

Abstract
Oxaliplatin is an integral component of colorectal cancer therapy, but its clinical use is associated with a dose-limiting peripheral neurotoxicity. We found that the organic cation transporter 2 (OCT2) is expressed on dorsal root ganglia cells within the nervous system where oxaliplatin is known to accumulate. Cellular uptake of oxaliplatin was increased by 16- to 35-fold in cells overexpressing mouse Oct2 or human OCT2, and this process was associated with increased DNA platination and oxaliplatin-induced cytotoxicity. Furthermore, genetic or pharmacologic knockout of Oct2 protected mice from hypersensitivity to cold or mechanical-induced allodynia, which are established tests to assess acute oxaliplatin-induced neurotoxicity. These findings provide a rationale for the development of targeted approaches to mitigate this debilitating toxicity.
Keywords: neuropathy, chemotherapy, solute carriers
Oxaliplatin is a third generation platinum drug that has been approved for the treatment of colorectal cancer. The clinical use of oxaliplatin is limited by the onset of a debilitating, dose-limiting peripheral neurotoxicity that often prevents further treatment. This side effect has two clinical presentations: acute toxicity is experienced by a majority of patients, and manifests as muscle cramps, jaw spasms, paraesthesia, and dysthesia, all of which can be exacerbated by exposure to cold temperatures, whereas chronic paraesthesias and dysthesias ensue later and may progress even after discontinuation of treatment. Clinical data have indicated an apparent lack of correlation between platinum-induced neurotoxicity and tumor response (1), suggesting that neurotoxicity could potentially be prevented without altering treatment. Nonetheless, previous efforts to prevent oxaliplatin-induced neurotoxicity, while maintaining treatment efficacy, have been rather unsuccessful. For example, past studies have suggested that use of thiol compounds and calcium or magnesium infusions can ameliorate oxaliplatin-induced neurotoxicity (2–4). However, the effectiveness of such interventions has been questioned, and concerns regarding their potential to alter treatment efficacy have been raised (5, 6). Similarly, the ability of oxaliplatin to alter Na+ currents and conductance have also been considered since the discovery that patients exposed to oxaliplatin had altered Na+ channel related parameters (7–9). Unfortunately, although this knowledge appears to serve as a reliable marker for neuropathy, the mechanism responsible for oxaliplatin-dependent alteration in Na+ channel function is unknown and therefore it is currently questionable whether targeting Na+ channels will be therapeutically advantageous to ameliorating platinum-induced neuropathy.
Within the nervous system, oxaliplatin appears to accumulate preferentially in the dorsal root ganglia (DRG) (10, 11). Because the extent to which oxaliplatin accumulates into DRG correlates with the degree of neurotoxicity (12), we hypothesized that understanding the mechanism of cellular uptake of oxaliplatin may shed light on the etiology of acute drug-induced neurotoxicity and may lead to the development of novel therapeutic interventions.
Results and Discussion
We sought to assess the role of the solute carrier family member 22a2 [Slc22a2; encoding the organic cation transporter 2 (Oct2)] in relation to oxaliplatin-induced adverse effects, considering that this protein has been implicated in renal disposition or nephrotoxicity associated with cisplatin and that the influence of oxaliplatin uptake by the human homolog transporter OCT2 has been reported to be far greater than the former platinum compound (13, 14). Using transfected mammalian cells (Fig. S1 A–E), we confirmed the efficiency of oxaliplatin transport by the human homolog transporter OCT2, which increased uptake by over 35-fold (Fig. 1A) and is in line with previous in vitro findings (14). Due to the necessity of measuring adverse effects in murine models, we assessed the ability of the mouse Oct2 transporter to promote uptake of oxaliplatin and found that its overexpression lead to ∼14-fold increased uptake compared with control cells (Fig. 1B), whereas the related transporter Oct1 (Slc22a1) did not significantly influence oxaliplatin uptake (Fig. 1C). Additionally, overexpression of OCT2 increased platinum-DNA adduct formation and increased sensitivity of HEK293 cells to oxaliplatin by ∼12-fold (Fig. 1 D and E). Uptake of platinum was dramatically reduced when cells were incubated in the presence of cimetidine, a known OCT2 competitive inhibitor (Fig. 1 F and G). It is important to note that cimetidine may directly interact with oxaliplatin and thereby reduce the effective concentration (15). Contrary to findings with cisplatin (16), transport of oxaliplatin by mouse Oct2 or human OCT2 was not saturable at concentrations as high as 1 mM (Fig. 1 H and I). The ability of Oct2 to facilitate uptake of oxaliplatin is of clinical interest, considering that we detected Oct2 protein expression in murine DRG (Fig. 2A) and confirmed the presence of OCT2 mRNA in human DRG (Fig. 2B). The data shown in Fig. 2A suggest a possibility that OCT2 might be enriched in satellite glia cells within DRG. However, in agreement with previous studies (13, 17, 18), we were unable to detect OCT2 expression in 48 colorectal samples or tumor cell lines (Fig. S2). Given that inhibition of Oct2 function with cimetidine has been shown to have no effect on platinum-based therapy (19), collectively, these findings suggest that OCT2 may represent a selective uptake transporter that, when inhibited, may reduce cellular toxicity without altering treatment efficacy.
