

ABSTRACT
LuxR-type transcription factors are the master regulators of quorum sensing in vibrios. LuxR proteins are unique members of the TetR superfamily of transcription factors because they activate and repress large regulons of genes. Here, we used chromatin immunoprecipitation and nucleotide sequencing (ChIP-seq) to identify LuxR binding sites in the Vibrio harveyi genome. Bioinformatics analyses showed that the LuxR consensus binding site at repressed promoters is a symmetric palindrome, whereas at activated promoters it is asymmetric and contains only half of the palindrome. Using a genetic screen, we isolated LuxR mutants that separated activation and repression functions at representative promoters. These LuxR mutants exhibit sequence-specific DNA binding defects that restrict activation or repression activity to subsets of target promoters. Altering the LuxR DNA binding site sequence to one more closely resembling the ideal LuxR consensus motif can restore in vivo function to a LuxR mutant. This study provides a mechanistic understanding of how a single protein can recognize a variety of binding sites to differentially regulate gene expression.
IMPORTANCE
Bacteria use the cell-cell communication process called quorum sensing to regulate collective behaviors. In vibrios, LuxR-type transcription factors control the quorum-sensing gene expression cascade. LuxR-type proteins are structural homologs of TetR-type transcription factors. LuxR proteins were assumed to function analogously to TetR proteins, which typically bind to a single conserved binding site to repress transcription of one or two genes. We find here that unlike TetR proteins, LuxR acts a global regulator, directly binding upstream of and controlling more than 100 genes. Again unlike TetR, LuxR functions as both an activator and a repressor, and these two activities can be separated by mutagenesis. Finally, the consensus binding motifs driving LuxR-activated and -repressed genes are distinct. This work shows that LuxR, although structurally similar to TetR, has evolved unique features enabling it to differentially control a large regulon of genes in response to quorum-sensing cues.
Introduction
Bacteria use the cell-cell communication process called quorum sensing to measure and respond to changes in the number and species relatedness of bacteria in the environment. Quorum sensing involves the production and detection of extracellular signaling molecules called autoinducers. At low cell density (LCD), autoinducer levels are below the concentration required for detection, and so bacteria behave as individuals. As bacteria grow to high cell density (HCD), autoinducers accumulate. When a critical-threshold concentration of autoinducers is achieved, bacteria detect them and express genes required for group behaviors, such as biofilm formation, virulence factor production, and bioluminescence (1). In the model quorum-sensing bacterium Vibrio harveyi, information encoded in autoinducers is funneled into the control of the production of LuxR, the master regulator of quorum-sensing gene expression.
LuxR is the founding member of a group of homologous proteins that control quorum-sensing responses in all vibrios, including HapR (Vibrio cholerae), SmcR (Vibrio vulnificus), LitR (Vibrio fischeri), and OpaR (Vibrio parahaemolyticus) (2). LuxR-type proteins are members of the TetR family of transcription factors, which are ubiquitous in bacteria and share a conserved helix-turn-helix (HTH) DNA binding motif in their N-terminal domains (3). TetR proteins function as dimers to recognize and bind DNA sequences possessing dyad symmetry. Many TetR proteins bind small molecules; for example, the canonical TetR protein binds tetracycline. In the unbound state, TetR binds to two DNA binding sites: one to autorepress its own expression and one to repress expression of tetA, which confers tetracycline resistance. Upon binding to tetracycline, TetR releases the DNA, allowing transcription. The crystal structures of two LuxR homologs, HapR and SmcR, have been solved, and they have identical domain folds and the same relative domain arrangements as the TetR protein QacR bound to DNA (2, 4, 5). No ligand has been identified for any LuxR-type protein.
Although LuxR possesses the HTH motif that places it in the TetR family, it is unique. First, LuxR can act as an activator and a repressor (6, 7), whereas TetR-type proteins are typically repressors (3). Second, LuxR directly and indirectly controls the expression of 625 genes (7). TetR-type proteins, by contrast, generally control only their own expression and that of the adjacent gene. Third, LuxR can bind to multiple binding sites in its target promoters (8). Typically, only one binding site is present in TetR-regulated promoters. Finally, unlike the conserved TetR binding motif, the LuxR, SmcR, and HapR binding site consensus sequences are degenerate, indicating that this is a general attribute of LuxR transcription factors (9, 10). Together, these features allow LuxR and its homologs to positively and negatively control large regulons of genes (7, 9).
Here, we used chromatin immunoprecipitation and nucleotide sequencing (ChIP-seq) to define the set of LuxR binding sites in the V. harveyi genome. We showed that LuxR directly activates 35 genes and represses 80 genes. We characterized LuxR binding and how that impinges on gene regulation at two representative promoters: one that is activated and one that is repressed. We isolated mutations in the DNA binding domain of LuxR that confer sequence-specific DNA binding defects. The binding defect of a repression-defective LuxR mutant can be suppressed by altering the LuxR DNA binding site sequence to one that more closely matches the ideal LuxR binding site. Our findings demonstrate that LuxR-type proteins recognize subtle variations in binding sites to distinguish between activated and repressed promoters.
RESULTS
LuxR directly regulates 115 genes.
Previous in vitro experiments coupled with bioinformatics analyses predicted 36 LuxR binding sites in the V. harveyi genome (11). However, LuxR is known to regulate 625 genes (7). We considered two possibilities to explain this discrepancy: (i) most LuxR-controlled genes are regulated indirectly, or (ii) the bioinformatic predictions underestimated the number of LuxR binding sites. To distinguish between these possibilities, we performed chromatin immunoprecipitation (ChIP) assays using FLAG-tagged LuxR. As a control, we verified that FLAG-LuxR behaves similarly to wild-type LuxR in vivo (see Fig. S1A in the supplemental material). We first measured FLAG-LuxR occupancy at promoter regions containing known LuxR binding sites (6, 11, 12). Our ChIP analyses revealed 10- to 100-fold enrichment of LuxR binding at the promoters of aphA, luxR, luxC, qrgB, and qrr4 but not at promoters of genes known not to be bound by LuxR (qrr1 and qrr5) (Fig. 1A). As a second control, we showed that a DNA binding-defective LuxR mutant (LuxR R17C) (13, 14) did not immunoprecipitate any promoter regions (Fig. 1A).
FIG 1 .
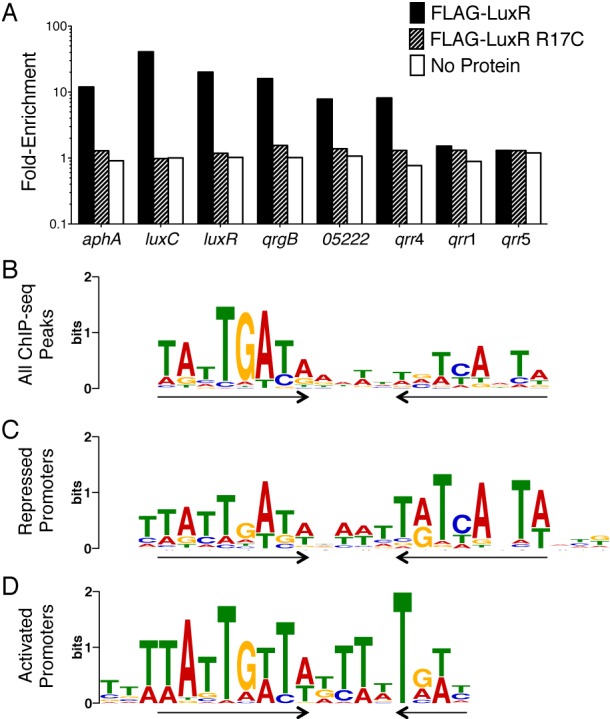
LuxR directly regulates 115 genes. (A) qRT-PCR analysis of LuxR occupancy at promoters in a V. harveyi ΔluxR strain (KM669) expressing FLAG-luxR (pAP116), FLAG-luxR R17C (pST012, a DNA binding mutant), and an empty vector (pSLS3, denoted as “no protein”). Fold enrichment at each promoter represents the ratio of immunoprecipitated DNA to input DNA. These data represent four independent experiments. (B to D) LuxR DNA binding motifs from MEME analyses of groups of ChIP-seq data peaks: all peaks (B), peaks present upstream of repressed promoters (C), or peaks present upstream of activated promoters (D). Inverted arrows denote the dyad symmetry.
LuxR binding was assessed globally by high-throughput sequencing of the FLAG-LuxR-bound DNA (ChIP-seq). We identified 1,165 LuxR binding peaks spanning 582 genomic regions. Quantitative Western blot analyses showed that there are approximately 6,500 dimers of LuxR in wild-type V. harveyi, and thus there is sufficient LuxR present to bind at 1,165 sites (see Fig. S1B in the supplemental material). MEME (multiple EM for motif elicitation) (15) analysis of the DNA sequences surrounding the LuxR binding peaks revealed a 20-bp consensus binding motif (Fig. 1B). This motif is nearly identical to the 21-bp LuxR consensus motif previously determined in vitro (11) and closely resembles the consensus motifs determined for other LuxR proteins (9, 10, 16). Although this motif exhibits dyad symmetry, LuxR has a stronger preference for nucleotide sequences on one side of the palindrome (Fig. 1B, left). Studies of other LuxR homologs identified motifs of 16 bp (HapR) to 22 bp (SmcR) (9, 10). We investigated the DNA substrate length required for LuxR binding using electrophoretic mobility shift assays (EMSAs) with DNA fragments of various lengths. LuxR binds to a 28-bp substrate with the highest affinity, and LuxR cannot bind to substrates shorter than 23 bp (see Fig. S1C in the supplemental material).
