

Abstract
Three bis-benzoxazine monomers based on the aniline derivatives of bisphenol A (BA-a), bisphenol F (BF-a), and 3,3′-thiodiphenol (BT-a) are examined using a variety of spectroscopic, chromatographic, and thermomechanical techniques. The effect on the polymerization of the monomers is compared using two common compounds, 3,3′-thiodiphenol (TDP) and 3,3′-thiodipropionic acid (TDA), at a variety of loadings. It is found that the diacid has a greater effect on reducing the onset of polymerization and increasing cross-link density and Tg for a given benzoxazine. However, the addition of >5 wt % of the diacid had a detrimental effect on the cross-link density, Tg, and thermal stability of the polymer. The kinetics of the polymerization of BA-a were found to be well described using an autocatalytic model for which values of n = 1.64 and m = 2.31 were obtained for the early and later stages of reaction (activation energy = 81 kJ/mol). Following recrystallization the same monomer yielded values n = 1.89, m = 0.89, and Ea = 94 kJ/mol (confirming the influence of higher oligomers on reactivity). The choice of additive (in particular the magnitude of its pKa) appears to influence the nature of the network formation from a linear toward a more clusterlike growth mechanism.
Introduction
Conventional phenolics are cross-linked products of low molecular weight precursors, typically formed through a condensation reaction. The versatility of their structures and the fact that they display desirable properties such as good heat resistance, flame-retardant properties, low dielectric constants, and the production of these materials is relatively inexpensive.1 However, there are some shortcomings associated with these materials in that they have poor toughness properties, they have a poor shelf life, and the process of polymerization of these materials produces byproducts and requires, in many cases, the use of a strong acid or base catalyst to effect cure.2 In the search for higher performance replacements for phenolic resins many materials have received attention, and published reports of the preparation of aromatic oxazines, or benzoxazines, date back some 60 years;3 although commercial exploitation of the corresponding polymers has only come about relatively recently, they are now receiving a great deal of academic and industrial interest.4
Poly(bis-benzoxazine)s (sometimes simply referred to as polybenzoxazines) are a family of thermosetting polymers that are made up through step growth ring-opening polyaddition from bis-benzoxazine monomers (Figure 1), which are in turn the products of the Mannich reaction between a bis-phenol, formaldehyde, and a primary amine.5 The monomer–oligomer ratio in the yield can also be influenced by using an excess of formaldehyde and amine during the synthesis, causing the products to form via a different mechanism and resulting in a greater proportion of monomer in the product.6 This, in turn, affects the properties of the resin before, during, and after cure (the presence of oligomers bearing hydroxyl groups in the chain is known to enhance the reactivity of the benzoxazine).
Figure 1.

Polymerization of bisbenzoxazines through ring-opening and cross-linking and representative network.
Commercial benzoxazines are currently being evaluated for use in the aerospace industry (in which they would replace or augment phenolic polymers in secondary applications such as interior paneling). Polybenzoxazines appear to incorporate the best properties from conventional phenolics and may find application in a number of their traditional niches, while improving on shelf life and offering the potential for greater toughness properties through their greater molecular flexibility; the relative cheapness of the monomer is also an important factor influencing their adoption. However, while polybenzoxazines display many benefits over conventional phenolics, the relatively low fracture toughness that is achieved by cured polymers (a KIC value of ca. 0.51–0.54 MPa·m0.5 is typical,7 although later linear polymers achieve higher KIC values) and the comparatively poor reactivity—Araldite MT35600 commences its reaction at 180–200 °C (DSC, 10 K/min) in the absence of additive (and 160 °C with the addition of 5 wt % 3,3′-thiodiphenol)—are still problems in advanced aerospace applications when compared with competitor resins.
Unlike many other commercial thermosetting resins, which evolve condensation products such as water or ammonia, benzoxazine monomers react relatively cleanly to form a polymer with few reaction byproducts,8 and although a number of studies have been published, the exact manner of the polymerization reaction to form a network has not been fully elucidated. In our ongoing research programmes, the ultimate aim is to examine and increase our understanding of the influence of the benzoxazine monomer structure on reactivity, fracture toughness, and thermal stability. In this paper we present the results of a study into the influence of the nature of the additive on the reaction mechanism. Significant differences in the mechanism of the network growth are observed via Raman spectroscopy when different initiators are employed, leading to significant differences in the thermal and mechanical properties that are observed.
Results and Discussion
Determination of Monomer Purity
The monomers were characterized fully using both spectroscopic and chromatographic techniques, as it is well-known that monomer purity (in particular, the oligomer content9) can have a significant effect on both cure mechanism and kinetics. Analysis using high performance liquid chromatography (HPLC) suggested that BA-a was markedly more pure (ca. 95%) than BF-a (ca. 80% and a mixture of isomers), but this was misleading. Subsequent discussion with the materials supplier confirmed that bis-benzoxazine monomers are prone to undergo degradation via ring-opening on the surface of the column during reverse-phase analysis, and so these data cannot be used to determine absolute purity. 1H and 13C NMR spectroscopic analyses were performed in parallel on the monomers (e.g., using DEPT-135, HSQC, and HMBC pulse sequences), and these supported the observation that BA-a was the more pure, containing a lower concentration of unreacted starting materials, but not disproportionately so. The NMR spectroscopic data are deposited as Supporting Information.
Thermal Behavior of Benzoxazine Monomers Determined Using DSC
The DSC data for the “uninitiated” polymerizations are displayed in Figure 2 and given in Table 1, from which it can be seen that BA-a offers a potentially relatively wide processing window (i.e., the gap between Tm = 43 °C and the onset of polymerization ca. 200 °C).
Figure 2.

