

Abstract
Flowers exist in exceedingly complex fitness landscapes, in which subtle variation in each trait can affect the pollinators, herbivores and pleiotropically linked traits in other plant tissues. A whole-genome approach to flower evolution will help our understanding of plant-pollinator interactions.
Keywords: Adaptation, coevolution, ecological genomics, hybridization, polyploidy, sensory systems, standing genetic variation, transcriptome
Plants are immobile, and therefore they cannot choose their own sex partners. To cope with this handicap, several lineages of plants have invented a trick to facilitate the directed transfer of gametes: harnessing animals as go-betweens. This is known in some gymnosperms and even mosses [1-4], but it is in the angiosperms where animal pollination is most commonly encountered and where, in many cases, it has evolved exquisite complexity. This means that the morphology of angiosperm sex organs (flowers), rather than fitting those of the opposite sex, must generate a lock-and-key fit with the animals that visit them. These visitors do not normally come for sex; instead they are paid for their services, typically by means of sugary nectar or surplus pollen. Plants advertise these rewards with showy displays to assist pollinators in finding them [5,6].
However, there are peculiar challenges that come with such an unusual sex life. Many animals, such as bees, butterflies, flies, birds and bats, might be opportunistically interested in nectar carbohydrates, and plants cannot know or see which ones are in the vicinity, nor can they accept or reject a visitor. They can only offer their commodities to a diverse army of potential visitors from where they stand, and try their luck. Flower structures can be used to limit the type of visitor to some extent [7,8], but such limitation is typically not absolute [9]. In addition, flowers can attempt to obtain some pollinator specificity by using advertising signals that appeal only to certain pollinators and not others. Colors, patterns, scents and even acoustic or electrostatic cues are all known to affect the behavior of different pollinators in different ways [10-18]. The most important factor that determines a pollinator's preference is often individual experience rather than innate predisposition [5,6]. Current genomic approaches will help verify this supposition, which, if true, should in some cases reveal relatively weak correlations between flower genotype and pollinator affinities. In terms of pollinators, a genomic approach can reveal the factors that enable flower visitors' ability to be generalists, such as the number and diversity of olfactory receptor genes, or genes for learning and memory [19].
A further challenge for plants is that some floral traits that are attractive to pollinators can also be of interest to herbivores [20], and in some cases, flowering plants may have the dual problem of attracting pollinators while deterring the same species in larval stages [21]. Finally, many flower traits are subject to extensive pleiotropies; for example, pigments that contribute to flower coloration are also used in other parts of the plant where they can have multiple important functions [22,23]. Flowers thus exist in exceedingly complex fitness landscapes, in which a large number of traits might all affect individual success, as well as the dynamics of speciation and plant-pollinator coevolution [24,25]. In this sense a genomic approach to understanding floral evolution has tremendous potential to move from the traditional question 'is this gene important?' (which carries the risk of generating a self-fulfilling prophecy) to a data-driven approach asking which genes and in which combinations are important and in what ways [26,27].
Paramount among these later approaches are genome-wide association studies (GWASs) and genomic selection (GS), which work to find correlations between genetic markers (such as single nucleotide polymorphisms (SNPs) and randomly amplified polymorphic (RAD) sequences) in linkage disequilibrium with phenotypic traits, and these approaches are being applied in both plant and animal breeding (for example, in maize [28,29], oil seed rape [30] and cows [31]). A GWAS compared 107 distinct phenotypes of Arabidopsis thaliana, revealing alleles of major effect for follow-up research [32]. Although A. thaliana is primarily self pollinating, comparisons between self- and insect-pollinated plants using these techniques, particularly when closely related, may reveal the genomic architecture important to plant-animal interactions. Several approaches in use for assessing genomic variability do not require de novo genome assembly; for example, sequenced restriction-site-associated DNA (sRAD) uses next-generation sequencing (NGS) approaches to find markers (frequently SNPs) for genotyping [33,34] or RNA-seq (alternatively called whole-transcriptome shotgun sequencing) to assess transcription profiles [33] across tissues or life stages. These approaches, whether applied to expressed gene regions or whole genomes, can accurately quantify genomic variation and help us to understand how much variation is available for selection to act on. The results may also have implications for conservation genetics, such as the question of which population is more variable and thus contributes most to the preservation of diversity.
We are still at the early stages of studying the interactions between plants and their pollinators using genomics-based approaches. In plant biology, most resources were initially devoted to sequencing genomes of autogamous (self-fertilizing) model species (the first being A. thaliana, with a small genome size [35]) or commercially important crop plants, some of which are not insect-pollinated [36,37]. Similarly, resources used to study insects were initially focused on species with pathogenic impacts on humans or crops, or those serving as model species. For the most part, species interactions between plants and animals have remained the domain of traditional genetics and quantitative trait mapping in combination with experiments in animal behavior. However, the genomes of several animal-pollinated plants, including some that have been models in evolutionary ecology, are now available or in progress, such as the bee-pollinated tomato Solanum sp. [38,39], the morning glory Ipomoea sp. [40], Petunia sp. [41], and Mimulus sp. [42]. These genera contain species with highly diverse ecology, and are pollinated by birds, bees and moths [43], which should therefore enable comprehensive insights into the genetic architecture underpinning floral traits that address different types of pollinators.
Today, the variety of tools available for genome-level analysis and the decreasing costs of NGS technologies open the door for the large-scale analysis of non-model organisms. The 5,000 insect genome project [44] and the 1000 plant transcriptome project [45] may have great potential for the understanding of pollinator-plant evolution. The scale of these projects reflects the orders of magnitude reduction in costs of sequencing, and they include the majority of model and non-model organisms of active interest around the world, including representatives from more than half (over 250) of all angiosperm plant families, and multiple representatives from all (approximately 30) major insect orders. These projects will enable questions to be addressed that cannot be achieved in studies targeted at individual species, and they have the potential for unprecedented insights into the phylogeny, evolution and patterns of diversification of angiosperms and their pollinators. For example, what is the nature of genetic change in plant speciation [24]? What is the relationship between prezygotic reproductive isolation via flower traits and associated pollinator behavior [46]? Did bee pollinators co-evolve and co-diversify with angiosperm flowers [47], and did butterfly diversity emerge much later [48]? In what ways do floral traits affect rates of plant species diversification, for example traits controlling bilateral floral symmetry and the morphology of nectar spurs [49]? Is the independent emergence of certain floral traits in different lineages (such as in transitions from predominantly bee-pollinated ancestors to principally hummingbird-pollinated flowers) mediated typically by parallel evolution (variation in homologous genes or indeed homologous mutations) or by convergence using different molecular-genetic pathways with similar phenotypic outcome [49]? Genome-wide studies using NGS approaches, combined with data mining and new statistical tests, will provide new insights into these questions, insights that would have been impossible using more traditional genetic approaches.
The possibility of sequencing genomes of multiple individuals of a plant population allows the quantification of intraspecific genetic variation for a large number of floral traits, and thus the raw material on which selection can act [26,27]. Moreover, comparing extant genome sequences with those from ancient plant material (for example, that conserved by permafrost [50]) will enable the monitoring of genomic changes across populations over time and the quantification of the relative contributions of new mutations and existing genetic variation to evolutionary change, and will help us to link these genomic changes to abiotic (for example, climate change) and biotic factors.
