

Abstract
Nuclear erythroid 2–related factor 2 (Nrf2) is an oxidative stress–mediated transcription factor with a variety of downstream targets aimed at cytoprotection. Nrf2 has recently been implicated as a new therapeutic target for the treatment of liver disease. Here, we focus on the most common liver diseases—nonalcoholic fatty liver disease/steatohepatitis, alcoholic liver disease, and drug-induced liver injury—and highlight areas in the development of these conditions where activation of Nrf2 may alleviate disease progression.
Over the past decade, nuclear erythroid 2–related factor 2 (Nrf2) has emerged as a significant transcription factor for the induction of a variety of detoxification enzymes, biotransformation enzymes, and xenobiotic efflux transporters. It has become evident that its activation is initiated by oxidative or electrophilic stress and aids in the detoxification and elimination of potentially harmful exogenous chemicals and their metabolites. In light of its role in detoxification and its ability to facilitate hepatoprotection, Nrf2 has more recently been considered as a target for new therapeutic approaches aimed at treating liver disease. This review focuses on the potential of Nrf2 enhancers, a relatively new class of therapeutics, in preventing several types of liver injury and disease. In doing this, we also review the relevant literature regarding Nrf2 activators and their efficacy in alleviating chronic and acute renal injury and disease so that speculative remarks can be made with regard to how these drugs might be beneficial in treating liver diseases.
Nrf2: BACKGROUND
Nrf2 was originally cloned from the complementary DNA library of a human leukemia cell line (K562) and identified as a member of the family of basic region leucine zipper (bZIP) transcription factors.1 The amino acid sequence of bZIP was found to be remarkably similar to that of the previously identified NF-E2 and Nrf1 because it includes a bZIP DNA-binding domain in the C-terminus and an acidic activation domain in the N-terminus.1 Nrf2 expression has been observed ubiquitously throughout human tissues, with relatively high expression in the key detoxification organs, specifically, the liver and kidney.1
Not long after the identification of this gene, it was determined that Nrf2 binds to and regulates the human antioxidant response element (ARE), a DNA motif located in the upstream promoter regions of many detoxification genes, and therefore mediates the expression of several antioxidant enzymes. The details of the mechanism of action for Nrf2 have been previously reviewed.2 Briefly, during homeostatic conditions Nrf2 is sequestered in the cytosol via physical attachment of the N-terminal domain of Nrf2 to Kelch-like ECH-associated protein 1 (Keap1), thereby causing inhibition of Nrf2 activity3 (Figure 1). Upon exposure to oxidative or electrophilic stress, Nrf2 dissociates from Keap1,4,5 translocates to the nucleus, and heterodimerizes with an array of transcriptional regulatory proteins. Common interactions include those with transcription regulators Jun and Fos,6,7 which are members of the activator protein 1 family of transcription factors. Following dimerization, these complexes then bind to AREs and either activate transcription, as is the case with the Jun–Nrf2 complex,6 or suppress transcription, as with the Fos–Nrf2 complex.7 The location of the ARE that is activated or inhibited will determine which genes are transcriptionally regulated by these factors. A detailed description of genes regulated by these mechanisms can be found in previous review articles.2,8 For the purpose of this review, emphasis is placed on the downstream targets of Nrf2 that are involved in certain cellular functions, including drug metabolism, reactive oxygen species (ROS) scavenging, regulation of glutathione levels, and alteration of stress protein and efflux transporter expression patterns. Given the effect that Nrf2 can have on these particular functions, it is not surprising that it could be considered a plausible target for drug therapies aimed at preventing cellular injury.
Figure 1.

A schematic representation of the mechanism of action of Nrf2. Nrf2 is sequestered in the cytoplasm via physical attachment to Keap1, causing inhibition of Nrf2 activity due to proteosomal degradation. During an oxidative stress event, Nrf2 dissociates from Keap1, translocates to the nucleus, and dimerizes with a variety of different transcription factors. Here, Jun is used as an example of one of those transcription factors that binds Nrf2. The heterodimer then binds to the antioxidant response element (ARE) in the promoter region of a variety of genes and activates transcription. Some of the genes that are transcriptionally regulated by Nrf2 are heme oxygenase 1 (Ho-1), NAD(P)H quinone oxidoreductase 1 (Nqo1), glutathione-S-transferase (Gst), glutamate–cysteine ligase (Glc), cytochrome P450s (CYPs), and multidrug-resistant proteins (MRPs). Nrf2, nuclear erythroid 2–related factor 2.
ROLE OF Nrf2 IN LIVER INJURY AND DISEASE
As a site of first-pass metabolism, the liver is inherently exposed to high concentrations of xenobiotics and other chemicals before delivery to the systemic circulation. Therefore, the liver is equipped with several defense mechanisms for protection against harmful chemicals and their potentially damaging metabolites. Although a variety of cytoprotective mechanisms are present in the liver, it remains highly susceptible to oxidative damage by reactive intermediates. This is a significant human health concern because (i) our ability to protect against oxidative stress seems to diminish with age, making elderly individuals more susceptible to liver injury and disease, and (ii) oxidative damage has been linked to several types of liver injury and disease, including nonalcoholic steatohepatitis (NASH),9 cirrhosis,10 acute hepatitis,11 and even hepatocellular carcinoma.12 In addition, oxidative stress–induced liver injury frequently creates an obstacle in the development of therapeutics because drug candidates often do not proceed to further trials owing to hepatotoxic effects. Therefore, the ability to prevent this toxicity or decrease the hepatic susceptibility to this toxicity would be extremely valuable.
It has been demonstrated throughout the literature that the alterations to phase I and II drug-metabolizing enzymes, efflux transporters, and other cytoprotective proteins that occur during periods of oxidative stress are, at least in part, coordinated by Nrf2. The development of Nrf2-null mice has provided a system for studying the role that Nrf2 plays in the pathogenesis of disease and the protection that it seemingly provides against hepatotoxicity. In addition, new pharmacological activators and inhibitors of Nrf2 are frequently emerging and are regularly used to provide insight to the functional roles of the Nrf2 signaling pathway.