Fig. 1.

Human and murine OCT2 expression as a mediator of oxaliplatin uptake and cytotoxicity. (A–C) Transport of oxaliplatin by human OCT2 (hOCT2) (A) mouse Oct2 (mOct2) (B) and mouse Oct1 (mOct1) (C) transfected in HEK293 cells (500 µM; 30-min incubations). (D) Platinum (Pt)-DNA adduct formation in HEK293 cells transfected with hOCT2 following exposure to oxaliplatin (500 µM; 30-min incubations). (E) Cell growth inhibition induced by various concentrations of oxaliplatin in HEK293 cells transfected with hOCT2 or VC. The inset shows the estimated concentration associated with 50% growth inhibition (IC50) and the associated 95% confidence intervals (95% CIs). (F) Inhibition of cellular uptake of oxaliplatin (500 µM; 30-min incubations) with cimetidine (1 mM; white bars) in transfected HEK293 cells. (G) Inhibition of 14C-oxaliplatin (2 µM) uptake with various concentrations of cimetidine in HEK293 cells transfected with hOCT2 or mOct2 (30-min incubations). The inset shows the estimated concentration associated with 50% inhibition of uptake (IC50) and the associated 95% CIs. (H) Concentration-dependence of oxaliplatin transport by mOct2 and hOCT2 transfected into HEK293 cells. (I) Concentration-dependence of cisplatin transport by hOCT2 transfected into HEK293 cells (16). Data represent the net difference in uptake observed in cells with or without the transporter. Km denotes the Michaelis–Menten constant, and Vmax denotes the maximum velocity. Data represent the mean of triplicate observations from experiments performed on at least three separate occasions, and are expressed as average percentage of uptake values in cells transfected with an empty vector (VC). Error bars represent SE. The asterisk denotes significant differences from VC (P < 0.05).
Fig. 2.

Identification of OCT2 as a transporter in dorsal root ganglia (DRG). (A) Detection of Oct2 by immunofluorescence in DRG of wild-type and Oct1/2(−/−) mice. Oct2 is depicted by green fluorescence, whereas DNA is depicted in blue (DAPI). (B) OCT2 expression detected in human DRG (Human 1 and Human 2) by RT-PCR (depicted by the 128-bp product). Oct2 expression was also detected in DRG of wild-type mice (lane 3; 306bp product) but not in DRG isolated from Oct1/2(−/−) mice.
We next assessed the pharmacokinetics and toxicity of oxaliplatin in mice with a genetic deletion of the Oct1 and Oct2 transporters [Oct1/2(−/−) mice]. Preliminary experiments in male wild-type mice indicated that oxaliplatin disposition was linear over a 10–40 mg/kg dose range and caused dose-dependent weight loss (Fig. S3). An i.p. dose of 40 mg/kg was selected for further study as the observed average systemic exposure, measured by the area under the curve in plasma, was remarkably similar to the average area under the curve reported in humans receiving a standard oxaliplatin dose of 130 mg/m2 (61.8 ± 6.24 µg⋅h/mL vs. 59.1 ± 11.4 µg⋅h/mL, respectively) (20). Furthermore, the percentage of the oxaliplatin dose excreted in urine under the applied conditions was similar in wild-type mice (40.0 ± 5.0%) and patients with cancer (between 33.1% and 53.8%) (20).