Here, we provide a historical perspective and a future outlook on the molecular and genomic basis of plant-pollinator interactions. We review what has been learned from traditional genetic approaches and the genomic search for quantitative trait loci (QTLs), the potential for new genomic approaches to document plant-pollinator interactions, and the application of these studies to understand the evolutionary and ecological mechanisms governing these interactions.
Traditional single and multi-locus genetic analyses
The influence of pollinator choice on the evolution of various sensory cues displayed by flowers is well documented (for example, [11-17,51]; Figure 1). Genetic analyses of plant-pollinator interactions can be roughly divided into studies that document candidate genes and quantify pollinator response to manipulation, those that explore genetic pathways and mechanisms of floral traits [52] and studies that seek to understand the evolutionary history of species interactions, such as adaptive radiations of plants and their pollinators [53].
Figure 1.
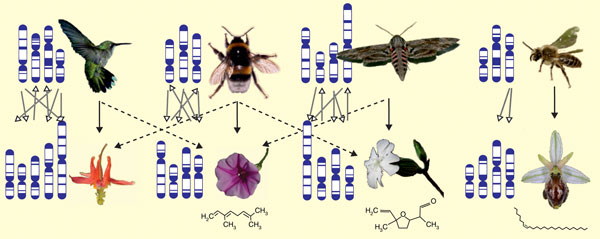
Interactions of different plants and pollinators; genomes are indicated symbolically. Left to right: the hummingbird-pollinated Aquilegia formosa, the bee-pollinated Ipomea pes-caprae, the moth-pollinated Silene latifolia and the sexual mimic Ophrys exaltata with its pollinator, a male Colletes cunicularius bee. Many of these affinities are neither fixed nor exclusive, as indicated by dashed diagonal arrows. Arrows between the chromosomes symbolize the many interactions of different loci distributed among the genomes, mediated by the phenotypes of plants and pollinators. Genomic approaches will probably lead to a better understanding of how interactions influence the evolution of molecular traits and their variability. The scent molecules β-ocimene, lilac aldehyde and (Z)-7-pentacosene (from left to right) are representative examples of the respective pollination systems bee pollination, moth pollination and sexual mimicry. Images reproduced, with permission, from [6] and the photographers: FP Schiestl (bumblebee, Silene, Ophrys), SD Johnson (sphingid moth), SA Hodges (Aquilegia), RARaguso (Ipomea), NJ Vereecken (Colletes bee).
Floral traits are sometimes controlled by a relatively small number of genes [54]. For example, in the common morning glory (Ipomoea purpurea), various aspects of pigmentation are controlled by single loci [55]: one locus determines pink versus blue flowers, another controls intensity, a third controls the patterning and degree of pigmentation, and a fourth locus has epistatic effects on the other three loci [56-58]. Pollinator selection for specific loci has been tested in various contexts. For example, red flowers are relatively poorly detected by insect pollinators that lack red receptors [59], whereas hummingbirds have such receptors and can thus detect and identify red flowers more easily [13,60,61]. In cases in which flower color is controlled by a small number of loci, crossing experiments can directly assess pollinator responses to a specific allele (Figure 1, 2). Among monkeyflowers, the predominantly hummingbird-pollinated species Mimulus cardinalis is orange and the typically bee-pollinated M. lewisii is pink. The difference in coloration is governed by variation at a single locus (YUP), which controls presence/absence of carotenoids in the petals. Substituting the YUP allele in each species with its counterpart from the other species (while leaving all other floral traits, including morphology, unaffected) caused hummingbirds to prefer M. lewisii flowers with altered ("bird-flower-like", orange) pigmentation over wild-type flowers (which they rarely visited). Bumblebees, conversely, visited M. cardinalis more frequently than wild-type flowers if the flowers had M. lewisii-like pigmentation, showing that a single major mutation affecting flower color may generate a profound shift in pollinator spectrum [59,62]. Genomic methods such as sRAD or homologous gene sequencing from target loci can be used to compare genotypes between individuals, species and so on within radiations in the search of genotypes (such as SNPs) that segregate with particular phenotypes.
Figure 2.

Hybridization, polyploidy and flower phenotypes. (a) Interspecific hybridization can generate a large range of characters in hybrids, including intermediate characters and potentially characters that fall outside the range found in the parents (transgressive characters). Hybrids usually backcross to one or both of the parents, providing gene flow between species. Herbivores and/or pollinators may select for particular combinations of characters. (b) F1 and F2 hybrids between bee-pollinated Mimulus lewisii and predominantly hummingbird-pollinated M. cardinalis. In F2 the different flowers show a range of intermediate characters. Adapted, with permission, from [59] (Copyright (1999) National Academy of Sciences, U.S.A.). (c) Synthetic allopolyploids of Nicotiana attenuata and Nicotiana obtusifolia generate flowers and seeds with a larger range of characters than is found within the range encompassed by the parental phenotypes. Adapted, with permission, from [110].
It is important to emphasize, however, that studies on pollinator 'attraction' or 'preference' for certain floral phenotypes should not be conducted in field conditions, because the preferred type of flower will most likely just be the one that is most similar to those experienced by the pollinators before the start of the experiment. For this reason, it is necessary to experiment with laboratory-reared pollinators that have no previous exposure to natural flowers before a preference test begins [63]. Using this approach on wild-type snapdragons (Antirrhinum majus) and various mutants that affect flower visual appearance and morphology, it has been possible to disentangle how visitation frequencies of various flower types emerge as a complex interplay between pollinator innate bias, flower detectability and learned preference [10,64].
Although many floral traits are controlled by a single locus, many more are multi-locus traits. Some floral scent cues that influence pollinators may be affected by a relatively small number of genetic loci, detected by screening for QTLs. For example, some floral traits in Petunia species are controlled by several loci of relatively large effect. F2 plants from crosses between P. axillaris (which produces scent cues) with P. exserta (which has no scent) led to the identification of two QTLs, one on chromosome II and another on chromosome VII, that together explained 60% of the variation in the production of benzenoid volatiles between the two species [65]. QTLs can, of course, correspond with large genetic intervals containing tens or even hundreds of genes, and so it is important to pinpoint the exact genes (and their number) to understand the ease with which an evolutionary transition between, for example, a scented and a scentless flower type can occur. To do that, the authors [65] developed introgression lines for high resolution mapping and localized one gene, ODORANT1, which encodes a MYB-type transcription factor that mapped to the QTL on chromosome VII. The breeding lines were then used to demonstrate that hawkmoths prefer scented plants, particularly when given choices between plants spaced over short distances [65]. QTLs have also been associated with many other aspects of pollination syndromes in P. axillaris (pollinated by nocturnal hawkmoths) and P. integrifolia (pollinated by diurnal bees), including corolla length, nectar volume, style/stamen arrangement and fragrance [66].
Many traits show continuous phenotypic distributions and environmental interactions, with examples in the shape and size of petals, corolla tubes, stamens and pistils, as well as placement and arrangement of anthers and stamens, nectar volume, and the number of ovules and pollen grains [67]. Frequently, traits are controlled not by a single or a few genes, but by multiple genes across the genome, each with small effects. To detect the causes and consequences of these effects, the genome of Petunia will be very useful because molecular markers (for example SNPs) can be mapped to the scaffold for GWAS studies. NGS approaches, analyzing many thousands of short sequences from across the genome or from the transcriptome, offer huge potential for GS, because they enable phenotype prediction by examining the combination of genetic markers that most strongly correlates with phenotype [28,31]. The method requires finding associations between genome-wide markers and a trait of interest in a population of individuals with known phenotype and then applying the derived knowledge to predict phenotype in new individuals [28]. The approach has already proved successful in enhancing milk production in cows, and lines have been adopted by the dairy industry worldwide [31], although the statistical approaches are currently limited to analyses of markers in populations of highly related individuals. For pollinator-floral evolution questions, the future will require analyses of more divergent populations, and for this the statistics will need to be extended.
