

Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by the extracellular accumulation of senile plaques composed of amyloid β (Aβ) and the intracellular accumulation of the microtubule-associated protein (MAP) tau into both non-filamentous and filamentous inclusions, such as neurofibrillary tangles (NFTs), neuropil threads (NT) and neuritic plaques (NP) [1, 2]. Tau’s primary role within neurons is thought to be the regulation and stabilization of microtubule dynamics [1, 2]. During neurodegeneration however, post-translational modifications occur in vivo, which cause tau to undergo conformational changes resulting in its disassociation from the microtubule and self-association into aggregates [2]. One or more species of these aggregates is believed to be responsible for AD pathogenesis.
The tau protein becomes highly phosphorylated in AD and this likely induces a conformational change causing its detachment from microtubules and its accumulation in aggregates [3]. Although there is much evidence linking tau to neurodegeneration, the precise mechanism of tau-mediated neurotoxicity remains to be elucidated. For many years, it was assumed that NFTs were the cause of neuronal toxicity, since they correlate very well with cognitive decline and neuronal loss [4, 5]. However, in some animal models over-expressing tau, neurodegeneration has been demonstrated in the absence of overt NFT pathology [6, 7]. Additionally, recent evidence suggests that memory function and neuronal loss can be restored in a tauopathy mouse model despite the ongoing accumulation of NFTs [5]. Moreover, NFTs have been suggested to persist in neurons for 20–30 years [8, 9] making them unlikely candidates for catalyzing immediate toxicity [10]. In fact, a large immunohistochemical study on cholinergic basal forebrain neurons in the Nucleus Basalis using an early tau marker demonstrated pretangle neurons and neuropil thread staining correlates extremely well with cognitive decline which occurs prior to the emergence of significant NFT pathology [11]. Finally, synaptic loss correlates better with cognitive decline than NFTs, again suggesting the possibility of a different mechanism for tau toxicity [12].
A novel antibody, TOC1, labels tau dimers and oligomers
If NFTs are not responsible for tau toxicity, what is the toxic species of tau? The existence of pre-fibrillar aggregates of recombinant tau in assembly assays in vitro has been known for some time [13]. Furthermore, the presence of tau-positive pre-tangle neurons lacking NFTs have been reported in AD brain tissue sections [14, 15]; however, these inclusions do not react with dyes such as Thiazine Red, a compound which binds to the β sheet conformation indicative of NFTs [16, 17]. The precise composition of these pre-fibrillar aggregates remains elusive [18–20]; also undetermined is their relationship to tau filaments that go on to form the NFTs. However, data from our lab indicate that these oligomers are toxic [16].
In order to study non-fibrillar tau aggregates (oligomers), our lab generated a monoclonal antibody that selectively recognizes tau dimers and higher order aggregates [16]. This antibody, named Tau Oligomeric Complex 1 (TOC1) was made against stable photochemically cross-linked tau dimers. The resultant stabilized small aggregates migrated as trimers on SDS-PAGE but were confirmed to be dimers using SELDI-TOF mass spectrometry [16]. Time course aggregation analysis revealed that dimerization precedes tau oligomerization which, in turn, is an earlier event than the formation of full length filaments. Employing recombinant full length hT40, we observe the presence of oligomers at approximately 15 minutes after the addition of the anionic inducer arachidonic acid, while filament formation requires five – six hours to attain steady state (Fig 1 A–C). Immunogold labeling and dot blot analysis (data not shown) of aggregated recombinant hT40, reveals that TOC1 selectively labels these dimers or oligomers and appears not to label filaments (Fig 1 G–I). TOC1’s immunoreactivity is greatly elevated in AD brains compared to healthy controls, but co-localizes best with early stage markers for AD pathogenesis such as pS422 [16]. Furthermore, TOC1 does not colocalize with MN423 or Thiazine red, both late stage markers for mature NFTs [16].
Figure 1. TOC1 and TNT1 preferentially detect aggregated tau.
