

Abstract
Oxidative damage has been associated with various neurodegenerative diseases including Parkinson's disease, amyotrophic lateral sclerosis (ALS), and Alzheimer's disease, as well as non-neurodegenerative conditions such as cancer and heart disease. The Keap1-Nrf2 system plays a central role in the protection of cells against oxidative and xenobiotic stress. The Nrf2 transcription function and its degradation by the proteasomal pathway (Keap1-Nrf2-Cul3-Roc1 complex) are regulated by the cytoplasmic repressor protein, Keap1 which possesses BTB, BACK (IVR region) and Kelch domains. The BTB-BACK domains are important for Keap1 homo-dimerization as well as to interact with Cullin-3 for Nrf2 degradation. The crystal structure of the Keap1-Kelch domain is known; however, that of the BTB-BACK domains are not yet determined. We present here, through molecular modeling studies, the analysis of Keap1-BTB dimerization, and of BTB-BACK domains role in complex with Cul3. The electrostatic charge distribution at the BTB dimer interface of Keap1 is significantly different from other known BTB containing protein structures. Another intriguing feature is also observed that the non-conserved residues at the BTB-BACK-Cul3 interface region may play critical role for differentiating Cul3 recognition by Keap1 from other adaptor proteins for their specific substrates proteasomal degradation.
Keywords: Nrf2, Keap1, BTB and IVR/BACK domains, Cul3, molecular modeling
Background
Oxidative and xenobiotic stresses including reactive oxygen species (ROS), electrophilic chemicals and heavy metals damage biological macromolecules and disrupt normal cellular functions (reviewed in [1] and references therein). These stress factors are responsible for the development of many diseases such as cancer, cardiovascular disease, diabetes and neurodegeneration. Human bodies possess cytoprotective mechanism for survival by defending oxidative and xenobiotic stress factors.
The Keap1-Nrf2 system is one of the most important cytoprotective system which has been developed over the course of evolution [2]. Nrf2 (nuclear factor (erythroid-derived 2)-like 2) is a basic region-leucine zipper (bZIP) transcription factor that plays essential role to express many cytoprotective genes in response to oxidative and electrophilic stresses [3, 4]. The Nrf2 transcription factor belongs to the Cap ‘n’ collar (CNC) family of transcription factors, and is composed of a conserved N-terminal regulatory domain, termed the Neh2 domain, two transactivation domain and a C-terminal b-ZIP domain (Figure 1A). Based on sequence homology, the sequence of Keap1 from human, rat and mouse are highly conserved between them. The (Kelchi-like ECH associated protein 1) sequence can be sub-divided into the N-terminal BTB domain, the intervening region (IVR), the double glycine repeat or Kelch repeat (DGR), and the C-term region (CTR) (Figure 1A). The DGR and CTR domains are collectively to name as DC-domain. The IVR region is also named to as the BACK domain.
Figure 1.
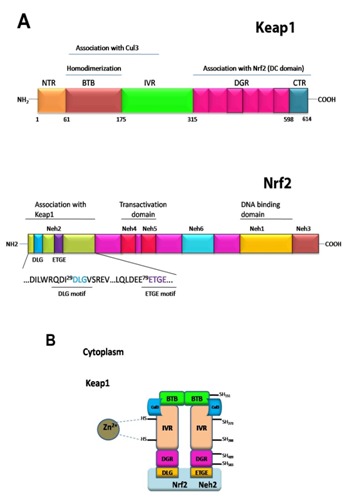
Schematic diagram of the Keap1-Nrf2 pathway. (A) Functional domains of Keap1 (top) and Nrf2 (bottom). (B) Cul3 based E3 ubiquitin ligase brings about ubiquitination of the substrate molecule Nrf2 via an adaptor protein, Keap1. Nrf2 is then presented to 26S proteasome for degradation.