In some cases, small genetic changes controlling floral traits not only generate a shift in the principal pollinators that visit the flowers, but also in the unwanted visitors, such as nectar-thieving ants [20]. In addition to sugars, which constitute the principal reward in most floral nectar, many other substances are also found in nectar. Bitter substances, such as caffeine or nicotine, repel visitors at high concentrations, but at low concentrations can be attractive [68] and indeed have beneficial effects on pollinator learning and memory for floral traits [69]. Different flower visitors can vary in their response thresholds for the repellent effect; for example, in Nicotiana attenuata flowers, moths responded more strongly to the presence of nicotine (repulsion) and benzyl acetone (attraction) in nectar than hummingbirds [20]. RNA interference on only two loci can block the production of both benzyl acetone and nicotine, resulting in fewer visits and nectar removal by pollinators, which has fitness effects (for example, altering seed set), but it also alters damage by herbivores, and nectar theft by ants [51]. This provides an example of how complex the interactions between selection and genetic associations can be in relationships between plants, pollinators and antagonists such as herbivores [67].
Beyond isolating floral mechanisms affecting pollinator behavior, genetic analysis provides a molecular phylogenetic context for the evaluation of correlated divergence between plants and their pollinators, as well as the demographic changes of populations. One of the earliest phylogeographic studies involved restriction site mapping and length polymorphism analysis of the honeybee Apis mellifera [70]. Phylogenetic perspectives using both genetic and genomic information serve to clarify the origin of particular traits (for example, [71]) as well as the timing of adaptive radiation. It is these radiations that may allow inter- and intraspecific comparisons, particularly with respect to closely related out-groups with different phenotypes.
Interspecific hybridization and polyploidy
When considering the consequences of interactions between angiosperms and their pollinators, it is vital to consider two predominant processes influencing angiosperm diversity: interspecific hybridization and polyploidy (or whole-genome multiplication; Figure 2). At least 25% of species form interspecific hybrids [72], resulting in introgression of characters between species and, rarely, the formation of homoploid hybrid taxa [24]. This process results in complex, reticulate patterns of evolution in many angiosperm groups, making it difficult or impossible to circumscribe species boundaries. In addition, polyploidy is widespread and is thought to have influenced all angiosperms, having occurred at the base of both the seed plants and angiosperms [73] and within most lineages thereafter, sometimes surprisingly frequently. For example, the apparently diploid species A. thaliana, a model species because of its small genome, has undergone three rounds of polyploidization since the divergence of angiosperms [74,75], and the existence of natural cultivars with double the expected number of chromosomes reveal that the process is ongoing. Indeed, an analysis of chromosome counts across multiple genera led Wood et al. [76] to predict that polyploidy accounts for 15% of angiosperm speciation events. Furthermore, many polyploids form in association with interspecific hybridization (allopolyploidy [77,78]).
A consequence of such a high prevalence of interspecific hybridization and polyploidy in the ancestry of angiosperms is that much diversity is generated. Recent advances in NGS approaches, including RNA-seq, reveal that these evolutionary processes can affect the genome and transcriptome, which in turn must govern changes to the metabolome and phenotype [74]. At the genome level, there is much variation in the outcome of allopolyploidy, from considerable genomic restructuring, even between progeny of the same cross, or little at all, depending on the example [74]. At the level of the transcriptome, there can be large-scale changes in both the nature (which genes are expressed) and in the total amount of RNA expressed [79] per cell, the significance of which is yet to be understood. Recurrent polyploidy in angiosperm evolution has also resulted in the evolution of genes in large multigene families [80]. With the evolution of gene duplicates, there can be relaxed selection on one of the gene copies, enabling it to diverge, potentially to acquire new functions. This can lead to individual gene members of the family having different metabolic roles, as observed through their differing patterns of gene expression [79]. For example, gene duplicates of anthocyanin-regulating transcription factors in Mimulus from Chile have evolved independently in separate tetraploid lineages to generate red floral pigmentation in flower lobes [81]. Interspecific hybridization and allopolyploidy provide the opportunity for new, transgressive characters to evolve. Transgressive characters [24] can be observed at all levels from the transcriptome to inflorescence morphology [79,82] and may arise through novel cis-trans interactions in the genome [83] or by 'mixing and matching' of different metabolic pathways found in the parents.
An example in which gene duplication has led to the evolution of a large plant gene family with key functions in plant-insect interactions is seen in the terpenoid synthase genes, encoding enzymes that synthesize volatile compounds called terpenoids that mediate interactions with mutualists and antagonists [84]. Minor structural changes in terpenoid synthase genes are known to change the product of the encoded enzymes, allowing rapid evolutionary change in volatile compounds [85]. Another example is the desaturase genes, which encode key compounds for pollinator attraction in the highly specific sexual mimicry systems in orchids of the genus Ophrys [18,86]. Ophrys species have large, variable families of desaturase genes with high allele diversity and species-specific expression patterns. Variation in structural genes, as well as changes in their expression levels, are likely mechanisms allowing rapid evolutionary responses to fluctuating pollinator communities. A major challenge for understanding the link between genotypic and phenotypic variation, however, is the functional characterization of gene copies and different alleles [18,87] and their respective roles in determining key traits in plant-insect interactions. This approach will require methods and statistics to be developed from GS studies to diverse natural situations, enabling the characterization of multiple genes with small effects, as already discussed.
Genomic tools to document plant-pollinator interactions
Although the term 'genomic' tends to imply the analysis of entire genomes and genome assembly, genomic methods themselves can be applied to other practical aspects of pollinator-plant interactions [40]. Molecular methods of documenting species interactions began with traditional immunological and genetic methods (reviewed in [88]) but progressed to genomic methodologies, in particular NGS (reviewed in [89]). One approach is to use NGS to amplify millions of homologous sequences from a target gene from DNA extracted from a mixed biomass and compare the resulting sequences with curated databases of sequences of known origin (for example, [90,91]) in a 'food forensics' approach [92]. The use of genomic sequencing platforms (such as Illumina, Roche 454 and Ion Torrent) for fragment analysis is one of the most common techniques. This approach enables diagnosis of specific and hard-to-observe interactions (such as nocturnal pollination) using targeted gene amplification in the context of 'ecological genomics' (Figure 3).
Figure 3.