To determine which LuxR binding sites contribute to LuxR regulation of gene expression, we analyzed the locations of LuxR binding peaks relative to genes in the LuxR regulon, which was defined previously by microarray analyses (7). We identified 227 LuxR peaks in promoter regions upstream of 115 LuxR-regulated genes (35 of these genes are activated by LuxR, and 80 genes are repressed by LuxR) (see Tables S1 and S2 in the supplemental material). Because LuxR both activates and represses transcription, we hypothesized that differences could exist in the consensus sequences for activated and repressed genes. To explore this, we separately analyzed peaks in the promoters of activated genes and repressed genes (see Tables S1 and S2). The LuxR binding motif in repressed promoters has clear dyad symmetry (Fig. 1C), whereas the motif present in activated promoters has a strong preference for one side, and the palindrome is incomplete on the other side (Fig. 1D). Thus, the combination of motifs from activated and repressed genes (see Fig. S1D) results in the overall consensus motif derived from the ChIP-seq (Fig. 1B).
LuxR: examples of activation and repression.
To examine how differences in LuxR binding motifs contribute to activation and repression, we characterized LuxR activity at one representative repressed promoter and one representative activated promoter. A typical case of a gene repressed by LuxR is represented by VIBHAR_05222, encoding a putative thioesterase. According to ChIP-seq analyses, the VIBHAR_05222 promoter (P05222) contains five LuxR binding peaks, although one peak was significantly stronger than the other four (Fig. 2A). To determine the precise locations of LuxR binding sites that are relevant for regulation, we used the position weight matrix (PWM) generated by our ChIP-seq data to scan for binding sites at the VIBHAR_05222 locus using the MAST (Motif Alignment and Search Tool) software tool (15, 17, 18). We identified only one binding site (Fig. 2A), and LuxR bound this site with high affinity (Kd = 0.7 nM) (Fig. 2A). We performed EMSAs with DNA fragments that tile across the entire promoter and verified that this is the only site bound by LuxR in vitro (see Fig. S1E in the supplemental material). The sizes of the DNA fragments analyzed during standard ChIP-seq analysis (~200 bp) are larger than the LuxR binding sites (28 bp). Because of this, at some promoters, such as P05222, a single LuxR binding site appears as a wide peak and is interpreted to be multiple peaks by the bioinformatics analysis.
FIG 2 .
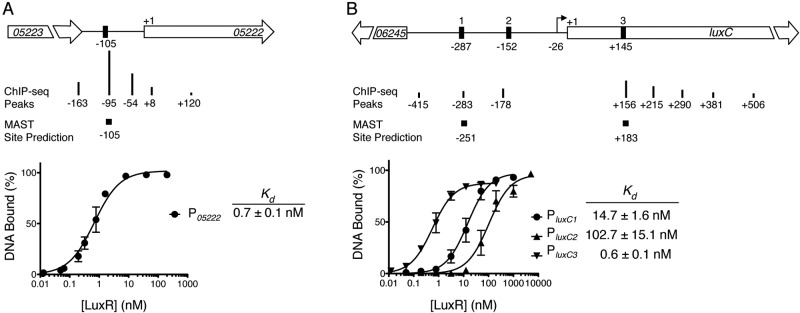
LuxR binding to activated and repressed promoters. Diagrams illustrating the locations and relative strengths of LuxR binding (ChIP-seq peaks) at the VIBHAR_05222 promoter region (A) or the luxC promoter region (B). Sites determined in vitro (black boxes) or by MAST are shown relative to the putative translation start codons (denoted as +1) of VIBHAR_05222 (A) or luxC (B). The transcription start site of luxC is shown with an arrow (12). DNA binding curves determined by quantitative EMSAs are shown for LuxR binding at the P05222 binding site (A) or PluxC binding sites 1 to 3 (B). Dissociation constants (Kd) are shown for each binding site. Error bars represent the standard deviations of three independent measurements.
To study a typical case of a gene activated by LuxR, we chose the promoter driving expression of the luciferase operon (luxCDABE, VIBHAR_06244-VIBHAR_06240), the most highly activated locus controlled by LuxR. Bioinformatic analysis of the ChIP-seq data identified eight LuxR binding peaks near luxC (PluxC). Scanning this region with the LuxR PWM by using MAST revealed only two sites, one of which is located within the luxC open reading frame (ORF) (denoted as sites 1 and 3 in Fig. 2B). Previous studies showed LuxR bound to an additional site that we do not identify by MAST (denoted as site 2 in Fig. 2B) (8, 12). LuxR bound all three sites in vitro, with the highest binding affinity for site 3 (Kd = 0.6 nM) and the weakest affinity for site 2 (Kd = 102.7 nM) (Fig. 2B). Others have shown that removing the high-affinity site 3 does not affect the level of LuxR activation in vivo, and thus site 3 is not required for PluxC activation (12). Similarly, we observed that a PluxC-gfp reporter construct lacking site 3 is fully activated by LuxR in vivo (see Fig. S1F in the supplemental material). Therefore, we focused on sites 1 and 2 to determine the site(s) required for activation in vivo. A randomized sequence that is not bound by LuxR (see Fig. S1G) was inserted in place of the LuxR binding site at position 1 or position 2. Randomizing site 1 did not alter activation, whereas randomizing site 2 eliminated activation (see Fig. S1F). Collectively, these results demonstrate that LuxR binds to multiple binding sites in vitro and in vivo, but only site 2 is necessary for luxC activation in vivo.
Mutations in luxR separate activation and repression at representative promoters.
Our ChIP-seq data indicated that LuxR binds distinct sequences in the promoters of activated and repressed genes (Fig. 1), suggesting that there could be different requirements for LuxR for activation and repression. To investigate the mechanisms underpinning LuxR activation and repression, we performed a screen to identify LuxR mutants that differentiate between activated and repressed promoters. We screened a library of random luxR mutants for two phenotypes: (i) LuxR variants that cannot activate but can repress (activation-defective mutants) and (ii) LuxR variants that can activate but cannot repress (repression-defective mutants). We used our two representative promoters, PluxC and P05222, to assay activation and repression, respectively. A gfp reporter was fused to PluxC and an mCherry reporter was fused to P05222 to facilitate simultaneous screening by fluorescence-assisted cell sorting (FACS).
First, we isolated activation-defective mutants; these mutants exhibit decreased PluxC activation (low GFP levels) but maintain wild-type P05222 repression (low mCherry levels, Fig. 3A). We obtained five mutants with substitutions at the N55 position, and of these, LuxR N55I and LuxR N55K exhibited the most severe phenotypes (see Fig. S2 in the supplemental material). Four mutants were partially defective for activation of PluxC, and they have the substitutions L139P, L139R, N55Y, and N142D (Fig. 3A) (see also Fig. S2). A reciprocal screen was used to identify repression-defective mutants; these variants exhibit decreased repression of P05222 (high mCherry levels) but maintain wild-type activation of PluxC (high GFP levels). We obtained two mutants: LuxR T52M/I24V is completely defective for repression, while LuxR A51T is partially defective (Fig. 3A). We separated the LuxR T52M/I24V mutations by constructing LuxR T52M and LuxR I24V. The LuxR T52M mutant was inactive at both the activated and repressed promoters, and the LuxR I24V mutant resembled wild-type LuxR (see Fig. S2), so both mutations are required for the phenotype. We showed that the phenotypes of the mutant proteins were not due to altered protein production (see Fig. S2). The amino acid substitutions are shown in an alignment of the DNA binding domains of LuxR homologs and E. coli TetR (Fig. 3B). Several of the mutations lie in the conserved HTH (N55I and other N55 substitutions, T52M/I24V and A51T), including a substitution of the T52 residue that is conserved between LuxR and TetR. We return to this point below.
FIG 3 .

LuxR mutants defective for activation or repression. (A) LuxR mutants with decreased activation of PluxC (activation-defective mutants) and decreased repression of P05222 (repression-defective mutants). E. coli strains containing an empty vector (pJV036, denoted as “No Protein”), expressing luxR (pJV239), or expressing luxR mutants (N55I [pJV240], L139P [pJV242], N142D [pJV261], T52M/I24V [pJV241], and A51T [pJV247]) were assayed for transcriptional activation of PluxC and repression of P05222. Fluorescence from each strain was measured using a reporter construct (pJV064) harboring P05222-mCherry and PluxC-gfp. Error bars represent the standard deviations of measurements for three biological samples. (B) Alignment of the predicted DNA binding domains of LuxR homologs from V. harveyi (LuxR, AAA27539), V. vulnificus (SmcR, AAF72582), V. cholerae (HapR; ABD24298), V. parahaemolyticus (OpaR, NP_798895), and V. fischeri (LitR, YP_205560) aligned to E. coli TetR (P0ACT4). Conserved residues (including in TetR) are shown in black, and similar residues are shown in gray. The black triangles indicate the locations of amino acid substitutions in activation- and repression-defective mutant LuxR proteins. Sequence alignments were assembled using the ClustalW software program (45) and viewed using the ESPript (46) and Boxshade programs.