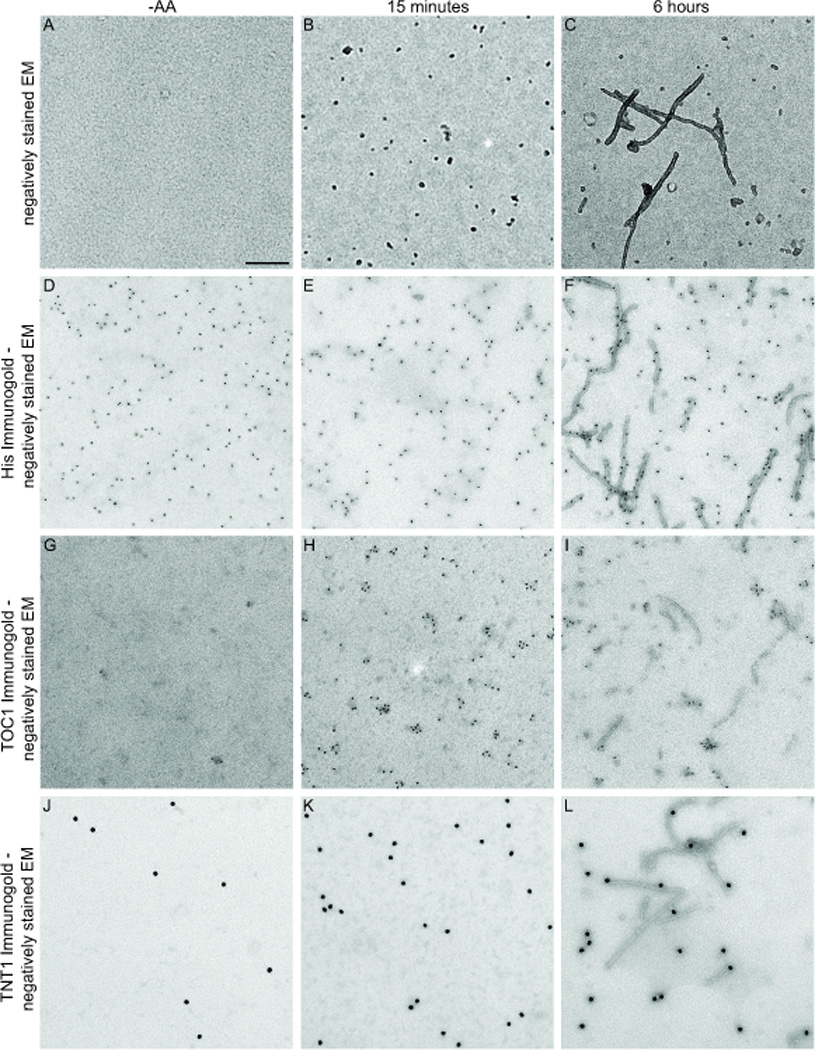
A–C. Negatively stained EM images of tau in the absence of arachidonic acid (−AA) or in its presence after 15 minutes or 6 hours of aggregation. D–F. His immunogold labeling with negatively stained EM. The histidine (his) antibody heavily labels all three tau preparations as recombinant tau contains an N-terminal Histidine tag. G–I. TOC1 immunogold labeling with negatively stained EM demonstrates that TOC1 preferentially detects tau oligomers. J–L. TNT1 immunogold labeling with negatively stained EM reveals that the PAD domain is exposed on all forms of recombinant tau but oligomers demonstrate greatest exposure. Scale bar = 200nm.
Toxicity in Fast Axonal Transport (FAT) is mediated by aggregated tau but not soluble tau monomers
Defects in axonal transport are prevalent in neurodegenerative diseases [19, 21–23]. Microtubules within the cell are used as molecular tracks for motor proteins to carry their cargo to precise destinations within the cell [24, 25]. There are two major classes of motor proteins: kinesin, which moves cargo towards the plus ends of microtubules in the direction of the synapse (anterograde transport) and dynein, which moves cargo towards the minus ends of the microtubules in the direction of the cell soma (retrograde transport) [17]. This fast axonal transport (FAT) is used by the cell to transport major membranous organelles such as mitochondria and synaptic vesicles [19]. There is increasing evidence that phosphorylation regulates this process in vivo, with much of this evidence stemming from data produced from studies performed on isolated axoplasm from the giant squid (Loligo pealii). Isolated squid axoplasm is particularly useful as it maintains its polarity, enabling direct investigations into the mechanisms controlling anterograde and retrograde transport [23, 26, 27].