As expected based on our previous work with cisplatin (13), loss of Oct1 and Oct2 function in mice resulted in decreased urinary excretion of oxaliplatin (Fig. 3A), without simultaneously affecting area under the curve (Fig. 3B) or substantially altering levels in liver or kidney (Fig. S4 B and C). Similar findings were obtained in female mice (Fig. S4 D–F). Furthermore, wild-type and Oct1/2(−/−) mice survived only 4 d after the administration of a 40 mg/kg dose of oxaliplatin due to excessive weight loss associated with the occurrence of lesions in the gastrointestinal tract and bone marrow hypocellularity, without any evidence of severe nephrotoxicity (Fig. 3 C–E). This finding suggests that differences in other oxaliplatin-related side effects can be properly assessed in Oct1/2(−/−) mice without considering altered mortality.
Fig. 3.

Oxaliplatin disposition and toxicity in Oct1/2(−/−) mice. (A and B) Time course of urinary excretion (A) and total plasma concentrations (Ct) and unbound plasma concentrations (Cu) (B) in male wild-type (urinary excretion: n = 29; plasma concentrations: n = 4) and Oct1/2(−/−) mice (urinary excretion: n = 25; plasma concentrations: n = 4) following a single i.p. administration of oxaliplatin (40 mg/kg). Data are presented as the mean (symbols or bars) and SE (error bars). The asterisk denotes a significant difference from wild-type mice (P < 0.05). (C and D) Cumulative overall survival (C) and overall toxicity (D) as determined from weight changes from baseline in male wild-type and Oct1/2(−/−) mice following a single i.p. administration of oxaliplatin (40 mg/kg). Data are presented as the mean (symbols) and SE (error bars) of six observations per group. (E) Representative histopathology in wild-type and Oct1/2(−/−) mice before (“untreated”) and 72-h after administration of oxaliplatin (40 mg/kg) (“treated”). Both in wild-type and Oct1/2(−/−) mice, the main lesions were damage to the small-intestinal epithelium (40× magnification) and bone marrow hypocellularity (10× magnification). The former was associated with crypt loss, implying the prior occurrence of crypt necrosis, the latter with severe myelosuppression.
To measure acute oxaliplatin-induced peripheral neuropathy, we used two well established tests that have been continuously shown to accurately and confidently monitor commonly altered behavioral changes in murine models associated with oxaliplatin-based treatment (21–24), namely a cold-plate assay to assess thermal sensitivity and a Von Frey Hairs test to assess mechanical allodynia. Wild-type and Oct1/2(−/−) mice showed no difference in sensitivity to cold before treatment with oxaliplatin (Fig. 4A). However, our study revealed that wild-type mice, following a single dose of oxaliplatin, experienced significant increased sensitivity to cold (Fig. 4B). In particular, average increases in paw lifts/licks compared with baseline were 29% (P = 0.0002) and 37% (P < 0.0001) at 24 or 48 h after administration of oxaliplatin, respectively (Fig. 5A and Fig. S5A). In contrast, Oct1/2(−/−) mice experienced no significant changes in sensitivity to cold at 24 (P = 0.48) or 48 h (P = 0.96) after administration of oxaliplatin (Fig. 5A and Fig. S5A).
Fig. 4.

Sensitivity to cold in response to oxaliplatin. (A) Baseline values for the number of paw lifts or licks performed by wild-type or Oct1/2(−/−) mice exposed to −4 °C for 5 min. (B) The number of paw lifts or licks performed by wild-type FVB mice exposed to various temperatures (−4 °C, 4 °C, and 30 °C) for 5 min both before and after treatment with oxaliplatin (40 mg/kg). Data are presented as the mean (symbols or bars) and SE (error bars) where n represents the number of events observed. P values above the bars denote statistical comparison between baseline and after treatment.
Fig. 5.

