

Abstract
Background
Bivalve mitochondrial genomes exhibit a wide array of uncommon features, like extensive gene rearrangements, large sizes, and unusual ways of inheritance. Species pertaining to the order Solemyida (subclass Opponobranchia) show many peculiar evolutionary adaptations, f.i. extensive symbiosis with chemoautotrophic bacteria. Despite Opponobranchia are central in bivalve phylogeny, being considered the sister group of all Autobranchia, a complete mitochondrial genome has not been sequenced yet.
Results
In this paper, we characterized the complete mitochondrial genome of the Atlantic awning clam Solemya velum: A-T content, gene arrangement and other features are more similar to putative ancestral mollusks than to other bivalves. Two supranumerary open reading frames are present in a large, otherwise unassigned, region, while the origin of replication could be located in a region upstream to the cox3 gene.
Conclusions
We show that S. velum mitogenome retains most of the ancestral conchiferan features, which is unusual among bivalve mollusks, and we discuss main peculiarities of this first example of an organellar genome coming from the subclass Opponobranchia. Mitochondrial genomes of Solemya (for bivalves) and Haliotis (for gastropods) seem to retain the original condition of mollusks, as most probably exemplified by Katharina.
Keywords: Solemya velum, Mitochondrial genome, Gene arrangement, Origin of replication, Mitogenomics
Background
Bivalves and mitochondrial DNA
In animals, the mitochondrial genome (mtDNA) is typically a small, circular and compact molecule, generally encoding for 37 genes: 13 protein-coding genes (PCGs), 2 rRNAs, and 22 tRNAs [1-3]. Even if striking exceptions to this standard are known [4-10], most differences among animal mtDNAs involve gene content and arrangement.
Mollusks have shown high variability in mitochondrial genome architecture [3,11-13], with respect to many genomic features, i.e. length, gene arrangement, strand assignment, gene duplications and losses, nucleotide composition, and more. Within mollusks, gastropods and bivalves show extensive variations, even with differences within the same family or genus [11,14,15]. Furthermore, a major peculiar trait of mitochondrial genome in some bivalve species is the presence of an alternative pattern of mitochondrial inheritance known as Doubly Uniparental Inheritance or DUI [16-20], that involves two separate lineages of mtDNAs. One mitochondrial genome (called F) is transmitted from the mother to the complete offspring, whereas the other one (called M) is transmitted from the father to sons, where it localizes in germline and, therefore, in sperm. Still, differences in gene order were also detected between the two DUI-related lineages within the same species [13,21,22].
Mitochondrial gene order has been shown to be a good marker for phylogenetic relationships, because rearrangements are random discrete events, and retro-mutation is very unlikely (see, f.i., [23-26]; and reference therein). This is even more true for arrangements of protein coding or rRNA genes, because they are much rarer than the ones involving tRNAs [1,11,13,27,28].
Tracing the “archaic” mtDNA gene arrangement of Mollusca, we can start from groups sequenced so far seeming to retain most ancestral features. Among them, chitons are aculiferan mollusks that are considered sister group to all conchiferans, which include scaphopods, cephalopods, gastropods, and bivalves (see, f.i., [29-31]; and reference therein). For PCGs and rRNAs, the gene arrangement of the chiton Katharina tunicata[32] is also shared by Haliotis, a primitive gastropod [33]; a single inversion separates it from the gene arrangement of Caenogastropoda (with the exception of D. gregarium; see [15,34]); few steps are required to transform the gene order of Katharina in that of Nautilus, a primitive cephalopod [35]. A single translocation also separates it from the aplacophoran C. nitidulum ([GenBank:EF211990]). Moreover, the gene arrangement of Katharina tunicata shows some outstanding similarities to lophophorates and even arthropods [27,32,36]. This clearly points out that Katharina may have the most “archaic” gene order known so far among Mollusca, and maybe the ancestral mollusk gene arrangement [13,28].
Many exceptions to typical gene content are known: f.i., it is well known that the atp8 gene has been reported as missing in several bivalve species, as discussed in ([37]; and reference therein). atp8 is present on the same strand in all Unionoida ([37], and reference therein); as a single exception, it is incomplete in the male mtDNA of Pyganodon grandis[22,38]. Moreover, it has been found in some heterodonts, like Loripes lacteus ([GenBank:EF043341]), Lucinella divaricata ([GenBank:EF043342]), Meretrix lamarckii[39], and Meretrix lusoria[40]; a putative atp8 has also been recently reported from the mytilid Musculista senhousia[18]. Conversely, it was not recovered in the mactrid Coelomactra antiquata[41].
Among the other exceptions to gene features and content, the rrnS gene is duplicated in some species of genus Crassostrea, while the rrnL gene is split in two separate fragments in all ostreids known to date ([14,42-44]; [GenBank:FJ841968]); finally, two versions of cox2 were found in the Musculista senhousia M mtDNA [18].
The taxonomic position of Solemya
Despite sharing a common bivalve shell, two major kind of bivalvian mollusks are known. The first group, the Opponobranchia, including most of the protobranchiate bivalves, is generally deemed to retain many ancestral features, while the second, the Autobranchia, i.e. lamellibranch bivalves with completely functional filter-feeding gills, is a big assemblage of species showing a more derived morphology, when compared to putative ancestral mollusks. Among bivalves, complete mitochondrial genomes are nowadays available for Autobranchia only, which show many differences with respect to the condition of K. tunicata.
In the subclass Opponobranchia there are basically two groups of living bivalves: Nuculida and Solemyida. The genus Solemya and relatives have edentulous or nearly edentulous, generally equivalved shells, with an homogeneous aragonitic ostracum, a glossy, thick and brown periostracum, and the mollusk shows a broad fringe, siphonate mantle and burrowing habits [45,46]. Most solemyids are involved in symbiosis with chemoautotrophic, gill-hosted, bacteria, enabling life in unusual habitats like deep-sea vents [46,47].
In this paper, we present the first complete mitochondrial genome of a representative of Opponobranchia, the Atlantic awning clam Solemya velum Say, 1822. This organelle genome was completely annotated and compared to other available bivalve and conchiferan complete mitochondrial genomes. Because of the sister-group relationship between Opponobranchia and Autobranchia, the characteristics of the Solemya mitogenome have proved useful to compare bivalve mtDNA features with those of other mollusks. In this paper we discuss our findings with particular reference to: (i) the presence of the atp8 gene; (ii) nucleotide composition; (iii) strand patterns; (iv) origin of replication; (v) supranumerary ORFs; (vi) gene arrangement.
Methods
DNA extraction, Long-PCR reactions and sequencing
Specimens of Solemya velum were collected by and commercially purchased at the Woods Hole Oceanographic Institution (Massachusetts, USA) in Summer 2003. A standard phenol:chloroform protocol was used to extract total genomic DNA from a pool of 5 individuals, because of the small dimensions of the obtained specimens.
The technique of Long-PCR amplification, paired with sequencing with primer-walking or shotgun cloning, was used to amplify the complete mitochondrial genome in several overlapping fragments. The Herculase® II Fusion Enzyme (Stratagene) kit was used to perform Long-PCR reactions up to 10,000 bp. The reaction conditions were set as follows: 10 μL 5× Herculase® II Fusion reaction buffer, nucleotides 250 μM each, primers 0.25 μM each, 0.5 μL Herculase® II Fusion, 5 μL template DNA, ddH2O up to 50 μL. Cycle conditions were set up as an initial denaturation step at 92°C for 2′, 40 cycles of denaturation at 92°C for 10′, annealing at 48-52°C for 30'', and extension at 68°C for 10', and a final extension step of 68°C for 8′. Primers used for Long-PCR were used to sequence long amplicons and new internal specific primers to complete primer-walking were designed with Primer3 online tool [48].
Routine PCR amplification was performed for amplicons <2,000 bp with GoTaq® Flexi DNA Polymerase (Promega) as in [49]. Amplicons were purified through PEG precipitation [50], or with Wizard® SV Gel and PCR Clean-Up System (Promega); when necessary, they were ligated into a pGEM® T-Easy Vector (Promega) and transformed into MAX Efficiency® DH5α™ Chemically Competent Cells (Invitrogen) as in [51]. Additional files 1 and 2 list all primers used for this study. All sequencing reactions were carried out through the Macrogen Europe (Amsterdam, The Netherland) facility. Only in the case of long-PCR with primers COI2F and Solemya_3a1894R (see Additional file 1), the 10,000 bp long amplicon was purified with 5% PEG precipitation [50] and sequenced via shotgun cloning. This was carried out by Macrogen Korea (Seoul, South Korea).
Sequence annotation
Protein-coding genes were annotated using the online ORF Finder tool [52]; the software Glimmer 3.02 [53] under iterated pipeline for assessing ORF features was used to confirm results; homology search was carried out with BLAST ([54,55]; and reference therein]). We investigated structures and putative functions of unknown ORFs through the @TOME 2.0 ([56]; and reference therein) and InterProScan ([57]; and reference therein) online tools: signal peptides were sought with SignalP [58], while similarities were detected using HHsearch [59], SP3[60] and Fugue [61].