Scanning DSC data for the thermally initiated polymerization of the benzoxazine monomers (10 K/min, nitrogen): (a) BA-a recrystallized, (b) BA-a as received, (c) BF-a as received, and (d) BT-a as received.
Table 1. Thermal Events for Bis-Benzoxazine Monomers As Determined by DSCa.
| ΔHp |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| monomer | additive | [additive] (mol %) | Tg1 (°C) | Top (°C) | Tmax (°C) | Tep (°C) | (J g–1) | (kJ mol–1/Bz) | Tg2 (°C) |
| BA-a | 39 | 203 | 240 | 283 | 299 | 69.2 | 157 | ||
| BA-ab | 106 | 218 | 257 | 292 | 304 | 70.3 | 162 | ||
| BA-a | TDP | 1 | 37 | 196 | 243 | 293 | 311 | 71.9 | 154 |
| BA-a | TDP | 2.5 | 38 | 186 | 233 | 295 | 318 | 73.6 | 160 |
| BA-a | TDP | 5 | 37 | 187 | 235 | 295 | 318 | 73.6 | 153 |
| BA-a | TDP | 10 | 36 | 178 | 226 | 295 | 324 | 74.9 | 153 |
| BA-a | TDP | 12 | 37 | 172 | 222 | 298 | 331 | 76.6 | 154 |
| BA-a | TDP | 25 | 38 | 154 | 214 | 294 | 322 | 74.5 | 155 |
| BA-a | TDA | 1 | 37 | 186 | 234 | 296 | 241 | 55.7 | 164 |
| BA-a | TDA | 3.1 | 37 | 172 | 225 | 298 | 358 | 82.8 | 164 |
| BA-a | TDA | 5 | 37 | 157 | 216 | 295 | 324 | 74.9 | 168 |
| BA-a | TDA | 10 | 38 | 111 | 196 | 274 | 365 | 84.4 | 174 |
| BA-a | TDA | 15 | 38 | 145 | 198 | 293 | 388 | 89.7 | 184 |
| BA-a | TDA | 31 | 39 | 133 | 188 | 268 | 292 | 67.5 | 187 |
| BF-a | 36.2 | 170 | 232 | 292 | 311 | 67.4 | 174 | ||
| BF-a | TDP | 12 | 34.6 | 132 | 218 | 295 | 300 | 65.2 | 164 |
| BF-a | TDA | 15 | 35.0 | 130 | 196 | 287 | 294 | 63.9 | 199 |
| BT-a | 173 | 221 | 278 | 350 | 79.2 | 173 | |||
| BT-a | TDP | 12 | 144 | 209 | 278 | 316 | 71.5 | 196 | |
| BT-a | TDA | 15 | 146 | 191 | 269 | 315 | 71.3 | 180 | |
Tg1 = possible glass transition or melting transition of monomer determined using a heating rate of 10 K min–1; Top = observed onset of polymerization; Tmax = temperature of exothermic peak maximum; Tep = observed end of polymerization; ΔHp = enthalpy of exothermic peak (both as J/g of monomer and kJ/mol–1 of benzoxazine ring); Tg2 = glass transition temperature of polymer determined using a heating rate of 10 K min–1; TDP = 3,3′-thiodiphenol; TDA = 3,3′-thiodipropionic acid.
Recrystallized.
The thermal data obtained in a previous paper10 showed that the onset of the thermal polymerization was lowest for the monomers formed from aniline and incorporating TDP and 4,4-biphenol (with relatively broad exotherms spanning 160–270 °C, Tmax 180 and 200 °C, respectively), with more flexible bridging groups or conjugated ring moieties. In the current work, the monomer based on 3,3′-thiodiphenol (BT-a) is also the most reactive of those studied. Narrower, more symmetrical exotherms were previously recorded at higher temperatures (Tmax = 215 and 225 °C) for the monomers based on dihydroxyphenyl ether and bisphenol A, respectively, with more flexible bridging groups and/or electron donating moieties. Conversely, the highest onset temperature (180 °C) was recorded for the monomer based on bisphenol AF6, with a highly electronegative bridge (i.e., a hexafluoroisopropyl moiety). It was notable that the enthalpies recorded for the polymerizations of the majority of the monomers were practically the same (147.9 ± 7.4 or 74 kJ/mol Bz ring) and compared favorably with literature values (of 144.7 or 72.4 kJ/mol Bz ring11).
The generation of kinetic information from thermal data in thermosetting polymers was reviewed extensively in the context of epoxy resins by Barton.12 Of these, the Kissinger method13 relates the peak maximum temperature, Tmax, obtained using dynamic DSC and the activation energy (Ea) for data obtained at different heating rates (Figure 3).
| 1 |
where rmax is the rate of reaction at the peak maximum derived from DSC and αmax is the conversion at the peak maximum.
Figure 3.

Scanning DSC data under nitrogen for BA-a at a variety of heating rates.
Using this method, the kinetics of the polymerizations were modeled using dynamic DSC at a variety of heating rates, from which an activation energy of 81 kJ mol–1 was calculated for the monomer BA-a (as received), which correlated well with literature values (Table 2). The data are presented for the “as-received” BA-a sample (Figure 4a) and following recrystallization (Figure 4b).
Table 2. Arrhenius Parameters for BA-a Monomer Showing the Effect of Purificationa.
| BA-a as obtained |
BA-a
recrystallized |
|||||
|---|---|---|---|---|---|---|
| heating rate (K min–1) | Ea (kJ mol–1) | n | m | Ea (kJ mol–1) | n | m |
| 5 | 81.4 | 3.04 | 1.30 | 93.7 | 1.95 | 1.17 |
| 8 | 2.74 | 1.30 | 1.73 | 0.85 | ||
| 10 | 2.31 | 1.64 | 1.90 | 0.90 | ||
| 12 | 2.23 | 1.30 | 1.69 | 0.89 | ||
| 15 | 2.14 | 1.39 | 1.75 | 0.80 | ||
Activation energy Ea was calculated using the Kissinger method. An autocatalytic model was used to calculate values for n and m.
Figure 4.

Kissinger (◆) and Ozawa (■) plots for the polymerization of (a) BA-a and (b) recrystallized BA-a.
However, the DSC data show the reaction mechanism is more complex with the reaction evolving from being chemically controlled to physically controlled as the viscosity increases throughout the reaction. Consequently, the kinetics were then determined using the following equation, which is better suited to an autocatalytic system than an nth-order model:
| 2 |
where α is the degree of conversion, β the heating rate, A the pre-exponential factor, R the gas constant, 8.314 J/(mol K), n and m the orders of reaction in the initial and later stages of reaction, respectively.
This equation was solved by multiple linear regression to yield values of A, m, and n using the activation energy previously determined; these values were found to differ significantly from those found in the literature. For example, Jubsilp et al.14 achieved average values of 1.7 for n and 0.8 for m for a BA-a benzoxazine and Ishida and Rodriguez11 achieved average values of 1.4 for n and 1.0 for m. Following recrystallization from ethanol, BA-a was reanalyzed using DSC, from which it was observed that the melting temperature, polymerization onset temperature, and peak maximum were all raised by some 15 K, suggesting the removal of one or more impurities (Figure 2a,b). When the same kinetic analysis was repeated on the recrystallized monomer BA-a, average values of 1.89 and 0.89 for n and m, respectively, were obtained which were more in line with the literature values for a 10 K/min heating rate. Therefore, commercial BA-a contains components that are affecting the cure kinetics of the benzoxazine, in the absence of the additive. This is supported by the value of Ea for recrystallized BA-a (94 kJ mol–1), which compares favorably with reported values (102–116 kJ mol–1) for an analogous material prepared in the laboratory.11 It is known that commercial benzoxazine monomers may be held at elevated temperature to yield soluble, low molecular weight oligomers,9 in which the presence of hydroxyl groups increase the reactivity. These data are consistent with the removal of higher molecular weight oligomers.
Examining the Effects of Additives Using DSC
Although bis-benzoxazines will polymerize through opening of the heterocyclic ring when subjected to heat, the use of initiators or catalysts is preferred since highly pure monomers are comparatively unreactive (onset temperature of 225 °C, at 10 K/min DSC scan, in the absence of deliberate catalysis/initiation). Samples were also analyzed using DSC in order to establish the relative effects of the additives, and the thermal data for the “uninitiated” polymerization of BA-a are displayed in Figure 5 along with the corresponding mixtures containing each of the additives (10 mol %).
Figure 5.