Mutations in luxR affect regulation of specific promoters.
To test the generality of the activation and repression defects for the LuxR mutant proteins, we measured their activities at other LuxR-regulated promoters in V. harveyi. First, we tested whether the activation-defective mutants were also defective at other activated promoters. In addition to eliminating activation of luxC, the LuxR N55I variant showed decreased activation of P05020 but wild-type activation of Pp08175 (Fig. 4). In contrast, the activation-defective mutants LuxR L139P and LuxR N142D activated P05020 but could not activate Pp08175 (Fig. 4; see also Fig. S2 in the supplemental material). All three activation-defective mutants fully repressed P05222 but did show defects in repression at other promoters (for example, PaphA) (Fig. 4; see also Fig. S2).
FIG 4 .
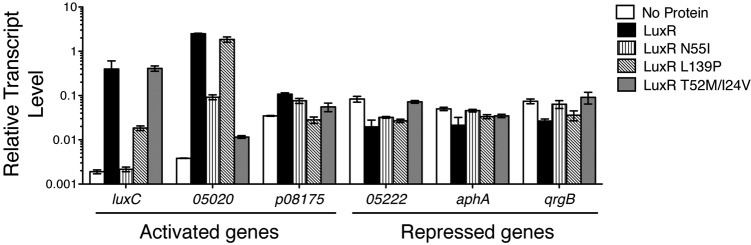
LuxR mutants are defective for regulating specific promoters in V. harveyi. Transcript levels of genes regulated by LuxR were assayed by qRT-PCR from a V. harveyi ΔluxR strain (KM669) containing an empty vector (pJV036, denoted as “No Protein”) or expressing luxR (pJV239), luxR N55I (pJV240), luxR L139P (pJV242), or luxR T52M/I24V (pJV241). Error bars represent the standard deviations of measurements of three biological samples. See also Fig. S2 in the supplemental material for all tested promoters and LuxR mutants.
We next tested the generality of the repression-defective mutants. The LuxR T52M/I24V substitution abolished repression at all promoters (Fig. 4; see also Fig. S2 in the supplemental material), and it showed a severe decrease in activation except at PluxC (Fig. 4), the promoter used in the screen. LuxR A51T, which was partially defective at repression of P05222, was also partially defective for repression of other promoters (see Fig. S2 ). Collectively, these results suggest that defects associated with mutations in the DNA binding domain of luxR (e.g., LuxR N55I, T52M/I24V, and A51T) are not specific for activation or repression but rather are specific to each promoter. Mutations residing outside the DNA binding domain (LuxR L139P and LuxR N142D), in contrast, are generally defective only for activation.
LuxR mutant proteins are defective for DNA binding at specific sequences.
The majority of the mutations we obtained in luxR reside in the DNA binding domain. Therefore, we hypothesized that the phenotypes of the LuxR activation- and repression-defective mutants could be due to changes in DNA binding capabilities at specific promoters. We examined DNA binding with a focus on the activation-defective mutants LuxR N55I and LuxR L139P and the repression-defective mutant LuxR T52M/I24V because those substitutions resulted in the strongest phenotypes in vivo. First, we examined DNA binding at our representative repressed promoter P05222 using EMSAs with purified proteins. The activation-defective mutants LuxR N55I and LuxR L139P both bound the P05222 binding site with only slightly lower affinity than wild-type LuxR (Fig. 5A), which is consistent with the observation that these substitutions did not affect repression of P05222. However, the repression-defective mutant LuxR T52M/I24V showed severely decreased binding affinity (Kd = 56.6 nM) compared to that of wild-type LuxR (Kd = 0.7 nM) (Fig. 5A). Thus, LuxR T52M/I24V is defective for repression at P05222 because it is impaired for DNA binding at this particular promoter.
FIG 5 .
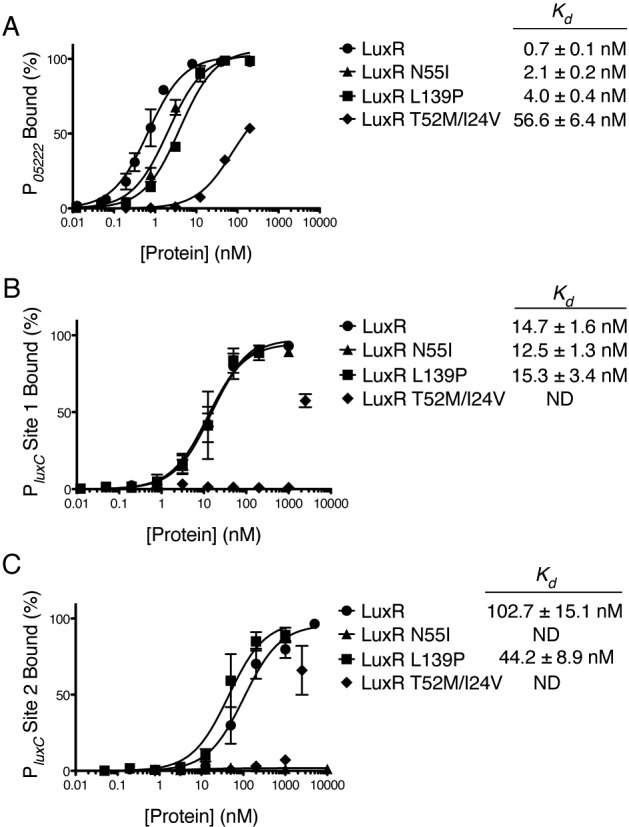
LuxR mutant proteins are defective for DNA binding. DNA binding curves from quantitative EMSAs are shown for LuxR, LuxR N55I, LuxR L139P, and LuxR T52M/I24V at the P05222 binding site (A), PluxC binding site 1 (B), and PluxC binding site 2 (C) (see Fig. S3 in the supplemental material for representative gels showing data used to calculate binding curves). Dissociation constants (Kd) are shown for each protein at each binding site at which a curve could be fit to the data. Error bars represent the standard deviations of three independent measurements.
We next tested DNA binding at sites 1 and 2 in PluxC, our representative activated promoter. The activation-defective mutant LuxR N55I bound site 1 with affinity similar to that of wild-type LuxR (Fig. 5B) and showed no binding to site 2 (Fig. 5C; see also Fig. S3 in the supplemental material). Thus, LuxR N55I is defective for activation of PluxC because it is unable to bind site 2, the site that is critical for activation of luxC. LuxR L139P, which is partially defective for activation at PluxC, has wild-type binding affinities at both sites 1 and 2 (Fig. 5B and C). Thus, the LuxR L139P phenotype is not due to a defect in DNA binding. The repression-defective mutant LuxR T52M/I24V did not bind site 1 (see Fig. S3) but bound site 2 with weak affinity (Fig. 5C; see also Fig. S3). Because strains containing LuxR T52M/I24V are capable of activating PluxC, similar to strains with wild-type LuxR, this result suggests that even modest binding at site 2 is necessary and sufficient for LuxR activation.
Mutagenesis of the 05222 promoter suppresses the LuxR T52M/I24V defect.
The above biochemical analyses of the LuxR mutants suggest that the activation- and repression-defective phenotypes of LuxR N55I and LuxR T52M/I24V stem from defects in binding to specific DNA sequences. We hypothesized that changing the binding sites in the corresponding promoters could suppress the mutant phenotypes. We first examined P05222. We randomly mutagenized the LuxR binding site at P05222 and screened for substitutions that suppressed the LuxR T52M/I24V repression defect but did not affect basal transcription in the absence of LuxR (Fig. 6A). Two substitutions restored repression to the level of that for wild-type LuxR (Fig. 6B). Importantly, these two substitutions (P05222 G → T and P05222 T → C) converted the P05222 sequence to one more closely resembling the ideal LuxR repressed gene consensus motif (Fig. 6A). Individually, each mutation partially increased LuxR T52M/I24V repression, whereas combining both mutations further increased LuxR T52M/I24V binding affinity and fully restored repression (Fig. 6B and C). The two mutations also increased the DNA binding affinity and repression activity of wild-type LuxR (Fig. 6B and C). Thus, suppression of the repression-defective phenotype occurs by increasing LuxR DNA binding affinity at the P05222 site. In a parallel screen, we attempted to identify substitutions in PluxC that suppressed the activation-defective phenotype of LuxR N55I. However, we were unsuccessful, perhaps because the LuxR binding site at this promoter could not be altered to suppress the N55I phenotype without affecting basal transcription rates.
FIG 6 .