Start codons of PCGs were set at the first start codon found by ORF Finder that did not overlap with an upstream gene; whenever a stop codon was overlapping with a following gene, it was moved backwards to the first suitable codon starting with T/TA (thus annotating a hypothetical truncated T--/TA- stop codon). In cases of neighboring PCGs, these in silico predictions were tested looking for a secondary structure with a possible cleavage signaling function in the connecting region, using the Mfold server [62] and a folding temperature of 14°C. tRNA genes were predicted with tRNAscan-SE 1.21 [63,64] and ARWEN 1.2 [65]. The Mfold server was used to predict the secondary structure of unassigned regions; all secondary structures were graphically edited with VARNA 3.7 [66].
Codon usage and nucleotide composition statistics were computed using MEGA 5.03 [67] and Microsoft Excel® 2007; repeated sequences were found with Spectral Repeat Finder v 1.1 [68]. The mitochondrial genome map was prepared using GenomeVx [69], setting cox1 as the starting point of the mtDNA and labeling its coding strand as “ + ”.
Phylogenetic analysis
Complete mitochondrial genomes of bivalves and other mollusks were downloaded from GenBank in November 2011 (Additional file 3). Summarizing, we included in our dataset 30 bivalves, 23 gastropods, 6 cephalopods, 1 scaphopod, 1 polyplacophoran, 1 chaetodermomorph, and the polychaete outgroup Platynereis dumerilii[70]. We assessed phylogenetic representativeness of this sample through the AvTD method as in [49]. We used the software PhyRe [71] and set the number of splits, merges, and moves to 2, shuffling at the family level. Sequences were managed through CLC Sequence Viewer 6.6.2 (CLC bio A/S), Microsoft Excel® 2007, and MEGA 5.03.
Each gene, with the exception of atp8, was separately translated into amminoacids and aligned with MAFFT 6 [72] and Muscle 3.8.31 [73,74], using the M-Coffee merging algorithm [75,76]. Gblocks [77,78] was used to select blocks of conserved positions suitable for phylogenetic analysis under default (stringent) conditions.
PartitionFinderProtein 1.0.1 [79], using the greedy option and Bayesian Information Criterion (BIC), tested the best partitioning scheme of our dataset, which was chosen for subsequent analysis, as well as the concatenated alignment and the completely partitioned model. Best-fitting amminoacid substitutions models were selected with ProtTest 3.2 ([80]; and reference therein), through Phyml [81] and BIC for model selection.
The software RAxML 7.2.8 [82,83] was used for maximum likelihood analyses, using both the fast (−x) and the standard (−b) bootstrap algorithm with 200 replicates. The PROTCAT model [84] was implemented for optimization of individual per-site substitution rates, using models suggested by ProtTest 3.2. Trees were graphically edited by PhyloWidget [85], Dendroscope [86], and Inkscape softwares.
Results
Genomic features
The complete mitochondrial genome of Solemya velum was found to be 15,660 bp long. It was deposited into GenBank database under Accession Number [GenBank:NC_017612]. All genes of the standard metazoan mitochondrial genome were found, including the atp8 gene (Figure 1). With the only exception of trnT, genes are organized in a large cluster on the “ + ” strand (from trnG to trnF) and in a slightly shorter cluster on the “-” strand (from trnE to atp6). 22 tRNAs are present: as usual for animal mtDNA, two serine-encoding tRNAs, trnS1(AGN) and trnS2(UCN), and two leucine-encoding tRNAs, trnL1(CUN) and trnL2(UUR) were found. The secondary structure of tRNA genes was predicted and is shown in Additional file 4; as expected [1,18,87-90], trnS1(AGN) presents a reduced DHU arm.
Figure 1.
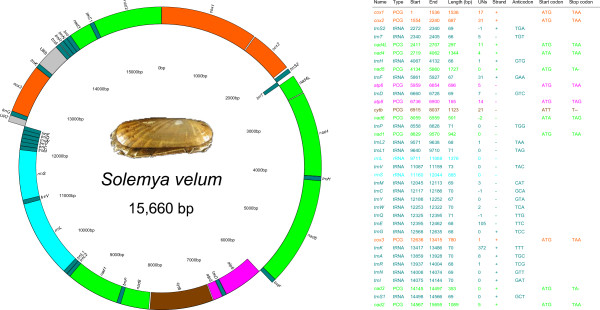
Genomic map and annotation of S. velum complete mitochondrial genome. Genes located on the external side of the map are on “ + ” strand (i.e. that encoding cox1), whereas genes on the internal side are on “-” strand. URs > 100 bp are shown and they were arbitrarily set on “ + ” strand. UNs, number of unassigned nucleotides after the gene: if negative, overlapping nucleotides with the following gene; PCG, Protein Coding Gene.
Nucleotide composition and A-T/G-C proportions were computed for each single gene and for PCGs, third codon positions, ribosomal genes, tRNAs, and URs taken as a whole (Additional file 5): the total A-T content of S. velum mitochondrial genome is 68.11%. A chi-square test with 1 d. f. demonstrated that the A-T composition of S. velum mtDNA is significantly different from that of other bivalves, gastropods, scaphopods (p < 0.005), and K. tunicata (Polyplacophora; p < 0.010); however, no significant difference was found with mtDNA A-T composition of C. nitidulum (Caudofoveata) and Cephalopoda (see raw data in Additional file 6).
A-T content and A-T/G-C skew are shown in Figure 2 for S. velum and three other mollusks for comparison: Katharina tunicata (Polyplacophora), Unio pictorum (Bivalvia: Palaeoheterodonta), and Meretrix petechialis (Bivalvia: Heterodonta). A-T content is often similar to that of K. tunicata (and M. petechialis), while skews, at the genomic level, are more similar to those of U. pictorum. We also plotted the A-C and the G-T content along the mitochondrial genome (using the “ + ” strand) with a sliding window of 151 bp (Figure 3). Overall, there is bias towards neither pair, but we evidenced a region of high A-C content spanning approximately from the atp6 to the rrnL gene, i.e. part of the mitochondrial genome where all genes are on the “-” strand.
Figure 2.

Compositional patterns of S. velum and other mollusks mitochondrial genomes. A-T content, A-T skew, and G-C skew are computed for each single gene and for many genomic regions following the legend below the chart.
Figure 3.
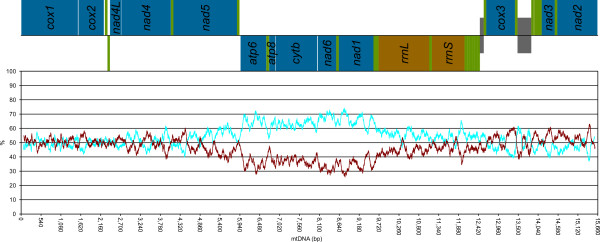
A-C and G-T content along the mitochondrial genome of S. velum. A-C (pale blue) and G-T (dark red) contents are computed on a sliding window of size 151 bp. A linear sketch of the complete genome as annotated in Figure 1 is depicted above the plot: blue, PCGs; green, tRNAs; brown, rRNAs; gray, URs >100 bp; genes on the “ + ” strand are above the black line and genes on the “-” strand are below it.
The most common start codon (Figure 1) is ATG (10 PCGs), but also alternative codons were detected, in accordance with previous findings in different invertebrates [1,91]. Most probably, truncated stop codon are used in three genes, namely nad3/nad5 (TA-) and cytb (T--). As already shown (f.i., [18,91,92]), these are common in metazoan mitochondrial genomes, with TAA stop codon subsequently restored by post-transcriptional polyadenilation. In five cases (Figure 1), two PCGs are not separated by any tRNA and are neighboring: in all cases, a stem-loop structure with a putative cleavage function of the polycistronic primary transcript has been found (Additional file 7). S. velum mtDNA contains 3,735 codifying codons, whose usage is shown in Additional file 8. Most used codon is UUA (Leu), while less used codon is CGC (Arg). The commonest amminoacid is leucine, while the rarest is cysteine.
The third base of the codon is most often an A (38.93%) or a T (38.32%), whereas C (12.39%) and G (10.36%) are less represented in this position. This may simply relate to the degeneracy of the mitochondrial code, but it is possible that natural selection is even looser in these positions because of a wobble effect: as already reported for other metazoans ([18]; and reference therein), only some codons in the PCGs do have their relative tRNA/anticodon on the mtDNA (see Additional file 8).
We found precise patterns of nucleotide content in four-fold degenerate third codon position along the mtDNA molecule (Figure 4). We used all the PCGs as a possible starting point for the analysis and best results were obtained when cox3 was used as the first gene in the analysis. All the correlations were found to be significant with the exception of C content (see caption to Figure 4 for details).