DSC data (10 K/min, under nitrogen) for BA-a (a) in the absence of catalyst, with 10 mol % TDP and with 10 mol % TDA.
The DSC thermograms for the acid-initiated reactions show several almost sequential thermal events, unlike the uninitiated system (which yields a near Gaussian exotherm with a high temperature tail covering several almost simultaneous events); this becomes more apparent when the heating rate is further reduced to 5 K/min (Figure 3).
The initial melt endotherm is visible at ca. 35–45 °C along with a more diffuse endotherm associated with the fusion of the additive in the mixture containing TDP. The monomer is most sensitive to the addition of the diacid (TDA), but although the onset of the polymerization and the peak maximum are moved significantly to a lower temperature, the end point of the exotherm does not move along with this, leading to a broad exotherm. The additive has the greatest impact on the initial stages of reaction (ring opening), but the later processes associated with cross-linking are not affected to the same degree. Speed of cure is a concern with polybenzoxazines, which are sluggish to react when highly pure, and this may hinder the more widespread use of these materials despite the interest in their thermomechanical and physical properties (especially FST).
Using ModelMaker (version 3.0.4, Cherwell Scientific Ltd.), peaks were fitted to the plots and activation energy determined to compare to those obtained using traditional methods. The curves chosen for peak fitting were 15 mol % TDA and 12 mol % TDP, as they gave the best improvement in temperature of onset, while still being able to discern the different reaction peaks. The best fit was obtained for the initiated systems when three peaks were used, especially in the TDP samples where the reactions are much more drawn out, but a fourth peak needed to be included in the first peak to prevent the fit distorting in the TDP plots, suggesting there may be more reactions occurring than are being observed. The 5 K/min experiments show that the initiated systems give very different models. In the uninitiated system the best peak fit was found using two peaks, which is consistent with two processes: ring-opening and bridge forming reactions. In the TDP plot (Figure 6) the initial stages of reaction are being drawn out, so that several consecutive reactions occur: i.e., the first (and smaller second) peak is associated with ring-opening; the third peak, which accounts for the bulk of the reaction, is associated with bridge forming, and the last peak is structural rearrangement or possible beginnings of degradation. Conversely, the TDA plot shows a more simultaneous reaction occurring where several reactions are occurring at once. The results of peak deconvoluting are debatable, but the models presented here represent the best fitting models (using regression coefficient) with the smallest number of peaks.
Figure 6.

Peak fitting for DSC data (5 K/min, under nitrogen) for BA-a (a) in the absence of catalyst and (b) with 12 mol % TDP and (c) with 10 mol % TDA.
Using the Kissinger method (1), the activation energy of for each peak was calculated (Table 3), and the activation energies for the second and third peaks in TDP are similar to those calculated for uninitiated BA-a. The activation energy for the second peak of TDA is slightly lower but of a similar magnitude, suggesting a similar reaction.
Table 3. Activation Energies for BA-a Monomer Using ModelMaker.
| activation
energy (kJ/mol) |
|||||
|---|---|---|---|---|---|
| initiator | initiator (mol %) | peak 1 | peak 2 | peak 3 | peak 4 |
| TDP | 12 | 61.68 | 83.67 | 83.15 | 149.16 |
| TDA | 15 | 59.34 | 77.23 | 90.27 | N/A |
| Kissinger 81.37 | Ozawa 87.65 | ||||
The activation energy of the first peak is some 20 kJ mol–1 lower than the subsequent peak in both instances; this is due to the initiator instigating the ring-opening reaction. It is apparent that the degree of catalysis and the nature of the catalyst have an influence on the progress of the reaction, although this is more marked in the case of the diacid (e.g., the reduction is visible in both the onset temperature and the peak maximum of the polymerization exotherm when compared with the uncatalyzed monomer and the formulations containing the diphenol) (Figure 7). Furthermore, the glass transition temperatures of the TDA-cured polybenzoxazines (Tg2, recorded during the DSC rescan experiment) are significantly higher (and proportionate to additive concentration), where the values are unchanged for the TDP-cured analogues. This supports the suggestion that the additives are giving rise to different polymerization mechanisms, since the cross-link density does not alter significantly for the TDP-initiated blends.
Figure 7.

Plots of glass transition temperature, Tmax, and ΔH (J/g) for BA-a initiated with (a) TDA and (b) TDP as a function of additive concentration.
Thermomechanical Characterization
Data from dynamic mechanical thermal analysis were acquired to explore the cross-link densities generated in the different network structures. Representative storage modulus curves for PBA-a when initiated with TDP and TDA are overlaid for comparison in Figure 8; similar trends in the data were observed for the other polybenzoxazines. The glass transition (α-transition) is clearly visible in the loss modulus data at ca. 125–225 °C (Tmax = 175 °C at 2 K/min) with a drop in the storage modulus of around 3500 MPa; a β-transition is visible between 50 and 100 °C. The polybenzoxazine that had been cured using a 5 wt % addition of TDP differs in several important respects: (i) the Tg occurs at slightly lower temperatures as the TDP content increases, (ii) the magnitude of the tan δ peak increases relative to the loss modulus peak as TDP (5 wt %) is added, but decreases slightly at 10 wt %; (iii) the magnitude of the β-transition increases as with TDP concentration. These observations (which are similar for the other polybenzoxazines studied) indicate the emergence of a different cured network with the use of the additive; the effect on modulus and damping behavior (Figure 9) is clearly demonstrated.
Figure 8.

Dynamic viscoelasticity data for thermally cured polybenzoxazines: (a) PBA-a, (b) PBA-a with 5 wt % TDP, (c) PBA-a with 10 wt % TDP, and (d) PBA-a with 10 wt % (TDA). N.B.: scale relates to plot (a) other data offset progressively downward by 1000 MPa for clarity.
Figure 9.