Under homeostatic/unstressed conditions, the cellular concentration of Nrf2 remains low, and it is repressed/modulated by Keap1 thus, Nrf2 is constantly ubiquitinated through Keap1 in the cytoplasm and subsequently undergoes proteasomal degradation [5–7]. Under stress condition, such as exposure to electrophiles or ROS, Keap1 loses repression activity and hence Nrf2 dissociates from Keap1 and translocate into the nucleus, and subsequently coordinately activates cytoprotective genes and exerts a protective function against xenobiotic and oxidative stress [8].
The Keap1 protein forms a homodimer through the N-terminal BTB domain [7]. Under normal conditions, the Keap1-Nrf2 complex forms in 2:1 ratio as revealed by biochemical and structural studies [9]. In the Keap1 homodimer, the C-terminal β-propeller domain (Keap1-DC) of each monomer is free from any intermolecular interactions, and is separated from each other. The Keap1-DC of each monomer associates with one molecule of Nrf2 [10]. The ETGE and DLG motifs in Neh2 of Nrf2 are key motifs for direct interactions with the Keap1-DC domain [11, 12], and thus, Nrf2 bridges two Keap1-DC of the Keap1 dimer, and appears to be favorable for the efficient ubiquitylationof Nrf2 [9].
The BTB-BACK domains of Keap1 are not only important for Keap1 homodimerization, but also serves as an adaptor for the Cullin 3-based ubiquitin E3 ligase for Nrf2 [11, [13–15] (Figure 1B). Cullin-RING ligases (CRLs) are the largest family of multisubunit E3 ubiquitin ligases and adopt a modular assembly that facilitates the ubiquitylation of divergent substrates. The CRL3 subclass utilizes Cul3, which combines exclusively with BTBcontaining proteins as substrate-specific adaptors [16]. Keap1 is a classic example which demonstrates the importance of Keap1 dimerization for its substrate ubiquitylation as it requires two β- propeller domains to interact with two distinct epitopes in Nrf2 simultaneously [9, 10, 12, 17]. The Cul3 binds to the BTB-BACK domains of Keap1, and also the ring-box (Rbx1/Roc1) to form a ternary complex of a core E3 ubiquitin ligase complex, which helps Nrf2 to undergo proteasomal degradation.
Though the tertiary structure of Keap1-DC is known, the tertiary structures of BTB domain and IVR region (BACK domain) of Keap1 are not yet determined. We present here the predicted structure of BTB-BACK domains of mKeap1 by molecular modeling studies. We have also predicted the structure of BTB-BACK domains of mKeap1 in complex with Cul3. Based on the modeling results, we discuss the homodimer formation of Keap1 and its interaction with Cul3.
Methodology
The NCBI-BLAST (Basic Local Alignment Search Tool) online program [18] was used to compare the target protein sequence against the protein database and to calculate sequence similarity between them. The mouse Keap1 sequence comprising the BTB domain (aa: 52-179) and the BTB-IVR domains (aa: 52-319) was used to obtain the protein sequences of similar structures, using the RCSB Protein Bank Database [19]. The intermolecular contacts were analyzed by using the program PDBsum [20]. The selected protein sequences, based on the sequence similarity and the size of predicted sequence, were then subjected to multiple-sequence alignment calculation using Clustal W [21]. The multiple-sequence alignment results were manually edited wherever necessary to obtain reasonable predicted comparable sequences/structures between the target Keap1 protein and the query proteins. The online STRAP program [22] was initially used to visually inspect the multiplesequence alignment results. The ESPript program [23] was used to produce figures of multiple-sequence alignment results. The PyMol program [24] was used to analyze protein structures as well as to produce figures.
Results and Discussion
The mouse Keap1 protein sequence corresponding to the Nterm region (Ser14 – Cys319) has taken for sequence comparison against the RCSB-PDB database using the program NCBI-BLAST [18]. The blast search has revealed two distinct results: (1) protein sequences corresponds to the intact BTBdomain (hereafter, intact BTB domain, aa: 52-179, mKeap1 numbering), and (2) protein sequences possess the intact BTB domain and the BACK domain (IVR region) (hereafter, BTBBACK domains, aa: 52-319, mKeap1 numbering).