Nrf2-null mice were first developed in 1996 by Chan et al.13 Many suspected that knocking out such a widespread protein would result in developmental complications; however, the Nrf2-null mice are visually indistinguishable from their heterozygous and wild-type littermates and exhibit normal growth and fertility.13 The effects of genetically eliminating Nrf2 became apparent when researchers observed an increased sensitivity to numerous xenobiotics, including benzo(a)pyrene and acetaminophen (APAP).14,15 As predicted, corresponding changes in expression patterns and activities of hepatic drug-metabolizing enzymes, detoxification enzymes, stress-related proteins, and efflux transporters were also observed in these mice.
One of the first notable effects of Nrf2 abolition was on the drug-metabolizing enzyme NAD(P)H quinone oxidoreductase 1 (Nqo1). Nqo1, which is widely and constitutively expressed, is cytoprotective against oxidative stress by scavenging superoxide, preserving various endogenous antioxidants in their reduced form, and catalyzing reductive metabolism of chemicals.16,17 Therefore, the induction of Nqo1 decreases cell susceptibility to oxidative stress; this is supported by evidence in the human population, in which the identified polymorphisms that result in low Nqo1 activity are associated with increased susceptibility to disease.18 It was also shown that Nrf2-null mice exhibit lower basal hepatic expression and activity of Nqo1,19 indicating that Nrf2 plays a role in Nqo1 expression. In support of this finding, it was later found that hepatic Nqo1 expression could be induced by chemical activation of Nrf2 in wild-type mice but not Nrf2-null mice.20
Another effect of genetic ablation of Nrf2 in mice is on the processes that regulate the availability of intracellular glutathione (GSH; L-γ-glutamyl-cysteinyl-glycine). GSH protects the cell against oxidative stress by detoxifying harmful chemicals, through either direct binding or enzymatic conjugation of GSH to the toxicant. Prolonged exposure to a toxicant will usually result in GSH depletion and, ultimately, oxidative stress. There are several ways in which Nrf2 factors into this detoxification process. First, it has been shown that untreated Nrf2-null mice have decreased levels of hepatocellular GSH,21 indicating that the mechanisms for making GSH within the cell are hindered in the absence of Nrf2. This effect seems to be due to decreased levels of the GSH-producing enzyme, glutamate–cysteine ligase, because livers from Nrf2-null mice have decreased expression of this enzyme.21 In addition to a lack of GSH in this mouse model, there is also a decreased ability to conjugate toxic chemicals with GSH. The enzyme glutathione-S-transferase is responsible for catalyzing this conjugation and has been shown to be present at lower levels in the liver of Nrf2-null mice.22
Heme oxygenase 1 (Ho-1), also known as heat shock protein 32, is another Nrf2 target gene that is activated during oxidative stress. Ho-1, which is ubiquitously expressed, combats cellular stress by initiating the oxidative cleavage of the pro-oxidant heme to subsequently form the antioxidants carbon monoxide and bilirubin. Because of the generation of these antioxidants, Ho-1 is believed to function as a cytoprotective enzyme and is therefore upregulated during oxidative stress. It has been demonstrated in mouse Hepa cells that the induction of Ho-1 expression is dependent on oxidative stress and transcriptional activation by Nrf2 of the AREs upstream of the Ho-1 promoter.23 In addition, macrophages from wild-type mice have induced Ho-1 upon exposure to a variety of toxicants, whereas those from Nrf2-knockout mice show no changes in Ho-1 expression.24 Perhaps most pertinent to this review are recent data showing that Ho-1 liver expression is induced by the Nrf2 activator 2-cyano-3,12 dioxooleana-1,9-dien-28-imidazolide (CDDO-Im) in wild-type mice but not in Nrf2-null mice,25 confirming that Nrf2 plays an important role in the regulation of this cytoprotective protein.
Other nonenzymatic proteins that are regulated, at least in part, by Nrf2 include a variety of membrane effiux transporters that play integral roles in the elimination of toxins and their metabolites in both the liver and kidney. Following conjugation by phase II detoxification enzymes, toxins are often transported across the plasma membrane of the cell by ATP-dependent efflux transporters such as those of the multidrug-resistance protein (MRP) family of transporters. Exposure to a toxicant that results in oxidative stress–induced activation of Nrf2 has been shown to affect the expression patterns of several of these MRPs, and their promoters have been shown to have the necessary ARE sequences to yield this effect.26–28 Several years ago, it was demonstrated that the Nrf2 activator oltipraz could sufficiently induce hepatic expression of Mrp2, Mrp3, and Mrp4 in wild-type but not in Nrf2-null mice.28 In support of those results, APAP-induced hepatotoxicity was also shown to cause induction of hepatic Mrp3 and Mrp4 in wild-type mice and was attenuated in Nrf2-knockout mice.29 In addition, it was found that hepatocyte-specific genetic ablation of Keap1 in mice increases nuclear Nrf2 and results in increases in a variety of transporters such as Mrp1, Mrp3, and organic anion transporter 1b2, with the most dramatic increases in Mrp4 and Mrp5 expression.30,31 Similar effects have also been seen recently in the mouse kidney, in which chemical-induced nephrotoxicity activates Nrf2 and upregulates Mrp2 and Mrp4.32 These studies, as well as others in this field, demonstrate the importance of efflux transporters in the detoxification process and their role in protection against oxidative stress.
Although significant research has been done to illustrate the role of Nrf2 in liver injury and disease in mouse models, the lack of clinical studies linking Nrf2 activation and liver disease makes it difficult to determine the relevance that this information will have to human liver pathologies. There are, however, a few studies that indicate that similarities may exist between these mouse models and human physiology outside the realm of liver injury and disease. For example, Nrf2-null mice have exacerbated acute kidney injury,33 and, in accordance, it has recently been shown that female patients with systemic lupus erythematosus, an autoimmune disease associated with oxidative stress and a G-653A mutation in the NRF2 gene, exhibit severe nephritis.34 Another example of the relevance that the mouse models have to human disease is with regard to the role that Nrf2 activation plays in skin cancer. It has been demonstrated that activation of Nrf2 in mice reduces skin tumor incidence,35 and a recent study from a variety of human cancer tissues revealed a high incidence of mutations in NRF2 in squamous cell carcinomas of the skin.36 Although these examples do not directly indicate the relevance of the Nrf2 mouse liver studies to human liver injury and disease, they provide some room for speculation with regard to the congruity that may exist between the species.