Dynamic viscoelasticity data (tan δ) for thermally cured polybenzoxazines: (a) PBA-a), (b) PBA-a with 5 wt % TDP, (c) PBA-a with 10 wt % TDP, and (d) PBA-a with 10 wt % (TDA). N.B.: scale relates to plot (a) other data offset progressively downward by 1.0 for clarity.
The breadth of the tan δ peaks show differences in the damping behavior of the polybenzoxazines, and the peak widths represent the temperature ranges over which the glass transition temperatures occur. Thus, the broadest tan δ peak (BA-a) can be attributed to more heterogeneous networks containing both highly and less densely cross-linked regions.15 This, in turn, results in a broad distribution of molecular mobilities or relaxation times (this is seen to be broadest for the “uninitiated” PBA-a). The cross-link density (ν) for each polybenzoxazine was calculated from the DMTA data using eq 3:16
| 3 |
where φ is taken as unity and Ge is the storage modulus strictly from a sample at equilibrium, but is taken at Te, where Te = Tg + 50 K.
This equation is technically most appropriate for lightly cross-linked materials so it should only be used as a comparison between similar materials. The cross-link density values are shown for the benzoxazines following polymerization in both the presence and absence of the additive (Table 4). Examination of the data for the neat polybenzoxazines indicates that the three commercial monomers develop very similar cross-link densities upon thermal cure, which is consistent with the structures differing only by the nature of the central bridge: isopropyl and methylene; the sulfur-bridged monomer BT-a develops a higher cross-link density. For comparison, values of cross-link density of (1.1–10.5) × 10–3 mol cm–3 have been reported depending on the type of benzoxazine and the functionality.17−20 Typically, BA-a is quoted as having a cross-link density of 1.1 × 10–3 mol dm–3, and the higher value found for the BA-a may be due to the higher oligomers or impurities as evidenced by the discrepancies in the reaction kinetics. However, it was not possible to obtain enough material to carry out DMTA on the polymer arising from the recrystallized monomer. Allen and Ishida16 obtained a cross-link density of 4–7 mol m–3 for polybenzoxazines based on long chain aliphatic diamines. Of the two additives employed in this work, TDA has the greatest impact on the cross-link density according to the DMTA data; e.g., it leads to an increase in the Tg of some 20 K (PBA-a), 30 K (PBF-a), and 20 K (PBT-a).
Table 4. Effect of Additive on Cross-Link Density of Polybenzoxazinesa.
| cross-link density (×10–3 mol cm–3) |
||||
|---|---|---|---|---|
| benzoxazine | pure resin | 5% TDP | 10% TDP | 5% TDA |
| PBA-a | 5.81 | 7.71 | 4.41 | 8.45 |
| PBF-a | 5.98 | 10.71 | 7.33 | |
| PBT-a | 8.51 | 12.80 | 7.40 | |
Values calculated from duplicate DMTA measurements.
Thermal Stability of Polybenzoxazine Degradation
Polybenzoxazine degradation can be broken down into three events,21 although the processes are not entirely discrete as evidenced by the breadth of the peaks. There is a lower temperature event (200–220 °C) for the initiated PBA-a systems that is much higher for the PBA-a alone (around 290 °C). This first degradation stage is associated with the breakdown of the Mannich bridge and loss of the pendant amino groups and is most prominent for the formulations containing the lowest concentrations of initiators, suggesting there are more pendant moieties in these systems. This might be due to the initiator preventing reaction at the arylamine ring or affecting the chain growth, although this growth in weight loss may also be due to loss of initiator from the polymer matrix. The second degradation peak is associated with the breakdown of the polymer chain, leading to release of substituted phenols, and is the largest contributor to the thermal degradation process. The observed rise in the cross-link density and Tg is also reflected in the measurement of initial thermal stability (Table 5) of the cured (but uninitiated) polybenzoxazines; e.g., the T5% rises from 258 °C (“uninitiated” PBA-a) to 294 °C (PBA-a + 5% TDA) and from 263 °C (“uninitiated” PBF-a) to 290 °C (PBF-a + 5% TDA). Although PBT-a is initially the more stable polymer, the mass loss tends to reverse beyond ca. 30%, suggesting a different degradation mechanism. Work is currently underway within our group to examine this aspect and will be reported in a future paper. The presence of 10 wt % TDA reduces thermal stability and leads to a more rapid loss in mass but provides an increase in Tg. The char yields are ordered in the following way: PBT-a > PBF-a ≫ PBA-a with the polymers containing the highest alkyl component providing the lowest char. This order of stability was confirmed by visual observations made on thermally cured and deliberately charred samples.22
Table 5. Effect of Additive on Thermal Stability and Glass Transition Temperatures of Cured Polybenzoxazinesa.
| temp (°C)
for a given mass loss (%) |
|||||
|---|---|---|---|---|---|
| sample | initiator (mol %) | 5% | 10% | char yield (%) | Tg (°C) |
| BA-a | 240 | 305 | 25 | 150 | |
| TDP (12) | 252 | 308 | 31 | 154 | |
| TDP (25) | 264 | 305 | 28 | 151 | |
| TDA (15) | 200 | 246 | 37 | 184 | |
| TDA (30) | 254 | 306 | 26 | 186 | |
| BF-a | 301 | 353 | 48 | 165 | |
| TDP (12) | 214 | 312 | 48 | 164 | |
| TDP (25) | 266 | 324 | 45 | 158 | |
| TDA (15) | 265 | 338 | 42 | 199 | |
| TDA (30) | 269 | 336 | 400 | 204 | |
| BT-a | 313 | 334 | 57 | 160 | |
| TDP (12) | 381 | 311 | 54 | 195 | |
| TDP (25) | 268 | 302 | 52 | 178 | |
| TDA (15) | 280 | 316 | 52 | 179 | |
| TDA (30) | 270 | 310 | 47 | 183 | |
All samples analyzed under nitrogen at a heating rate of 10 K/min. Char yield determined as residue remaining at 800 °C.
The introduction of TDP is beneficial at the 5 wt % level of incorporation: the initial stability (T5%) is raised for both PBA-a and PBF-a but has no apparent effect on PBT-a. At higher mass losses (e.g., 30%), the stability of both PBF-a and PBT-a is elevated, but the effect on PBA-a is to reduce stability. TDP is less influential at 10 wt % (where it leads to a reduction in both cross-link density and thermal stability for all the polymers, accompanied by the release of volatiles during cure). TDA can be incorporated at 5 and 10 wt % and leads to enhanced thermal stability in PBA-a, but a reduction in the char yield of PBF-a and also PBT-a. The differences in cross-link density observed, coupled with the thermal behavior, may reflect a different initiation mechanism as the pKa = 6.6 for TDP23 is greater than for TDA (pKa = 4.11).24 Wang and Ishida17 also saw improved char yield and Tg when Lewis acids were used to initiate the ring-opening reaction of BA-a and, when considering the first-derivative plot of the weight change from TGA data, suggested that a different degradation mechanism occurred in the polybenzoxazines that arose from initiated and uninitiated mechanisms. This was attributed by the authors to the formation of a different polymer architecture caused by the presence of the initiator. Dunkers and Ishida25 discuss the use of both strong and weak protonic catalysts and found that a slightly different polymerization mechanism occurs depending on the pKa of the acid. For acids with pKa in the range 0.70–4.43, this was explained by the formation of a stable iminium ion at early conversions, but generally the stronger the acid, the more rapidly the monomer was consumed and more rapidly converted to the tetra-substituted ring of the product, which is consistent with our findings.
Monitoring the Polybenzoxazine Cure Using Vibrational Spectroscopy
Thermal analysis provides information about physical changes brought about during the thermal treatment of the monomer, but (without hyphenation) no chemical information. Consequently, the cure of monomer BA-a was monitored initially in situ using Raman spectroscopy using a heated cell (ramped rapidly to 180 °C and held isothermally for 120 min; spectra being taken at intervals of 7.5 min), but while a stacked spectral plot could be produced as a function of cure time (Figure 10) the fluorescence background produced by the thermal degradation at an impurity level caused the signal to be increasingly swamped between 1800 and 3400 cm–1 so that any data in this region could not be used quantitatively. It is apparent that the vibrational spectra of the monomers and propagating species are quite complex and assignments in the literature are occasionally contradictory, but arguably, the most diagnostically useful bands in the spectra are those associated with the constitution of the benzoxazine ring, given that the ring opens to form an oligomer during polymerization. The band corresponding to the presence of the C–N–C asymmetric stretch will be present only in the benzoxazine and can be found at 1070–1200 cm–1 and the symmetric stretch at 800–860 cm–1. Similarly, the bands for C–O–C stretching can be found at ca. 1200 cm–1 for the asymmetric stretch and ca. 1030 cm–1 for the symmetric stretch.
Figure 10.