Intact BTB domain:
The NCBI-BLAST has revealed 12 protein structures, which possess an intact BTB domain, show significant sequence and structural similarity with the proposed BTB region of mKeap1 (Figure 2). Multiple-sequence alignment of these proteins with mKeap1 showed certain highly conserved amino acids between them such as His59, Asn68, Cys77, Asp78, His96, Ser142, Phe139, Tyr141 and Thr142 (Figure 2A). A conserved triplet motif, VLA (Val99, Leu100 and Ala101), which lie in α2, was also observed in the sequence analysis.
Figure 2.
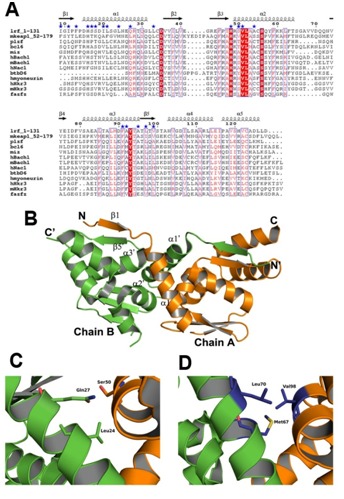
Comparison analysis of the intact BTB-dimer. (A) Sequence alignment of mKeap1 (aa: 52-179; KEAP1_MOUSE) with sequences of selected known crystal structures. The dimer interface residues are marked by blue stars. The secondary structural features from hLrf (PDB Id: 2NN2) are shown above the alignments. The first eight amino acids in hLrf are not included as this region is absent in the crystal structure. The colors reflect the similarity (red boxes and white characters for conserved residues; red characters for similarity in a group; blue frames for similarity across groups). The sequence was aligned and rendered by Clustal W [21] and ESPript [23], respectively. (B) The dimer arrangement of hLrf-BTB (PDB Id: 2NN2). The dimer interface region is only labeled for clarity. (C) A representative figure showing hydrophilic intermolecular interactions in the hLrf-BTB domain. (D) The corresponding region in mKeap1 showing hydrophobic environment due to Met67, Leu70 and Val98.
The predicted protein structures are from human Lrf (PDB Id: 2NN2), human Bcl6 (PDB Id: 1R28), human Miz1 (PDB Id: 3M52), human PlzF (PDB Id: 1BUO), human Fazf (PDB Id: 3M5B), human myoneurin (PDB Id: 2VPK), human Hkr3 (PDB Id: 3B84) and mouse Hkr3 (PDB Id: 2YY9) belong to the zinc finger family of transcription factors. Human Bach1 (PDB Id: 2IHC) and mouse Bach1 (PDB Id: 2Z8H) belong to the leucine zipper family of transcription factor, and human BtbD6 (PDB Id: 2VKP) is an adaptor protein for Cul3 ligase. Members of the BTB family share a secondary structure topology in the core BTB domain, and differ in the peripheral secondary structure elements [25]. The core BTB domain as seen among the known structures of BTB domain containing protein consists of five conserved α-helices (α1 – α5), and three common β-strands (β1 – β3) (Figure 2B).
The proteins used for sequence alignment also form functional homodimers through their respective BTB domains. Thus, in order to predict the dimer interface residues of Keap1-BTB, the crystal structure of the BTB dimer of human Lrf (PDB Id: 2NN2) [26] was used as a reference structure for discussion, since it has highest sequence identity of 31% with mKeap1 compared to other proteins. As seen from the hLrf structure, the dimer interactions are found between α1 helix of chain A and α2 and α3 of chain B, and vice-versa. Also, an antiparallel β-sheet conformation occurs between β1 strand of chain A and β5 strand of chain B, and vice-versa (Figure 2B). The dimer interface residues of the BTB domain of hLrf were obtained by using the PDBsum database [20]. The corresponding probable dimer interface residues of mKeap1-BTB were identified from sequence alignment. These dimer interface residues were found to be located in the predicted α1, α2, α3 helices, β1 and β5 strands Table 1 (see supplementary material), (Figure 2B).