Nrf2 ENHANCERS: A NEW THERAPEUTIC CLASS
Over the past few years, many naturally occurring as well as synthetic compounds have been shown to activate Nrf2 in a variety of systems. The chemical inducers of Nrf2 have been organized into 10 classes according to chemical structure (see ref. 8). Several of the more commonly used Nrf2 activators and their effects in vivo are outlined in Table 1. Some of these compounds have even progressed to human clinical trials and seem to be promising therapeutics for treatment of conditions such as multiple sclerosis, muscular dystrophy, skin cancer, and chronic kidney disease (CKD). Because there have not been clinical trials on the effects of Nrf2 activation on liver injury and disease, this review employs information obtained regarding its effects on the development of other disease states to draw conclusions about how Nrf2 activation may alleviate different types of liver injury and disease.
Table 1.
Nfr2 activators and their effect on human and rodent tissues in vivo
| Nrf2 activator | In vivo effects |
|---|---|
| Protandim | Rodents |
| ↓ Fibrosis in MD40 | |
| Humans | |
| ↑ SOD and catalase activity39 | |
| ↓ Lipid peroxidation39 | |
| Dimethyl fumarate (BG-12) | Humans |
| ↓ MRI activity and lesions associated with MS37 | |
| Oltipraz | Rodents |
| ↑ Hepatic Mrp2, Mrp3, and Mrp4 expression28 | |
| CDDO-Im | Rodents |
| Prevents cisplatin-induced hepatotoxicity32 | |
| Prevents APAP-induced hepatotoxicity25 | |
| Prevents hepatic lipid accumulation59 | |
| Prevents ConA-mediated acute inflammatory liver injury | |
| ↑ Ho-1 expression in liver25 | |
| CDDO-Me (bardoxolone methyl) | Rodents |
| ↓ Inflammation | |
| Induces renal PPAR- and Ho-1 expression47 | |
| Prevents renal ischemia-induced increases in BUN47 | |
| Humans | |
| ↑ GFR41,43 | |
| ↓ Serum creatinine41,43 |
APAP, acetaminophen; BUN, blood urea nitrogen; CDDO-Im, 2-cyano-3,12 dioxooleana-1,9-dien-28-imidazolide; GFR, glomerular filtration rate; Ho-1, heme oxygenase 1; MRI, magnetic resonance imaging; MS, multiple sclerosis; PPAR-γ, peroxisome proliferator–activated receptor-γ; SOD, superoxide dismutase.
For example, in a phase IIb clinical trial, the compound BG-12 (dimethyl fumarate) (Biogen Idec) was administered to patients with relapsing–remitting multiple sclerosis and was shown to reduce brain magnetic resonance imaging activity and lesions associated with multiple sclerosis as compared with the patients who received placebo.37 Another compound, Protandim (LifeVantage), has demonstrated efficacy in several diseases. This is a dietary supplement that is of particular interest because it consists of five low-dose natural Nrf2 activators that activate Nrf2 and induce Ho-1 through multiple kinase pathways, including PI-3, p38, mitogen-activated protein kinase, and protein kinase Cδ, in a mouse β-cell line (MIN6) and a human neuroblastoma cell line (SK-N-MC).38 Individually, these components would be difficult to administer orally and would probably be toxic at therapeutic doses38; therefore, using them in concert allows for a therapeutic response while avoiding the problems associated with using the components individually at higher doses. The synergistic effect of these five components has been shown in a human clinical trial to yield significant elevations in the levels of antioxidant enzymes superoxide dismutase 1 and catalase, as well as a decline in levels of lipid peroxidation markers, indicating an overall reduction in oxidative stress.39 In addition, in a skin carcinogenesis mouse model, a reduction in skin tumor incidence was revealed in mice given a Protandim-supplemented diet as compared with those fed a nonsupplemented diet.35 Furthermore, in a Duchenne muscular dystrophy mouse model, Protandim demonstrated the ability to reduce fibrosis,40 an effect that could be valuable in a disease in which fatality often occurs as a result of heart and diaphragm fibrosis. Currently, Protandim is being sold as an over-the-counter dietary supplement, although the concept of activating Nrf2 from multiple pathways could have more promising therapeutic value in the future.
Arguably, the most exciting Nrf2 activator that is currently being tested is bardoxolone methyl (methyl 2-cyano-3,12-di-oxooleana-1,9(11)dien-28-oate) (Reata Pharmaceuticals), often referred to simply as bardoxolone. This novel synthetic triterpenoid was originally advanced to clinical trials for its potential as an anticancer agent; however, it was in these trials that clinicians observed reduced serum creatinine levels and improved glomerular filtration rates (GFRs) in oncology patients receiving bardoxolone.41 In addition, the observations with regard to renal function were even more pronounced in a subset of patients who also had established CKD. These findings, along with the previously described role of oxidative stress in CKD,42 prompted further research in the area of CKD treatment with bardoxolone.
In a recent study by Pergola et al.41 CKD patients with type 2 diabetes were given bardoxolone to examine its effects on renal function. The end-point evaluations revealed a steady increase in GFR (with no change in blood pressure), which became statistically significant after 4 weeks of treatment with bardoxolone and continued to increase further during the following 4 weeks when a higher dose was received. In addition, serum creatinine levels decreased with a corresponding increase in creatinine clearance at day 56 as compared with baseline. There was no evidence of liver toxicity in these patients, as demonstrated by no change in bilirubin or serum lactic dehydrogenase levels, or in cynomolgus monkeys treated with bardoxolone that revealed no adverse liver histopathology (C.J. Meyer, personal communication). Although this study did not address whether the effect is maintained without the development of serious adverse effects over a long period of time, it was the first to show that bardoxolone and the activation of Nrf2 might slow progression of renal disease in humans. As a follow-up to this study, the same researchers released the results of a longer study that confirmed that treatment with bardoxolone improved GFR in CKD patients at 24 and 52 weeks,43 indicating that this effect can be sustained over longer periods of time and that bardoxolone continues to be a potential treatment for CKD. However, several experts in the field argued that the increases in GFR could be due to changes in glomerular hemodynamics—specifically, increases in intra-glomerular pressure—rather than amelioration of fibrosis.44–46 In response to these criticisms, the author reiterated that the increase in GFR persisted 4 weeks after discontinuing treatment with bardoxolone, indicating that the results are not consistent with the suggested changes in intraglomerular hydrostatic pressure.43 It is clear that longer studies are necessary to confirm these results and provide convincing evidence that bardoxolone can restore kidney function by reducing inflammation and fibrosis in patients with CKD.