Stacked Raman spectra for the polymerization of BA-a acquired in situ (comprising 16 spectra, sampling rate 1 every 7.5 min).
The peaks cannot be assigned unequivocally to pure monomer or dimer/oligomer as the same functional groups appear in both species (and some studies have reached tentative assignments),26,27 but while the basic bands can be assigned with confidence the data from several published studies (Table 6) are sometimes contradictory on key points. Consequently, it was decided to employ chemometrics techniques (particularly principal components analysis, PCA) as our group has already demonstrated the benefits of this approach in unrelated areas of polymer chemistry. PCA is a multivariate analysis technique28 that utilizes all the data and uses a holistic approach to determine similar and dissimilar spectra. The extraction of the principal components (PCs), which account for the variance in data, enables the particular analysis to be simplified but makes no suppositions about the chemical meaning of the data. This technique has been shown29 to yield useful information in the correlation of infrared spectra of cyanate ester/bismaleimide blends with thermomechanical properties and has also been employed30 to probe the mechanism by which our previous complexes have undergone dissociation.
Table 6. Assignments for Selected Key Bands Apparent in the PCA of the Raman Vibrational Spectra.
| peak range (cm–1) | proposed assignment | reference source |
|---|---|---|
| 1615–1620 | C–C stretch in benzene ring | (31, 32) |
| 1584–1590 | C–C stretch in benzene ring | (31) |
| 1575 | C–C stretch in benzene ring | (31) |
| 1486–1489 | C–H bend in CH2 | (27, 33) |
| 1451–1455 | CH2 scissoring (ring) | (26, 27) |
| 1434 | CH2 scissoring (ring) | (26, 27) |
| 1308–1325 | CH2 wag | (27, 33) |
| 1285–1288 | C–H bridge | (31, 33) |
| 1205–1215 | C–O–C or possibly C–N–C asymmetric stretch (ring closed) | (27) |
| 1192 | C–N–C asymmetric stretch (ring closed) | (27) |
| 1172–1174 | C–H bending (ring) | (33) |
| 1118 | Wilson 18b (ring closed) | (27) |
| 1104 | C–N–C symmetric stretch (ring closed) | (27) |
| 1049–1052 | C–O–C symmetric stretch of ring or during cure | (33) (ring), (27) (curing) |
| 1033 | C–C stretching vibration of ring | (33) |
| 999 | C–C bending vibration of ring | (27) |
| 860 | C–N–C symmetric stretch (ring closed) | (27) |
| 745–751 | benzoxazine ring breathing | (27) |
Figure 11 shows the PCA data arising from the raw Raman spectra for the time course of uncatalyzed BA-a in a series of in situ measurements. The data show that two principal components (PCs) account for the variation in the data. A plot of PC1 (accounting for 97%) vs PC2 (accounting for 3%) shows regular incremental changes in the data until spectrum 7 (i.e., after 52 min at 180 °C), at which point there is a large change in the plot, indicating that a significant change in the curing polymer is occurring at this point. Most of the spectral changes observed during the experiment occur in regions associated with the aromatic rings and the phenolic moiety. The main band changes are reductions at 700–800 cm–1 (C–H out-of-plane bending vibrations), 990–1050 cm–1 (C–H in-plane vibrations), and 1170–1250 cm–1 (C–N–C stretch) and increases at 1260–1300 cm–1 (C–O stretch), 1400–1500 cm–1 (C–H deformations), and 1530–1630 cm–1 (skeletal vibrations such as quadrant stretching bands). These changes in the bands associated with aryl moieties upon heating is consistent with the monomer undergoing ring-opening leading to changes in the rotational freedom of the phenyl ring.
Figure 11.

PCA data from cure of BA-a as received (top) and recrystallized (below). Raman data from in situ experiment showing (left) scores for PC2 versus PC1 and (right) loadings for PC1. N.B.: the first data point (monomer) has been omitted from this treatment.
The scores plot of PC-2 versus PC-1 indicates that the spectra are very similar and show very little difference during the first five acquisitions. Spectrum 6 begins to differ, and then there is a progressive change in the spectral characteristics, with PC-1 becoming increasingly influential, up to spectrum 9. From spectrum 10 onward until the end of the experiment there is a change in the trend, perhaps related to the change from chemical to physical control (and from chain extension to cross-linking). Similar plots were obtained for the range of bis-benzoxazine monomers and support the observation that similar changes are observed in key peaks in the spectra during cure (this, in turn, suggests that the “uninitiated” thermal cure mechanism is largely independent of the nature of the bis-phenol of the benzoxazines studied here).
Unfortunately, the samples could only be cured up to 180 °C for 2 h in the in situ Raman experiment, due to instrumental limitations making it impossible to monitor reactions cured/postcured at 200 °C. Consequently, in order to validate the two experimental methods, it was decided to perform an ex situ analysis (i.e., curing individual samples in a fan-assisted oven to differing degrees of cure following the cure schedule—2 h at 180 °C—and analyzing these samples individually with respect to cure time) before comparing the results of the two methods. The data may be compared directly since the sampling intervals are the same and it is clear that there is no significant effect on the pathway of the reaction by changing the sampling method. There is inevitably a slight delay/variation in this method associated with removal of the samples from the oven, quenching them in liquid nitrogen before introducing them into the Raman spectrometer. For comparison, the data for the uninitiated BA-a (as received) and following recrystallization (Figure 11, bottom) are presented, showing graphically the differences in the scores plot (marked by a reduction in the significance of PC-2 is evident and a more symmetrical presentation of the data for the latter).
Examining the Effect of the Nature of the Additive on Polymerization Using Vibrational Spectroscopy
The bis-benzoxazine monomers were each formulated with TDP or TDA at 10 mol % addition, and ex situ analysis was performed using the same conditions (heated at 2 K/min). Using the same data treatment, the PCA results arising from these spectra are presented for the polymerization reaction initiated with TDP (Figure 12) and TDA (Figure 13). These treatments indicate that the initiated systems obey very different kinetics during the early stages of the reaction; e.g., the changes in the scores plots occur over a markedly shorter time scale during both the early and later stages of reaction when compared with the uninitiated blend (confirming that the effect of the additives is to induce a faster rate of cure). Furthermore, in the case of TDP only one PC is required to describe the data (whereas in the case of TDA two PCs are needed: PC-1 covers >98% and PC-2 >1%). However, it is possible to state that the final networks yielded different spectra in the presence of the additives, possibly due to a different termination mechanism, increased cross-link density, and consequent differences in thermo-oxidative stability (discussed below in the context of the TGA data).
Figure 12.