OCT2 regulation of oxaliplatin-induced neuropathy. (A) Sensitivity to cold associated with a single dose of oxaliplatin (40 mg/kg) in wild-type and Oct1/2(−/−) mice, as determined by a cold-plate test. Data are presented as the number of paw lifts or licks at baseline and following exposure to a temperature of −4 °C for 5 min at 24 h [wild type: n = 25; Oct1/2(−/−): n = 17] or 48 h [wild type: n = 25; Oct1/2(−/−): n = 16] after drug administration. (B) Mechanical allodynia associated with a single dose of oxaliplatin (40 mg/kg) in wild-type and Oct1/2(−/−) mice, as determined by a Von Frey Hairs test. Data are presented as the force required to promote paw withdrawal in grams (g) at baseline and following 24 h [wild type: n = 11; Oct1/2(−/−): n = 11] or 48 h [wild type: n = 11, Oct1/2(−/−): n = 10] after drug administration. (C) Change in sensitivity to cold in wild-type (n = 7) and Oct1/2(−/−) (n = 7) mice 24 h after receiving oxaliplatin (5 mg/kg) alone or in combination with cimetidine (30 mg/kg, i.v. bolus). (D) Change in mechanical allodynia in wild-type (n = 8) and Oct1/2(−/−) (n = 7) mice 48 h after receiving oxaliplatin (5 mg/kg) alone or in combination with cimetidine (30 mg/kg, i.v. bolus). (E) Change in sensitivity to cold in wild-type mice (n = 7) 24 h after receiving oxaliplatin (5 mg/kg) alone (n = 7) or in combination with cimetidine (i.v. bolus) at a concentration of 5 mg/kg (n = 7) or 30 mg/kg (n = 9). Bars represent the mean and error bars are the SE. The asterisk denotes significant difference from baseline and between strains (P < 0.05).
In addition to experiencing cold hypersensitivity, wild-type mice also showed increased sensitivity to mechanical stimulation after administration of oxaliplatin. The force to induce a response was decreased by 20% (P = 0.0098) and 40% (P = 0.0003) compared with baseline at 24 and 48 h following administration of oxaliplatin, respectively (Fig. 5B and Fig. S5B). Meanwhile, Oct1/2(−/−) mice exposed to oxaliplatin did not experience a significant change in sensitivity to mechanical allodynia at 24 (P = 0.59) or 48 h (P = 0.40) (Fig. 5B and Fig. S5B). The contribution of Oct2 in regulating oxaliplatin-induced neurotoxicity was further validated by demonstrating that the concurrent administration of cimetidine (30 mg/kg), a known inhibitor of Oct2 (17), protected wild-type mice from oxaliplatin-induced thermal sensitivity (Fig. 5C and Fig. S5C) or mechanical allodynia (Fig. 5D and Fig. S5D). Furthermore, protection of cold hypersensitivity by cimetidine was dose dependent, where coadministration of 5 mg/kg cimetidine was partially protective and 30 mg/kg cimetidine completely ameliorated oxaliplatin-induced sensitivity to cold (Fig. 5E and Fig. S5E). These phenotypic changes occurred in the absence of any detectable changes in the expression of other transporter genes (Table S1) of putative relevance to oxaliplatin (Fig. 6), including the ATP-binding cassette transporter family c2 [Abcc2; Multidrug resistance-associated protein 2 (Mrp2)] (25), the organic cation transporter, novel, type 1 [Octn1, encoded by the solute carrier family member 22a4 (Slc22a4)] (26), the copper-transporting ATPase 1 (Atp7a) and the copper transporter 1 [Ctr1, encoded by the solute carrier family member 31a1 (Slc31a1)] (27, 28). Additionally, both sensitivity to mechanical stimuli and cold occurred in the absence of any histological damage (Fig. S6).
Fig. 6.
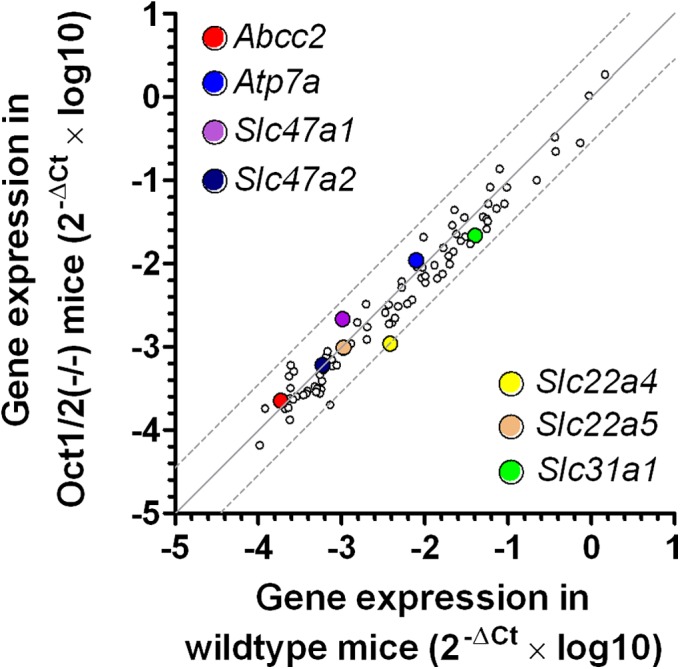
Transporter genes in dorsal root ganglia of Oct1/2(−/−) mice. Comparative expression of 84 transporter genes at baseline in dorsal root ganglia of wild-type mice and Oct1/2(−/−) mice (n = 3 each). Each symbol represents an average reading for a single gene, the solid line is the line of identity, and the dotted lines are the 95% CIs. The colored symbols represent transporter genes previously implicated in oxaliplatin toxicity.