Among the predicted dimer interface residues of mKeap1, it was seen that 40% was conserved, 10% was semi-conserved and 50% was non-conserved. The dimer interacting residues of hLrf in the crystal structure were mutated to corresponding mKeap1 residues using PyMol [24]. When we checked residue by residue in the dimer interface region, more hydrophilic patches were seen on the surface of mKeap1 when compared to hLrf (not shown). Moreover, as shown in the Figures 2C & 2D, the hydrophilic environment in hLrf contributed by Gln27 and Ser50 (Lrf numbering) is replaced with hydrophobic environment in mKeap1 contributed by the non-conserved residues (Leu70 and Val98). From this analysis, we speculate that besides the interactions due the conserved residues, variations in charge distribution in the dimer interface of mKeap1 may play essential role to form a stable Keap1 dimer as well as to make a complex with Cul3 for Nrf2 ubiquitination.
BTB-BACK domains:
Only two proteins, human Gigaxonin (KLHL16) (PDB Id: 3HVE) [27] and KLHL11 (PDB Id: 3I3N) [28] were resulted from the BLAST search (Figure 3A). Both KLHL11 and KLHL16 adaptors recognize Cul3 of CRL3 subclass for the substrates proteasomal degradation.
Figure 3.
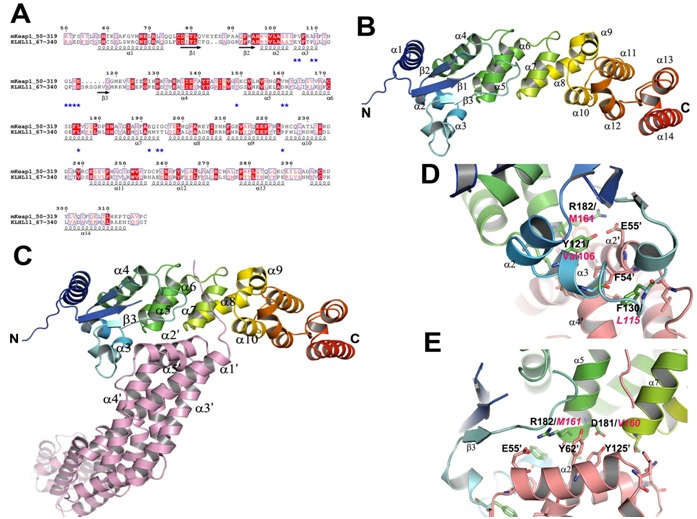
Comparison analysis of the BTB-BACK domains and its complex with Cul3. (A) Sequence alignment of mKeap1 (aa: 50- 319; KEAP1_MOUSE) with hKLHL11 (aa: 67-340; Q13618). The potential interface residues responsible for making complex with Cul3 are shown in blue stars. The secondary structural features from hKLHL11 (PDB Id: 4AP2) are shown above the alignments. The colors reflect the similarity (red boxes and white characters for conserved residues; red characters for similarity in a group; blue frames for similarity across groups). The sequence was aligned and rendered by Clustal W [21] and ESPript [23], respectively. (B) A cartoon ribbon diagram of the BTB-BACK domains (PDB Id: 4AP2). Only one chain of the homodimer is shown for clarity. The loop connecting β1 and β2 is absent in the crystal structure. (C) A cartoon ribbon diagram of the BTB-BACK domains in complex with Cul3 (salmon) (PDB Id: 4AP2), and labeled only the interacting secondary structure elements. (D) & (E) Showing representative diagrams at the Cul3 interface region. The corresponding residues in mKeap1are labeled in magenta.