PCA data from in situ cure of BA-a (as received) with 10 mol % TDP. Raman data showing (top left) scores for PC2 versus PC1 and (top right) loadings for PC1. N.B.: the first and second data points were omitted from this treatment.
Figure 13.

PCA data from in situ cure of BA-a (as received) with 10 mol % TDA. Raman data showing (top left) scores for PC2 versus PC1 and (top right) loadings for PC1. N.B.: the first, second, and third data points were omitted from this treatment.
The difference in the scores plots suggests differences in the mechanisms. From an overlay plot of PC1 for the different samples (Figure 14), there are spectral regions (1550–1700 cm–1) that overlay well in terms of intensity, suggesting that the observed differences in intensity are real. This would indicate that during the initial stages of the polymerization reaction the greatest reductions are in the bands due to aromatic substitution. For instance, reductions are observed in the C–H out-of-plane deformation at 750 and 793 cm–1 attributed by Dunkers and Ishida27 to 1,2,4-trisubstituted rings and 999 cm–1 (mono-, 1,3-, or 1,3,5-trisubstitution perhaps in the arylamine).
Figure 14.

Overlay of plots of PC1 for BA-a, BA-a in the presence of TDA (10 mol %), and BA-a in the presence of TDP (10 mol %).
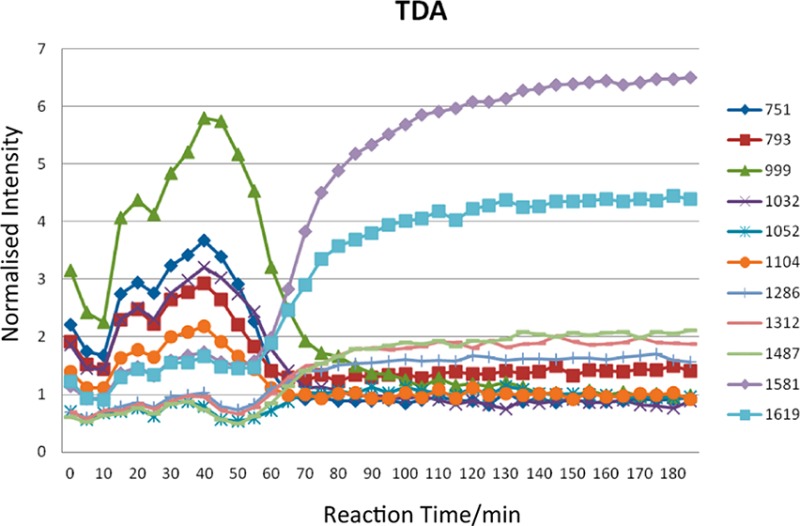
This is accompanied by reductions in the bands at 1031 cm–1 (from C–C ring stretch or C–O–C stretch27) and 1102 (possibly C–N–C stretch) and 1208 cm–1 (C–N stretch), 1300–1310 cm–1 (C–O stretch), and 1450–1500 cm–1 (CH2 scissoring). Over the same period, increases in band intensity are observed for 1050 (C–O stretch) and 1114 cm–1 (C–N stretch) and 1518 and 1619 cm–1 (aromatic ring breathing), while bands appear to shift between 1150 and 1250 cm–1. To explore the mechanisms further, selected individual spectral bands within PC1 for the thermal polymerization of BA-a in the absence of additive and with both TDP and TDA were monitored as a function of reaction time (Figure 15) over a total period of 185 min.
Figure 15.