In addition to CKD, bardoxolone has recently been studied with regard to its ability to prevent acute kidney injury (AKI) in an ischemic mouse model.47 Previous studies had shown that AKI could be partially prevented in rats by activation of Nrf2 with sulforaphane48 and that ischemic AKI is exacerbated in Nrf2-knockout mice.33 In conjunction with those results, the recent study by Wu et al.47 showed that pretreatment with bardoxolone prevents the increase in blood urea nitrogen concentration that normally occurs following renal ischemia and reperfusion. In addition, histopathology reveals less renal injury, inflammation, and apoptosis in ischemia and reperfusion mice pretreated with bardoxolone as compared with ischemia and reperfusion mice that did not receive the drug. The proposed mechanism for the prevention of AKI in these mice is through increases in Nrf2, peroxisome proliferator–activated receptor-γ, and Ho-1 expression, because this study revealed significant increases in mRNA abundance of these genes following bardoxolone pretreatment followed by ischemia and reperfusion. There was a corresponding increase in Nrf2 protein expression in these mice—primarily in the glomerular endothelial cells, suggesting that bardoxolone prevents AKI by protecting glomerular function. Although this supports the previously reported claim that activation of Nrf2 mitigates AKI, this study and several of the previous studies report decreases in tubular injury and necrosis rather than decreases in glomerular damage, indicating that the mechanism of action of Nrf2 in AKI remains unclear and requires further examination.
As new pharmacological Nrf2 activators emerge, so do concerns regarding their use in a complex biological system. The first concern regarding Nrf2 activators is the specificity of the compounds being developed. Although researchers can confirm activation of Nrf2 with each of these drugs, whether or not they are exclusive to Nrf2 remains undetermined in most cases. Although many have been efficacious in providing protection against oxidative damage in a variety of systems, there is a problematic lack of comparability between the compounds because often they are not tested under identical conditions or even in the same physiological systems. The importance of the variability between different methods of Nrf2 induction is evidenced by a few studies in which the observed changes in gene expression patterns by activation of Nrf2 can vary from one method of enhancement to another. For example, in a comparison between hepatocyte-specific Keap1-knockout mice and wild-type mice treated with CDDO-Im, an Nrf2 activator, both groups showed increases in nuclear Nrf2 localization, but most of the changes in Nrf2 target gene expression between the two models were different.30 In a similar and more recent study, knockdown of Keap1 by small interfering RNA and treatment with sulforphane, another Nrf2 activator, revealed only a 14% similarity in Nrf2 target gene expression changes.49 This indicates either that these mechanisms of activation differ in the degree to which they activate Nrf2 or that they have other unidentified targets in addition to Nrf2. If any of the Nrf2-enhancing compounds have other targets, as was suggested in a recent review,8 understanding the downstream effects of those pathways is essential for predicting other potential beneficial or adverse effects associated with therapeutic use of those Nrf2 activators. One example reported in the literature of an alternative effect of Nrf2 activators is inflammation. It has been shown in primary mouse peritoneal macrophages that several Nrf2 activators have anti-inflammatory effects that are only partially dependent on Nrf2, indicating that these compounds are activating other anti-inflammatory pathways for the purpose of cytoprotection.50 Identification of the molecular targets of these pathways is of great importance with regard to liver injury and disease, in which inflammation contributes significantly to the concurrent damage. In addition, understanding the differences in downstream targets of these Nrf2 activators is a critical step toward their safe and effective use as therapeutics for any disease.
COULD Nrf2 ENHANCERS BE USED TO TREAT LIVER INJURY AND DISEASE?
Although all the clinical studies with Nrf2 enhancers thus far have been focused on the attenuation of renal injury and disease, there is a large amount of literature and a variety of animal studies suggesting that Nrf2 activation could prevent or potentially alleviate liver injury and disease as well. However, there are still significant gaps in this field of research, both in basic science and in the clinic, that need to be filled before Nrf2 activators can be considered a viable therapy option for liver injury and disease in humans. That aside, in order to draw inferences regarding whether Nrf2 activators may be viable therapeutics for liver injury and disease, it is necessary to first briefly review the pathophysiological elements underlying common chronic and acute liver diseases. This review focuses on nonalcoholic fatty liver disease (NAFLD), NASH, alcoholic liver disease, and acute drug-induced liver injury (DILI) because these pathologies have a high prevalence in the United States and many other countries.
NAFLD and NASH
NAFLD is the most common cause of chronic liver disease in North America, affecting one-third of the general population,51 and is therefore a major public health concern. Many years of research indicate that the progression of NAFLD depends on multiple mechanisms operating simultaneously to produce cell injury, apoptosis, inflammation, fibrosis, and, ultimately, NASH. Following the accumulation of triglycerides in the liver, impairment of mitochondrial respiratory chain activity results in the overproduction of ROS52 and the depletion of mitochondrial glutathione.53 In addition, continual ROS generation may diminish insulin sensitivity and further prevent the export of triglyerides,54,55 exacerbating the underlying pathogenesis. Other characteristics of NASH include reduced superoxide dismutase56 and catalase activity57 and increased lipid peroxidation within the hepatocytes. Lipid peroxidation has been shown to impair nucleotide and protein synthesis, thereby inducing apoptosis, inflammation, and liver fibrosis.51
It has been proposed that Nrf2 plays a role in NASH because genetic deletion of Nrf2 in mice results in rapid onset and progression of the disease.58 There are several stages within this disease progression at which activation of Nrf2 may exert a potential therapeutic effect. The first is in the initial stages of the disease, when lipids are accumulating in the hepatocytes. Nrf2 activation with CDDO-Im has been shown to effectively prevent hepatic lipid accumulation in wild-type mice but not in Nrf2-disrupted mice,59 an effect that may be dependent on a decrease in gene expression of fatty acid synthesis enzymes by Nrf2 activation. Another way in which Nrf2 activation may be protective against NAFLD and NASH is through several preventive effects on inflammation. Several chemotherapeutic agents have been shown in a variety of cell culture and rodent systems to induce Nrf2 and cause simultaneous repression of nuclear factor-κB,60 an important cell-signaling molecule for the inflammatory response. Also, as stated previously, pharmacological activation of Nrf2 results in increased gene expression of Ho-1 and Nqo1, which in turn have inhibitory effects on inflammation. Ho-1 has been shown in mice to prevent phosphorylation of nuclear factor-κB by its endogenous substrate tumor necrosis factor-α,61 indicating that it has inhibitory effects on the initiation of the inflammatory response. Under normal conditions, lipopolysaccharide can be used to induce the expression of tumor necrosis factor-α and cause inflammation through activation of nuclear factor-κB; however, Nqo1 and Ho-1 overexpression, such as would occur with Nrf2 activation, has been shown in human monocytes to prevent the lipopolysaccharide induction of tumor necrosis factor-α expression,62 thereby preventing inflammation. Another mechanism for potential treatment of NAFLD and NASH is through activation of superoxide dismutase and catalase, antioxidant enzymes with decreased activity in this disease state. The pharmaceutical agent and Nrf2 activator Protandim has been shown in human trials to increase erythrocyte superoxide dismutase and catalase activity by 30 and 54%, respectively.39 Finally, it is possible that activation of Nrf2 could play a role in regulating transforming growth factor-β (TGF-β), a profibrotic signaling factor in plasma, and therefore contributes to alleviation of fibrosis in NASH. A recent study demonstrated that sulforaphane attenuates hepatic fibrosis through Nrf2-mediated inhibition of TGFβ signaling in a human hepatic stellate cell line.63 This effect was ultimately due to the suppression of hepatic stellate cell activation and fibrogenic gene expression,63 indicating that activation of Nrf2 has an antifibrotic effect in liver. This finding confirms a previous report that TGF-β is significantly decreased, and liver fibrosis is therefore suppressed, upon overexpression of Nrf2 in mouse embryonic fibroblast cells.64 On the basis of the available literature, it is possible that Nrf2 activators could have therapeutic effects for the treatment of NAFLD and NASH and potentially be used to slow the progression of these disease states, as summarized in Figure 2.
