded interactions are indicated by dashes. Coordinates of the complex deposited as 4J03 with the Protein Data Bank. The image was produced using PYMOL.
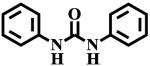
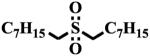
To test if the observed inhibition of sEH is specific to the sulfoxide group of fulvestrant, we measured the potency of several sulfoxides (Table 1). Of the three sulfoxides tested (1–3), two of them (2 and 3) gave significant inhibition of sEH, confirming that sulfoxides in general could inhibit sEH. Interestingly, replacement of aryl groups by alkyl groups in compounds 1 to 3 yields more potent inhibitors. The difference in potency between compounds 1 and 3 is probably due to steric effects by rigid benzyl group, because the catalytic tunnel is relatively constricted around the catalytic residues.3,5 The most potent compound tested 3 is around 100-fold less potent than fulvestrant, suggesting that the hydrophobic interactions from the steroid rings and the pentafluoropentane play a significant part in the binding of fulvestrant. Compared to 1, the corresponding amide 4 and urea 5 are significantly more potent, suggesting that the amide and urea form a stronger network of interactions with the enzyme than the sulfoxide. This is probably due to the absence of hydrogen bonds to stabilize the dipole-dipole interaction of the sulfur atom with the catalytic carboxylic acid, as such bonds do for the urea/amide pharmacophore.3 Replacement of the sulfur atom of 3 by a carbon, to yield the ketone 6, resulted in a total loss of potency. This underlines the necessity of a strong dipole moment on the central pharmacophore to establish strong hydrogen bonds with the enzyme residues. Finally, we tested if the degree of oxidation of the sulfur atom influences the potency of the compound. Both the thio-ether 7 and the sulfone 8 displayed a greater loss of potency (> 100-fold) compared to sulfoxide 3. Sulfones are more polar than corresponding sulfoxides,28 thus, the charge on the sulfur atom of 8 should be greater than the charge on the sulfur atom of 3 leading to greater inhibition. The fact that 8 is less potent than 3 suggests that some steric constrains probably impede the sulfone from binding efficiently into the active site of sEH.
Table 1.
Inhibition of the human sEH by a series of related chemicals
| Structure | IC50 (nM)a | |
|---|---|---|
|
|
1 | 70,000± 6,000 |
|
|
2 | 6,800 ± 300 |
|
|
3 | 1,350 ± 40 |

|
4 | 11,000 ± 1,000 |
|
5 | 400 ± 50 |
|
|
6 | > 100,000 |
|
|
7 | > 100,000 |
|
8 | > 100,000 |
The IC50s were measured using CMNPC as substrate.23 Results are mean ± standard deviation of at least three separate measurements.
Taken together, the results obtained clearly show that sulfoxides represent a new class of competitive sEH inhibitors, with alkyl sulfoxides being the most potent. In this class of compound, fulvestrant was found to have a low nanomolar KI for the human sEH. Fulvestrant is an estrogen receptor antagonist used for the treatment of hormone receptor positive metatastic breast cancer.30 It is beyond the scope of this paper to determine if sEH inhibition account for part of fulvestrant efficacy. A human pharmaco-kinetic study showed a blood concentration around 10 nM for several weeks following intramuscular treatment.31,32 At such a concentration (slightly below the KI), it is unlikely that all the sEH is inhibited; however a significant portion of it should be. Because sulfones are not inhibitors of sEH, development of sulfoxides as sEH inhibitors will be limited by their stability and sensitivity to oxidation. Interestingly, fulvestrant is very stable in humans.31
Supplementary Material
Acknowledgments
This work was partially funded by NIEHS grant ES02710 and NIEHS Superfund Basic Research Program grant P42 ES04699. BDH is a George and Judy Marcus Senior Fellow of the American Asthma Foundation. This work was supported in part by NIH R01 HL 107887 (MEN). Structural data used in this publication were collected at the Gulf Coast Protein Crystallography Beamline at the Center for Advanced Microstructures and Devices. This beamline is supported by the National Science Foundation grant DBI-9871464 with co-funding from the National Institute for General Medical Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Methods for the library screening as well as for protein crystallization and structure determination are given in Supplementary material.