Variation in intensity of selected spectral bands (cm–1) as a function of reaction time for BA-a (a) in the absence of additive, (b) in the presence of TDP (5 mol %), and (c) in the presence of TDA (5 mol %).
The profiles for BA-a when thermally initiated or in the presence of TDP are quite similar, albeit with the initiated polymerization occurring earlier. At the point at which crossover occurs the sample appears to be undergoing some molecular change, e.g., gelation causing a change in the kinetics, as this is the same point at which the parabola changes gradient in the scores plot (60–80 min for the TDP-initiated reaction; 100–120 min for the thermally initiated reaction). This demonstrates the sluggish nature of the reaction in the absence of initiators or catalysts; the observations would have been retarded still further on the purified BA-a monomer. There are reductions in the bands at 793, 999, 1032, and 1104 cm–1 (associated with the ring-closed benzoxazine structure) from about 85 min in the TDP-initiated BA-a and from 35 to 115 min in the BA-a alone. All the aforementioned bands appear to drop at a similar rate, while the intensity of the band at 751 cm–1 decreases more rapidly and earlier in the polymerization reaction. Conversely, the bands at 1052, 1312, and 1487 cm–1, which are attributed to aromatic ring functions, increase at approximately the same rate, and bands at 1619 and 1581 cm–1 initially increase at the same rate, before 1581 cm–1 increases its growth. These observations imply that the bands associated with 1581 and 751 cm–1 are related, as are those at 999 and 1619 cm–1. When compared with BA-a alone the differences in the growth of the band at 1581 and 1619 cm–1 are much more marked for the TDP initiated system, with 1581 cm–1 increasing more rapidly and to a greater intensity, whereas in BA-a alone the increase in 1619 cm–1 is greater than TDP, relative to many of the other bands.
A similar analysis of the spectral data relating to the BA-a initiated with TDA yields quite different results. For instance, the bands at 999 and 1619 cm–1 are negatively correlated, as are 1581 and 751 cm–1. This observation is confirmed by the PC1 loadings plots as these are the most intense peaks (either positive or negative) and have similar loadings although 1581 cm–1 is slightly greater. The increase in the degree of substitution of the newly formed phenolic ring can be monitored by observing the reduction in the intensities of the characteristic phenolic bands at 999 and 1032 cm–1; the latter is assigned to the C–O–C stretching mode of the benzoxazine ring.33 Thus, these may relate to the changing nature of the benzene ring substitution through benzoxazine ring-opening and bridge-forming. Dunkers and Ishida26,27 also observe a band at 1581 cm–1 in a monofunctional benzoxazine with alkylamine groups, but this apparently shifts to 1600 cm–1 in the corresponding dimer, suggesting that this band is related to the closed ring structure alone; the band at 1620 cm–1 is reported in both instances. The same authors note that the oxazine ring typically gives rise to a band around 960–920 cm–1 due to an out-of-plane B2 mode with medium to strong intensity in the IR spectrum.
The changes in peak intensities show a different pattern for the TDA initiated monomer (Figure 15c): the bands at 751, 793, 1032, and 1104 cm–1 increase at a similar rate, but 999 cm–1 appears to increase more rapidly and over a longer period of time. The bands at 1312, 1487, and 1052 cm–1 increase at the same rate, but 1052 cm–1 reaches a much lower ultimate intensity. In fact, in BA-a, TDP, and TDA, the difference in intensity between 1312 and 1487 cm–1 and 1052 cm–1 increase from nearly 0 to 1 arbitrary unit. The bands at 1312 and 1487 cm–1 are most likely to reflect aromatic functions or possibly the methylene groups in the bridges, but 1052 cm–1 is probably a C–O–C stretching mode.25 Examining Figure 15c, the relationship between 1581 and 1619 cm–1 differs from 751 and 999 cm–1; 1581 and 751 cm–1 appear to be associated, as are 1619 and 999 cm–1 (unlike both BA-a and TDP).
The thermal initiation and the TDP initiation of BA-a conform to a similar polymerization mechanism, but initiation shifts the reaction to a lower temperature regime and the resulting polymer networks display similar cross-link density and glass transition temperatures (spanning a wider range–implying a more heterogeneous network). The initiation of BA-a with TDA progresses via a different mechanism involving the paired bands at 999 and 1581 cm–1 and 751 and 1619 cm–1, which are all aromatic vibration modes. The TDA works more efficiently, leading to the opening of more benzoxazine rings, and propagation is less linear (more cluster-like) prior to cross-linking. The result is that there are more points of reaction leading to a higher cross-link density and consequent higher Tg. The effect of additive concentration [TDA] is initially really marked but reaches an apparent plateau at 10–15 mol %.
Conclusions
A combination of thermal and spectroscopic analysis has proven successful in offering support to the proposed ring-opening mechanisms associated with benzoxazine monomers. The presence of the additive improves the temperature of polymerization, by encouraging the ring-opening reaction at a lower temperature, and its presence also appears to improve the thermal stability of the final polymer. Spectral and thermal data suggest both initiated and uninitiated blends undergo the same mechanism: suggesting a step growth mechanism whereby the rings open and an active center reacts so there may be several centers active at one time. The selection of an initiator based on its pKa appears to be able to influence both the nature of the network architecture and potentially the final properties of the polybenzoxazine as the Tg (determined from DMTA) is elevated when 3,3′-thiodipropionic acid (TDA) is used in preference to 3,3′-thiodiphenol. TDA is found to be the superior initiator, offering a more rapid polymerization mechanism and a higher glass transition temperature in the cured polybenzoxazines.
Experimental Section
Materials
The monomers based on bisphenol A (BA-a), a mixture of 4,4′-, 2,4′-, and 2,2′-bisphenol F (BF-a), 3,3′-thiodiphenol (BT-a), and additives 3,3′-thiodiphenol (TDP) and 3,3′-thiodipropionic acid (TDA) were all supplied by Huntsman Advanced Materials (Basel, Switzerland) and having been characterized using 1H NMR, Raman spectroscopy, and elemental analysis were generally used as received without further purification. Monomer (BA-a) was recrystallized from ethanol (96%) prior to DSC analysis when examining the kinetics of reaction. The structures and designations for the monomers studied are given in Table 7; the corresponding polybenzoxazine of BA-a is designated PBA-a.
Table 7. Bis-benzoxazine Monomer Structures.