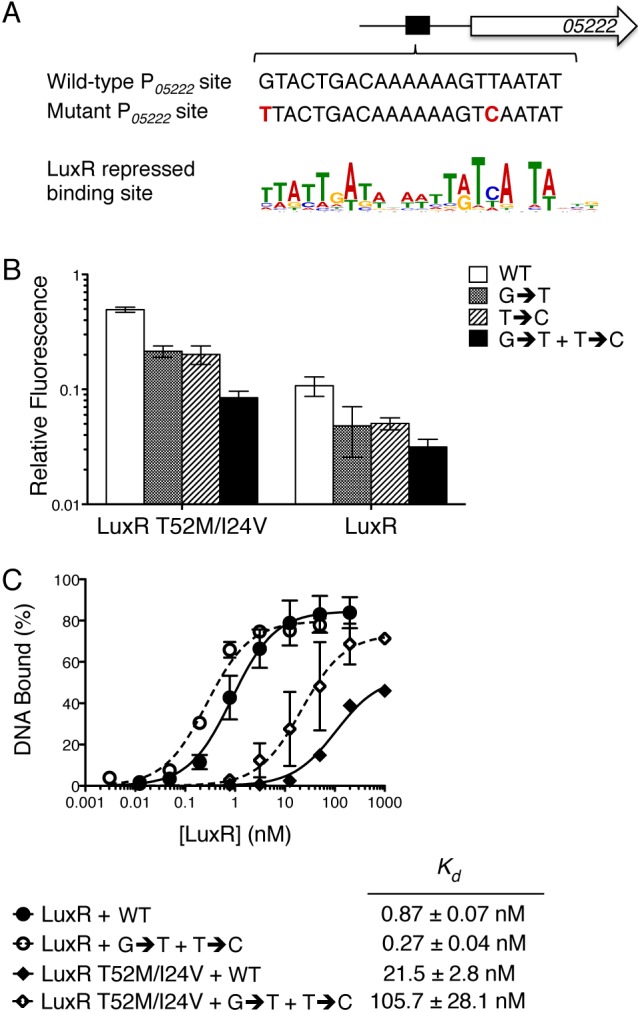
The LuxR T52M/I24V defect is suppressed by altering the corresponding DNA binding site. (A) The P05222 binding site and the two suppressor mutations (G → T and T → C) aligned with the LuxR binding site consensus motif from repressed promoters. (B) E. coli strains expressing luxR (pJV239), luxR T52M/I24V (pJV241), or containing an empty vector (pJV036) were assayed for transcriptional regulation of P05222. Fluorescence was measured from P05222-mCherry with either the wild-type P05222 site (WT, pJV141), mutation G → T (pJV230), mutation T → C (pJV227), or mutations G → T and T → C (pJV233). Fluorescence was normalized to the empty vector control. Error bars represent the standard deviations of measurements of three biological samples. (C) DNA binding curves from quantitative EMSAs for LuxR and LuxR T52M/I24V bound to either the P05222 binding site (WT) or the P05222 mutant binding site containing two mutations (G → T + T → C). Dissociation constants (Kd) are shown for each protein at each site. Error bars represent the standard deviations of three independent measurements.
DISCUSSION
The master transcription factor LuxR precisely controls the expression of more than 600 genes in the V. harveyi quorum-sensing regulon. LuxR directly regulates approximately one-fifth of these genes (115 genes). Quorum-sensing regulons of similar sizes are known in other vibrios, such as V. vulnificus, in which the LuxR homolog SmcR binds to 121 promoters (9). Four of the genes directly controlled by LuxR are predicted to be transcription factors, which likely control second-tier genes in the LuxR regulon.
Our ChIP-seq studies provide a global view of LuxR binding. We identified a significant number of LuxR binding sites in promoters of genes for which we do not observe regulation by LuxR. We propose that LuxR regulates these genes during growth under conditions that are not mimicked by our laboratory experiments. Previous studies of transcription factors in other organisms, such as Drosophila melanogaster and Candida albicans, also found that the number of protein binding sites is significantly larger than the number of genes displaying regulation (19–22). More than half of the LuxR binding peaks were present within ORFs (671 out of 1,165 LuxR binding peaks), another feature consistent with findings of these earlier studies. For example, LuxR bound within the ORFs of VIBHAR_05222 and luxC, the two representative promoters we characterized, although none of these sites is necessary for LuxR regulation of the gene in vivo. It is noteworthy that among the 75 LuxR-regulated genes with binding peaks within the ORF, 50 of these also harbor additional binding peaks upstream of their start codons. It is therefore possible that binding sites within ORFs paired with binding sites in the promoter are important for LuxR transcriptional regulation under some conditions.
Unlike canonical TetR proteins with stringent consensus motifs, the consensus motif of LuxR-type proteins is degenerate and asymmetric (9–11, 16). One-half of the dyad is more strongly conserved than the other half, as observed from 1,165 binding site sequences. However, we discovered how the unusual asymmetric nature of the LuxR consensus motif is generated. As with other TetR-type proteins, LuxR binding sites in repressed promoters contain a palindrome of roughly equal symmetry. In contrast, the LuxR binding sequences present in activated promoters are nonpalindromic, containing only the left half of the repressed promoter site. Thus, the combination of these two distinct sites results in a skewed palindrome in vitro and in vivo (11). We propose that LuxR has evolved the flexibility to tolerate minor changes to the DNA binding sequence at activated and repressed promoters, which could underpin why LuxR proteins have the capability to control the expression of hundreds of quorum-sensing genes. A model showing these ideas is presented in Fig. 7.
FIG 7 .
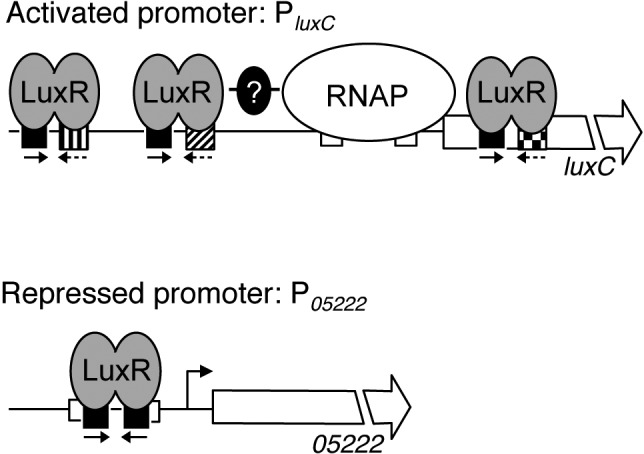
Model for LuxR transcriptional control. At activated promoters (e.g., PluxC), LuxR interacts with RNA polymerase (denoted RNAP) and/or other transcription factors to promote transcription. At repressed promoters (e.g., P05222), LuxR binding occludes RNA polymerase, which prevents transcription. LuxR binding site sequences at repressed promoters are symmetrical palindromes (denoted by black boxes with inverted arrows), whereas binding site sequences at activated promoters contain only one-half of the palindrome. Furthermore, LuxR binding site sequences at each activated promoter vary, resulting in distinct LuxR binding affinities (denoted by the different fill patterns for the binding site boxes in the PluxC promoter).
To study the mechanism that drives LuxR activation and repression activity, we performed a screen to identify LuxR variants that only activate or only repress. None of the resulting LuxR mutants was specifically defective for either activation or repression. Two activation-defective mutants, LuxR L139P and LuxR N142D, were defective only at activating luxC and VIBHAR_p08175. However, LuxR 139P did not exhibit any defects in DNA binding. We predict that the region of LuxR containing amino acids L139 and N142 may be critical for RNA polymerase interaction and thus required for activation of luxC. Several of our LuxR mutants exhibited defects in DNA binding only at specific LuxR binding sites, and each of these mutant LuxR proteins harbored substitutions in the DNA binding domain. This finding suggests that modifications to the amino acids in the conserved HTH motif restrict DNA sequence recognition to specific sequences. Two such LuxR mutants, carrying N55I and T52M/I24V, exhibited the strongest defects in gene regulation, suggesting that these amino acids likely play the most important roles in DNA sequence recognition, at least at the two promoters we examined.
A common theme that we observe is that when repression is considered, LuxR functions similarly to TetR. First, the LuxR consensus motif at repressed promoters is a symmetrical palindrome. Second, LuxR bound a single site at the example repressed promoter P05222, which is reminiscent of TetR proteins that bind a single operator at a promoter. Third, similar to what we show for LuxR, altering the conserved residues in the DNA binding domain of TetR residues decreases its repression activity (23). For example, the TetR T40M substitution (compare with LuxR T52M) reduced TetR repression activity 153-fold in vivo. Finally, we found that the phenotype of a repression-defective mutant, LuxR T52M/I24V, could be suppressed by altering the DNA sequence in P05222. In an analogous experiment, tet operator sequence variants also suppress defective repression phenotypes of TetR proteins with substitutions at T40 (23). Thus, LuxR likely functions similarly to TetR at repressed promoters by binding to a single palindromic operator site via specific interactions with residues in the DNA binding domain. LuxR is not like TetR when one considers activated promoters. At activated promoters, LuxR recognizes a different consensus motif and binds three sites in the example activated promoter PluxC. Multiple LuxR binding sites commonly exist in each promoter (59% of the LuxR-bound regions contain 2 or more sites; see Fig. S4 in the supplemental material; for example, PluxC). LuxR may facilitate DNA looping by binding to multiple sites, a mechanism that has been proposed for the LuxR homolog SmcR (24).