Figure 2.

A schematic representation of the changes that occur during the development of NAFLD/NASH (shown in yellow boxes) from the initial accumulation of triglycerides in the liver to the inflammation and fibrosis that would be seen in steatohepatitis. In addition, this schematic depicts the areas in which Nrf2 activation may positively affect this pathway and prevent the development and/or progression of this disease (shown in green circles). These deductions are based on many findings from the literature cited in the text of this review. NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; Nrf2, nuclear erythroid 2–related factor 2; ROS, reactive oxygen species; SOD, superoxide dismutase.
Alcoholic liver disease
Approximately one in five alcoholics will develop alcoholic hepatitis, and one in three will develop cirrhosis.51 Given that alcoholism affects ~16 million people in the United States,51 alcoholic liver disease is a significant health concern. Because the large majority of alcohol metabolism takes place in the liver, it is also the main site of damage by excessive alcohol consumption. The metabolism of alcohol takes place via three main enzymatic pathways: oxidation of ethanol by alcohol dehydrogenase in hepatocytes, microsomal oxidation catalyzed by cytochrome P450 2E1 (CYP2E1), and nonoxidative metabolism catalyzed by fatty acid ethyl ester synthase.51 There are many mechanisms by which alcohol induces liver injury. First, ethanol metabolism by alcohol dehydrogenase results in acetaldehyde, which, by itself, is a weak profibrogenic factor. Generation of acetaldehyde induces expression of collagen I genes in hepatic stellate cells51 and therefore contributes to fibrosis. In addition, acetaldehyde recruits TGFβ in order to further promote and enhance fibrogenesis in human hepatic stellate cells.65 Other important downstream effects of increased acetaldehyde production include GSH depletion, lipid peroxidation, and the generation of ROS and acetaldehyde adducts.66 Second, ethanol metabolism by CYP2E1 occurs during chronic alcohol consumption, when alcohol dehydrogenase reaches saturation, and results in the generation of additional acetaldehydes, ROS, and free radicals. Because of its high rate of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, CYP2E1 generates large amounts of O2•- and H2O2, which are ROS that undoubtedly contribute to oxidative stress. In addition, clinical studies have shown that chronic ethanol consumption triggers induction of CYP2E1,67 exacerbating its effects on oxidative stress in hepatocytes. It has been demonstrated that rats fed alcohol have significantly less oxidative stress and liver injury when administered CYP2E1 inhibitors,51 indicating that this drug-metabolizing enzyme plays an important role in the development of this pathology.
It remains unclear whether Nrf2 plays a major role in the pathogenesis of this disease state because there seem to be conflicting results in this area of research. Many believe that activation of Nrf2 is critical in combating the oxidative stress caused by ROS generated during the normal catalytic cycle of CYP2E1. This is supported by preclinical studies showing that ethanol-induced CYP2E1 expression also results in upregulation of Nrf2 and its targets, namely Ho-1.68 In addition, it has been demonstrated that ethanol-induced ROS generation and lipid peroxidation are increased in cells in which Nrf2 has been knocked down with small interfering RNA.68 Perhaps the most convincing evidence that Nrf2 plays a role in protecting against ethanol-induced damage arises from the research done with Nrf2-null mice. It has been reported that Nrf2-null mice have increased liver-associated mortality when fed doses of ethanol as compared with wild-type mice.69 This detrimental effect of alcohol on Nrf2-null mice was shown to be the result of increased lipogenesis, depletion of total and mitochondrial glutathione, and a Kupffer cell–mediated aggravated inflammatory response.69 In addition, it has recently been shown that many of the genes that encode enzymes involved in the pentose phosphate pathway, which generates most of the cellular NADPH, are induced by Nrf2 activation.70 That study also shows that Nrf2-null mice have lower NADPH concentrations in the liver, rendering them less capable of reducing oxidative stress. These studies strongly indicate that Nrf2 is involved in protection against ethanol-induced oxidative stress. However, there are opposing studies which argue that, although Ho-1 induction is critical for this protection, the mechanisms by which Ho-1 is activated during this type of injury are independent of Nrf2. One study reports a decrease in Nrf2 liver expression in rats fed chronically with alcohol.71 That study suggests that ATF4, another regulator of AREs, is a mechanism for the upregulation of oxidative stress–protecting genes.71 In partial agreement with this study, Yeligar et al.72 also suggest that ethanol-induced Ho-1 This not primarily regulated by Nrf2 in Kupffer cells. They show that Ho-1 is also largely induced by hypoxia-inducible factor-1α in Kupffer cells from ethanol-fed rats and that the upregulation of Ho-1 prevents excessive formation of inflammatory cytokines in ethanol-induced liver injury.72 It is clear that further research is required in this field to determine the role that Nrf2 activation may play in alleviating alcoholic liver disease, especially because the effect of previously discussed Nrf2 pharmacological activators has seemingly not been tested in any animal model as a preventive measure for this disease.