A schematic of genomic methods as applied to document plant-pollinator interactions. The development of high-throughput sequencing platforms for genome analysis coupled with the establishment of public databases of standardized marker regions for the express purpose of taxonomic identification (for example, BOLDSYSTEMS [111] and the National Center for Biotechnology Information [112]) has enabled the development of a branch of ecological genomics devoted to documentation of species interactions. An unobserved flower visitation event can be conclusively demonstrated by sequencing plant markers from the mixed pollen sample carried by the pollinator (either in the gut or on the animal). The resulting markers can be compared with public or private collections of taxonomically validated references for species-level documentation of the ecological event. This enables large-scale measurements of species' interactions to be largely automated. The resulting databases can be used to quantitatively measure a variety of ecologically and evolutionarily important events, such as the relative specialization or generalization of specific plant-animal pairs, the selection pressure of one group of species on another, the economic impact of a particular ecosystem service (for example, pollination of a particular crop of interest) or the response of an ecological system to disruption. Photographs reproduced with permission of EL Clare, MB Fenton (bee on flower) and J Nagel (bat with pollen on its fur).
The goal of these analyses is to accurately diagnose species-level interactions that cannot easily be documented in the wild. The most recent investigations have focused on predator-prey or herbivorous interactions rather than mutualistic interactions, in part because reference taxonomic databases of molecular information for animals have developed faster than those for plants. Interactions among species form the basis of ecosystem functioning and underlie evolutionary and ecological principles of conservation biology. However, directly measuring biological diversity is much simpler than characterizing the interactions between taxa [93]. These relationships between species form the building blocks of food webs, and exploring the mechanisms structuring these interaction patterns is crucial in understanding their spatial-temporal variation and predicting their responses to disturbance. Knowing precisely which pollinators visit which plants in the wild is also vital to our measurements of selection, particularly when trying to clarify whether a particular pollinator is a true specialist or whether their behavior is flexible.
We expect there to be swift progress in this field since a plant barcoding community consortium [94] proposed that a combination of genomic regions (including the plastid regions rbcL and matK) should be used as the core barcode for land plants, to be supplemented by the plastid intergenic spacer trnH-psbA or the nuclear ribosomal internal transcribed spacers (ITS). The final selection of a standard plant barcode is leading to the rapid acquisition of databases of plant barcodes for taxonomic and biodiversity analysis (Box 1). The application of these databases to study mutualistic interactions between plants and animals is, however, in its infancy. In Hawaiian solitary bees, molecular identification of the pollen carried (using the ITS region) linked these grains to the local flora and revealed preferential foraging for pollen from native species [95]. Similar extensions using trnL and other official plant barcode genes for the study of plant-animal interactions are under way by several research groups ([96,97], and the authors) with the potential to rapidly diagnose thousands of individual pollination events simultaneously without the need for laborious morphological identification of pollen (Figure 3).
Genomes of plants and pollinators
The accumulation of complete genome sequences is increasing quickly providing larger numbers of genome scaffolds (reference genomes) for the assembly of related taxa, an approach that will be exploited for our understanding of non-model species. As an example, the genome of the honeybee A. mellifera has been sequenced and is available as version 4 (about 236 megabases with 7.5X coverage, N50 contig = 41 kb, N50 scaffolds = 362 kb) [19]. A survey of this genome revealed a higher than expected number of odor receptor genes and novel genes for nectar and pollen utilization, as well as more loci involved in learning and memory than have been observed in Drosophila [19]. The number of olfactory receptor genes is more than twice as high as in other insects that have been examined, perhaps reflecting the importance of pheromones in the orchestration of social organization in honeybees but also underpinning the sophisticated olfactory learning abilities that bees show in floral visitation. Olfactory cues are also potentially important to bat pollinators [98]. Olfactory receptors show substantial divergence in bats compared with other terrestrial mammals [99], although this is seen in both pollinating and non-pollinating species.
There are also significant phylogenetic insights that emerged from the honeybee genome, and comparison with other insect genomes. A survey of 185 genes from sequenced insect genomes suggests that the Hymenoptera share a common ancestor with all other holometabolous insects (beetles, moths and flies) [71]. These authors propose [71] that previous phylogenetic hypotheses, in which this arrangement was not recovered, were based on poorly resolved analyses based on single loci or small numbers of loci. The split between Hymenoptera and other holometabolous insects is estimated to have occurred nearly 300 million years ago [71]. It is likely that no study of any single or small number of genes could reliably reveal this ancestral relationship. Only the analysis of dozens of protein alignments derived from genome-level scans provides sufficient support to resolve the relationship. Well resolved phylogenies, particularly depicting ancient branching points, allow better characterization of evolutionary pressures involved in adaptive radiations, demographic shifts and major leaps in phenotypic evolution. This is particularly true when there is convincing evidence that diversification may be based, at least in part, on the selective pressure of two (or more) species involved in a mutualistic relationship.
The genomes of several insect-pollinated plant species have now been sequenced or are currently being sequenced (see [38-42] and [100-103]). An interesting model for studying the genomics of adaptive divergence is the genus Mimulus. Sequences of the whole genome and of expressed sequence tags (ESTs) of Mimulus guttatus (version 1.0 early release: about 322 Mb arranged in 2,216 scaffolds, about 301 Mb arranged in 17,831 contigs, N50 scaffold = 81, N50 contig = 1,770) are being assembled and are now available for browsing on the Phytozome website [42], containing approximately 321.7 Mb assembled (of 362 Mb [104], arranged in 2,216 scaffolds). There are extensive genetic resources for Mimulus, including ESTs, polymorphic markers, linkage maps, bacterial artificial chromosome libraries, seed stock and centralized repositories of this information for public use ([105], reviewed in [106]). Mimulus is a model system for studying adaptive phenotypic traits by analysis of pollinator selection, as it contains extensive phenotypic and genomic variation explored in both laboratory and field contexts. Furthermore, the potential for hybridization between species forms a continuum from high compatibility to reproductive isolation (reviewed in [106]), enabling the examination of interactions between floral traits and reproductive barriers. Although model species allow an understanding of evolutionary mechanisms of one particular system in detail, important insights will be gained by comparative approaches spanning the diversity of different plant-pollinator systems. This promises insights into animal sensory systems, floral signals and morphology that may co-evolve or be shaped by pre-existing biases [6]. The availability of powerful sequencing and bioinformatics tools will allow us to study non-model systems, including those with very large genomes, allowing insights through comparative approaches [66,100-103].
One obvious next step is to ask questions about parallel phenotypic diversification. Genome studies have the potential to uncover whether parallel phenotypic changes are caused by similar underlying genomic architecture (for example, parallel phenotypes associated with homologous genes and mutations), or achieved through many alternative genetic mechanisms [27]. The exploration of parallel cases of divergence increases statistical power because evolutionary trends can be examined in replicate [27] and genomic approaches will add more dimensions through the analysis of hundreds or thousands of candidate genes and co-adapted gene complexes. Floral phenotypes are sometimes characterized by phylogenetically convergent adaptations to the requirements and preferences of certain groups of pollinators (pollination syndromes); for example, bat-pollinated flowers from multiple lineages often contain certain sulfuric components in their scent, to which bats seem to be innately attracted [98]. There are also differences in the color perception of different classes of pollinators, such as with respect to differences in red sensitivity between bees and hummingbirds (see above) [61,107]. Convergence in regulatory mechanisms was discovered for switches in floral color from blue pigmentation in bee-pollinated flowers to red pigmentation in principally hummingbird-visited flowers [108].