Figure 4.
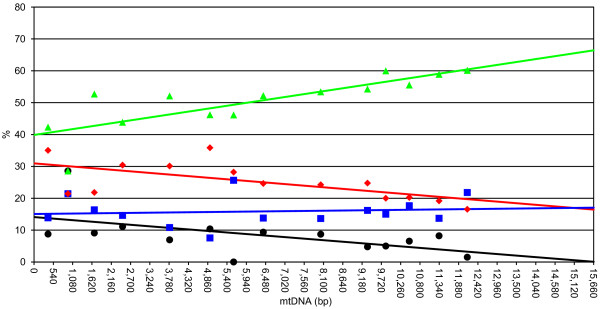
Location of the origin of replication of the H strand. A (green), C (blue), G (black), and T (red) content at four-fold degenerate codons of PGCs are shown. Percent contents of each PCG are plotted at the midpoint of the ORF using the first nucleotide of cox3 ORF as the starting point. We also included ORF117 in this analysis (see text for further details). Equations are as follows. %A, y = 0.0017 × + 39.88, r2 = 0.64, p < 0.005;%C, y = 0.0001 × 15.07, r2 = 0.01, p = 0.71;%G, y = − 0.0009 × 14.09, r2 = 0.30, p < 0.05;%T, y = − 0.0009 × 30.96, r2 = 0.39, p < 0.05.
Unassigned regions
Relatively few unassigned regions (URs) are present in the mitochondrial genome of S. velum (4.12% of the genome length; Figure 1; Additional files 5 and 9). Most of them are between 11 and 31 bp, but the largest ones are UR7 (105 bp) and UR8 (372 bp), between the trnE/trnG and trnK/trnA gene pairs, respectively.
The putative secondary structures of UR7 and UR8 are shown in Figure 5. UR7 folds as a double hairpin; UR8 folds in a more complex pattern, with several stem-and-loop substructures. A repeated 17-bp long motif was found in this region (5′-ACCAGCCGGTTTTTCTA-3′), starting at bases 220 and 337 of UR8 sequence. Both UR7 and UR8 have a high A-T content (84.76% and 70.43%, respectively), making of UR7 the A-T-richest region in the genome.
Figure 5.

UR secondary structures. Putative secondary structures of largest URs of S. velum mitochondrial genome were inferred by software Mfold. The Gibbs energy (dG) is shown at the bottom of each structure. a, UR7; b, UR8.
Two small ORFs of 117 and 195 nucleotides were found within UR8 and were called ORF117 and ORF195, respectively. ORF117 starts at nucleotide 13,524 (start codon: ATA) and ends at 13,640 (stop codon: TAA) on the “ + ” strand; according to Glimmer results, ORF117 has a “raw” scoring value of 3.05, which is comparable with that of other PCGs, like nad4L and nad6 (2.58 and 3.19, respectively). A BLAST search could find a possible homolog within the putative control region of Haliotis rubra[33], which was also confirmed by Glimmer. The A-T content of ORF117 is 59.83% and the most used codon is UUU (Phe), with 7 hits.
ORF195 starts at nucleotide 13,846 (start codon: ATT) and ends at 13,652 (stop codon: TAG) on the “-” strand. ORF195 was not confirmed by Glimmer, but SignalP could retrieve a weak similarity with a signal peptide in the first 29 amminoacids of the putative translated protein; the A-T content of ORF195 is 73.33% and the most used codon is AAA (Lys), with 12 hits.
InterProScan with TMHMM 2.0 online tool could not identify any domain within ORF117, while a transmembrane domain was found within ORF195 (amminoacids 15–32). Using the @TOME 2.0 server, HHsearch, SP3 and Fugue could find some similarities between ORF117 and the DNA-binding domains of some proteins ([PDB:1TNS], [PDB:1C20], [PDB:3GNA], [PDB:1G4D]); HHsearch and Fugue also showed homology of ORF195 with two membrane-linked proteins ([PDB:1FME], [PDB:1MEQ]).
Phylogenetic analysis
The Phylogenetic Representativeness of our sample of mollusk mitogenomes is shown in Additional file 10. Using the original mollusk master list as published by [93], the AvTD of our sample is lying exactly on the mean AvTD of 100 random subsamples of equal size (Additional file 10a, diamond) and it is above the highest random result (Additional file 10b, diamond) if a change in underlying taxonomy is simulated.
The overall length of concatenated alignment, after Gblocks masking, was of 1,782 amminoacids and the nad4L gene was completely excluded from the analysis, lacking suitable blocks. The software PartitionFinderProtein selected a 3-blocks model: the first cluster was atp6-cytb-nad2-nad3-nad4-nad5; the second one was cox1-cox2-cox3-nad1; the nad6 gene was given its own partition. All models selected by ProtTest and piped to RAxML are listed in Additional file 11.
The six ML searches converged on similar trees: following the partitioning scheme selected by PartitionFinderProtein and using the complete bootstrap procedure we obtained the tree shown in Figure 6. Katharina tunicata is the sister taxon of all other mollusks; a node with low bootstrap support (BS = 20.5) separates Solemya + (Haliotis + Caenogastropoda) from (Graptacme + Cephalopoda) + (Heterobranchia + Autobranchia). In this scenario, both Bivalvia and Gastropoda seem polyphyletic, but deep nodes are weakly supported (13.0 < BS < 44.5). Conversely, Cephalopoda, Heterobranchia, Amarsipobranchia sensu[49], Palaeoheterodonta, and Caenogastropoda were recovered as monophyletic with high BS.
Figure 6.

Maximum likelihood (ML) tree. The tree shown is the consensus tree of 200 standard bootstrap replicates and the bootstrap proportion (BP) is shown for each node. Bivalves are shown in red; the asterisk indicates Solemya velum.
Gene order
As expected from the high plasticity of molluscan (and particularly bivalvian) mitochondrial genomes, many differences were found between S. velum and other mollusks’ gene arrangement. However, we found that Katharina tunicata and Haliotis rubra (Gastropoda: Vetigastropoda) share the same gene order (recall that we excluded tRNAs for this analysis) and, more interestingly, that this gene order is highly similar to that of S. velum, the only difference being an event of inversion of the atp8-atp6-nad5-nad4-nad4L cluster (Figure 7). If tRNAs were taken into account, this inversion would hold true (even if the cluster is slightly different due to trnT, trnH, trnF, and trnD). The same inversion event can be partially recovered in the unpublished partial mitochondrial genome of the opponobranchiate Nucula nucleus (Bivalvia: Nuculida), which is available in GenBank under the Accession Number [GenBank:EF211991].
Figure 7.
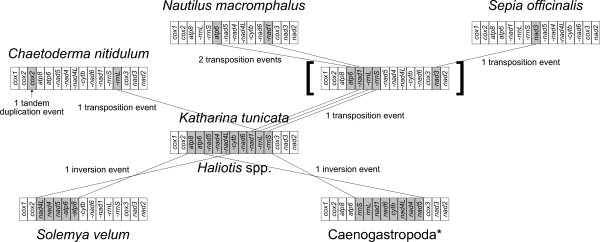
Gene rearrangements. Reconstruction of relationships among gene arrangements of Katharina tunicata/Haliotis rubra/Haliotis tuberculata, Solemya velum, C. nitidulum, Caenogastropoda, N. macromphalus, and S. officinalis is shown, after the exclusion of tRNAs. Genes involved in the rearrangements are shaded in gray. If a minus sign (“-”) is present, the gene is encoded on the “-” strand, otherwise it is encoded on the “ + ” strand. The asterisk is to signal that D. gregarium is an exception to the displayed common gene arrangement of Caenogastropoda; square brackets refer to a possible gene arrangement ancestral to N. macromphalus (subclass Nautiloida) and S. officinalis.
Discussion
Gene content
The Solemya mtDNA contains all genes of the standard metazoan mitochondrial genome (Figure 1). It is tempting to conclude that the loss/degeneracy of atp8 is restricted to Amarsipobranchia, given the presence of this gene in palaeoheterodonts and in S. velum (and, following the GenBank partial mitochondrial genome, also in Nucula nucleus). Whether the absence of atp8 gene is real or just an outcome of incorrect annotations (see, f.i., [18,37]), its presence in a bivalve like Solemya further supports, if necessary, that the ancestral bivalve condition is the retention of a fully functional atp8 gene.
Moreover, it is common in metazoans to find neighboring atp6 and atp8 on the same strand [1] and it has been suggested that uncleaved transcripts may be co-translated [94,95]. Tough this arrangement is not found in many phyla, like Plathyhelminthes, Nematoda, Annelida, Sipunculida, Brachiopoda, and Mollusca (and atp8 itself is also lacking in some of them; [13]), nevertheless atp6 and atp8 are neighboring in S. velum (Tab 3). The same association can be found in basal mollusks, like C. nitidulum and K. tunicata, and in other conchiferans, like cephalopods (with the exception of N. macromphalus), Caenogastropoda, and Heterobranchia (albeit on the opposite strand).