DILI
Xenobiotic agents can instigate liver injury in a variety of ways, depending on the toxicant. In general, they can initiate a liver-damaging inflammatory response by direct activation of Kupffer cells or they can initiate tissue injury by mechanisms not initiated by inflammation, including reactive-intermediate formation, protein adduct accumulation, and alterations in drug-metabolizing enzymes. This reactive intermediate–dependent toxicity typically initiates covalent bonding, lipid peroxidation, and ultimately oxidative stress. Secondary to those initial responses is the activation of an inflammatory response that will contribute to the progression of the injury. Perhaps the most relevant example of DILI in our society is APAP-induced hepatotoxicity; APAP is the leading cause of acute liver failure in the United States and the most commonly used analgesic and antipyretic agent in the world. It has been widely used as a model toxicant for studying the underlying mechanisms of DILI. APAP metabolism results in the generation of a highly reactive electrophile, depletion of glutathione, protein adduct formation, and, ultimately, oxidative stress. The oxidative stress that occurs with APAP toxicity suggests a role of Nrf2 in this toxicological event. This is supported by preclinical studies showing that Nrf2 accumulates in the nuclei as rapidly as 60 min following APAP exposure73 and that Nrf2-null mice have enhanced liver injury and mortality with APAP exposure as compared with wild-type mice.14,29,74 Furthermore, it has been demonstrated that the Nrf2 activator CDDO-Im is protective against APAP hepatotoxicity by the induction of Ho-1, Nqo1, and glutamate-cysteine ligase catalytic subunit in wild-type but not Nrf2-null mice.25 In addition, activation of Nrf2 by hepatocyte-specific Keap1 knockout in mice has revealed an increase in NADPH concentration in liver, which would be protective against oxidative stress.70 APAP toxicity also causes induction of hepatic membrane transporters Mrp2, Mrp3, and Mrp4 in an Nrf2-dependent manner in mice,28,29 indicating that they are important for detoxification of APAP and/or its metabolites. Pharmacological activation of Nrf2 may facilitate prevention of liver injury by enhancing efflux pathways, a mechanism that would presumably be important for the elimination of many different toxicants because these transporters have very broad substrate specificity. Apart from APAP toxicity, there have been other examples in the literature that indicate that Nrf2 activation may prevent DILI. For example, it was recently shown that Nrf2-null mice are more susceptible to hepatotoxicity by the toxic cleaning solvent 1-bromopropane.75 The hepatotoxic effects of this chemical, including decreased glutathione, increased necrosis, and increased malondialdehyde levels (a measure of oxidative stress), are all exacerbated in the absence of Nrf2.75 These studies indicate that activation of Nrf2 could potentially protect the liver against DILI, but more research is necessary to confirm this hypothesis.
CONCLUDING REMARKS
To our knowledge, based on the information currently available in the literature, Nrf2 activators have great potential as therapeutic agents against both chronic liver injuries, such as NAFLD and NASH, and acute liver injury, such as DILI. An overview of the ways in which Nrf2 activators might help alleviate the three liver diseases discussed in the previous section is provided in Figure 3. However, some of the mechanisms by which activation of Nrf2 facilitates the prevention or alleviation of these conditions are probably different. For example, the effects of Nrf2 activation on the inflammatory response are evidently more important for preventing the progression of a chronic injury to a more severe disease state, such as in NAFLD, NASH, and chronic alcohol consumption. By contrast, the effects of Nrf2 activation on toxicant elimination are clearly more important in acute liver injury cases, in which enhanced efflux of the toxicant itself or endogenous mediators of injury is critical, such as that induced by APAP and other drugs. For this reason, it is critical to understand why variability in gene activation occurs with different methods of Nrf2 activation and, more importantly, how these differences can be used to our advantage when trying to develop a therapeutic against one specific type of liver disease. Also, the more information that is acquired with regard to the transcriptional regulatory cofactors (e.g., Maf, Jun, Fos) with which Nrf2 forms a heterodimer, the more likely it is that we will be able to develop a drug able to differentially control Nrf2 gene expression targets in a manner that is ideal for treating a particular liver disease.
Figure 3.
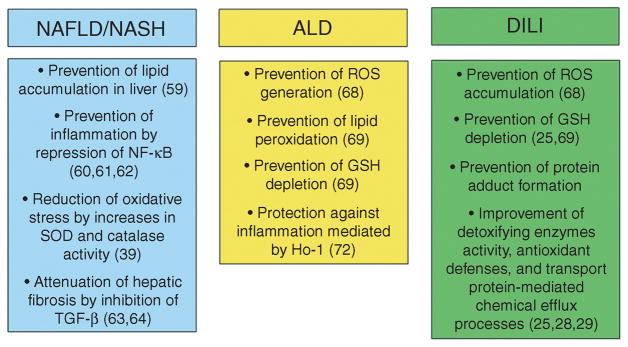
Overview of ways in which Nrf2 activation may affect the progression of three common liver pathologies. This schematic outlines the various ways by which an Nrf2-enhancing therapeutic may help to alleviate nonalcoholic fatty liver disease/steatohepatitis (NAFLD/NASH), alcoholic liver disease (ALD), and drug-induced liver injury (DILI), based on the current literature as cited in the text. GSH, glutathione; NF- B, nuclear factor- B; ROS, reactive oxygen species; SOD, superoxide dismutase; TGF-β, transforming growth factor-β.
Footnotes
CONFLICT OF INTEREST
The authors declared no conflict of interest.