Previous work from our laboratory employed this model system to investigate the effects of aggregated tau on axonal transport. We discovered that aggregated tau inhibits FAT only in the anterograde direction at physiological tau levels while tau monomers had no effect on FAT in either direction, even at concentrations of tau > 10-fold higher than normal physiological levels [19, 24]. Further investigation illustrated that this inhibition occurs via activation of a signaling cascade involving protein phosphatase 1 (PP1) and glycogen synthase kinase 3 (GSK3) [24]. Deletion analyses demonstrates that FAT inhibition requires a small stretch of amino acids (aa 2–18) located within the N-terminus that we have termed that Phosphatase Activation Domain (PAD) [21]. When the PAD region is removed from the tau protein, no inhibition of FAT occurs, indicating that PAD is both necessary and sufficient for anterograde FAT inhibition [21].
Tau reportedly exerts an effect on axonal transport by interfering and reducing the attachment frequency of the motor proteins to the microtubules [28]. However, work from our group suggests that tau’s conformation plays an important role in its ability to affect axonal transport. Within monomers, we suspect that the PAD is hidden, being masked conformationally in something akin to the ‘paperclip’ conformation, whose existence was suggested by a FRET study [29]. This would explain why monomeric tau doesn’t inhibit anterograde FAT. However, when tau aggregates, this conformation is altered, exposing PAD and allowing activation of the PP1-GSK3 signaling pathway facilitating FAT inhibition [21].
To further investigate the role of the PAD in FAT, our lab generated a monoclonal antibody directed against its 17 aa termed ‘Tau-N-Terminal 1’ (TNT1). Immunogold labeling of recombinant tau demonstrates that TNT1 detects predominantly, oligomers and filaments, although a limited reaction with monomers is apparent (Fig 1 J–L). However, in disease, TNT1 largely reacts with AD extracts containing soluble tau, exhibiting little reactivity to tau from extracts obtained from non-demented control subjects; this indicates that even soluble tau is altered in AD brains [21]. TNT1 also co-localizes in tissue with the phospho-epitope defined by the AT8 antibody, indicating that PAD exposure represents an early event in AD pathology. Previous data demonstrates that aggregated tau favors an Alz-50 conformation in which the C-terminus vacates its usual position in close proximity to the microtubule binding regions (MTBR’s) and allows the N-terminus to bind to the repeat regions [30]. In pathological tau, it seems that the N-terminus remains in this conformation and the PAD remains exposed, causing aberrant activation of the PP1-GSK3 signaling cascade leading to inhibition of FAT [21]. However, the aggregated tau used in this initial investigation, included a mixture of both fibrillar and oligomeric tau. Therefore, it is difficult to elucidate which species is producing the toxic effects within the cell. Since we now appreciate that NFT’s are most likely not the toxic form of tau, this allows us speculate that these pre-fibrillar oligomers, recognized by both TOC1 and TNT1, may constitute a key player in neurodegeneration.
Heat Shock Protein 70 protects against FAT Inhibition through interaction with tau oligomers
Following our studies with TNT1 and PAD, we used the cell-free squid axoplasm model described above to investigate the effects of the molecular chaperone Hsp70 on tau toxicity [31]. This family of proteins has been implicated in AD, Parkinson’s disease and Huntington’s disease for some time [32, 33]. Previous data indicates that Hsp70 prevents tau toxicity by preserving tau in its soluble form and preventing it from aggregating by binding to exposed hydrophobic residues [33]. It has also been demonstrated that Hsp70 can facilitate the degradation of pre-formed aggregates [33]. However, our data indicated that Hsp70 preferentially bound to oligomeric as opposed to fibrillar tau aggregates and prevented anterograde FAT inhibition [31]. This indicated that tau oligomers represent the main toxic species responsible for neurodegeneration associated with AD. Furthermore, when Hsp70 was pre-incubated with the PAD peptide and introduced to the squid axoplasm, inhibition of FAT was still observed [31]. This is noteworthy, as this suggests that Hsp70 does not directly interact with PAD.