References and notes
- 1.Spector AA. J. Lipid Res. 2009;50:S52. doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imig JD. Physiol. Rev. 2012;92:101. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morisseau C, Hammock BD. Annu. Rev. Pharmacol. Toxicol. 2005;45:311. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 4.Morisseau C, Hammock BD. Annu. Rev. Pharmacol. Toxicol. 2013;53:37. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomez G, Morisseau C, Hammock BD, Christianson DW. Biochemistry. 2004;43:4716. doi: 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- 6.Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Proc. Natl. Acad. Sci. USA. 1999;96:8849. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen HC, Hammock BD. J. Med. Chem. 2012;55:1789. doi: 10.1021/jm201468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eldrup AB, Soleymanzadeh F, Taylor SJ, Muegge I, Farrow NA, Joseph D, McKellop K, Man CC, Kukulka A, De Lombaert S. J. Med. Chem. 2009;52:5880. doi: 10.1021/jm9005302. [DOI] [PubMed] [Google Scholar]
- 9.Xie Y, Liu Y, Gong G, Smith DH, Yan F, Rinderspacher A, Feng Y, Zhu Z, Li X, Deng S-X, Branden L, Vidoic D, Chung C, Schürer S, Morisseau C, Hammock BD, Landry DW. Bioorg. Med. Chem. let. 2009;19:2354. doi: 10.1016/j.bmcl.2008.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen HC, Ding FX, Deng Q, Xu S, Tong X, Zhang X, Chen Y, Zhou G, Pai LY, Alonso-Galicia M, Roy S, Zhang B, Tata JR, Berger JP, Colletti SL. Bioorg. Med. Chem. Lett. 2009;19:5716. doi: 10.1016/j.bmcl.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Xing L, McDonald JJ, Kolodziej SA, Kurumbail RG, Williams JM, Warren CJ, O'Neal JM, Skepner JE, Roberds SL. J. Med. Chem. 2011;54:1211. doi: 10.1021/jm101382t. [DOI] [PubMed] [Google Scholar]
- 12.Moser D, Achenbach J, Klingler FM, Estella B, Hahn S, Proschak E. Bioorg. Med. Chem. Lett. 2012;22:6762. doi: 10.1016/j.bmcl.2012.08.066. [DOI] [PubMed] [Google Scholar]
- 13.Asokukumar K, Prathyush LT, Umamaheshwari M, Sivashanmugam T, subhadradevi V, Jagannath P, Madeswaran A, Salesheir F. J. Chil. Chem. Soc. 2012;57:1442. [Google Scholar]
- 14.Tang L, Ma WH, Ma YL, Ban SR, Feng XE, Li QS. Bioorg. Med. Chem. Lett. 2013;23:2380. doi: 10.1016/j.bmcl.2013.02.048. [DOI] [PubMed] [Google Scholar]
- 15.Wang SB, Pang XB, Zhao Y, Wang YH, Zhang L, Yang XY, Fang LH, Du GH. J. Asian. Nat. Prod. Res. 2012;14:1084. doi: 10.1080/10286020.2012.723200. [DOI] [PubMed] [Google Scholar]
- 16.Liu JY, Park SH, Morisseau C, Hwang SH, Hammock BD, Weiss RH. Mol. Cancer. Ther. 2009;8:2193. doi: 10.1158/1535-7163.MCT-09-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu JY, Qiu H, Morisseau C, Hwang SH, Tsai HJ, Ulu A, Chiamvimonvat N, Hammock BD. Toxicol. Appl. Pharmacol. 2011;255:200. doi: 10.1016/j.taap.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Proc. Natl. Acad. Sci. U S A. 2005;102:9772. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu JY, Yang J, Inceoglu B, Qiu H, Ulu A, Hwang SH, Chiamvimonvat N, Hammock BD. Biochem. Pharmacol. 2010;79:880. doi: 10.1016/j.bcp.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang SH, Wagner KM, Morisseau C, Liu JY, Dong H, Wecksler AT, Hammock BD. J. Med. Chem. 2011;54:3037. doi: 10.1021/jm2001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inceoglu B, Wagner K, Schebb NH, Morisseau C, Jinks SL, Ulu A, Hegedus C, Rose T, Brosnan R, Hammock BD. Proc. Natl. Acad. Sci. U S A. 2011;108:5093. doi: 10.1073/pnas.1101073108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolf NM, Morisseau C, Jones PD, Hock B, Hammock BD. Anal. Biochem. 2006;315:71. doi: 10.1016/j.ab.2006.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Anal. Biochem. 2005;343:66. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morisseau C, Schebb NH, Dong H, Ulu A, Aronov PA, Hammock BD. Biochem. Biophy. Res. Comm. 2012;419:796. doi: 10.1016/j.bbrc.2012.02.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morisseau C, Sahdeo S, Cortopassi G, Hammock BD. Anal. Biochem. 2013;434:105. doi: 10.1016/j.ab.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morisseau C, Merzlikin O, Lin A, He G, Feng W, Padilla I, Denison MS, Pessah IN, Hammock BD. Environ. Health. Persp. 2009;117:1867. doi: 10.1289/ehp.0900834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen D, Whitcomb R, MacIntyre E, Tran V, Do ZN, Sabry J, Patel DV, Anandan SK, Gless R, Webb HKJ. Clin. Pharmacol. 2012;52:319. doi: 10.1177/0091270010397049. [DOI] [PubMed] [Google Scholar]
- 28.Vandermeeren L, Leyssens T, Peeters D. J. Mol. Struct: THEOCHEM. 2007;804:1. [Google Scholar]
- 29.Morisseau C, Inceoglu B, Schmelzer K, Tsai HJ, Jinks SL, Hegedus CM, Hammock BD. J. Lipid. Res. 2010;51:3481. doi: 10.1194/jlr.M006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Croxtall JD, McKeage K. Drugs. 2011;71:363. doi: 10.2165/11204810-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 31.Robertson JF, Erikstein B, Osborne KC, Pippen J, Come SE, Parker LM, Gertler S, Harrison MP, Clarke DA. Clin. Pharmacokinet. 2004;43:529. doi: 10.2165/00003088-200443080-00003. [DOI] [PubMed] [Google Scholar]
- 32.McCormack P, Sapunar F. Clin. Breast. Cancer. 2008;8:347. doi: 10.3816/CBC.2008.n.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.