References
- 1.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aleksunes LM, Manautou JE. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol Pathol. 2007;35:459–473. doi: 10.1080/01926230701311344. [DOI] [PubMed] [Google Scholar]
- 3.Itoh K, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 5.Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med. 2004;36:1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 6.Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–3156. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 7.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci USA. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 9.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Aldaba-Muruato LR, Moreno MG, Shibayama M, Tsutsumi V, Muriel P. Protective effects of allopurinol against acute liver damage and cirrhosis induced by carbon tetrachloride: modulation of NF-kappaB, cytokine production and oxidative stress. Biochim Biophys Acta. 2012;1820:65–75. doi: 10.1016/j.bbagen.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 11.Ivanov AV, Smirnova OA, Ivanova ON, Masalova OV, Kochetkov SN, Isaguliants MG. Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS ONE. 2011;6:e24957. doi: 10.1371/journal.pone.0024957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagen TM, et al. Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc Natl Acad Sci USA. 1994;91:12808–12812. doi: 10.1073/pnas.91.26.12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enomoto A, et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci. 2001;59:169–177. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- 15.Ramos-Gomez M, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci USA. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross D. Quinone reductases multitasking in the metabolic world. Drug Metab Rev. 2004;36:639–654. doi: 10.1081/dmr-200033465. [DOI] [PubMed] [Google Scholar]
- 17.Siegel D, et al. NAD(P)H:quinone oxidoreductase 1: role as a superoxide scavenger. Mol Pharmacol. 2004;65:1238–1247. doi: 10.1124/mol.65.5.1238. [DOI] [PubMed] [Google Scholar]
- 18.Smith MT, et al. Low NAD(P)H:quinone oxidoreductase activity is associated with increased risk of leukemia with MLL translocations in infants and children. Blood. 2002;100:4590–4593. doi: 10.1182/blood-2001-12-0264. [DOI] [PubMed] [Google Scholar]
- 19.Itoh K, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 20.Ishii T, Itoh K, Yamamoto M. Roles of Nrf2 in activation of antioxidant enzyme genes via antioxidant responsive elements. Meth Enzymol. 2002;348:182–190. doi: 10.1016/s0076-6879(02)48637-5. [DOI] [PubMed] [Google Scholar]
- 21.Chan JY, Kwong M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim Biophys Acta. 2000;1517:19–26. doi: 10.1016/s0167-4781(00)00238-4. [DOI] [PubMed] [Google Scholar]
- 22.Chanas SA, et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J. 2002;365:405–416. doi: 10.1042/BJ20020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong P, et al. Activation of the mouse heme oxygenase-1 gene by 15-deoxy-Delta(12,14)-prostaglandin J(2) is mediated by the stress response elements and transcription factor Nrf2. Antioxid Redox Signal. 2002;4:249–257. doi: 10.1089/152308602753666307. [DOI] [PubMed] [Google Scholar]
- 24.Ishii T, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 25.Reisman SA, Buckley DB, Tanaka Y, Klaassen CD. CDDO-Im protects from acetaminophen hepatotoxicity through induction of Nrf2-dependent genes. Toxicol Appl Pharmacol. 2009;236:109–114. doi: 10.1016/j.taap.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayashi A, Suzuki H, Itoh K, Yamamoto M, Sugiyama Y. Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein 1 in mouse embryo fibroblasts. Biochem Biophys Res Commun. 2003;310:824–829. doi: 10.1016/j.bbrc.2003.09.086. [DOI] [PubMed] [Google Scholar]
- 27.Vollrath V, Wielandt AM, Iruretagoyena M, Chianale J. Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem J. 2006;395:599–609. doi: 10.1042/BJ20051518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maher JM, et al. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology. 2007;46:1597–1610. doi: 10.1002/hep.21831. [DOI] [PubMed] [Google Scholar]
- 29.Aleksunes LM, et al. Induction of Mrp3 and Mrp4 transporters during acetaminophen hepatotoxicity is dependent on Nrf2. Toxicol Appl Pharmacol. 2008;226:74–83. doi: 10.1016/j.taap.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yates MS, et al. Genetic versus chemoprotective activation of Nrf2 signaling: overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis. 2009;30:1024–1031. doi: 10.1093/carcin/bgp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng Q, et al. Constitutive activation of nuclear factor-E2-related factor 2 induces biotransformation enzyme and transporter expression in livers of mice with hepatocyte-specific deletion of Kelch-like ECH-associated protein 1. J Biochem Mol Toxicol. 2011;25:320–329. doi: 10.1002/jbt.20392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aleksunes LM, Goedken MJ, Rockwell CE, Thomale J, Manautou JE, Klaassen CD. Transcriptional regulation of renal cytoprotective genes by Nrf2 and its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J Pharmacol Exp Ther. 2010;335:2–12. doi: 10.1124/jpet.110.170084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu M, et al. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009;76:277–285. doi: 10.1038/ki.2009.157. [DOI] [PubMed] [Google Scholar]
- 34.Córdova EJ, Velázquez-Cruz R, Centeno F, Baca V, Orozco L. The NRF2 gene variant, −653G/A, is associated with nephritis in childhood-onset systemic lupus erythematosus. Lupus. 2010;19:1237–1242. doi: 10.1177/0961203310367917. [DOI] [PubMed] [Google Scholar]
- 35.Liu J, et al. Protandim, a fundamentally new antioxidant approach in chemoprevention using mouse two-stage skin carcinogenesis as a model. PLoS One. 2009;4:e5284. doi: 10.1371/journal.pone.0005284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim YR, et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol. 2010;220:446–451. doi: 10.1002/path.2653. [DOI] [PubMed] [Google Scholar]
- 37.Kappos L, et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;372:1463–1472. doi: 10.1016/S0140-6736(08)61619-0. [DOI] [PubMed] [Google Scholar]
- 38.Velmurugan K, Alam J, McCord JM, Pugazhenthi S. Synergistic induction of heme oxygenase-1 by the components of the antioxidant supplement Protandim. Free Radic Biol Med. 2009;46:430–440. doi: 10.1016/j.freeradbiomed.2008.10.050. [DOI] [PubMed] [Google Scholar]