Conclusion
In summary, the findings discussed support our hypothesis that tau oligomers may be the toxic form of tau in neurodegenerative disease. Our novel tau antibody TOC1 selectively recognizes dimers and higher order oligomers over full-length filaments and co-labels with other early stage antibodies such as pS422, which suggests that these oligomers appear early in AD pathology [16]. These data raise the possibility that oligomers may be a pre-fibrillar intermediate that has been alluded to in the literature [13]. Utilizing the cell-free squid axoplasm as a model along with another novel antibody, TNT1 to investigate FAT, we illustrated that full preparations (filaments and oligomers) of aggregated recombinant hT40 could inhibit anterograde FAT only. Together these studies indicate that anterograde FAT inhibition is likely caused by a conformational change occurring within tau that results in an aberrant exposure of the PAD located within the N-terminus of tau, and that such a conformational change also occurs early in AD vulnerable neurons. Further use of the squid axoplasm model established that the chaperone, Hsp70 likely protects the cell against the toxic effects of tau aggregation by interacting with oligomers rather than filaments [31], allowing us to speculate that, in the presence of Hsp70, the conformation of oligomers is altered, obscuring the PAD and thereby reducing its toxicity (Figure 2). Hence, we hypothesize that tau oligomers represent the toxic species in AD and perhaps in other tauopathies, also.
Figure 2. Role of tau in the regulation of fast axonal transport.

A. In healthy neurons, tau interacts with microtubules and is in its “paperclip” conformation with the PAD unexposed. This leads to regulation of local cargo delivery through PAD-mediated activation of the PP1-GSK3 signaling cascade. Cargo is delivered along the microtubules via the motor protein kinesin. Cargo may be delivered at suitable locations once tau becomes extended exposing the PAD domain. B. In diseased states, when tau becomes aggregated or post-translationally modified, the PAD domain is exposed leading to dissociation from the microtubules, which promotes abnormal activation of the PP1-GSK3 cascade. This ultimately results in increased inhibition of anterograde FAT. C. Hsp70 binds to monomeric tau that has dissociated from the microtubule, therefore preventing tau aggregation. Hsp70 can also interact with tau oligomers, potentially preventing the extension of oligomers into filaments and/or inactivating oligomer toxicity by conformationally obscuring the PAD domain.