Our biochemical analyses suggest that the DNA sequence of the LuxR binding site is not the only factor that specifies activation or repression, because LuxR mutants that activated PluxC, for example, could not activate all LuxR-activated promoters. Likewise, LuxR mutants that repressed P05222 could not repress all LuxR-repressed promoters. In addition, DNA binding affinity alone cannot account for all LuxR transcription activity because LuxR had the weakest affinity for PluxC site 2, which is the critical binding site in that promoter in terms of regulation. It is also peculiar that LuxR exhibits the strongest affinity for PluxC site 3 but this site is not required for activation in vivo. Thus, features such as the DNA sequence, number and relative locations of LuxR binding sites, and productive LuxR interactions with RNA polymerase and other transcription factors likely combine to dictate whether LuxR activates or represses a given promoter and to what extent. For example, we know that the cAMP receptor protein (CRP) and MetR regulate PluxC (25), and LuxR may interact with these proteins to activate transcription (Fig. 7). We are currently exploring LuxR interactions with RNA polymerase and other transcription factors at PluxC as a model to understand the mechanism of LuxR activation.
Our studies of LuxR gene regulation support a role for LuxR as a dual-function global transcriptional regulator. In many ways, our analysis suggests that LuxR is more similar to CRP than to TetR. CRP is an activator and repressor of transcription of >200 genes (26–31). CRP binds to a conserved 22-bp operator sequence (32–35), and the positioning of the CRP site dictates its mode of action (36–38). CRP DNA binding affinity varies between promoters, with the highest affinities corresponding to those sites that are most similar to the consensus site (32, 35, 39). Finally, activation-defective mutants of CRP have been identified, and they contain substitutions in amino acids that interact with RNA polymerase (40, 41). While these general parallels suggest that LuxR functions similarly to CRP, there are two striking contrasts: first, there is no known ligand that controls LuxR activity, and second, LuxR repression requires a symmetrical palindrome, whereas LuxR activation requires only a half-site. We are currently determining how the position of LuxR binding sites with respect to RNA polymerase at promoters correlates with activation or repression.
As the master regulator of quorum-sensing gene expression, LuxR controls the timing of expression of hundreds of genes in response to changes in cell density. The concentration of LuxR increases as autoinducers accumulate (7). This LuxR protein concentration gradient enables LuxR to control promoters via different binding affinities at various cell densities, producing a temporal pattern of gene expression (7). Absent other modulatory features, promoters containing the highest-affinity binding sites will be regulated first during the transition from LCD to HCD. For example, VIBHAR_05222 is repressed 2-fold by LuxR at LCD and 7-fold at HCD. Thus, because LuxR has a high binding affinity for the binding site at P05222, it is repressed even by the low concentrations of LuxR present at LCD. In contrast, luxC is one of the final genes to be activated in response to quorum sensing (data not shown), which fits with our observation that LuxR has a weak affinity for PluxC site 2. Thus, DNA binding affinity, coupled to other features, results in a finely choreographed pattern of gene expression. We propose that LuxR, although structurally similar to TetR, has evolved unique characteristics that enable it to differentially control the genes in the quorum-sensing regulon in response to quorum-sensing cues.
MATERIALS AND METHODS
Bacterial strains and media.
Escherichia coli strains S17-1 λpir, DH10B (Electromax; Invitrogen), BL21(DE3) (Invitrogen), and derivatives were grown with aeration at 37°C in Luria-Bertani (LB) medium with 40 µg ml−1 kanamycin and 10 µg ml−1 chloramphenicol. V. harveyi strain BB120 (BAA-1116) and derivatives were grown with aeration at 30°C in Luria marine (LM) medium with 100 µg ml−1 kanamycin (Sigma) and 10 µg ml−1 chloramphenicol (Sigma). Plasmids were transferred from E. coli to V. harveyi by conjugation (42).
Molecular methods.
E. coli strains S17-1 λpir and DH10B were used for cloning. PCR reactions used iProof DNA polymerase (Bio-Rad). Restriction enzymes, T4 polynucleotide kinase, calf intestinal phosphatase (CIP), and T4 DNA ligase were purchased from New England Biolabs (NEB). Oligonucleotides were purchased from Integrated DNA Technologies. The Genemorph II EZClone Domain mutagenesis kit (Stratagene) was used for random mutagenesis. The QuikChange mutagenesis kit (Stratagene) was used to introduce mutations into plasmids. Cloning procedures and sequences of PCR primers are available upon request. All plasmid constructs were confirmed by sequencing by Genewiz, Inc. RNA isolation, cDNA synthesis, and quantitative real-time PCR (qRT-PCR) reactions were carried out as described elsewhere (42). Samples were normalized to the internal standard hfq, and the standard curve method or the ΔΔCT method was used for data analysis.
For quantitative Western blot analyses of LuxR production in wild-type cells, V. harveyi BB120 cells were collected at an optical density at 600 nm (OD600) of 1.0 (HCD), and cell counts were determined by viability plating (see Fig. S1B in the supplemental material). For quantitative Western blots of mutant LuxR proteins, cells were collected from V. harveyi ΔluxR mutant strains expressing wild-type or mutant luxR following induction (see Fig. S2 and see below). Pellets were resuspended at 0.01 OD600 units per μl in lysis buffer (Bugbuster [Novagen], 50 µg ml−1 lysozyme, and 1 U ml−1 Benzonase [Novagen]). Lysates were serially diluted to determine the linear range for quantification of LuxR bands, and proteins were analyzed by SDS-PAGE on 12% polyacrylamide gels. Protein transfer, membrane blocking, and incubations with anti-LuxR primary antibodies, anti-mouse secondary antibodies, and anti-rabbit secondary antibodies were performed as described elsewhere (43). Anti-beta′ antibodies raised against E. coli beta′ (RpoC) were purchased from Neoclone and used at a 1:10,000 dilution. Chemiluminescence was carried out using a SuperSignal West Pico kit (Pierce). Protein bands were quantified using ImageQuant software.
Inducible expression of luxR.
luxR was expressed under control of the Ptac promoter from a plasmid (pJV239) constitutively expressing lacIq. Overnight E. coli cultures containing this plasmid and derivatives were diluted 1:1,000 and grown at 30°C for 16 h, and samples were analyzed by FACS or qRT-PCR. Overnight V. harveyi cultures containing the plasmid and derivatives were diluted 1:1,000 and grown to an approximate OD600 of 0.2, 10 µM IPTG was added, and the cultures were grown for 3 additional hours. FLAG-luxR was expressed under the control of the Ptactheo promoter (pJV057) as previously described (7). To induce FLAG-luxR expression, overnight cultures of E. coli strains containing this construct were diluted 1:1,000 in the presence of 1 mM IPTG and 10 µM theophylline, and samples were measured by FACS or harvested for RNA isolation after 16 h of growth at 30°C.
ChIP-seq.
Plasmids expressing either FLAG-luxR (pAP116) or FLAG-luxR R17C (pST012; a DNA binding-defective luxR mutant) and empty vector controls (pSLS3 or pJV139) were conjugated into a V. harveyi ΔluxR strain (KM669). The ChIP protocol is based on previously published methods (44) and the Affymetrix ChIP assay protocol with several modifications. Overnight cultures were diluted 1:50,000 and grown for 16 h at 30°C. Fifty OD600 units of cells were cross-linked and washed as previously described (44), and cells were lysed in 1 ml of lysis buffer (1× protease inhibitors [Sigma], 50 µg ml−1 lysozyme, 1× Bugbuster, 1% Triton X-100, and 1 mM phenylmethylsulfonyl fluoride [PMSF]) for 20 min at room temperature on a rotator. Following lysis, the DNA was sheared by sonication to an average size of 100 to 1,000 bp. The supernatant was clarified at 13,000 rpm for 10 min at 4°C. Immunoprecipitation reaction mixtures contained a 200-µl aliquot of input sample, 800 µl of IP buffer (50 mM HEPES-KOH, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and 1 mM PMSF), and 40 µl EZ-view anti-FLAG agarose beads (Sigma) (equilibrated in Tris-buffered saline [TBS]) and were carried out for 2 h at room temperature on a rotator. Following immunoprecipitation, beads were collected and washed (Affymetrix ChIP assay protocol). Immunoprecipitated complexes were eluted, and cross-links were reversed as described elsewhere (Affymetrix ChIP assay protocol). Samples (input DNA and IP DNA) were analyzed by qRT-PCR to assess the quality of the immunoprecipitation from four independent experiments. DNA from representative ChIP samples was prepared for sequencing using the Illumina ChIP-seq sample prep kit and verified by qRT-PCR. The following ChIP samples were sequenced: the input DNA and IP DNA from FLAG-luxR (pAP116), IP DNA from FLAG-luxR R17C (pST012), and input DNA and IP DNA from an empty vector control (pJV139). See the supplemental material for a description of ChIP-seq data analysis procedures.