- 39.Nelson SK, Bose SK, Grunwald GK, Myhill P, McCord JM. The induction of human superoxide dismutase and catalase in vivo: a fundamentally new approach to antioxidant therapy. Free Radic Biol Med. 2006;40:341–347. doi: 10.1016/j.freeradbiomed.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 40.Qureshi MM, et al. The dietary supplement Protandim decreases plasma osteopontin and improves markers of oxidative stress in muscular dystrophy mdx mice. J Diet Suppl. 2010;7:159–178. doi: 10.3109/19390211.2010.482041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pergola PE, et al. Effect of bardoxolone methyl on kidney function in patients with T2D and stage 3b-4 CKD. Am J Nephrol. 2011;33:469–476. doi: 10.1159/000327599. [DOI] [PubMed] [Google Scholar]
- 42.Haugen E, Nath KA. The involvement of oxidative stress in the progression of renal injury. Blood Purif. 1999;17:58–65. doi: 10.1159/000014377. [DOI] [PubMed] [Google Scholar]
- 43.Pergola PE, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365:327–336. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 44.Rogacev KS, Bittenbring JT, Fliser D. Bardoxolone methyl, chronic kidney disease, and type 2 diabetes. N Engl J Med. 2011;365:1745–6. doi: 10.1056/NEJMc1110239. reply, 1746. [DOI] [PubMed] [Google Scholar]
- 45.Upadhyay A, Sarnak MJ, Levey AS. Bardoxolone methyl, chronic kidney disease, and type 2 diabetes. N Engl J Med. 2011;365:1746. doi: 10.1056/NEJMc1110239. reply, 1746–1746; reply, 1747. [DOI] [PubMed] [Google Scholar]
- 46.McMahon GM, Forman JP. Bardoxolone methyl, chronic kidney disease, and type 2 diabetes. N Engl J Med. 2011;365:1746. doi: 10.1056/NEJMc1110239. reply, 1746–1746; reply, 1747. [DOI] [PubMed] [Google Scholar]
- 47.Wu QQ, et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARgamma, and HO-1. Am J Physiol Renal Physiol. 2011;300:F1180–F1192. doi: 10.1152/ajprenal.00353.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoon HY, Kang NI, Lee HK, Jang KY, Park JW, Park BH. Sulforaphane protects kidneys against ischemia-reperfusion injury through induction of the Nrf2-dependent phase 2 enzyme. Biochem Pharmacol. 2008;75:2214–2223. doi: 10.1016/j.bcp.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 49.Agyeman AS, et al. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res Treat. 2012;132:175–187. doi: 10.1007/s10549-011-1536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu H, Dinkova-Kostova AT, Talalay P. Coordinate regulation of enzyme markers for inflammation and for protection against oxidants and electrophiles. Proc Natl Acad Sci USA. 2008;105:15926–15931. doi: 10.1073/pnas.0808346105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arias I, et al. The Liver: Biology and Pathobiology. 5. Wiley and Sons; Hoboken, NJ: 2009. [Google Scholar]
- 52.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 53.Marí M, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 54.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 55.Pan M, Cederbaum AI, Zhang YL, Ginsberg HN, Williams KJ, Fisher EA. Lipid peroxidation and oxidant stress regulate hepatic apolipoprotein B degradation and VLDL production. J Clin Invest. 2004;113:1277–1287. doi: 10.1172/JCI19197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nobili V, et al. Glutathione metabolism and antioxidant enzymes in patients affected by nonalcoholic steatohepatitis. Clin Chim Acta. 2005;355:105–111. doi: 10.1016/j.cccn.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 57.Videla LA, et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci. 2004;106:261–268. doi: 10.1042/CS20030285. [DOI] [PubMed] [Google Scholar]
- 58.Sugimoto H, et al. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2010;298:G283–G294. doi: 10.1152/ajpgi.00296.2009. [DOI] [PubMed] [Google Scholar]
- 59.Shin S, et al. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur J Pharmacol. 2009;620:138–144. doi: 10.1016/j.ejphar.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW. When NRF2 talks, who’s listening? Antioxid Redox Signal. 2010;13:1649–1663. doi: 10.1089/ars.2010.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seldon MP, et al. Heme oxygenase-1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of NF-kappaB RelA phosphorylation at serine 276. J Immunol. 2007;179:7840–7851. doi: 10.4049/jimmunol.179.11.7840. [DOI] [PubMed] [Google Scholar]
- 62.Rushworth SA, MacEwan DJ, O’Connell MA. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J Immunol. 2008;181:6730–6737. doi: 10.4049/jimmunol.181.10.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oh CJ, et al. Sulforaphane attenuates hepatic fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-beta/Smad signaling. Free Radic Biol Med. 2012;52:671–682. doi: 10.1016/j.freeradbiomed.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 64.Choi HK, et al. Inhibition of liver fibrosis by solubilized coenzyme Q10: Role of Nrf2 activation in inhibiting transforming growth factor-beta1 expression. Toxicol Appl Pharmacol. 2009;240:377–384. doi: 10.1016/j.taap.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 65.Svegliati-Baroni G, et al. Early response of alpha2(I) collagen to acetaldehyde in human hepatic stellate cells is TGF-beta independent. Hepatology. 2005;42:343–352. doi: 10.1002/hep.20798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–S74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 67.Girre C, Lucas D, Hispard E, Menez C, Dally S, Menez JF. Assessment of cytochrome P4502E1 induction in alcoholic patients by chlorzoxazone pharmacokinetics. Biochem Pharmacol. 1994;47:1503–1508. doi: 10.1016/0006-2952(94)90524-x. [DOI] [PubMed] [Google Scholar]
- 68.Gong P, Cederbaum AI. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology. 2006;43:144–153. doi: 10.1002/hep.21004. [DOI] [PubMed] [Google Scholar]
- 69.Lamlé J, et al. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury. Gastroenterology. 2008;134:1159–1168. doi: 10.1053/j.gastro.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 70.Wu KC, Cui JY, Klaassen CD. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci. 2011;123:590–600. doi: 10.1093/toxsci/kfr183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bardag-Gorce F, Oliva J, Lin A, Li J, French BA, French SW. Proteasome inhibitor up regulates liver antioxidative enzymes in rat model of alcoholic liver disease. Exp Mol Pathol. 2011;90:123–130. doi: 10.1016/j.yexmp.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yeligar SM, Machida K, Kalra VK. Ethanol-induced HO-1 and NQO1 are differentially regulated by HIF-1alpha and Nrf2 to attenuate inflammatory cytokine expression. J Biol Chem. 2010;285:35359–35373. doi: 10.1074/jbc.M110.138636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goldring CE, et al. Activation of hepatic Nrf2 in vivo by acetaminophen in CD-1 mice. Hepatology. 2004;39:1267–1276. doi: 10.1002/hep.20183. [DOI] [PubMed] [Google Scholar]
- 74.Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci USA. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu F, et al. Increased susceptibility of Nrf2-null mice to 1-bromopropane-induced hepatotoxicity. Toxicol Sci. 2010;115:596–606. doi: 10.1093/toxsci/kfq075. [DOI] [PubMed] [Google Scholar]