Conclusions
We expect further genomics studies to yield insights into timing and sequence of convergent evolutionary processes, allowing us to address the long-standing question as to whether some species act as 'models' that drive selection for phenotypic similarities in other (mimic) species [6]. As in the honeybee phylogenetic example [71], thousands of loci increase the chance that true phylogenetic signal will overcome stochastic noise from loci with complicated selective histories. The exploration of convergence will provide novel insights into phylogenetic tracking as pollinators and plants co-diversify. This is particularly powerful when comparisons are made with a close relative that does not share the trait of interest (an outgroup) that can provide a basis to study adaptive evolution. Better phylogenetic resolution of deep branching structure in combination with an understanding of the genetic architecture of phenotypic plasticity and divergence will be a powerful tool.
The existence of convergent solutions in floral evolution can involve considerable underlying genetic architectural variation (many routes to a solution), as suggested for replicate adaptive radiations [27]. There can be a surprising amount of genomic variation in natural populations on which natural selection may act [109], and one principal goal must be to quantify such variation for more species and populations. Plant-pollinator interactions are a perfect example of cases in which rapid adaptive radiations in both plant phenotype and pollinator choice set up parallel selective regimes. These are fundamental questions that can be addressed only by the comparison of genomes from parallel radiations of interacting species, which represent evolutionary replicates.
Box 1. DNA barcoding
DNA (or molecular) barcoding can broadly refer to any system whereby genomic data are used to assist in, or as a surrogate for, systematic identification of living organisms. Such methods were first applied in organismal groups that present great difficulties for traditional taxonomic approaches because of their microscopic size, such as Plasmodium parasites [113] or soil nematodes [114]. These cases showed the possibility of using short DNA sequences to essentially substitute for morphological identification, as sequences may be clustered into molecular operational taxonomic units (MOTUs) and used as a fast and practical means to identify strains or assess diversity. DNA barcoding was later proposed as a more formalized system [90] for linking sequence data to species identifications using standardized protocols. Barcoding in this strict sense uses a small number of universal marker genes (mitochondrial cytochrome c oxidase subunit I (COI) for animals, ITS for fungi, rbcL, matK and trnH-psbA or ITS for plants) and a single reference data repository, BOLD [111], in which sequence data are associated with validated taxonomic identifications. Among the advantages of this system are that a DNA sequence from any unknown (obtainable from fragmentary tissue samples, pollen grains, seed fragments, hair, feces, and so on) can be readily assigned to a species or other level of identification by matching against this database.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Elizabeth L Clare, Email: e.clare@qmul.ac.uk.
Florian P Schiestl, Email: florian.schiestl@systbot.uzh.ch.
Andrew R Leitch, Email: a.r.leitch@qmul.ac.uk.
Lars Chittka, Email: l.chittka@qmul.ac.uk.
Acknowledgements
We thank Ian Baldwin, Toby Bradshaw, Teresa Crease, M Brock Fenton, Robin Floyd, Sian Lewis, Randy Mitchell and Nick Waser for comments and literature pointers.
References
- Rosenstiel TN, Shortlidge EE, Melnychenko AN, Pankow JF, Eppley SM. Sex-specific volatile compounds influence microarthropod-mediated fertilization of moss. Nature. 2012;489:431–433. doi: 10.1038/nature11330. [DOI] [PubMed] [Google Scholar]
- Wetschnig W, Depisch B. Pollination biology of Welwitschia mirabilis Hook.f. (Welwitschiaceae, Gnetopsida). Phyton. 1999;39:167–183. [Google Scholar]
- Norstog KJ, Stevenson DW, Niklas KJ. The role of beetles in the pollination of Zamia furfuracea L. fil. (Zamiaceae). Biotropica. 1986;18:300–306. doi: 10.2307/2388573. [DOI] [Google Scholar]
- Kato M, Inoue T, Nagamitsu T. Pollination biology of Gnetum (Gnetaceae) in a lowland mixed dipterocarp forest in Sarawak. Am J Bot. 1995;82:862–868. doi: 10.2307/2445972. [DOI] [Google Scholar]
- Chittka L, Raine NE. Recognition of flowers by pollinators. Curr Opin Plant Biol. 2006;9:428–435. doi: 10.1016/j.pbi.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Schiestl FP, Johnson SD. Pollinator-mediated evolution of floral signals. Trends Ecol Evolution. 2013;28:307–315. doi: 10.1016/j.tree.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Hermann K, Kuhlemeier C. The genetic architecture of natural variation in flower morphology. Curr Opin Plant Biol. 2011;14:60–65. doi: 10.1016/j.pbi.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Schiestl FP, Schlüter PM. Floral isolation, specialized pollination, and pollinator behavior in orchids. Annu Rev Entomol. 2009;54:425–446. doi: 10.1146/annurev.ento.54.110807.090603. [DOI] [PubMed] [Google Scholar]
- Waser NM, Chittka L, Price MV, Williams NM, Ollerton J. Generalization in pollination systems, and why it matters. Ecology. 1996;77:1043–1060. doi: 10.2307/2265575. [DOI] [Google Scholar]
- Dyer AG, Whitney HM, Arnold SEJ, Glover BJ, Chittka L. Mutations perturbing petal cell shape and anthocyanin synthesis influence bumblebee perception of Antirrhinum majus flower colour. Arthropod Plant Interact. 2007;1:45–55. doi: 10.1007/s11829-007-9002-7. [DOI] [Google Scholar]
- Muchhala N. Adaptive trade-off in floral morphology mediates specialization for flowers pollinated by bats and hummingbirds. Am Nat. 2007;169:494–504. doi: 10.1086/512047. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Papaj DR. "X" marks the spot: The possible benefits of nectar guides to bees and plants. Funct Ecol. 2011;25:1293–1301. doi: 10.1111/j.1365-2435.2011.01885.x. [DOI] [Google Scholar]
- Lunau K, Papiorek S, Eltz T, Sazima M. Avoidance of achromatic colours by bees provides a private niche for hummingbirds. J Exp Biol. 2011;214:1607–1612. doi: 10.1242/jeb.052688. [DOI] [PubMed] [Google Scholar]
- Gegear RJ. Multicomponent floral signals elicit selective foraging in bumblebees. Naturwissenschaften. 2005;92:269–271. doi: 10.1007/s00114-005-0621-5. [DOI] [PubMed] [Google Scholar]
- Whitney HM, Kolle M, Andrew P, Chittka L, Steiner U, Glover BJ. Floral iridescence, produced by diffractive optics, acts as a cue for animal pollinators. Science. 2009;323:130–133. doi: 10.1126/science.1166256. [DOI] [PubMed] [Google Scholar]