The crystal structures of the BTB-BACK domains of hKLHL11 both apo-form and in complex with Cul3 were recently determined (PDB Ids: 3I3N and 4AP2) [28]. The hKLHL11 structure reveals the entire domain of BACK besides the BTB domain; whereas the KLHL16 structure lacks two helices at the N-terminal side of the BACK domain, which is functionally important for the Cul3 complex formation. Hence, we have taken the hKLHL11-Cul3 complex structure for our comparison studies.
The homodimer of the BTB-BACK domains of hKLHL11 is an elongated shape with overall dimensions of 150 × 35 × 25 Å (Figure 3B). In the complex structure, it forms heterotetrameric assemblies with each subunit in hKLHL11 homodimer binding one molecule of Cul3. The BACK domain mainly consists of β- helical secondary structures. The two N-terminal helices (α7 and α8 of hKLHL11) form the 3-box motif and subsequently create an antiparallel four helix bundle configuration by interacting with α5 and α6 of the BTB domain (Figure 3B). The helical bundle plays critical role in making complex with Cul3. The remaining helices, α9-α14 at the C-terminus creates a distinct sub-domain and packing perpendicular to the 3-box. This arrangement produces a cleft of 16 Å deep and 18 Å wide between the BTB and BACK domains which is responsible for Cul3 interaction.
In the complex structure, the complex is formed by α2' and α5' of the first Cullin repeat with the BTB and 3-box domains of hKLHL11 (Figure 3C). The contact surface area of the Cul3NTD interface is 1508 Å2. A shallow cleft in the BTB surface forms via an induced fit mechanism facilitated by conformational changes in the α3-β4 loop. This loop is disordered in the apo-form, but change into an α-helix when interacts with Cul3; Ser131 of hKLHL11 hydrogen bonds with Glu132 of Cul3 α2' helix. Moreover, Phe130 is shifted about 5 Å to insert into a deep hydrophobic pocket produced between Cul3 helices α2' and α4' (Figure 3D). The backbone carbonyl of His213 (α7) in hKLH11 makes a hydrogen bond with Lys68' of Cul3. Phe246 (α10) of hKLHL11 also contributes a hydrogen bond with Thr24 of Cul3.
The sequence comparison between mKeap1 and hKLHL11 corresponding to the BTB-BACK domains showed 16.6% sequence identity, and 66.8% non-conserved residues between them. The interacting residues in the hKLHL11-Cul3 complex were analyzed using the PDBsum database [20]. The corresponding residues in mKeap1 were identified from the sequence alignment Table 2 (see supplementary material).
The side chain of Asp181 of KLHL11 interacts with the sidechains of Tyr125' and Tyr62' of Cul3 (Figure 3E). Glu55' of Cul3 makes a salt-bridge and a hydrogen bond with Arg18 and Tyr121 of hKLHL11, respectively (Figures 3D & 3E). Intriguingly, these electrostatic interactions contributed by Tyr121, Asp181, and Arg182 in hKLHL11 may be absent in the Keap1-Cul3 complex as these residues are replaced with hydrophobic residues Val106, Val160 and Met161, respectively, in Keap1 (Figures 3D & 3E). Another interesting feature is also observed in the complex that Leu184 of hKLHL11 hydrophobically interacts with Met124' of Cul3. But, in mKeap1, the corresponding residue is replaced with a hydrophilic residue, Glu163 (Figure 3A). Hence, we speculate that in the Keap1-Cul3 complex, electrostatic surface charge distribution may be different at the Keap1-Cul3 interface when compare to the hKLHL11-Cul3 complex. This unique difference may play critical role for differentiating Cul3 recognition by Keap1 and KLHL11 adaptors for their substrates proteasomal degradation.