Collectively, our findings indicate that Oct2 function is required in the onset of acute oxaliplatin-induced peripheral neurotoxicity. Considering that Oct2 may serve as the initial regulator for accumulation of platinum based agents, this protein may also contribute to other forms of platinum-induced neuropathy phenotypes, such as the chronic conditions experienced by patients treated with oxaliplatin or cisplatin. Studies are currently underway to answer these questions. Moreover, it remains to be seen whether the loss of Oct2 function ameliorates other markers of neurotoxicity, such as altered nerve conductance. Nonetheless, targeting Oct2 appears to be an ideal approach in ameliorating platinum-induced toxicities without sacrificing treatment efficacy, especially because it has been shown that inhibition of Oct2 function with cimetidine has no effect on platinum-based therapy (17, 19). Considering the known functional similarities of mouse Oct2 and human OCT2, we expect that specific inhibitors of OCT2 can be exploited therapeutically as modulators of oxaliplatin-induced neurotoxicity.
Methods
Plasmid Construction.
The original cDNA of human OCT2 (hOCT2), mouse Oct1 (mOct1), and mOct2 were purchased from Origene (SC126370, MC200831, and MC205995). The reconstructed cDNA with or without Flag-tag were subcloned into the PMIG II vector, engineered from a murine stem cell virus (MSCV)-Internal ribosome entry site (IRES)-GFP and kindly provided by Dario Vignali (St. Jude Children’s Research Hospital, Memphis, TN).
Transfection and Infection.
The empty vector (PMIG II) or vectors encoding Flag-tagged cDNA of hOCT2, mOct1, and mOct2 were cotransfected with pEQ-Pam3(-E) and pVSVg into 293T cells using Fugene transfection reagent (Roche). Virus-containing medium was collected at 48 and 72 h and used to infect HEK293 cells with 6 µg/mL polybrene. Cells expressing GFP, following 10 d of infection, were sorted by FACS to ∼95% purity.
Western Blot.
Whole cell pellets were lysed in M-PER mammalian protein extraction reagent (Pierce) with a protease inhibitor (Roche 04693124001), and proteins were subjected to Western blot analysis using a standard protocol. The mouse anti-Flag antibody (dilution, 1:1,000; F1804) was obtained from Sigma, and a HRP-conjugated antibody specific to mouse IgG was from Santa Cruz Biotechnolgy.
Cellular Accumulation Studies.
Uptake experiments were performed with oxaliplatin or tetraethylammonium as described (16), in the presence or absence of cimetidine with results normalized to uptake values in cells transfected with an empty vector. Platinum–DNA adducts were determined as described (16), and cell growth inhibitory potential by oxaliplatin was evaluated using an MTT assay following a continuous 72-h exposure.
Pharmacokinetic Analyses in Mice.
Adult male or female Oct1/2(−/−) mice and age-matched wild-type mice (Taconic), both on an FVB background, were housed in a temperature-controlled environment with a 12-h light cycle, and given a standard diet and water ad libitum. Experiments were approved by the Institutional Animal Care and Use Committee of St. Jude Children’s Research Hospital. Oxaliplatin was diluted in normal saline and administered by the i.p. route at a dose of 10–40 mg/kg. Plasma was collected at 15, 45, 240, and 480 min, and liver, kidney, and spleen from each animal were collected at 72 h. Urine was collected from animals housed in metabolic cages for 72 h after oxaliplatin administration. Samples were analyzed by flameless atomic absorption spectrometry as described (13). Drug concentrations were determined using a linear-least squares regression analysis through linear calibration curves prepared in drug free nitric acid (0.2%). Noncompartmental parameters were calculated using PK Solutions 2.0 (Summit Research Services).