Genome features
Mean A-T content in main molluscan classes ranges between 63.51% (bivalves) and 74.12% (scaphopod G. eborea): S. velum has a high A-T content (68.11%), being significantly more similar to aculiferans and cephalopods than to other bivalves (Additional file 6): actually, the A-T content of Autobranchia is between 55.20% (M. yessoensis; [96]) and 69.70% (V. philippinarum, [GenBank:NC_003354]). Irrespective of the functional constraints and gene features, we found an unbiased, when not low, G-T content in the “ + ” strand (Figure 3). Most commonly, the leading strand is G-T rich ([35,97-103]; but see [104-106]). The G-T content is not particularly high in any region of the S. velum “ + ” strand, and it even drops to very low values where the molecule encodes PCGs on the “-” strand (Figure 3).
All genes are located on the same strand in Amarsipobranchia, and most of them in Palaeoheterodonta. Contrastingly, in Solemya velum, genes are evenly distributed among “ + ” and “-” strands, with 18 and 19 genes, respectively. Even if a H-biased distribution of genes is found in other lophotrochozoans, like annelids, brachiopods, bryozoans and platyhelminths (see, f.i., [13,27]), an even distribution is the commonest situation among Mollusca (Additional file 6; but see [15]) and, notably, as for A-T content, S. velum is quite similar to Caudofoveata, Cephalopoda, Polyplacophora, and Scaphopoda. This gene distribution on both strands rises a stimulating question on strand assignment: which is the leading (heavy; antisense) strand in S. velum? Patterns evidenced in S. velum resemble those of N. macromphalus[35]. Contrarily, a strand with a sharper G-T predominance has been signaled, f.i., in some gastropods [15] and in Katharina[35]. It seems that mtDNAs with most genes on the same strand (e.g., Caenogastropoda, Amarsipobranchia) tend to have higher G-T values than mtDNAs with genes evenly distributed on either strand (e.g., Cephalopoda, Palaeoheterodonta).
Control region and origins of replication
The animal mtDNA control region (CR) should contain or neighbor the origins of replication (ORs). [107] and, specifically for bivalves, Breton and colleagues ([22]; and reference therein) proposed several parameters to annotate the CR, like (i) UR length, (ii) evidence for secondary structure with T-rich loops, (iii) high A-T content and (iv) repetitive elements and palindromes. The best candidates for S. velum control region are the unassigned regions UR7 and UR8 (see Additional file 9), but, as expected, they give no BLAST hits with putative CRs of other mollusks. Among the two largest URs, UR8 is the longest unassigned region. Both URs evidenced complex putative secondary structures (Figure 5); again, both URs have a high A-T content, but while the A-T content of UR8 is 70.43% (somewhat near the overall genome score of 68.11%), it is up to 84.76% for UR7, much more than other putative CRs of mollusks [108]. On the other side, the only 17 bp-long repeated motif found in these URs was found in UR8. So, based on the above mentioned characteristics, it is not possible to unambiguously assign the CR function to either UR.
[102] suggested that mutations at four-fold degenerate sites should be completely neutral, being positions under no or limited selection. Therefore, in absence of selective constraints, the heavy (antisense) strand would accumulate G and T at these sites, while the light (sense) strand would accumulate A and C. Consequently, A-T and G-C skews at the four-fold degenerate codon sites are known to be significantly correlated with the single-strand duration during duplication, and therefore with the position of each PCG with respect to the OR of that strand [22,102]. Precise linear patterns of the percentages of each of the 4 nucleotides were found when cox3 was used as the starting point (Figure 4), being the correlation significant for 3 nucleotides out of 4. The slope was positive for A and C, and negative for G and T. This is very similar to the findings of [102] for mammals and [15] for vermetid gastropods. These data finally point to three conclusions: (i) the strand we call “ + ” (i.e., the one encoding cox1) is the heavy (antisense) strand, while the strand we call “-” is the light (sense) strand; (ii) the CR of S. velum mtDNA is located immediately before cox3 in the UR7 region; (iii) as UR7 is located at the H/L switch, we suggest this to be the OR of both strands, working in either direction.
The region of the putative OR of the H strand encompasses UR7 and a cassette of tRNAs on the “-” strand, namely trnM, trnC, trnY, trnW, trnQ, and trnE, a situation already signaled in the family Vermetidae [15] and in the unionid Inversidens japanensis[22]. As shown by [109], tRNAs on one strand can sometimes work as OR in the opposite one by forming alternative secondary structures other than conventional cloverleaves. Further comparison with other bivalves is tricky because all genes are on the same strand within Amarsipobranchia, but, similarly, the CR of Mytilus spp. is located after a group of 7 tRNAs and the rrnL gene [21]; the major non-coding region (MNR) of Mimachamys nobilis is located after a cassette of 8 tRNAs [96]; the CR of Meretrix spp. is located after 7 consecutive tRNAs [39,40,110,111].
The supranumerary ORFs within UR8
If UR7 is the putative CR on Solemya mtDNA, the function of UR8 remains unknown. Its length and secondary structure may have some kind of signaling function, but it is quite noteworthy that two ORFs were found here: ORF117 and ORF195. Are they functional or not? Remarkably, they span over the almost complete UR8, leaving only small unassigned nucleotide stretches of 37, 11, and 12 bp, similar to other intergenic spacers in S. velum mtDNA (see Additional file 9).
ORF117 has a Glimmer “raw” score comparable to other PCGs of S. velum mtDNA (i.e., nad4L and nad6). Although it is nested in the A-T rich UR8 (70.43%), it has a lower A-T content (59.83%), so that its composition is actually different from the rest of the UR8. Moreover, notwithstanding the low (for S. velum) A-T content, the most used codon is UUU (Phe), which is also among the most used ones in all PCGs of this mtDNA, and therefore its codon composition is similar to that of other PCGs. The nucleotide pattern at four-fold degenerate sites is also consistent with other PCGs (Figure 4). Finally, ORF117 is almost completely located in a region of UR8 with few secondary structures (Figure 5). The presence of an homolog of ORF117 in the putative CR of Haliotis rubra, a species that, as above mentioned, seems to retain most ancestral features of molluscan mtDNA [33], may be related to a common origin of this ORF.
Conversely, ORF195 is not found by Glimmer and has a higher A-T content than ORF117 (73.33%); consistently, the most used codon is AAA (Lys). However, possible homology of ORF195 with membrane proteins is confirmed to some extent by the finding of a large transmembrane domain; the presence of a signal peptide constitutes a further in silico evidence favoring the functionality of this ORF.
It is not easy to assign to a protein a functional role only relying on bioinformatics data: expectedly, given the low homology scores and the short length of both ORFs, many different kinds of proteins and ligands were suggested by tools hosted on the @TOME 2.0 server. The presence of supranumerary ORFs in mitochondrial genomes has been reported elsewhere (f.i., [11,22,112,113]; and references therein) and they mostly are of obscure function, but they generally share either a DNA-binding motif or a transmembrane region.
The commonest hit of ORF117 was with DNA-binding domains of other polypeptides, and many of them were top-ranked using the alignments scores as a sorting criterion. The putative transmembrane region of ORF195 is 19 amminoacids long and it is found in the N-terminal part of the peptide; it is followed by 12 positively charged amminoacids (either K or R) out of 32 in the C-terminal half of the protein. Interestingly, this architecture is the same described by [22] for supranumerary sex-linked ORFs in unionid mitochondrial genomes. Breton and colleagues suggest a possible role for these ORFs, which must be involved in the complex machinery of the DUI mechanism. Present findings may confirm their claim that natural selection is working on maintaining the structure, rather than the sequence, of transmembrane supranumerary mitochondrial ORFs [22].
The presence of a putative transmembrane signaling peptide in ORF195 and the DNA-binding signal in ORF117 may suggest a regulatory role for both these proteins; moreover, their presence in the S. velum mtDNA might constitute an evidence of the ancestral presence of such supranumerary ORFs in all bivalves. However, it has to be noted that this remains an in silico analysis and that some features of ORF195 could be randomly due to the high A-T content (f.i., the signal peptide): therefore, detailed analyses of mRNA gene expression are required to shed light on these issues.
Phylogenetic analysis and the usefulness of mitochondrial markers
The position of Solemya is unexpected, being nested within Gastropoda in our tree. However, node support is evidently very low (44.5), so our analysis does not point to a diphyly of Bivalvia, but rather to a polytomy at the base of the tree, including Solemya, Katharina and Haliotis. We may suggest that the lineage leading to Opponobranchia arose so early in the molluscan radiation that little or no phylogenetic signal of this event may be retrievable in Solemya mtDNA. The structural similarities of Solemya mtDNA to Haliotis and Katharina, and the huge differences to other bivalvian mitochondrial genomes, may have affected the clustering, hence the incorrect weak relationship to Gastropoda, which has rather to be considered an artifact.