Keap1 is a cysteine rich protein. Mouse Keap1 contains 25 cysteine residues and human Keap1 contains 27 cysteine residues. Out of these cysteines, a few have been implicated to play important role as sensors viz. Cys151, Cys273 and Cys288. Cys151 is present in the BTB domain of Keap1. It is evident from the analysis that Cys151 is exposed to the solvent region and hence, it has free access to the cellular environment. Also, it is located at the N-terminal side of α5 which is a key secondary element for Cul3 interaction (not shown). Any disturbance in this region caused by adduction of Cys151 might bring about disruption of Cul3 interactions, thereby, preventing ubiquitination of Nrf2.
Conclusions
The Keap1 protein, being the master regulator of Nrf2, has become an important therapeutic target for regulating the Nrf2 transcription function. Besides the Keap1-DC domain which is essential for Nrf2 binding, the BTB domain of Keap1 is an important domain to form functional homo-dimerization, essential for Nrf2 ubiquitination via Cul3 ligase complex. The BTB-BACK domains of Keap1 are essentially interacts with Cul3. The important dimer interacting residues, Cul3 interacting residues, electrostatic charge distribution at the dimer interface as well as at the Cul3 binding site have been mapped in the comparative analysis. The information gained in the present study will be helpful for further biochemical analysis of the Keap1-Cul3 complex as well as to aid in designing inhibitor molecules to inhibit the Nrf2 degradation pathway.
Conflict of Interest
The authors declare that they have no conflict of interest.
Supplementary material
Acknowledgments
We thank Prof. P. Satishchandra, Director/Vice Chancellor to provide the finance support for computational facility.
Footnotes
Citation:Chauhan et al, Bioinformation 9(9): 450-455 (2013)
References
- 1.Calkins MJ, et al. Antioxid Redox Signal. 2009;11:497. doi: 10.1089/ars.2008.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitsuishi Y, et al. Front Oncol. 2012;2:200. doi: 10.3389/fonc.2012.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itoh K, et al. Biochem Biophys Res Cummun. 1997;236:313. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 4.Ishii T, et al. J Biol Chem. 2000;275:16023. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 5.Sekhar KR, et al. Oncogene. 2002;21:6829. doi: 10.1038/sj.onc.1205905. [DOI] [PubMed] [Google Scholar]
- 6.McMahon M, et al. J BiolChem. 2003;278:592. [Google Scholar]
- 7.Kobayashi A, et al. Mol Cell Biol. 2004;24:7130. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itoh K, et al. J BiolChem. 2004;279:31556. [Google Scholar]
- 9.Ogura T, et al. Proc Natl Acad Sci USA. 2010;107:2842. doi: 10.1073/pnas.0914036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong KI, et al. Mol Cell Biol. 2006;26:2887. doi: 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Padmanabhan B, et al. Mol Cell. 2006;21:689. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 12.Tong KI, et al. Mol Cell Biol. 2007;27:7511. doi: 10.1128/MCB.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cullinan SB, et al. Mol Cell Biol. 2004;24:8477. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang DD, et al. Mol Cell Biol. 2004;24:10941. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furukawa M, Xiong Y. Mol Cell Biol. 2005;25:162. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pintard L. EMBO J. 2004;23:1681. doi: 10.1038/sj.emboj.7600186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMahon M, et al. J Biol Chem. 2006;28:24756. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 18. http://blast.ncbi.nlm.nih.gov/Blast.cgi.
- 19. http://www.rcsb.org.
- 20.Laskowski RA, et al. Nucleic Acids Res. 2009;37:D355. doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson JD, et al. Nucleic Acids Research. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. http://www.bioinformatics.org/strap/
- 23.Gouet P, et al. Bioinformatics. 1999;15:305. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 24. http://pymol.org.
- 25.Stogios PJ, et al. Genome Biol. 2005;6:R82. doi: 10.1186/gb-2005-6-10-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stogios PJ, et al. Protein Sci. 2007;16:336. doi: 10.1110/ps.062660907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhuang M, et al. Mol cell. 2009;36:39. doi: 10.1016/j.molcel.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canning P, et al. J Biol Chem. 2013;288:7803. doi: 10.1074/jbc.M112.437996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.