References
- 1.Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta. 2005;1739(2–3):216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 2.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative Tauopathies. Annual Review of Neuroscience. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 3.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8(9):663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 4.Guilloz AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Archives of Neurology. 2003;60(5):729–736. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- 5.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25(22):5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila. neurodegeneration without neurofibrillary tangles. Science. 2001;293(5530):711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 8.Guillozet-Bongaarts AL, Cahill ME, Cryns VL, Reynolds MR, Berry RW, Binder LI. Pseudophosphorylation of tau at serine422 inhibits caspase cleavage. in vitro evidence and implications for tangle formation in vivo. Journal of Neurochemistry. 2006;97(4):1005–1014. doi: 10.1111/j.1471-4159.2006.03784.x. [DOI] [PubMed] [Google Scholar]
- 9.Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. Journal of Neuropathology & Experimental Neurology. 1999;58(2):188–197. doi: 10.1097/00005072-199902000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Kordower JH, Chu Y, Stebbins GT, DeKosky ST, Cochran EJ, Bennett D, Mufson EJ. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol. 2001;49(2):202–213. [PubMed] [Google Scholar]
- 11.Vana L, Kanaan NM, Ugwu IC, Wuu J, Mufson EJ, Binder LI. Progression of Tau Pathology in Cholinergic Basal Forebrain Neurons in Mild Cognitive Impairment and Alzheimer's Disease. Am J Pathol. 2011 doi: 10.1016/j.ajpath.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer's disease. Hum Mol Genet. 2010;19(R1):R12–R20. doi: 10.1093/hmg/ddq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, Miyasaka T, Murayama S, Ikai A, Takashima A. Granular tau oligomers as intermediates of tau filaments. Biochemistry. 2007;46(12):3856–3861. doi: 10.1021/bi061359o. [DOI] [PubMed] [Google Scholar]
- 14.Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci. 2007;27(14):3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedhoff P, von Bergen M, Mandelkow E, Davies P. A nucleated assembly mechanism of alzheimer paired helical filaments. Proc Natl Acad Sci U S A. 1998;95(26):15712–15717. doi: 10.1073/pnas.95.26.15712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ, et al. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem. 2011;286(26):23063–23076. doi: 10.1074/jbc.M111.237974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279(5350):519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- 18.Friedhoff P, Schneider A, Mandelkow EM, Mandelkow E. Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein tau monitored by fluorescence in solution. Biochemistry. 1998;37(28):10223–10230. doi: 10.1021/bi980537d. [DOI] [PubMed] [Google Scholar]
- 19.Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr, Brown H, Tiwari A, Hayward L, et al. Axonal transport defects in neurodegenerative diseases. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29(41):12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010;49(47):10039–10041. doi: 10.1021/bi1016233. [DOI] [PubMed] [Google Scholar]
- 21.Kanaan NM, Morfini GA, Lapointe NE, Pigino GF, Patterson KR, Song Y, Andreadis A, Fu Y, Brady ST, Binder LI. Pathogenic Forms of Tau Inhibit Kinesin-Dependent Axonal Transport through a Mechanism Involving Activation of Axonal Phosphotransferases. J Neurosci. 2011;31(27):9858–9868. doi: 10.1523/JNEUROSCI.0560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gotz J. Tau and transgenic animal models. Brain Res Brain Res Rev. 2001;35(3):266–286. doi: 10.1016/s0165-0173(01)00055-8. [DOI] [PubMed] [Google Scholar]
- 23.Kanaan NM, Morfini G, Pigino G, Lapointe NE, Andreadis A, Song Y, Leitman E, Binder LI, Brady ST. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport. implications for filament toxicity. J Neurosci Res. 2009;87(2):440–451. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330(6001):198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brady ST, Pfister KK, Bloom GS. A monoclonal antibody against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc Natl Acad Sci U S A. 1990;87(3):1061–1065. doi: 10.1073/pnas.87.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson GV, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. Journal of cell science. 2004;117(Pt 24):5721–5729. doi: 10.1242/jcs.01558. [DOI] [PubMed] [Google Scholar]
- 28.Seitz A, Kojima H, Oiwa K, Mandelkow EM, Song YH, Mandelkow E. Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO Journal. 2002;21(18):4896–4905. doi: 10.1093/emboj/cdf503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45(7):2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- 30.Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50's selectivity for Alzheimer's disease pathology. J Biol Chem. 1996;271(51):32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- 31.Patterson KR, Ward SM, Combs B, Voss K, Kanaan NM, Morfini G, Brady ST, Gamblin TC, Binder LI. Heat Shock Protein 70 Prevents both Tau Aggregation and the Inhibitory Effects of Preexisting Tau Aggregates on Fast Axonal Transport. Biochemistry. 2011;50(47):10300–10310. doi: 10.1021/bi2009147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arawaka S, Machiya Y, Kato T. Heat shock proteins as suppressors of accumulation of toxic prefibrillar intermediates and misfolded proteins in neurodegenerative diseases. Current pharmaceutical biotechnology. 2010;11(2):158–166. doi: 10.2174/138920110790909713. [DOI] [PubMed] [Google Scholar]
- 33.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100(2):721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]