- Simon R, Holderied MW, Koch CU, Von Helversen O. Floral acoustics: conspicuous echoes of a dish-shaped leaf attract bat pollinators. Science. 2011;333:631–633. doi: 10.1126/science.1204210. [DOI] [PubMed] [Google Scholar]
- Clark D, Whitney H, Sutton G, Robert D. Detection and learning of floral electric fields by bumblebees. Science. 2013;340:66–69. doi: 10.1126/science.1230883. [DOI] [PubMed] [Google Scholar]
- Xu S, Schlüter PM, Grossniklaus U, Schiestl FP. The genetic basis of pollinator adaptation in a sexually deceptive orchid. PLoS Genet. 2012;8:e1002889. doi: 10.1371/journal.pgen.1002889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006;443:931–949. doi: 10.1038/nature05260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D, Baldwin IT. Making sense of nectar scents: the effects of nectar secondary metabolites on floral visitors of Nicotiana attenuata. Plant J. 2006;49:840–854. doi: 10.1111/j.1365-313X.2006.02995.x. [DOI] [PubMed] [Google Scholar]
- Kessler D, Diezel C, Baldwin IT. Changing pollinators as a means of escaping herbivores. Curr Biol. 2010;20:237–242. doi: 10.1016/j.cub.2009.11.071. [DOI] [PubMed] [Google Scholar]
- Chittka L, Döring TF. Are autumn foliage colors red signals to aphids?. PLoS Biol. 2007;5:1640–1644. doi: 10.1371/journal.pbio.0050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessinger C, Rausher MD. Lessons from flower colour evolution on targets of selection. J Exp Bot. 2012;63:5741–5749. doi: 10.1093/jxb/ers267. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Willis JH. Plant speciation. Science. 2007;317:910–914. doi: 10.1126/science.1137729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauw A, Stofberg J, Waterman R. Flies and flowers in Darwins's race. Evolution. 2009;63:268–279. doi: 10.1111/j.1558-5646.2008.00547.x. [DOI] [PubMed] [Google Scholar]
- Strasburg JL, Sherman NA, Wright KM, Moyle LC, Willis JH, Rieseberg LH. What can patterns of differentiation across plant genomes tell us about adaptation and speciation?. Philos Trans R Soc Lond B Biol Sci. 2012;367:364–373. doi: 10.1098/rstb.2011.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmer KR, Meyer A. Adaptation in the age of ecological genomics: insights from parallelism and convergence. Trends Ecol Evol. 2011;26:298–306. doi: 10.1016/j.tree.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Wallace JG, Larsson SJ, Buckler ES. Entering the second century of maize quantitative genetics. Heredity. 2013. doi: 10.1038/hdy.2013.6. [DOI] [PMC free article] [PubMed]
- Weber A, Briggs W, Rucker J, Baltazar M, Sanchez-Gonzales J, Feng P, Buckler ES, Doebley J. The genetic architecture of complex traits in teosinte (Zea mays ssp. parviglumis): new evidence from association mapping. Genetics. 2008;180:1221–1232. doi: 10.1534/genetics.108.090134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancroft I. Association genetics and more from crop transcriptome sequences. Information Systems for Biotechnology News. 2013. http://www.isb.vt.edu/news/2013/Jan/bancroft.pdf
- Hayes B, Lewin H, Goddard M. The future of livestock breeding: genomic selection for efficiency, reduced emissions intensity and adaptation. Trends Genet. 2013;29:206–214. doi: 10.1016/j.tig.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Atwell S, Huang YS, Vilhjálmsson BJ, Willems G, Horton M, Li Y, Meng D, Platt A, Tarone AM, Hu TT, Jiang R, Muliyati NW, Zhang X, Amer MA, Baxter I, Brachi B, Chory J, Dean C, Debieu M, De Meaux J, Ecker JR, Faure N, Kniskern JM, Jones JDG, Michael T, Nemri A, Roux F, Salt DE, Tang C, Todesco M. et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010;465:627–631. doi: 10.1038/nature08800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011;107:1–15. doi: 10.1038/hdy.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Feuillet C, Leach JE, Rogers J, Schnable PS, Eversole K. Crop genome sequencing: lessons and rationales. Trends Plant Sci. 2010;16:77–88. doi: 10.1016/j.tplants.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Edwards D, Batley J. Plant genome sequencing: applications for crop improvement. Plant Biotechnol J. 2010;8:2–9. doi: 10.1111/j.1467-7652.2009.00459.x. [DOI] [PubMed] [Google Scholar]
- Moyle LC. Ecological and evolutionary genomics in the wild tomatoes (Solanum sect. Lycopersicon). Evolution. 2008;62:2995–3013. doi: 10.1111/j.1558-5646.2008.00487.x. [DOI] [PubMed] [Google Scholar]
- Consortium TTG. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucom RS, Chang S-M, Kniskern JM, Rausher MD, Stinchcombe JR. Morning glory as a powerful model in ecological genomics: tracing adaptation through both natural and artificial selection. Heredity. 2011;107:377–385. doi: 10.1038/hdy.2011.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossolini E, Klahre U, Brandenburg A, Reinhardt D, Kuhlemeier C. High resolution linkage maps of the model organism Petunia reveal substantial synteny decay with the related genome of tomato. Genome. 2011;54:327–340. doi: 10.1139/g10-116. [DOI] [PubMed] [Google Scholar]
- Phytozone: Mimulus guttatus v1.1 (Monkey flower). http://www.phytozome.net/mimulus
- Cronk Q, Ojeda I. Bird-pollinated flowers in an evolutionary and molecular context. J Exp Bot. 2008;59:715–727. doi: 10.1093/jxb/ern009. [DOI] [PubMed] [Google Scholar]
- Robinson GE, Hackett KJ, Purcell-Miramontes M, Brown SJ, Evans JD, Goldsmith MR, Lawson D, Okamuro J, Robertson HM, Schneider DJ. Creating a buzz about insect genomes. Science. 2011;331:1386. doi: 10.1126/science.331.6023.1386. [DOI] [PubMed] [Google Scholar]
- The 1KP Project. http://www.onekp.com
- Yuan Y-W, Sagawa JM, Young RC, Christensen BJ, Bradshaw HD. Genetic dissection of a major anthocyanin QTL contributing to pollinator-mediated reproductive isolation between sister species of Mimulus. Genetics. 2013;194:255–263. doi: 10.1534/genetics.112.146852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinal S, Danforth BN. Bees diversified in the age of eudicots. Proc Biol Sci. 2013;280:1471–2954. doi: 10.1098/rspb.2012.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheat CW, Vogel H, Wittstock U, Braby MF, Underwood D, Mitchell-Olds T. The genetic basis of a plant-insect coevolutionary key innovation. Proc Natl Acad Sci USA. 2007;104:20427–20431. doi: 10.1073/pnas.0706229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittall JB, Hodges SA. Pollinator shifts drive increasingly long nectar spurs in columbine flowers. Nature. 2007;447:706–709. doi: 10.1038/nature05857. [DOI] [PubMed] [Google Scholar]
- Yashina S, Gubin S, Maksimovich S, Yashina A, Gakhova E, Gilichinsky D. Regeneration of whole fertile plants from 30,000-y-old fruit tissue buried in Siberian permafrost. Proc Natl Acad Sci USA. 2012;109:4008–4013. doi: 10.1073/pnas.1118386109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D, Gase K, Baldwin IT. Field experiments with transformed plants reveal the sense of floral scents. Science. 2008;321:1200–1202. doi: 10.1126/science.1160072. [DOI] [PubMed] [Google Scholar]