Fluorescent reporter assays.
LuxR transcriptional regulation of P05222 and PluxC was measured in E. coli strains carrying pJV064, containing a transcriptional fusion of mCherry to P05222 and a transcriptional fusion of gfp to PluxC. Overnight E. coli DH10B cultures containing pJV239 (or derivatives) and pJV064 were diluted 1:1,000 and grown for 16 h at 30°C. GFP and mCherry fluorescence was measured either on a 1420 Victor2 multilabel counter (Wallac) or on a Becton Dickinson FACSAria cell sorter using FACS Diva software.
Genetic screens.
Mutant luxR libraries were generated by random mutagenesis of the luxR or FLAG-luxR ORF. For the wild-type luxR screen, 796,100 luxR mutants (average, 1.8 mutations per clone) were pooled, and DNA was extracted and transformed into E. coli DH10B containing pJV064. Colonies (338,500) were pooled, and the cultures were diluted 1:1,000 and grown at 30°C for 16 h. The FLAG-luxR screen was performed similarly except the mutant library culture was grown in medium containing 1 mM IPTG and 10 µM theophylline. Mutant clones (86,850) were obtained (average, 3.5 mutations per clone), DNA was extracted and transformed into DH10B carrying pJV064, and 580,000 colonies were pooled for screening. In all experiments, mutants were sorted into two groups using FACS: (i) high mCherry fluorescence (no repression) and high GFP fluorescence (wild-type activation), and (ii) low mCherry fluorescence (wild-type repression) and low GFP fluorescence (no activation). Positive clones were retested and sequenced. All mutants showed identical phenotypes in the presence or absence of the FLAG tag.
To screen for mutations in P05222, an oligonucleotide containing the binding site (GTACTGACAAAAAAGTTAATAT) was purchased from IDT with 3% randomization and inserted in place of the wild-type site in pJV064. Twenty-two thousand four hundred ten mutant clones (average, 2.1 mutations per clone) were pooled, and DNA was extracted and transformed into E. coli DH10B containing the plasmid expressing luxR T52M/I24V (pJV241). Forty-six thousand colonies were pooled, and the culture was diluted 1:100 and grown for 3 h. Populations of cells were sorted using FACS to obtain cells exhibiting high GFP fluorescence (wild-type activation) and low mCherry fluorescence (wild-type repression). Clones were sequenced and analyzed in the presence of wild-type luxR, luxR T52M/I24V, or an empty vector (pJV036).
EMSAs.
LuxR proteins were purified and EMSA substrates (see Table S3 in the supplemental material) were annealed as previously described (7). Labeling reactions (100 fmol of double-stranded DNA [dsDNA] substrate, 10 µCi of [γ-32P]ATP, T4 polynucleotide kinase [NEB], and kinase buffer) were incubated for 30 min at 37°C, and the labeled substrates were purified on ProbeQuant G-50 Micro columns (GE Healthcare). EMSA reactions (12 µl) were performed as previously described (7) with two modifications. Reaction mixtures contained 0.1 nM dsDNA substrate and the LuxR protein in dilution buffer (20 mM imidazole, pH 7.5, 300 mM NaCl, 0.5 mM EDTA, 1 mM dithiothreitol [DTT], and 5% glycerol). DNA binding measurements were determined using ImageQuant software (GE Healthcare) and GraphPad Prism software.
SUPPLEMENTAL MATERIAL
Supplemental materials and methods. Download
LuxR binding at the promoters of activated and repressed genes. (A) luxC transcript levels were measured by qRT-PCR in V. harveyi ΔluxR (KM669) containing an empty vector (pJV036), expressing luxR (pJV239), or expressing FLAG-luxR (pJV274). Error bars represent the standard deviations of measurements of three biological samples. (B) Quantitative Western blot analysis of cell lysates from wild-type V. harveyi (BB120). Purified LuxR protein (0.42 pmol, 0.85 pmol, 1.69 pmol, and 3.38 pmol LuxR) mixed with cell lysates from a V. harveyi ΔluxR (KM669) strain was used as a standard control. Cell counts were determined by viability plating. Error bars represent the standard deviations of measurements of three biological samples. LuxR antibodies detect a nonspecific band that runs slightly faster than LuxR. (C) EMSAs containing LuxR (100 nM) bound to P05222 binding site substrates. The lengths of the dsDNA substrates are shown above the lanes. Sequences of selected substrates (labeled 1 to 7) are shown on the right. Substrates correspond to annealed oligonucleotides as follows (from left to right): JCV811/JCV812 (1), JCV813/JCV814 (2), JCV877/JCV878, JCV815/JCV816, JCV879/JCV880, JCV881/JCV882, JCV883/JCV884 (3), JCV841/JCV842, JCV849/JCV850 (4), JCV851/JCV852 (5), JCV843/JCV844 (6), JCV875/JCV876 (7), and JCV885/JCV886. All substrate sequences are listed in Table S3. (D) The LuxR binding consensus motif generated from the subset of ChIP-seq peaks near LuxR-regulated genes (105 regions, both activated and repressed genes). (E) Binding was assessed by EMSAs with substrates (A to E) from the P05222 promoter region as shown in the diagram in the presence (+) or absence (−) of 100 nM LuxR. Substrates correspond to annealed oligonucleotides as follows: JCV645/JCV646 (A), JCV647/JCV648 (B), JCV649/JCV650 (C), JCV651/JCV652 (D), or JCV653/JCV654 (E). The fragments correspond to the region of P05222 that is required for transcription activity in vivo (site −163 is not required). (F) Activation of PluxC was measured in E. coli strains containing an empty vector (pJV036, white bars) or expressing luxR (pJV239, black bars). Fluorescence was measured by FACS from PluxC-gfp (containing PluxC from −367 through +63 relative to the transcription start site) with either a wild-type sequence (pJV064), binding site 1 replaced with the randomized binding site sequence from panel G (BS 1*, pJV284), or binding site 2 replaced with the randomized binding site sequence from panel G (BS 2*, pJV285). Error bars represent the standard deviations of measurements of three biological samples. (G) LuxR binding was assessed by EMSAs with the wild-type binding site in the aphA promoter (BS; JCV428/JCV358) and with a randomized aphA promoter binding site sequence (BS*; JCV429/JCV360) in the presence (+) or absence (−) of 100 nM LuxR. Download
LuxR mutants are defective for activation or repression. E. coli strains containing an empty vector (no protein, pJV036), expressing luxR (WT, pJV239), or expressing the specified luxR mutants were assayed for transcriptional regulation of PluxC and P05222. Fluorescence was measured by FACS using pJV064 with P05222-mCherry (gray bars) and PluxC-gfp (black bars). Error bars represent the standard deviations of measurements of three biological samples. (A) Regulation of PluxC and P05222 by LuxR N55I (pJV240), N55Y (pJV249), N55S (pJV253), N55K (pJV254), and N55A (pJV281) mutants compared to results for wild-type LuxR. (B) Regulation of PluxC and P05222 by LuxR L139P (pJV242) and L139R (pJV260) mutants compared to that for wild-type LuxR. (C) Regulation of PluxC and P05222 by LuxR (pJV057) and the T52M (pJV103), I24V (pJV207), and T52M/I24V (pJV120) mutants. (D) Quantitative Western blot analysis of V. harveyi ΔluxR strain KM669 expressing luxR (pJV239) or luxR carrying N55I (pJV240), luxR T52M/I24V (pJV241), luxR L139P (pJV242), luxR A51T (pJV247), or luxR N142D (pJV261). LuxR antibodies detect a nonspecific band that runs slightly faster than LuxR. The background was calculated from results for KM669 containing an empty vector (denoted as “−”; pJV036). LuxR protein bands were normalized to the beta′ loading control. Purified LuxR was loaded as an additional control. Error bars represent the standard deviations of measurements of three biological samples. (E) Transcript levels of genes directly regulated by LuxR were assayed by qRT-PCR from a V. harveyi ΔluxR strain (KM669) containing an empty vector (pJV036, denoted as “no protein”) or expressing luxR (pJV239), luxR N55I (pJV240), luxR L139P (pJV242), luxR N142D (pJV261), luxR T52M/I24V (pJV241), or luxR A51T (pJV247). Error bars represent the standard deviations of measurements of three biological samples. Download
LuxR mutant proteins are defective for DNA binding at P05222, PluxC site 1, and PluxC site 2. EMSAs with LuxR, LuxR N55I, LuxR L139P, and LuxR T52M/I24V at P05222 (A), PluxC site 1 (B), and PluxC site 2 (C) are shown. The protein concentrations are shown below each gel. Download
Determination of the LLR and α parameters for ChIP-seq peak calling and the distribution of ChIP-seq peaks. (A and B) Empirical false discovery rate (FDR) assessment of the log-likelihood ratio (LLR) (A) or regularization factor (α) (B) parameter used to predict LuxR binding sites from ChIP-seq data. (C) Distribution of the number of peaks among the 582 merged genomic regions. Download
Motifs derived from ChIP-Seq peak sequences in the promoters of LuxR-regulated genes.