The diphyly of Bivalvia is not a new find however (see, f.i., [114-116]), even if most recent phylogenomic studies could strongly retrieve bivalves as monophyletic [29-31]. On this aspect, [117] misinterpreted our previous results [49,51], as this is the first time we obtain bivalves as diphyletic with mitochondrial DNA; this is simply because in previous works, focusing on internal relationships of the Class, we invariably forced bivalves to be monophyletic [49,51]. It is remarkable that consistency with bivalves’ lower-level taxonomy was always maintained by our previous mtDNA analyses, a consistency which is actually lacking in [117]. On the other hand, mtDNA fails to retrieve strong phylogenetic signal for the most basal molluscan phylogenetic events, thus retrieving controversial results. Other molecular markers are needed on the issue.
Mitochondrial gene arrangement may better help in tracing basal phylogenetic relationships [23-26]. Remarkably, Solemya gene order connects to the Katarina one by a single gene inversion event (Figure 7), once again pointing out that Solemya belongs to a group of mollusks maintaining archaic mtDNA features. The same inversion can be traced in N. nucleus: the presence of the inversion can be extrapolated by the sequenced regions and therefore can be considered very likely. This may be taken as evidence for a sister group relationship between Nuculida and Solemyida, thus supporting again the monophyly of Opponobranchia. Given the questioned status of Nuculanoidea (f.i., [30,49,51,117,118]; and reference therein), it would be very interesting to obtain the complete mitochondrial genome of a species belonging to this superfamily and to compare it with the one of S. velum. On the other hand, gene orders of autobranchiate bivalves known so far are so highly derived and hardly connectible (if not at all) to this archaic condition that the gene order of S. velum is useless in tracing phylogenetic relationships between Opponobranchia and other bivalves. Only the invention of a slow-evolving autobranch bivalve mtDNA (if it exists) would help to trace Bivalvia deep phylogenetic relationships based on mtDNA gene arrangements.
Conclusions
In previous paragraphs we extensively discussed many features of the mitochondrial genome of S. velum, in terms of gene/nucleotide content, strand identification, putative control region, and gene arrangement. All evidences gathered from different (and partially independent) sources point towards the same conclusion: S. velum retains most of the ancestral mtDNA features of conchiferans, like H. rubra does within gastropods. The large similarities found with K. tunicata, an outgroup of conchiferans, on one side, and the great differences found with other known bivalves, on the other side, lead us to polarize genomic characters and conclude that the mtDNA of S. velum has been probably “freezed” in a condition very similar to that of the most recent common ancestor of Opponobranchia and Autobranchia.
If this is true, the wide discontinuity between S. velum and Autobranchia is intriguing: in facts, no mtDNA representing an intermediate state is known to date, the only exceptions being some genomic features of Palaeoheterodonta (f.i., nucleotide skews and the proportion of unassigned nucleotides; unpublished observation). In the branch leading to Autobranchia, mitochondrial genome evolved on its own, like for the translocation of most – if not all – genes on a single strand; the decrease in A-T content; a possible tendency towards heavy changes in atp8 gene; multiple and lineage-specific events of gene rearrangement. The evolution of Autobranchia seems to be tightly coupled with a dramatic increase of gene rearrangement events. Which factors triggered this boost of genomic evolution, while the main cladogenetic event leading to the Opponobranchia-Autobranchia split was taking place in the lower Cambrian [51]? An exhaustive answer is probably beyond the scope of this paper, but it is tempting to investigate whether the appearance of DUI played a main role in this burst [16,17]: further research on DUI evolution and the characterization of mitochondrial inheritance in Opponobranchia can surely shed more light on this issue. The recent discovery of DUI in the nuculanid Ledella ultima[118] is very interesting in this regard and has still to be evaluated in the light of the controversial phylogenetic position of the group.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FP participated in the design of the study and in molecular lab work, analyzed the data and drafted the manuscript; AR carried out the molecular lab work and analyzed the data; MP conceived the study and helped to draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Primers used in this study for Long-PCR reactions. Primers that were used for the same experiment as a forward/reverse couple were marked with the same letter. The position of the primer annealing site on the complete molecule is reported in the “Target” column [119-121].
Primers used in this study for routine PCR reactions. Primers that were used for the same experiment as a forward/reverse couple were marked with the same letter. The position of the primer annealing site on the complete molecule is reported in the “Target” column [120-123].
The dataset used for this study.
Secondary structures of tRNAs.
Nucleotide composition of Solemya velum mitochondrial genome.
Mitogenomic features of taxa used in this study. %UNs, percentage of unassigned nucleotides over the total length of the genome; H, number of genes on the putative H strand; L, number of genes on the putative L strand; aa, number of amminoacids encoded by the totality of protein coding genes (excluding stop codons). The rrnS gene is duplicated in C. gigas; length of either copy is reported.
Secondary structures of regions between two consecutive PCGs.
Codon usage in Solemya velum mitochondrial genome. The total frequency of each amminoacid is reported under the three-letter/one-letter name; underlined codons correspond to anticodons of mitochondrial tRNAs. All truncated (TA-/T--) stop codons were attributed to TAA. RSCU, Relative Synonymous Codon Usage.
Unassigned regions longer than 10 bp.
Phylogenetic Representativeness. Test is reported as in [49] for (a) the original master list of mollusks taken from [93] and (b) a set of 100 shuffled master lists simulating taxonomical revisions. In both cases, AvTD is plotted on the left axis in the upper part of the chart and VarTD on the right one in the lower part. Sample size is plotted on x-axis; the greatest AvTD value (upper thick continue line), the AvTD mean (thin continue line), the AvTD 95% lower confidence limit (lower thick continue line), the VarTD 95% upper confidence limit (upper thick dashed line), the VarTD mean (thin dashed line), and the lowest VarTD value (lower thick dashed line) are shown. All these lines (with the exception of greatest AvTD and lowest VarTD) are shown as two-tailed 95% confidence limits for shuffling test (b). Sample used for this work is shown as a black diamond (AvTD)/circle (VarTD).
Amminoacid substitution models selected by ProtTest 3.2 [80] and piped to RAxML 7.2.8 [82,83].
Contributor Information
Federico Plazzi, Email: federico.plazzi@unibo.it.
Anisa Ribani, Email: anisa.rib@hotmail.it.
Marco Passamonti, Email: marco.passamonti@unibo.it.
Aknowledgements
We would like to thank Jeffrey L. Boore for providing specimens of S. velum used for this study and for the stimulating discussions on mitochondrial genomics with MP. This work was supported by the University and Research Italian Ministry (MIUR PRIN07, grant number 2007NSHJL8_002, and PRIN09, grant number 2009NWXMXX_002) and the “Canziani Bequest” fund (University of Bologna, grant number A.31.CANZELSEW). FP was financially supported by Fondazione del Monte di Bologna e Ravenna.