- Sapir Y. Pollination genetics: using molecular genetic underlying floral traits to study pollination ecology in an evolutionary context. Israel J Plant Sci. 2009;57:141–149. doi: 10.1560/IJPS.57.3.141. [DOI] [Google Scholar]
- Darwin C. The Effects of Cross and Self-Fertilization in the Vegetable Kingdom. London: John Murray; 1877. [Google Scholar]
- Bradshaw HD, Wilbert SM, Otto KG, Schemske DW. Genetic mapping of floral traits associated with reproductive isolation in monkeyflowers (Mimulus). Nature. 1995;376:762–765. doi: 10.1038/376762a0. [DOI] [Google Scholar]
- Clegg MT, Durbin ML. Flower color variation: a model for the experimental study of evolution. Proc Natl Acad Sci USA. 2000;97:7016–7023. doi: 10.1073/pnas.97.13.7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen DJ, Giannasi DE, Ennos RA, Clegg MT. Stem color and pleiotropy of genes determining flower color in the common morning glory. J Hered. 1984;75:113–116. [Google Scholar]
- Epperson BK, Clegg MT. Instability at a flower color locus in the morning glory. J Hered. 1987;78:346–352. [Google Scholar]
- Epperson BK, Clegg MT. Unstable white flower colour genes and their derivatives in morning glory. J Hered. 1992;83:405–409. [Google Scholar]
- Schemske DW, Bradshaw HD. Pollinator preference and the evolution of floral traits in monkeyflowers (Mimulus). Proc Natl Acad Sci USA. 1999;96:11910–11915. doi: 10.1073/pnas.96.21.11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudash MR, Hassler C, Stevens PM, Fenster CB. Experimental floral and inflorescence trait manipulations affect pollinator preference and function in a hummingbird-pollinated plant. Am J Bot. 2011;98:275–282. doi: 10.3732/ajb.1000350. [DOI] [PubMed] [Google Scholar]
- Chittka L, Waser NM. Why red flowers are not invisible to bees. Israel J Plant Sci. 1997;45:169–183. doi: 10.1080/07929978.1997.10676682. [DOI] [Google Scholar]
- Bradshaw HD, Schemske DW. Allele substitution at a flower colour locus produces a pollinator shift in monkeyflowers. Nature. 2003;426:176–178. doi: 10.1038/nature02106. [DOI] [PubMed] [Google Scholar]
- Raine N, Ings T, Dornhaus A, Saleh N, Chittka L. Adaptation, genetic drift, pleiotropy, and history in the evolution of bee foraging behaviour. Adv Study Behav. 2006;36:305–354. [Google Scholar]
- Whitney HM, Chittka L, Bruce TJA, Glover BJ. Conical epidermal cells allow bees to grip flowers and increase foraging efficiency. Curr Biol. 2009;19:948–953. doi: 10.1016/j.cub.2009.04.051. [DOI] [PubMed] [Google Scholar]
- Klahre U, Gurba A, Hermann K, Saxenhofer M, Bossolini E, Guerin PM, Kuhlemeier C. Pollinator choice in Petunia depends on two major genetic loci for floral scent production. Curr Biol. 2011;21:730–739. doi: 10.1016/j.cub.2011.03.059. [DOI] [PubMed] [Google Scholar]
- Stuurman J, Hoballah ME, Broger L, Moore J, Basten C, Kuhlemeier C. Dissection of floral pollination syndromes in Petunia. Genetics. 2004;168:1585–1599. doi: 10.1534/genetics.104.031138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner JK. In: The Ecology and Evolution of Flowers. Harder LD, Barrett SCH, editor. Oxford: Oxford University Press; 2006. Ecological genetics of floral evolution. pp. 260–277. [Google Scholar]
- Singaravelan N, Ne'eman G, Inbar M, Izhaki I. Feeding responses of free-flying honeybees to secondary compounds mimicking floral nectars. J Chem Ecol. 2005;31:2791–2804. doi: 10.1007/s10886-005-8394-z. [DOI] [PubMed] [Google Scholar]
- Wright GA, Baker DD, Palmer MJ, Stabler D, Mustard JA, Power EF, Borland AM, Stevenson PC. Caffeine in floral nectar enhances a pollinator's memory of reward. Science. 2013;339:1202–1204. doi: 10.1126/science.1228806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Brown WM. Polymorphisms in mitochondrial DNA of European and Africanized honeybees (Apis mellifera). Experientia. 1988;44:257–260. doi: 10.1007/BF01941730. [DOI] [PubMed] [Google Scholar]
- Savard J, Tautz D, Richards S, Weinstock GM, Gibbs RA, Werren JH, Tettelin H, Lercher MJ. Phylogenomic analysis reveals bees and wasps (Hymenoptera) at the base of the radiation of Holometabolous insects. Genome Res. 2006;16:1334–1338. doi: 10.1101/gr.5204306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J. Hybridization as an invasion of the genome. Trends Ecol Evol. 2005;20:229–237. doi: 10.1016/j.tree.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, Soltis DE, Clifton SW, Schlarbaum SE, Schuster SC, Ma H, Leebens-Mack J, dePamphilis CW. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473:97–100. doi: 10.1038/nature09916. [DOI] [PubMed] [Google Scholar]
- Leitch AR, Leitch IJ. Genomic plasticity and the diversity of polyploid plants. Science. 2008;320:481–483. doi: 10.1126/science.1153585. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Albert VA, Leebens-Mack J, Bell CD, Paterson AH, Zheng C, Sankoff D, Depamphilis CW, Wall PK, Soltis PS. Polyploidy and angiosperm diversification. Am J Bot. 2009;96:336–348. doi: 10.3732/ajb.0800079. [DOI] [PubMed] [Google Scholar]
- Wood TE, Takebayashi N, Barker MS, Mayrose I, Greenspoon PB, Rieseberg LH. The frequency of polyploid speciation in vascular plants. Proc Natl Acad Sci USA. 2009;106:13875–13879. doi: 10.1073/pnas.0811575106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggs RJA, Soltis PS, Mavrodiev E V, Symonds VV, Soltis DE. Does phylogenetic distance between parental genomes govern the success of polyploids?. Castanea. 2008;73:74–93. doi: 10.2179/0008-7475(2008)73[74:DPDBPG]2.0.CO;2. [DOI] [Google Scholar]
- Paun O, Forest F, Fay MF, Chase MW. Hybrid speciation in angiosperms: parental divergence drives ploidy. New Phytol. 2009;182:507–518. doi: 10.1111/j.1469-8137.2009.02767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover CE, Gallagher JP, Szadkowski EP, Yoo MJ, Flagel LE, Wendel JF. Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol. 2012;196:966–971. doi: 10.1111/j.1469-8137.2012.04365.x. [DOI] [PubMed] [Google Scholar]
- Leitch AR, Leitch IJ. Ecological and genetic factors linked to contrasting genome dynamics in seed plants. New Phytol. 2012;194:629–646. doi: 10.1111/j.1469-8137.2012.04105.x. [DOI] [PubMed] [Google Scholar]
- Cooley AM, Modliszewski JL, Rommel ML, Willis JH. Gene duplication in Mimulus underlies parallel floral evolution via independent trans-regulatory changes. Curr Biol. 2011;21:700–704. doi: 10.1016/j.cub.2011.03.028. [DOI] [PubMed] [Google Scholar]