Distribution of ChIP-seq peaks in the promoter regions of LuxR-regulated genes.
Oligonucleotides used in EMSAs.
Distribution of sequence reads per chromosome.
LuxR binding peak positions determined by CSDeconv analysis.
ACKNOWLEDGMENTS
We thank Colleen O’Loughlin, Zhuo Li, Amanda Izzo, and Anne McCabe for assistance with experiments. We thank Matthew Bochman and Steven Rutherford for insightful comments. We appreciate Donna Storton, Jessica Buckles, and Lance Parsons for assistance with Illumina sequencing.
This work was supported by the Howard Hughes Medical Institute, National Institutes of Health (NIH) grant 5R01GM065859 and National Science Foundation (NSF) grant MCB-0343821 to B.L.B., NIH grant GM07225 to I.B.Z., and NIH fellowship F32GM089019 to J.C.V.K.
Footnotes
Citation van Kessel JC, Ulrich LE, Zhulin IB, Bassler BL. 2013. Analysis of activator and repressor functions reveals the requirements for transcriptional control by LuxR, the master regulator of quorum sensing in Vibrio harveyi. mBio 4(4):e00378-13. doi:10.1128/mBio.00378-13.
REFERENCES
- 1. Rutherford ST, Bassler BL. 2012. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold. Spring Harb. Perspect. Med. 2:11. 10.1101/cshperspect.a012427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kim Y, Kim BS, Park YJ, Choi WC, Hwang J, Kang BS, Oh TK, Choi SH, Kim MH. 2010. Crystal structure of SmcR, a quorum-sensing master regulator of Vibrio vulnificus, provides insight into its regulation of transcription. J. Biol. Chem. 285:14020–14030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ramos JL, Martínez-Bueno M, Molina-Henares AJ, Terán W, Watanabe K, Zhang X, Gallegos MT, Brennan R, Tobes R. 2005. The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69:326–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Silva RS, Kovacikova G, Lin W, Taylor RK, Skorupski K, Kull FJ. 2007. Crystal structure of the Vibrio cholerae quorum-sensing regulatory protein HapR. J. Bacteriol. 189:5683–5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schumacher MA, Miller MC, Grkovic S, Brown MH, Skurray RA, Brennan RG. 2002. Structural basis for cooperative DNA binding by two dimers of the multidrug-binding protein QacR. EMBO J. 21:1210–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Waters CM, Bassler BL. 2006. The Vibrio harveyi quorum-sensing system uses shared regulatory components to discriminate between multiple autoinducers. Genes Dev. 20:2754–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Kessel JC, Rutherford ST, Shao Y, Utria AF, Bassler BL. 2013. Individual and combined roles of the master regulators AphA and LuxR in control of the Vibrio harveyi quorum-sensing regulon. J. Bacteriol. 195:436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Swartzman E, Meighen EA. 1993. Purification and characterization of a poly(dA-dT) lux-specific DNA-binding protein from Vibrio harveyi and identification as LuxR. J. Biol. Chem. 268:16706–16716 [PubMed] [Google Scholar]
- 9. Lee DH, Jeong HS, Jeong HG, Kim KM, Kim H, Choi SH. 2008. A consensus sequence for binding of SmcR, a Vibrio vulnificus LuxR homologue, and genome-wide identification of the SmcR regulon. J. Biol. Chem. 283:23610–23618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsou AM, Cai T, Liu Z, Zhu J, Kulkarni RV. 2009. Regulatory targets of quorum sensing in Vibrio cholerae: evidence for two distinct HapR-binding motifs. Nucleic Acids Res. 37:2747–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pompeani AJ, Irgon JJ, Berger MF, Bulyk ML, Wingreen NS, Bassler BL. 2008. The Vibrio harveyi master quorum-sensing regulator, LuxR, a TetR-type protein is both an activator and a repressor: DNA recognition and binding specificity at target promoters. Mol. Microbiol. 70:76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miyamoto CM, Smith EE, Swartzman E, Cao JG, Graham AF, Meighen EA. 1994. Proximal and distal sites bind LuxR independently and activate expression of the Vibrio harveyi lux operon. Mol. Microbiol. 14:255–262 [DOI] [PubMed] [Google Scholar]
- 13. Hammer BK, Bassler BL. 2003. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol. Microbiol. 50:101–104 [DOI] [PubMed] [Google Scholar]
- 14. Svenningsen SL, Waters CM, Bassler BL. 2008. A negative feedback loop involving small RNAs accelerates Vibrio cholerae’s transition out of quorum-sensing mode. Genes Dev. 22:226–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2:28–36 [PubMed] [Google Scholar]
- 16. Zhang Y, Qiu Y, Tan Y, Guo Z, Yang R, Zhou D. 2012. Transcriptional regulation of opaR, qrr2-4 and aphA by the master quorum-sensing regulator OpaR in Vibrio parahaemolyticus. PLoS One 7:e34622. 10.1371/journal.pone.0034622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37:W202–W208. 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bailey TL, Gribskov M. 1998. Combining evidence using p-values: application to sequence homology searches. Bioinformatics 14:48–54 [DOI] [PubMed] [Google Scholar]
- 19. Tuch BB, Mitrovich QM, Homann OR, Hernday AD, Monighetti CK, De La Vega FM, Johnson AD. 2010. The transcriptomes of two heritable cell types illuminate the circuit governing their differentiation. PLoS Genet. 6:e1001070. 10.1371/journal.pgen.1001070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zordan RE, Miller MG, Galgoczy DJ, Tuch BB, Johnson AD. 2007. Interlocking transcriptional feedback loops control white-opaque switching in Candida albicans. PLoS Biol. 5:e256. 10.1371/journal.pbio.0050256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Georlette D, Ahn S, MacAlpine DM, Cheung E, Lewis PW, Beall EL, Bell SP, Speed T, Manak JR, Botchan MR. 2007. Genomic profiling and expression studies reveal both positive and negative activities for the Drosophila Myb MuvB/dREAM complex in proliferating cells. Genes Dev. 21:2880–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li XY, MacArthur S, Bourgon R, Nix D, Pollard DA, Iyer VN, Hechmer A, Simirenko L, Stapleton M, Luengo Hendriks CL, Chu HC, Ogawa N, Inwood W, Sementchenko V, Beaton A, Weiszmann R, Celniker SE, Knowles DW, Gingeras T, Speed TP, Eisen MB, Biggin MD. 2008. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol. 6:e27. 10.1371/journal.pbio.0060027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baumeister R, Helbl V, Hillen W. 1992. Contacts between Tet repressor and tet operator revealed by new recognition specificities of single amino acid replacement mutants. J. Mol. Biol. 226:1257–1270 [DOI] [PubMed] [Google Scholar]
- 24. Jeong HS, Kim SM, Lim MS, Kim KS, Choi SH. 2010. Direct interaction between quorum-sensing regulator SmcR and RNA polymerase is mediated by integration host factor to activate vvpE encoding elastase in Vibrio vulnificus. J. Biol. Chem. 285:9357–9366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chatterjee J, Miyamoto CM, Zouzoulas A, Lang BF, Skouris N, Meighen EA. 2002. MetR and CRP bind to the Vibrio harveyi lux promoters and regulate luminescence. Mol. Microbiol. 46:101–111 [DOI] [PubMed] [Google Scholar]
- 26. Aiba H. 1983. Autoregulation of the Escherichia coli crp gene: CRP is a transcriptional repressor for its own gene. Cell 32:141–149 [DOI] [PubMed] [Google Scholar]
- 27. Musso RE, Di Lauro R, Adhya S, de Crombrugghe B. 1977. Dual control for transcription of the galactose operon by cyclic AMP and its receptor protein at two interspersed promoters. Cell 12:847–854 [DOI] [PubMed] [Google Scholar]
- 28. Grainger DC, Hurd D, Harrison M, Holdstock J, Busby SJ. 2005. Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc. Natl. Acad. Sci. U. S. A. 102:17693–17698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zheng D, Constantinidou C, Hobman JL, Minchin SD. 2004. Identification of the CRP regulon using in vitro and in vivo transcriptional profiling. Nucleic Acids Res. 32:5874–5893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gama-Castro S, Jiménez-Jacinto V, Peralta-Gil M, Santos-Zavaleta A, Peñaloza-Spinola MI, Contreras-Moreira B, Segura-Salazar J, Muñiz-Rascado L, Martínez-Flores I, Salgado H, Bonavides-Martínez C, Abreu-Goodger C, Rodríguez-Penagos C, Miranda-Ríos J, Morett E, Merino E, Huerta AM, Treviño-Quintanilla L, Collado-Vides J. 2008. RegulonDB (version 6.0): gene regulation model of Escherichia coli K-12 beyond transcription, active (experimental) annotated promoters and Textpresso navigation. Nucleic Acids Res. 36:D120–D124. 10.1093/nar/gkn491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shimada T, Fujita N, Yamamoto K, Ishihama A. 2011. Novel roles of cAMP receptor protein (CRP) in regulation of transport and metabolism of carbon sources. PLoS One 6:e20081. 10.1371/journal.pone.0020081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Berg OG, von Hippel PH. 1988. Selection of DNA binding sites by regulatory proteins. II. The binding specificity of cyclic AMP receptor protein to recognition sites. J. Mol. Biol. 200:709–723 [DOI] [PubMed] [Google Scholar]