References
- Boore JL. Animal mitochondrial genome. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger G, Gray MW, Lang BF. Mitochondrial genomes: anything goes. Trends Genet. 2003;19:709–716. doi: 10.1016/j.tig.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Simison WB, Boore JL. In: Phylogeny and evolution of the mollusca. Ponder W, Lindberg DR, editor. Berkeley: University of California Press; 2008. Molluscan evolutionary genomics; pp. 447–461. [Google Scholar]
- Armstrong MR, Blok VC, Phillips MS. A multipartite mitochondrial genome in the potato cyst nematode Globodera pallida. Genetics. 2000;154:181–192. doi: 10.1093/genetics/154.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron SL, Yoshizawa K, Mizukoshi A, Whiting MF, Johnson KP. Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera) BMC Genomics. 2011;12:394. doi: 10.1186/1471-2164-12-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson T, Blok VC, Dowton M. Sequence and characterization of six mitochondrial subgenomes from Globodera rostochiensis: multipartite structure is conserved among close nematode relatives. J Mol Evol. 2007;65:308–315. doi: 10.1007/s00239-007-9007-y. [DOI] [PubMed] [Google Scholar]
- Shao R, Kirkness EF, Barker SC. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009;19:904–912. doi: 10.1101/gr.083188.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga K, Welch DBM, Tanaka Y, Sakakura Y, Hagiwarak A. Two circular chromosomes of unequal copy number make up the mitochondrial genome of the rotifer Brachionus plicatilis. Mol Biol Evol. 2008;25:1129–1137. doi: 10.1093/molbev/msn058. [DOI] [PubMed] [Google Scholar]
- Watanabe KI, Bessho Y, Kawasaki M, Hori H. Mitochondrial genes are found on minicircle DNA molecules in the mesozoan animal Dicyema. J Mol Biol. 1999;286:645–650. doi: 10.1006/jmbi.1998.2523. [DOI] [PubMed] [Google Scholar]
- Wei D-D, Shao R, Yuan M-L, Dou W, Barker SC, Wang J-J. The multipartite mitochondrial genome of liposcelis bostrychophila: insights into the evolution of mitochondrial genomes in bilateral animals. PLoS One. 2012;7:e33973. doi: 10.1371/journal.pone.0033973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101:301–320. doi: 10.1038/hdy.2008.62. [DOI] [PubMed] [Google Scholar]
- Grande C, Templado J, Zardoya R. Evolution of gastropod mitochondrial genome arrangements. BMC Evol Biol. 2008;8:61. doi: 10.1186/1471-2148-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallès Y, Boore JL. Lophotrochozoan mitochondrial genomes. Integr Comp Biol. 2006;46:544–557. doi: 10.1093/icb/icj056. [DOI] [PubMed] [Google Scholar]
- Milbury CA, Gaffney PM. Complete mitochondrial DNA sequence of the eastern oyster crassostrea virginica. Mar Biotechnol. 2005;7:697–712. doi: 10.1007/s10126-005-0004-0. [DOI] [PubMed] [Google Scholar]
- Rawlings TA, MacInnis MJ, Bieler R, Boore JL, Collins TM. Sessile snails, dynamic genomes: gene rearrangements within the mitochondrial genome of a family of caenogastropod molluscs. BMC Genomics. 2010;11:440. doi: 10.1186/1471-2164-11-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton S, Doucet-Beaupré H, Stewart DT, Hoeh WR, Blier PU. The unusual system of doubly uniparental inheritance of mtDNA: isn’t one enough? Trends Genet. 2007;23:465–474. doi: 10.1016/j.tig.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Passamonti M, Ghiselli F. Doubly uniparental inheritance: two mitochondrial genomes, one precious model for organelle DNA inheritance and evolution. DNA Cell Biol. 2009;28:1–10. doi: 10.1089/dna.2008.0807. [DOI] [PubMed] [Google Scholar]
- Passamonti M, Ricci A, Milani L, Ghiselli F. Mitochondrial genomes and doubly uniparental inheritance: new insights from musculista senhousia sex-linked mitochondrial DNAs (bivalvia mytilidae) BMC Genomics. 2011;12:442. doi: 10.1186/1471-2164-12-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouros E. An unusual type of mitochondrial-DNA inheritance in the blue mussel Mytilus. Proc Natl Acad Sci USA. 1994;91:7463–7467. doi: 10.1073/pnas.91.16.7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouros E. Mitochondrial-DNA inheritance - Reply. Nature. 1994;368:818. doi: 10.1038/368818a0. [DOI] [PubMed] [Google Scholar]
- Breton S, Burger G, Stewart DT, Blier PU. Comparative analysis of gender associated complete mitochondrial genomes in marine mussels (Mytilus spp.) Genetics. 2006;172:1107–1119. doi: 10.1534/genetics.105.047159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton S, Doucet-Beaupré H, Stewart DT, Piontkivska H, Karmakar M, Bogan AE, Blier PU, Hoeh WR. Comparative mitochondrial genomics of freshwater mussels (bivalvia: unionoida) with doubly uniparental inheritance of mtDNA: gender-specific open reading frames and putative origins of replication. Genetics. 2009;183:1575–1589. doi: 10.1534/genetics.109.110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore JL, Brown WM. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev. 1998;8:668–674. doi: 10.1016/S0959-437X(98)80035-X. [DOI] [PubMed] [Google Scholar]
- Dreyer H, Steiner G. The complete sequence and gene organization of the mitochondrial genome of the gadilid scaphopod Siphonondentalium labatum (Mollusca) Mol Phylogenet Evol. 2004;31:605–617. doi: 10.1016/j.ympev.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Rokas A, Holland PWH. Rare genomic changes as a tool for phylogenetics. TREE. 2000;15:454–459. doi: 10.1016/s0169-5347(00)01967-4. [DOI] [PubMed] [Google Scholar]
- Stechmann A, Schlegel M. Analysis of the complete mitochondrial DNA sequence of the brachiopod Terebratulina retusa places Brachiopoda within the protostomes. Proc R Soc Lond B. 1999;266:2043–2052. doi: 10.1098/rspb.1999.0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang KH, Hwang UW. Complete mitochondrial genome of Bugula neritina (Bryozoa, Gymnolaemata, Cheilostomata): phylogenetic position of Bryozoa and phylogeny of lophophorates within the Lophotrochozoa. BMC Genomics. 2009;10:167. doi: 10.1186/1471-2164-10-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Jameson D, Tang B, Higgs PG. The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J Mol Evol. 2006;63:375–392. doi: 10.1007/s00239-005-0246-5. [DOI] [PubMed] [Google Scholar]
- Kocot KM, Cannon JT, Todt C, Citarella MR, Kohn AB, Meyer A, Santos SR, Schander C, Moroz LL, Lieb B, Halanych KM. Phylogenomics reveals deep molluscan relationships. Nature. 2011;477:452–456. doi: 10.1038/nature10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SA, Wilson NG, Goetz FE, Feehery C, Andrade SCS, Rouse GW, Giribet G, Dunn CW. Resolving the evolutionary relationships of molluscs with phylogenomic tools. Nature. 2011;480:364–367. doi: 10.1038/nature10526. [DOI] [PubMed] [Google Scholar]
- Wilson NG, Rouse GW, Giribet G. Assessing the molluscan hypothesis Serialia (Monoplacophora + Polyplacophora) using novel molecular data. Mol Phylogenet Evol. 2010;54:187–193. doi: 10.1016/j.ympev.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Boore JL, Brown WM. Complete DNA sequence of the mitochondrial genome of the black chiton, Katharina tunicata. Genetics. 1994;138:423–443. doi: 10.1093/genetics/138.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard BT, Kerr LJ, McKiernan JM, Jansen ES, Hanna PJ. Mitochondrial DNA sequence and gene organization in the Australian blacklip abalone Haliotis rubra (Leach) Mar Biotechnol. 2005;7:645–658. doi: 10.1007/s10126-005-0013-z. [DOI] [PubMed] [Google Scholar]
- Cunha RL, Grande C, Zardoya R. Neogastropod phylogenetic relationships based on entire mitochondrial genomes. BMC Evol Biol. 2009;9:210. doi: 10.1186/1471-2148-9-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore JL. The complete sequence of the mitochondrial genome of Nautilus macromphalus (Mollusca: Cephalopoda) BMC Genomics. 2006;7:182. doi: 10.1186/1471-2164-7-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfenbein KG, Boore JL. The mitochondrial genome of phoronis architecta—comparisons demonstrate that phoronids Are lophotrochozoan protostomes. Mol Biol Evol. 2004;21:153–157. doi: 10.1093/molbev/msh011. [DOI] [PubMed] [Google Scholar]