- Karrenberg S, Lexer C, Rieseberg LH. Reconstructing the history of selection during homoploid hybrid speciation. Am Nat. 2007;169:725–737. doi: 10.1086/516758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu Rev Plant Biol. 2007;58:377–406. doi: 10.1146/annurev.arplant.58.032806.103835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubourg S, Lecharny A, Bohlmann J. Genomic analysis of the terpenoid synthase (AtTPS) gene family of Arabidopsis thaliana. Mol Genet Genom. 2002;267:730–745. doi: 10.1007/s00438-002-0709-y. [DOI] [PubMed] [Google Scholar]
- Dudareva N, Klempien A, Muhlemann JK, Kaplan I. Biosynthesis, function and metabolic engineering of plant volatile organic compounds. New Phytol. 2013;198:16–32. doi: 10.1111/nph.12145. [DOI] [PubMed] [Google Scholar]
- Schlüter PM, Xu S, Gagliardini V, Whittle E, Shanklin J, Grossniklaus U, Schiestl FP. Stearoyl-acyl carrier protein desaturases are associated with floral isolation in sexually deceptive orchids. Proc Natl Acad Sci USA. 2011;108:5696–5701. doi: 10.1073/pnas.1013313108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta AK, Akhtar TA, Widmer A, Pichersky E, Schiestl FP. Identification of white campion (Silene latifolia) guaiacol O-methyltransferase involved in the biosynthesis of veratrole, a key volatile for pollinator attraction. BMC Plant Biol. 2012;12:158. doi: 10.1186/1471-2229-12-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symondson WOC. Molecular identification of prey in predator diets. Mol Ecol. 2002;11:627–641. doi: 10.1046/j.1365-294X.2002.01471.x. [DOI] [PubMed] [Google Scholar]
- Pompanon F, Deagle BE, Symondson WOC, Brown DS, Jarman SN, Taberlet P. Who is eating what: diet assessment using next generation sequencing. Mol Ecol. 2012;21:1931–1950. doi: 10.1111/j.1365-294X.2011.05403.x. [DOI] [PubMed] [Google Scholar]
- Hebert PDN, Cywinska A, Ball SL, deWaard JR. Biological identifications through DNA barcodes. Proc Biol Sci. 2003;270:313–321. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnasingham S, Hebert PDN. BOLD The barcode of life data systems http://www.barcodinglife.org. Mol Ecol Notes. 2007;7:355–364. doi: 10.1111/j.1471-8286.2007.01678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd RM, Clare EL. Food forensics: New technology uses DNA to reveal what's on the menu for bats. BATS. 2009;27:6–8. [Google Scholar]
- McCann K. Protecting biostructure. Nature. 2007;446:29. doi: 10.1038/446029a. [DOI] [PubMed] [Google Scholar]
- CBOL plant working group. A DNA barcode for land plants. Proc Natl Acad Sci USA. 2009;106:12794–12797. doi: 10.1073/pnas.0905845106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EE, Sidhu CS, LeVan KE, Holway DA. Pollen foraging behaviour of solitary Hawaiian bees revealed through molecular pollen analysis. Mol Ecol. 2010;19:4823–4829. doi: 10.1111/j.1365-294X.2010.04849.x. [DOI] [PubMed] [Google Scholar]
- Valentini A, Miquel C, Nawaz MA, Bellemain E, Coissac E, Pompanon F, Gielly L, Cruaud C, Nascetti G, Wincker P, Swenson JE, Taberlet P. New perspectives in diet analysis based on DNA barcoding and parallel pyrosequencing: the trnL approach. Mol Ecol Resour. 2009;9:51–60. doi: 10.1111/j.1755-0998.2008.02352.x. [DOI] [PubMed] [Google Scholar]
- Soininen EM, Valentini A, Coissac E, Miquel C, Gielly L, Brochmann C, Brysting AK, Sønstebø JH, Ims RA, Yoccoz NG, Taberlet P. Analysing diet of small herbivores: the efficiency of DNA barcoding coupled with high-throughput pyrosequencing for deciphering the composition of complex plant mixtures. Front Zool. 2009;6:16. doi: 10.1186/1742-9994-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Helversen O, Winkler L, Bestmann HJ. Sulphur-containing "perfumes'' attract flower-visiting bats. J Comp Physiol A. 2000;186:143–153. doi: 10.1007/s003590050014. [DOI] [PubMed] [Google Scholar]
- Hayden S, Bekaert M, Crider TA, Mariani S, Murphy WJ, Teeling EC. Ecological adaptation determines functional mammalian olfactory subgenomes. Genome Res. 2010;20:1–9. doi: 10.1101/gr.099416.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapley J, Reger J, Feulner PGD, Smadja C, Galindo J, Ekblom R, Bennison C, Ball AD, Beckerman AP, Slate J. Adaptation genomics: the next generation. Trends Ecol Evol. 2010;25:705–712. doi: 10.1016/j.tree.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Hodges SA, Derieg NJ. Adaptive radiations: From field to genomic studies. Proc Natl Acad Sci USA. 2009;106:9947–9954. doi: 10.1073/pnas.0901594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C-C, Chung Y-L, Chen T-C, Lee Y-L, Kuo Y-T, Tsai W-C, Hsiao Y-Y, Chen Y-W, Wu W-L, Chen H-H. An overview of the Phalaenopsis orchid genome through BAC end sequence analysis. BMC Plant Biol. 2011;11:1–11. doi: 10.1186/1471-2229-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombarely A, Rosli HG, Vrebalov J, Moffett P, Mueller LA, Martin GB. A draft genome sequence of Nicotiana benthamiana to enhance molecular plant-microbe biology research. Mol Plant Microbe Interact. 2012;25:1523–1530. doi: 10.1094/MPMI-06-12-0148-TA. [DOI] [PubMed] [Google Scholar]
- Plant DNA C-values database. http://data.kew.org/cvalues/
- mimulusevolution.org. http://www.mimulusevolution.org
- Wu CA, Lowry DB, Cooley AM, Wright KM, Lee YW, Willis JH. Mimulus is an emerging model system for the integration of ecological and genomic studies. Heredity. 2008;100:220–230. doi: 10.1038/sj.hdy.6801018. [DOI] [PubMed] [Google Scholar]
- Ollerton J, Cranmer L, Stelzer RJ, Sullivan S, Chittka L. Bird pollination of Canary Island endemic plants. Naturwissenschaften. 2009;96:221–232. doi: 10.1007/s00114-008-0467-8. [DOI] [PubMed] [Google Scholar]
- Des Marais DL, Rausher MD. Parallel evolution at multiple levels in the origin of hummingbird pollinated flowers in Ipomoea. Evolution. 2010;64:2044–2054. doi: 10.1111/j.1558-5646.2010.00972.x. [DOI] [PubMed] [Google Scholar]
- Caruso CM. The ecological genetics of floral traits. Heredity. 2006;97:86–87. doi: 10.1038/sj.hdy.6800853. [DOI] [PubMed] [Google Scholar]
- Anssour S, Krugel T, Sharbel T, Saluz H, Bonaventure G, Baldwin I. Phenotypic, genetic and genomic consequences of natural and synthetic polyploidization of Nicotiana attenuata and Nicotiana obtusifolia. Ann Bot. 2009;103:1207–1217. doi: 10.1093/aob/mcp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLDSYSTEMS. http://boldsystems.org/
- NCBI Taxonomy. http://www.ncbi.nlm.nih.gov/
- Arnot DE, Roper C, Bayoumi RAL. Digital codes from hypervariable tandemly repeated DNA sequences in the Plasmodium falciparum circumsporozoite gene can genetically barcode isolates. Mol Biochem Parasitol. 1993;61:15–24. doi: 10.1016/0166-6851(93)90154-P. [DOI] [PubMed] [Google Scholar]
- Floyd R, Abebe E, Papert A, Blaxter M. Molecular barcodes for soil nematode identification. Mol Ecol. 2002;11:839–850. doi: 10.1046/j.1365-294X.2002.01485.x. [DOI] [PubMed] [Google Scholar]