Gene Expression Analysis.
RNA from mouse DRG [thoracic position 8 (T8) to lumbar position 5 (L5)], was extracted using the RNEasy mini kit (Qiagen). Samples were obtained from three animals per group and analyzed using real-time PCR or the Mouse Transporter RT2 Profiles PCR array system on a 96-well format (SABiosciences). Each array contained a panel of primer sets for 84 transporter genes, including ABC transporters and solute carriers. Expression of the Slc47a1, Slc47a2, and Slc22a5 transporter genes was determined using TaqMan probes (Applied Biosystems). Relative gene expression was determined using the ΔΔCt method, and normalized to the housekeeping gene, Gapdh.
RNA from HEK293 cells transfected with vector control or hOCT2 was extracted as described above. Samples obtained were analyzed using or the Human Transporter RT2 Profiles PCR array system on a 96-well format (SABiosciences). Each array contained a panel of primer sets for 84 transporter genes, including ABC transporters and solute carriers.
Human DRG isolated from the lumbar position 4 (L4) were obtained from the National Disease Research Interchange (NDRI) and RNA was extracted as described above. Expression of SLC22A2 was measured using gene-specific primers (forward: 5′-ATGCCCACCACCGTGGACGAT-3′; reverse: 5′-AGGAAGACGATGCCCACGTA-3′). Research involving human tissue was approved by the institutional review board of St. Jude Children's Research Hospital and the NDRI obtained informed consent from donors of human tissue.
Amplified products were later visualized after separation of bands on a 2% (wt/vol) agarose gel.
Expression of SLC22A2 was assessed in 48 tissues covering four disease stages and normal colorectal tissue using the TissueScan Colon Cancer Tissue qPCR Panel V array (Origene) and TaqMan probes specific to SLC22A2 and GAPDH (Applied Biosystems).
Immunofluorescence of Oct2 in Mouse DRG.
Expression of Oct2 in DRG was investigated using a specific antibody (29). L4 DRG were extracted from male FVB wild-type or Oct1/2(−/−) mice that were perfused with 4% (wt/vol) paraformaldehyde and fixed overnight at 4 °C. Cryosections (7-µm) were incubated for 1 h at room temperature in PBS containing 10% (wt/vol) normal goat serum (pH 7.4) and then 90 min at room temperature with an Oct2-specific antibody (1/100 dilution). After washing in PBS, the sections were incubated for 45 min at room temperature with PBS-diluted secondary antibodies (Alexa Fluor 488 goat anti-rabbit-Ig, 1/1,000; Invitrogen). Sections were rinsed with PBS and mounted with Crystal Mount (GeneTex) and evaluated on an Observer Z1 microscope with apotome (Zeiss). Due to the lack of suitable commercially available antibodies, the detection of OCT2 in human DRG has remained elusive.
Toxicity Studies in Mice.
Thermal sensitivity associated with a single i.p. dose of oxaliplatin (40 or 5 mg/kg) in male wild-type mice and age-matched male Oct1/2(−/−) mice was assessed by a cold-plate test (21). The number of paw lifts and licks when exposed to a temperature of −4, 4, or 30 °C for 5 min was obtained for each mouse at 120 and 24 h before receiving oxaliplatin to determine the number of baseline events. Data were recorded as the percentage change in the number of paw lifts or paw licks compared with baseline values when the animals were exposed to the same temperature 24 or 48 h after the administration of oxaliplatin. Investigators performing the experiments were blinded to the mouse genotypes. Mechanical allodynia was determined by a Von Frey Hairs test (21). Mice were left to acclimate on a mesh platform and in 100 × 60 mm cylindrical tubes before the force necessary for a rigid Von Frey hair filament (IITC Life Science) to induce paw withdrawal of the hind limbs was assessed for each mouse at 120 and 24 h before receiving oxaliplatin to determine baseline events. Paw withdrawal was assessed in triplicate on each hind paw with 5-min intervals between repeating the test on each mouse and alternating from the left paw to the right paw. Data were also recorded as the percent change of force (in grams) necessary to promote paw withdrawal before and after the administration of oxaliplatin. In select experiments, the administration of oxaliplatin (5 mg/kg) was preceded by i.v. injection of cimetidine at a dose of 30 or 5 mg/kg.
Histological Evaluation of Mouse Tissues.