- Breton S, Stewart DT, Hoeh WR. Characterization of a mitochondrial ORF from the gender-associated mtDNAs of mytilus spp. (Bivalvia: mytilidae): identification of the “missing” ATPase 8 gene. Mar Genom. 2010;3:11–18. doi: 10.1016/j.margen.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Doucet-Beaupré H, Breton S, Chapman EG, Blier PU, Bogan AE, Stewart DT, Hoeh WR. Mitochondrial phylogenomics of the bivalvia (mollusca): searching for the origin and mitogenomics correlates of doubly uniparental inheritance of mtDNA. BMC Evol Biol. 2010;10:50. doi: 10.1186/1471-2148-10-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhang S, Xiao G, Liu B. Complete mtDNA of the Meretrix lamarckii (Bivalvia: Veneridae) and molecular identification of suspected M. lamarckii based on the whole mitochondrial genome. Marine Genomics. 2011;4:263–271. doi: 10.1016/j.margen.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhang S, Li Y, Liu B. Complete mtDNA of Meretrix lusoria (Bivalvia: Veneridae) reveals the presence of an atp8 gene, length variation and heteroplasmy in the control region. Comp Biochem Physiol Part D Genomics Proteomics. 2010;5:256–264. doi: 10.1016/j.cbd.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Meng X, Zhao N, Shen X, Hao J, Liang M, Zhu X, Cheng H, Yan B, Liu Z. Complete mitochondrial genome of coelomactra antiquata (mollusca: bivalvia): the first representative from the family mactridae with novel gene order and unusual tandem repeats. Comp Biochem Phys D. 2012;7:175–179. doi: 10.1016/j.cbd.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Danic-Tchaleu G, Heurtebise S, Morga B, Lapegue S. Complete mitochondrial DNA sequence of the European flat oyster Ostrea edulis confirms Ostreidae classification. BMC Res Notes. 2011;4:400. doi: 10.1186/1756-0500-4-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Xu X, Yu Z, Wei Z, Xia J. Comparison of seven Crassostrea mitogenomes and phylogenetic analyses. Mol Phylogenet Evol. 2010;57:448–454. doi: 10.1016/j.ympev.2010.05.029. [DOI] [PubMed] [Google Scholar]
- Yu H, Li Q. Mutation and selection on the wobble nucleotide in tRNA anticodons in marine bivalve mitochondrial genomes. PLoS One. 2011;6:e16147. doi: 10.1371/journal.pone.0016147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell ND. Classification of the Bivalvia. Am Mus Novit. 1965;2206:1–25. [Google Scholar]
- Taylor JD, Glover EA, Williams ST. Ancient chemosynthetic bivalves: systematics of Solemyidae from eastern and southern Australia (Mollusca: Bivalvia) Memoirs Queensland Museum - Nature. 2008;54:75–104. [Google Scholar]
- Taviani M, Angeletti L, Ceregato A. Chemosynthetic Bivalves of the Family Solemyidae (Bivalvia, Protobranchia) in the Neogene of the Mediterranean Basin. J Paleontol. 2011;85:1067–1076. doi: 10.1666/10-119.1. [DOI] [Google Scholar]
- Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Plazzi F, Ceregato A, Taviani M, Passamonti M. A molecular phylogeny of bivalve mollusks: ancient radiations and divergences as revealed by mitochondrial genes. PLoS One. 2011;6:e27147. doi: 10.1371/journal.pone.0027147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis JT, Schleif R. Size fractionation of double-stranded DNA by precipitation with polyethylene glycol. Nucleic Acid Res. 1975;2:383–389. doi: 10.1093/nar/2.3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plazzi F, Passamonti M. Towards a molecular phylogeny of Mollusks: Bivalves’ early evolution as revealed by mitochondrial genes. Mol Phylogenet Evol. 2010;57:641–657. doi: 10.1016/j.ympev.2010.08.032. [DOI] [PubMed] [Google Scholar]
- Tatusov T, Tatusov R. ORF Finder. http://www.ncbi.nlm.nih.gov/projects/gorf/
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Ye J, McGinnis S, Madden TL. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 2006;34:W6–W9. doi: 10.1093/nar/gkl164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons J-L, Labesse G. @TOME-2: a new pipeline for comparative modeling of protein-ligand complexes. Nucleic Acids Res. 2009;37:W485–W491. doi: 10.1093/nar/gkp368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- Zhou H, Zhou Y. SPARKS 2 and SP3 servers in CASP6. Proteins. 2005;58:321–328. doi: 10.1002/prot.20732. [DOI] [PubMed] [Google Scholar]
- Shi J, Blundell TL, Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent Gap penalties. J Mol Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. TRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattner P, Brooks A, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33(suppl 2):W686–W689. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett D, Canbäck B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–175. doi: 10.1093/bioinformatics/btm573. [DOI] [PubMed] [Google Scholar]
- Darty K, Denise A, Ponty J. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, Issac B, Raghava GP, Ramaswamy R. Spectral repeat finder (SRF): identification of repetitive sequences using Fourier transformation. Bioinformatics. 2004;20:1405–1412. doi: 10.1093/bioinformatics/bth103. [DOI] [PubMed] [Google Scholar]
- Conant GC, Wolfe KH. GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics. 2008;24:861–862. doi: 10.1093/bioinformatics/btm598. [DOI] [PubMed] [Google Scholar]
- Boore JL, Brown WM. Mitochondrial genomes of Galathealinum, Helobdella, and Platynereis: sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa. Mol Biol Evol. 2000;17:87–106. doi: 10.1093/oxfordjournals.molbev.a026241. [DOI] [PubMed] [Google Scholar]
- Plazzi F, Ferrucci RR, Passamonti M. Phylogenetic representativeness: a new method for evaluating taxon sampling in evolutionary studies. BMC Bioinformatics. 2010;11:209. doi: 10.1186/1471-2105-11-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti S, Armougom F, Wallace IM, Higgins DG, Jongeneel CV, Notredame C. The M-Coffee web server: a meta-method for computing multiple sequence alignments by combining alternative alignment methods. Nucleic Acids Res. 2007;35:W645–W648. doi: 10.1093/nar/gkm333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace IM, O’Sullivan O, Higgins DG, Notredame C. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 2006;34:1692–1699. doi: 10.1093/nar/gkl091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29:1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. Proceedings of 20th international parallel and distributed processing symposium. Rhodes; 2006. Phylogenetic models of rate heterogeneity: a high performance computing perspective. http://ieeexplore.ieee.org/xpls/abs_all.jsp?arnumber=1639535&tag=1. [Google Scholar]
- Jordan GE, Piel WH. PhyloWidget: Web-based visualizations for the tree of life. Bioinformatics. 2008;15:1641–1642. doi: 10.1093/bioinformatics/btn235. [DOI] [PubMed] [Google Scholar]
- Huson DH, Richter DC, Rausch C, Dezulian T, Franz M, Rupp R. Dendroscope – An interactive viewer for large phylogenetic trees. BMC Bioinformatics. 2007;8:460. doi: 10.1186/1471-2105-8-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Liu D-F, Wang N-X, Zhu C-D, Jiang G-F. The mitochondrial genome of the butterfly Papilio xuthus (Lepidoptera: Papilionidae) and related phylogenetic analyses. Mol Biol Rep. 2010;37:3877–3888. doi: 10.1007/s11033-010-0044-z. [DOI] [PubMed] [Google Scholar]
- Garey GR, Wolstenholme DR. Platyhelminth mitochondrial DNA: evidence for early evolutionary origin of a tRNA that contains a dihydrouridine-arm replacement loop, and of serine-specifying AGA and AGG codons. J Mol Evol. 1989;28:374–387. doi: 10.1007/BF02603072. [DOI] [PubMed] [Google Scholar]
- Kim I, Cha SY, Yoon MH, Hwang JS, Lee SM, Sohn HD, Jin BR. The complete nucleotide sequence and gene organization of the mitochondrial genome of the oriental mole cricket, Gryllotalpa orientalis (Orthoptera: Gryllotalpidae) Gene. 2005;353:155–168. doi: 10.1016/j.gene.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Sheffield NC, Song H, Cameron SL, Whiting MF. A comparative analysis of mitochondrial genomes in coleoptera (arthropoda: insecta) and genome descriptions of Six New beetles. Mol Biol Evol. 2008;25:2499–2509. doi: 10.1093/molbev/msn198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serb JM, Lydeard C. Complete mtDNA sequence of the North American freshwater mussel, Lampsilis ornata (Unionidae): an examination of the evolution and phylogenetic utility of mitochondrial genome organization in Bivalvia (Mollusca) Mol Biol Evol. 2003;20:1854–1886. doi: 10.1093/molbev/msg218. [DOI] [PubMed] [Google Scholar]
- Fernandez-Silva P, Enriquez JA, Montoya J. Replication and transcription of mammalian mitochondrial DNA. Exp Physiol. 2003;88:4156. doi: 10.1113/eph8802514. [DOI] [PubMed] [Google Scholar]
- Millard V. Classification of Mollusca: A Classification of World Wide Mollusca. Volume 3. 2. South Africa; 2001. [Google Scholar]
- Boore JL, Staton JL. The mitochondrial genome of the sipunculid Phascolopsis gouldii supports its association with Annelida rather than Mollusca. Mol Biol Evol. 2002;17:540–552. doi: 10.1093/oxfordjournals.molbev.a004065. [DOI] [PubMed] [Google Scholar]
- Watkins RF, Beckenbach AT. Partial sequence of a sponge mitochondrial genome reveals sequence similarity to cnidaria in cytochrome oxidase subunit II and the large ribosomal RNA subunit. J Mol Evol. 1999;48:542–554. doi: 10.1007/PL00006497. [DOI] [PubMed] [Google Scholar]