- 33. Ebright RH, Cossart P, Gicquel-Sanzey B, Beckwith J. 1984. Molecular basis of DNA sequence recognition by the catabolite gene activator protein: detailed inferences from three mutations that alter DNA sequence specificity. Proc. Natl. Acad. Sci. U. S. A. 81:7274–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ebright RH, Cossart P, Gicquel-Sanzey B, Beckwith J. 1984. Mutations that alter the DNA sequence specificity of the catabolite gene activator protein of E. coli. Nature 311:232–235 [DOI] [PubMed] [Google Scholar]
- 35. Busby S, Ebright RH. 1999. Transcription activation by catabolite activator protein (CAP). J. Mol. Biol. 293:199–213 [DOI] [PubMed] [Google Scholar]
- 36. Kolb A, Busby S, Buc H, Garges S, Adhya S. 1993. Transcriptional regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 62:749–795 [DOI] [PubMed] [Google Scholar]
- 37. Xiong XF, de la Cruz N, Reznikoff WS. 1991. Downstream deletion analysis of the lac promoter. J. Bacteriol. 173:4570–4577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aiba H. 1985. Transcription of the Escherichia coli adenylate cyclase gene is negatively regulated by cAMP-cAMP receptor protein. J. Biol. Chem. 260:3063–3070 [PubMed] [Google Scholar]
- 39. Kolb A, Busby S, Herbert M, Kotlarz D, Buc H. 1983. Comparison of the binding sites for the Escherichia coli cAMP receptor protein at the lactose and galactose promoters. EMBO J. 2:217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Irwin N, Ptashne M. 1987. Mutants of the catabolite activator protein of Escherichia coli that are specifically deficient in the gene-activation function. Proc. Natl. Acad. Sci. U. S. A. 84:8315–8319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eschenlauer AC, Reznikoff WS. 1991. Escherichia coli catabolite gene activator protein mutants defective in positive control of lac operon transcription. J. Bacteriol. 173:5024–5029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rutherford ST, van Kessel JC, Shao Y, Bassler BL. 2011. AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev. 25:397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shao Y, Bassler BL. 2012. Quorum-sensing non-coding small RNAs use unique pairing regions to differentially control mRNA targets. Mol. Microbiol. 83:599–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grainger DC, Overton TW, Reppas N, Wade JT, Tamai E, Hobman JL, Constantinidou C, Struhl K, Church G, Busby SJ. 2004. Genomic studies with Escherichia coli MelR protein: applications of chromatin immunoprecipitation and microarrays. J. Bacteriol. 186:6938–6943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 46. Gouet P, Courcelle E, Stuart DI, Métoz F. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental materials and methods. Download
LuxR binding at the promoters of activated and repressed genes. (A) luxC transcript levels were measured by qRT-PCR in V. harveyi ΔluxR (KM669) containing an empty vector (pJV036), expressing luxR (pJV239), or expressing FLAG-luxR (pJV274). Error bars represent the standard deviations of measurements of three biological samples. (B) Quantitative Western blot analysis of cell lysates from wild-type V. harveyi (BB120). Purified LuxR protein (0.42 pmol, 0.85 pmol, 1.69 pmol, and 3.38 pmol LuxR) mixed with cell lysates from a V. harveyi ΔluxR (KM669) strain was used as a standard control. Cell counts were determined by viability plating. Error bars represent the standard deviations of measurements of three biological samples. LuxR antibodies detect a nonspecific band that runs slightly faster than LuxR. (C) EMSAs containing LuxR (100 nM) bound to P05222 binding site substrates. The lengths of the dsDNA substrates are shown above the lanes. Sequences of selected substrates (labeled 1 to 7) are shown on the right. Substrates correspond to annealed oligonucleotides as follows (from left to right): JCV811/JCV812 (1), JCV813/JCV814 (2), JCV877/JCV878, JCV815/JCV816, JCV879/JCV880, JCV881/JCV882, JCV883/JCV884 (3), JCV841/JCV842, JCV849/JCV850 (4), JCV851/JCV852 (5), JCV843/JCV844 (6), JCV875/JCV876 (7), and JCV885/JCV886. All substrate sequences are listed in Table S3. (D) The LuxR binding consensus motif generated from the subset of ChIP-seq peaks near LuxR-regulated genes (105 regions, both activated and repressed genes). (E) Binding was assessed by EMSAs with substrates (A to E) from the P05222 promoter region as shown in the diagram in the presence (+) or absence (−) of 100 nM LuxR. Substrates correspond to annealed oligonucleotides as follows: JCV645/JCV646 (A), JCV647/JCV648 (B), JCV649/JCV650 (C), JCV651/JCV652 (D), or JCV653/JCV654 (E). The fragments correspond to the region of P05222 that is required for transcription activity in vivo (site −163 is not required). (F) Activation of PluxC was measured in E. coli strains containing an empty vector (pJV036, white bars) or expressing luxR (pJV239, black bars). Fluorescence was measured by FACS from PluxC-gfp (containing PluxC from −367 through +63 relative to the transcription start site) with either a wild-type sequence (pJV064), binding site 1 replaced with the randomized binding site sequence from panel G (BS 1*, pJV284), or binding site 2 replaced with the randomized binding site sequence from panel G (BS 2*, pJV285). Error bars represent the standard deviations of measurements of three biological samples. (G) LuxR binding was assessed by EMSAs with the wild-type binding site in the aphA promoter (BS; JCV428/JCV358) and with a randomized aphA promoter binding site sequence (BS*; JCV429/JCV360) in the presence (+) or absence (−) of 100 nM LuxR. Download
LuxR mutants are defective for activation or repression. E. coli strains containing an empty vector (no protein, pJV036), expressing luxR (WT, pJV239), or expressing the specified luxR mutants were assayed for transcriptional regulation of PluxC and P05222. Fluorescence was measured by FACS using pJV064 with P05222-mCherry (gray bars) and PluxC-gfp (black bars). Error bars represent the standard deviations of measurements of three biological samples. (A) Regulation of PluxC and P05222 by LuxR N55I (pJV240), N55Y (pJV249), N55S (pJV253), N55K (pJV254), and N55A (pJV281) mutants compared to results for wild-type LuxR. (B) Regulation of PluxC and P05222 by LuxR L139P (pJV242) and L139R (pJV260) mutants compared to that for wild-type LuxR. (C) Regulation of PluxC and P05222 by LuxR (pJV057) and the T52M (pJV103), I24V (pJV207), and T52M/I24V (pJV120) mutants. (D) Quantitative Western blot analysis of V. harveyi ΔluxR strain KM669 expressing luxR (pJV239) or luxR carrying N55I (pJV240), luxR T52M/I24V (pJV241), luxR L139P (pJV242), luxR A51T (pJV247), or luxR N142D (pJV261). LuxR antibodies detect a nonspecific band that runs slightly faster than LuxR. The background was calculated from results for KM669 containing an empty vector (denoted as “−”; pJV036). LuxR protein bands were normalized to the beta′ loading control. Purified LuxR was loaded as an additional control. Error bars represent the standard deviations of measurements of three biological samples. (E) Transcript levels of genes directly regulated by LuxR were assayed by qRT-PCR from a V. harveyi ΔluxR strain (KM669) containing an empty vector (pJV036, denoted as “no protein”) or expressing luxR (pJV239), luxR N55I (pJV240), luxR L139P (pJV242), luxR N142D (pJV261), luxR T52M/I24V (pJV241), or luxR A51T (pJV247). Error bars represent the standard deviations of measurements of three biological samples. Download
LuxR mutant proteins are defective for DNA binding at P05222, PluxC site 1, and PluxC site 2. EMSAs with LuxR, LuxR N55I, LuxR L139P, and LuxR T52M/I24V at P05222 (A), PluxC site 1 (B), and PluxC site 2 (C) are shown. The protein concentrations are shown below each gel. Download
Determination of the LLR and α parameters for ChIP-seq peak calling and the distribution of ChIP-seq peaks. (A and B) Empirical false discovery rate (FDR) assessment of the log-likelihood ratio (LLR) (A) or regularization factor (α) (B) parameter used to predict LuxR binding sites from ChIP-seq data. (C) Distribution of the number of peaks among the 582 merged genomic regions. Download
Motifs derived from ChIP-Seq peak sequences in the promoters of LuxR-regulated genes.
Distribution of ChIP-seq peaks in the promoter regions of LuxR-regulated genes.
Oligonucleotides used in EMSAs.
Distribution of sequence reads per chromosome.
LuxR binding peak positions determined by CSDeconv analysis.