After collection, tissues were fixed overnight in 10% (wt/vol) neutral-buffered formalin. Next, the tissues were processed routinely, embedded in paraffin, sectioned (4 µm), and stained with hematoxylin and eosin. Microscopic evaluation (10–40× magnification) was performed by an experienced veterinary pathologist blinded to the composition of the groups.
Statistical Considerations.
Data are presented as mean with SE, unless stated otherwise. Statistical calculations were done using a Student t test in NCSS 2004 (Number Cruncher Statistical System). P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Sharyn Baker for critical review of the manuscript and Steve Zatechka for technical assistance. This work was supported in part by the American Lebanese Syrian Associated Charities, US Public Health Service Cancer Center Support Grant 3P30CA021765, National Cancer Institute Grant 5R01CA151633-01, the Interdisciplinary Center for Clinical Research (IZKF) Münster (Grant Cia2/013/13), and the Network Enabled Drug Design (NEDD) Lombardy Region grant. We acknowledge use of tissues procured by the National Disease Research Interchange with support from National Institutes of Health Grant 5 U42 RR006042. None of the funding bodies had a role in the preparation of the manuscript.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. R.H.E. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305321110/-/DCSupplemental.
References
- 1.McWhinney SR, Goldberg RM, McLeod HL. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 2009;8(1):10–16. doi: 10.1158/1535-7163.MCT-08-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cascinu S, et al. Neuroprotective effect of reduced glutathione on oxaliplatin-based chemotherapy in advanced colorectal cancer: A randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2002;20(16):3478–3483. doi: 10.1200/JCO.2002.07.061. [DOI] [PubMed] [Google Scholar]
- 3.Gamelin L, et al. Prevention of oxaliplatin-related neurotoxicity by calcium and magnesium infusions: A retrospective study of 161 patients receiving oxaliplatin combined with 5-Fluorouracil and leucovorin for advanced colorectal cancer. Clin Cancer Res. 2004;10(12 Pt 1):4055–4061. doi: 10.1158/1078-0432.CCR-03-0666. [DOI] [PubMed] [Google Scholar]
- 4.Ishibashi K, Okada N, Miyazaki T, Sano M, Ishida H. Effect of calcium and magnesium on neurotoxicity and blood platinum concentrations in patients receiving mFOLFOX6 therapy: A prospective randomized study. Int J Clin Oncol. 2010;15(1):82–87. doi: 10.1007/s10147-009-0015-3. [DOI] [PubMed] [Google Scholar]
- 5.Hochster HS, Grothey A, Childs BH. Use of calcium and magnesium salts to reduce oxaliplatin-related neurotoxicity. J Clin Oncol. 2007;25(25):4028–4029. doi: 10.1200/JCO.2007.13.5251. [DOI] [PubMed] [Google Scholar]
- 6.Dong M, Xing PY, Liu P, Feng FY, Shi YK. [Assessment of the protective effect of calcium-magnesium infusion and glutathione on oxaliplatin-induced neurotoxicity] Zhonghua Zhong Liu Za Zhi. 2010;32(3):208–211. [PubMed] [Google Scholar]
- 7.Park SB, et al. Oxaliplatin-induced neurotoxicity: Changes in axonal excitability precede development of neuropathy. Brain. 2009;132(Pt 10):2712–2723. doi: 10.1093/brain/awp219. [DOI] [PubMed] [Google Scholar]
- 8.Park SB, et al. Acute abnormalities of sensory nerve function associated with oxaliplatin-induced neurotoxicity. J Clin Oncol. 2009;27(8):1243–1249. doi: 10.1200/JCO.2008.19.3425. [DOI] [PubMed] [Google Scholar]
- 9.Park SB, et al. Dose effects of oxaliplatin on persistent and transient Na+ conductances and the development of neurotoxicity. PLoS ONE. 2011;6(4):e18469. doi: 10.1371/journal.pone.0018469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonald ES, Randon KR, Knight A, Windebank AJ. Cisplatin preferentially binds to DNA in dorsal root ganglion neurons in vitro and in vivo: A potential mechanism for neurotoxicity. Neurobiol Dis. 2005;18(2):305–313. doi: 10.1016/j.nbd.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 11.Screnci D, et al. Relationships between hydrophobicity, reactivity, accumulation and peripheral nerve toxicity of a series of platinum drugs. Br J Cancer. 2000;82(4):966–972. doi: 10.1054/bjoc.1999.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holmes J, et al. Comparative neurotoxicity of oxaliplatin, cisplatin, and ormaplatin in a Wistar rat model. Toxicol Sci. 1998;46(2):342–351. doi: 10.1006/toxs.1998.2558. [DOI] [PubMed] [Google Scholar]