- Wu X, Xu X, Yu Z, Kong X. Comparative mitogenomic analyses of three scallops (Bivalvia: Pectinidae) reveal high level variation of genomic organization and a diversity of transfer RNA gene sets. BMC Research Notes. 2009;2:69. doi: 10.1186/1756-0500-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beletskii A, Bhagwat AS. Transcription-induced mutations: increase in C to T mutations in the nontranscribed strand during transcription in Escherichia coli. Proc Natl Acad Sci USA. 1996;93:13919–13924. doi: 10.1073/pnas.93.24.13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francino MP, Ochman H. Strand asymmetries in DNA evolution. Trends Genet. 1997;13:240–245. doi: 10.1016/S0168-9525(97)01118-9. [DOI] [PubMed] [Google Scholar]
- Frederico LA, Kunkel TA, Shaw BA. A sensitive genetic assay for the detection of cytosine deamination: Determination of rate constant and the activation energy. Biochemistry. 1990;29:2532–2537. doi: 10.1021/bi00462a015. [DOI] [PubMed] [Google Scholar]
- Hassanin A, Léger N, Deutsch J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Syst Biol. 2005;54:277–298. doi: 10.1080/10635150590947843. [DOI] [PubMed] [Google Scholar]
- Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353–358. doi: 10.1007/BF01215182. [DOI] [PubMed] [Google Scholar]
- Reyes A, Gissi C, Pesole G, Saccone C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol. 1998;15:957–966. doi: 10.1093/oxfordjournals.molbev.a026011. [DOI] [PubMed] [Google Scholar]
- Sancar A, Sancar GB. DNA repair enzymes. Annu Rev Biochem. 1998;57:29–67. doi: 10.1146/annurev.bi.57.070188.000333. [DOI] [PubMed] [Google Scholar]
- Faith JJ, Pollock DD. Likelihood analysis of asymmetrical mutation bias gradients in vertebrate mitochondrial genomes. Genetics. 2003;165:735–745. doi: 10.1093/genetics/165.2.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodakis GC, Cao L, Mizi A, Kenchington EL, Zouros E. Nucleotide content gradients in maternally and paternally inherited mitochondrial genomes of the mussel Mytilus. J Mol Evol. 2007;65:124–136. doi: 10.1007/s00239-005-0298-6. [DOI] [PubMed] [Google Scholar]
- Saccone C, Gissi C, Reyes A, Larizza A, Sbisà E, Pesole G. Mitochondrial DNA in metazoa: degree of freedom in a frozen event. Gene. 2002;286:3–12. doi: 10.1016/S0378-1119(01)00807-1. [DOI] [PubMed] [Google Scholar]
- Brugler MR, France SC. The mitochondrial genome of a deep-sea bamboo coral (Cnidaria, Anthozoa, Octocorallia, Isidiidae): genome structure and putative origins of replication are not conserved among octocorals. J Mol Evol. 2008;67:125–136. doi: 10.1007/s00239-008-9116-2. [DOI] [PubMed] [Google Scholar]
- Arunkumar KP, Nagaraju J. Unusually long palindromes are abundant in mitochondrial control regions of insects and nematodes. PLoS One. 2006;1:e110. doi: 10.1371/journal.pone.0000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seligmann H, Krishnan NM, Rao BJ. Possible multiple origins of replication in primate mitochondria: alternative role of tRNA sequences. J Theor Biol. 2006;241:321–332. doi: 10.1016/j.jtbi.2005.11.035. [DOI] [PubMed] [Google Scholar]
- He C, Wang J, Gao X, Song W, Li H, Li Y, Liu W, Su H. The complete mitochondrial genome of the hard clam Meretrix meretrix. Mol Biol Rep. 2011;38:3401–3409. doi: 10.1007/s11033-010-0449-8. [DOI] [PubMed] [Google Scholar]
- Ren J, Shen X, Sun M, Jiang F, Yu Y, Chi Z, Liu B. The complete mitochondrial genome of the clam Meretrix petechialis (Mollusca: Bivalvia: Veneridae) Mitochondr DNA. 2009;20:78–87. doi: 10.1080/19401730902964425. [DOI] [PubMed] [Google Scholar]
- Pont-Kingdon G, Okada NA, Macfarlane JL, Beagley CT, Watkins-Sims CD, Cavalier-Smith T, Clark-Walker GD, Wolstenholme DR. Mitochondrial DNA of the coral Sarcophyton glaucum contains a gene for a homologue bacterial MutS: a possible case of gene transfer from the nucleus to the mitochondrion. J Mol Evol. 1998;46:419–431. doi: 10.1007/PL00006321. [DOI] [PubMed] [Google Scholar]
- Shao Z, Shannon G, Chaga OY, Lavrov DV. Mitochondrial genome of the moon jelly Aurelia aurita (Cnidaria, Scyphozoa): a linear DNA molecule encoding a putative DNA-dependant DNA polymerase. Gene. 2006;381:92–101. doi: 10.1016/j.gene.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Giribet G, Distel DL. In: Molecular systematics and phylogeography of mollusks. Lydeard C, Lindberg DR, editor. Washington: Smithsonian Books; 2003. Bivalve phylogeny and molecular data; pp. 45–90. [Google Scholar]
- Giribet G, Okusu A, Lindgren AR, Huff SW, Schrödl M, Nishiguchi MK. Evidence for a clade composed of molluscs with serially repeated structures: Monoplacophorans are related to chitons. Proc Natl Acad Sci USA. 2006;103:7723–7728. doi: 10.1073/pnas.0602578103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giribet G, Wheeler W. On bivalve phylogeny: A high-level analysis of the Bivalvia (Mollusca) based on combined morphology and DNA sequence data. Invert Biol. 2002;121:271–324. [Google Scholar]
- Sharma PP, González VL, Kawauchi GY, Andrade SCS, Guzmán A, Collins TM, Glover EA, Harper EM, Healy JM, Mikkelsen PM, Taylor JD, Bieler R, Giribet G. Phylogenetic analysis of four nuclear protein-encoding genes largely corroborates the traditional classification of Bivalvia (Mollusca) Mol Phylogenet Evol. 2012;65:64–74. doi: 10.1016/j.ympev.2012.05.025. [DOI] [PubMed] [Google Scholar]
- Boyle EE, Etter RJ. Heteroplasmy in a deep-sea protobranch bivalve suggests an ancient origin of doubly uniparental inheritance of mitochondria in Bivalvia. Mar Biol. 2013;160:413–422. doi: 10.1007/s00227-012-2099-y. [DOI] [Google Scholar]
- Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. Incorporating molecular evolution into phylogenetic analysis, and a New compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu Rev Ecol Evol Syst. 2006;37:545–579. doi: 10.1146/annurev.ecolsys.37.091305.110018. [DOI] [Google Scholar]
- Palumbi SR, Martin A, Romano S, McMillan WO, Stice L, Grabowski G. The simple fool’s guide to PCR. Hawaii: Kewalo Marine Laboratory and University of Hawaii; 1996. [Google Scholar]
- Stewart FJ, Baik AHY, Cavanaugh CM. Genetic subdivision of chemosynthetic endosymbionts of Solemya velum along the Southern New England Coast. Appl Environ Microb. 2009;75:6005–6007. doi: 10.1128/AEM.00689-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M. Phylogenetic analysis of the subclass Pteriomorpha (Bivalvia) from mtDNA COI sequences. Mol Phylogenet Evol. 2003;27:429–440. doi: 10.1016/S1055-7903(03)00013-7. [DOI] [PubMed] [Google Scholar]
- Burger G, Lavrov DV, Forget L, Lang BF. Sequencing complete mitochondrial and plastid genomes. Nat Protoc. 2007;2:603–614. doi: 10.1038/nprot.2007.59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used in this study for Long-PCR reactions. Primers that were used for the same experiment as a forward/reverse couple were marked with the same letter. The position of the primer annealing site on the complete molecule is reported in the “Target” column [119-121].
Primers used in this study for routine PCR reactions. Primers that were used for the same experiment as a forward/reverse couple were marked with the same letter. The position of the primer annealing site on the complete molecule is reported in the “Target” column [120-123].
The dataset used for this study.
Secondary structures of tRNAs.
Nucleotide composition of Solemya velum mitochondrial genome.
Mitogenomic features of taxa used in this study. %UNs, percentage of unassigned nucleotides over the total length of the genome; H, number of genes on the putative H strand; L, number of genes on the putative L strand; aa, number of amminoacids encoded by the totality of protein coding genes (excluding stop codons). The rrnS gene is duplicated in C. gigas; length of either copy is reported.
Secondary structures of regions between two consecutive PCGs.
Codon usage in Solemya velum mitochondrial genome. The total frequency of each amminoacid is reported under the three-letter/one-letter name; underlined codons correspond to anticodons of mitochondrial tRNAs. All truncated (TA-/T--) stop codons were attributed to TAA. RSCU, Relative Synonymous Codon Usage.
Unassigned regions longer than 10 bp.
Phylogenetic Representativeness. Test is reported as in [49] for (a) the original master list of mollusks taken from [93] and (b) a set of 100 shuffled master lists simulating taxonomical revisions. In both cases, AvTD is plotted on the left axis in the upper part of the chart and VarTD on the right one in the lower part. Sample size is plotted on x-axis; the greatest AvTD value (upper thick continue line), the AvTD mean (thin continue line), the AvTD 95% lower confidence limit (lower thick continue line), the VarTD 95% upper confidence limit (upper thick dashed line), the VarTD mean (thin dashed line), and the lowest VarTD value (lower thick dashed line) are shown. All these lines (with the exception of greatest AvTD and lowest VarTD) are shown as two-tailed 95% confidence limits for shuffling test (b). Sample used for this work is shown as a black diamond (AvTD)/circle (VarTD).
Amminoacid substitution models selected by ProtTest 3.2 [80] and piped to RAxML 7.2.8 [82,83].