- 13.Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther. 2009;86(4):396–402. doi: 10.1038/clpt.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokoo S, et al. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem Pharmacol. 2007;74(3):477–487. doi: 10.1016/j.bcp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Buss I, Kalayda GV, Marques-Gallego P, Reedijk J, Jaehde U. Influence of the hOCT2 inhibitor cimetidine on the cellular accumulation and cytotoxicity of oxaliplatin. Int J Clin Pharmacol Ther. 2009;47(1):51–54. doi: 10.5414/cpp47051. [DOI] [PubMed] [Google Scholar]
- 16.Filipski KK, Loos WJ, Verweij J, Sparreboom A. Interaction of Cisplatin with the human organic cation transporter 2. Clin Cancer Res. 2008;14(12):3875–3880. doi: 10.1158/1078-0432.CCR-07-4793. [DOI] [PubMed] [Google Scholar]
- 17.Franke RM, et al. Influence of Oct1/Oct2-deficiency on cisplatin-induced changes in urinary N-acetyl-beta-D-glucosaminidase. Clin Cancer Res. 2010;16(16):4198–4206. doi: 10.1158/1078-0432.CCR-10-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burger H, et al. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2) Br J Pharmacol. 2010;159(4):898–908. doi: 10.1111/j.1476-5381.2009.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katsuda H, et al. Protecting cisplatin-induced nephrotoxicity with cimetidine does not affect antitumor activity. Biol Pharm Bull. 2010;33(11):1867–1871. doi: 10.1248/bpb.33.1867. [DOI] [PubMed] [Google Scholar]
- 20.Graham MA, et al. Clinical pharmacokinetics of oxaliplatin: A critical review. Clin Cancer Res. 2000;6(4):1205–1218. [PubMed] [Google Scholar]
- 21.Ta LE, Low PA, Windebank AJ. Mice with cisplatin and oxaliplatin-induced painful neuropathy develop distinct early responses to thermal stimuli. Mol Pain. 2009;5:9. doi: 10.1186/1744-8069-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renn CL, et al. Multimodal assessment of painful peripheral neuropathy induced by chronic oxaliplatin-based chemotherapy in mice. Mol Pain. 2011;7:29. doi: 10.1186/1744-8069-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakurai M, et al. Oxaliplatin-induced neuropathy in the rat: Involvement of oxalate in cold hyperalgesia but not mechanical allodynia. Pain. 2009;147(1-3):165–174. doi: 10.1016/j.pain.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Melisi D, et al. Oral poly(ADP-ribose) polymerase-1 inhibitor BSI-401 has antitumor activity and synergizes with oxaliplatin against pancreatic cancer, preventing acute neurotoxicity. Clin Cancer Res. 2009;15(20):6367–6377. doi: 10.1158/1078-0432.CCR-09-0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theile D, Grebhardt S, Haefeli WE, Weiss J. Involvement of drug transporters in the synergistic action of FOLFOX combination chemotherapy. Biochem Pharmacol. 2009;78(11):1366–1373. doi: 10.1016/j.bcp.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Jong NN, Nakanishi T, Liu JJ, Tamai I, McKeage MJ. Oxaliplatin transport mediated by organic cation/carnitine transporters OCTN1 and OCTN2 in overexpressing human embryonic kidney 293 cells and rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 2011;338(2):537–547. doi: 10.1124/jpet.111.181297. [DOI] [PubMed] [Google Scholar]
- 27.Ip V, Liu JJ, Mercer JF, McKeage MJ. Differential expression of ATP7A, ATP7B and CTR1 in adult rat dorsal root ganglion tissue. Mol Pain. 2010;6:53. doi: 10.1186/1744-8069-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu JJ, et al. Neuronal expression of copper transporter 1 in rat dorsal root ganglia: Association with platinum neurotoxicity. Cancer Chemother Pharmacol. 2009;64(4):847–856. doi: 10.1007/s00280-009-1017-6. [DOI] [PubMed] [Google Scholar]
- 29.Ciarimboli G, et al. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am J Pathol. 2010;176(3):1169–1180. doi: 10.2353/ajpath.2010.090610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.