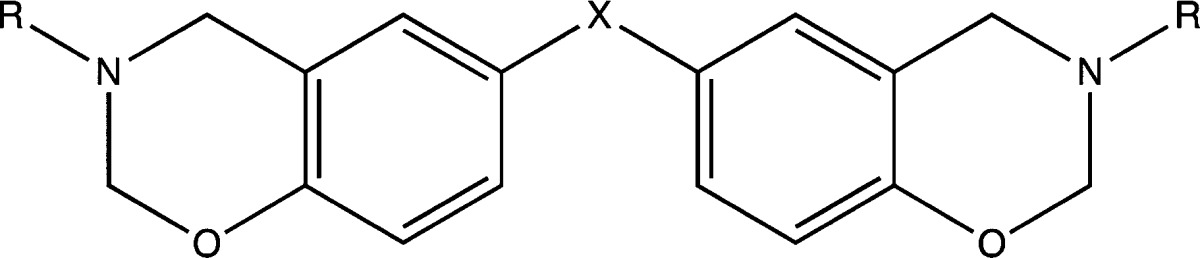
| abbreviations | R | X | IUPAC chemical names |
|---|---|---|---|
| BA-a | Ph | (CH3)2C | 2,2-bis(3,4-dihydro-3-phenyl-2H-1,3-benzoxazine)propane |
| BF-a | Ph | CH2 | 2,2-bis(3,4-dihydro-3-phenyl-2H-1,3-benzoxazine)methane |
| BT-a | Ph | S | 3,3-bis(3,4-dihydro-3-phenyl-2H-1,3-benzoxazine) sulfide |
Blending and Cure of Polymer Samples for Thermomechanical Analyses
Where additives were used, these were first ground (together with the monomer) and then heated to 90–100 °C (to melt the additive) and degassed for 2–3 h under vacuum depending on the level of foaming. Where additives were not used, monomers were first degassed at 100–120 °C (2–3 h depending on the level of foaming) and cured in aluminum dishes (55 mm diameter, depth 10 mm) in a fan-assisted oven: heating at 2 K/min to 180 °C (2 h isothermal) + heating at 2 K/min to 200 °C (2 h isothermal) followed by a gradual cool (3 K/min) to room temperature. Cured samples were cut to the correct token size for analysis using a diamond saw. For the dynamic cure study, the BA-a monomer was first degassed at ca. 100 °C (1 h) and cured in aluminum dishes (55 mm diameter, depth 10 mm) and transferred to a fan-assisted oven set at 90 °C. The samples were allowed to equilibrate before being heated at 2, 8, or 15 K/min to 180 °C (2 h isothermal) + heating at 2 K/min to 200 °C (2 h isothermal) followed by a gradual cool (3 K/min) to room temperature. Cured samples were cut to the correct token size for analysis using a diamond saw. Producing flat, void-free plaques for DMTA provided a big challenge as the benzoxazines were difficult to degas, and the addition of the initiators compounded the problem as they seemed to undergo degradation during cure to produce gas bubbles; this was particularly problematic at higher concentrations of TDA.
Instrumentation
Vibrational spectra were obtained using a PerkinElmer system 2000 FT-NIR-Raman spectrometer operating at 250–500 mW (Nd:YAG laser) and a PerkinElmer FTIR system 2000 spectrometer. Samples that were analyzed in situ during the cure process using Raman spectroscopy employed a heated cell that was ramped rapidly from 50 to 180 °C and held isothermally for 120 min; spectra being taken at intervals of 7.5 min. In contrast, samples analyzed ex situ were heated in a fan-assisted oven that was ramped rapidly to 180 °C and held isothermally for 120 min; samples being taken at intervals of 7.5 min and quenched in liquid nitrogen. Samples for IR analysis were cast from chloroform (GPR) on to KBr disks and the solvent removed in a fan oven (50 °C, 30 min). For each measurement, 10 spectra were obtained at a resolution of 4 cm–1 and coadded to produce the final spectrum. Chemometrics analysis (PCA) was carried out on the spectral data using The Unscrambler X, v10.1 software (Camo, Oslo). Nuclear magnetic resonance (NMR) spectra (including DEPT-135, HSQC and HMBC pulse sequences to assign individual environments) were obtained for 1H (500 MHz) and 13C (75 MHz) using a Bruker DRX500 FT-NMR at 298 K in CDCl3 with TMS. High performance liquid chromatography (HPLC) was undertaken using a Varian 920-LC using C18 column and UV detection (λ = 254 nm). Samples were dissolved in HPLC grade CHCl3, and the mobile phase (flow rate 0.7 mL/min, 40 °C) was 100% CHCl3/100% methanol gradient (both HPLC grade). Differential scanning calorimetry (DSC) was undertaken using a TA Instruments Q1000 running TA Q Series Advantage software on samples (4.0 ± 0.5 mg) in hermetically sealed aluminum pans. Experiments were conducted at a heating rate of 5, 8, 10, 12, and 15 K/min from room temperature to 300 °C (heat/cool/heat) under flowing nitrogen (50 cm3/min.). In order to gauge the reactivity of the monomer in the bulk, dynamic DSC analysis was performed on all of the systems. Thermogravimetric analysis (TGA) was performed on a TA Q500 on milled, cured resin samples (5.5 ± 0.5 mg) in a platinum crucible from 20 to 1000 °C at 10 K/min in air and nitrogen (40 cm3/min). Dynamic mechanical thermal analysis (DMTA) (in single cantilever mode at a frequency of 1 Hz) was carried out on cured neat resin samples (2 mm × 10 mm × 17 mm) using a TA Q800 in static air.
Acknowledgments
We thank Cytec and the Engineering and Physical Sciences Research Council (EPSRC) for funding this work in the form of a studentship (L.T.M.).
Supporting Information Available
Spectroscopic data associated with the characterization of the monomers. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Su Y.-C.; Chang F.-C. Polymer 2003, 44, 7989–7996. [Google Scholar]
- Espinosa M. A.; Galiá M.; Cádiz V. Polymer 2004, 45, 6103–6109. [Google Scholar]
- Holly F. W.; Cope A. C. J. Am. Chem. Soc. 1944, 66, 1875–1879. [Google Scholar]
- Handbook of Polybenzoxazine Resins; Ishida H., Agag T., Eds.; Elsevier: New York, 2011. [Google Scholar]
- Ishida H.; Ohba O. Polymer 2005, 46, 5588–5595. [Google Scholar]
- Kim H. J.; Brunovska Z.; Ishida H. Polymer 1999, 40, 1815–1822. [Google Scholar]
- Kimure H.; Matsumoto A. J. Appl. Polym. Sci. 1998, 68, 1903–1910. [Google Scholar]
- Sudo A.; Sudo R.; Nakayama H.; Arima K.; Endo T. Macromolecules 2008, 41, 9030–9034. [Google Scholar]
- Hirai Y.; Aizawa T.; Yoshimura Y. European Patent Application 0659832, 1994.
- Hamerton I.; Howlin B. J.; Mitchell A. L.; McNamara L. T.; Takeda S. React. Funct. Polym. 2012, 72, 736–744. [Google Scholar]
- Ishida H.; Rodriguez Y. Polymer 1995, 36, 3151–3158. [Google Scholar]
- Barton J. M. Adv. Polym. Sci. 1985, 72, 111–154. [Google Scholar]
- Kissinger H. Anal. Chem. 1957, 21, 1702–1706. [Google Scholar]
- Jubsilp C.; Damrongsakkul S.; Takeichi T.; Rimdusit S. Thermochim. Acta 2006, 447, 131–140. [Google Scholar]
- Kannurpatti A. R.; Anseth J. W.; Bowman C. N. Polymer 1998, 39, 2507–2513. [Google Scholar]
- Allen D. J.; Ishida H. J. Appl. Polym. Sci. 2006, 101, 2798–2809. [Google Scholar]
- Wang Y.-X.; Ishida H. Polymer 1999, 40, 4563–4570. [Google Scholar]
- Wang Y.-X.; Ishida H. J. Appl. Polym. Sci. 2002, 86, 2953–2966. [Google Scholar]
- Shen S. B.; Ishida H. J. Polym. Sci., Part B: Polym. Phys. 1999, 37, 3257–3268. [Google Scholar]
- Ishida H.; Allen D. J. J. Polym. Sci., Part B: Polym. Phys. 1996, 34, 1019–1030. [Google Scholar]
- Hemvichian K.; Ishida H. Polymer 2002, 43, 4391–4402. [Google Scholar]
- Hamerton I.; Howlin B. J.; Thompson S.; Stone C. A. Examining the thermal stability and degradation mechanisms of polybenzoxazines and the physical properties of their chars, submitted 2013.
- http://chemweb.unp.ac.za/chemistry/Physical_Data/pKa_values.htm (accessed 20th July, 2011).
- http://www.drugfuture.com/chemdata/3-3-Thiodipropionic-Acid.html (accessed 20th July, 2011).
- Dunkers J.; Ishida J. J. Polym. Sci., Part A: Polym. Chem. 1999, 37, 1913–1921. [Google Scholar]
- Dunkers J.; Ishida J. Spectrochim. Acta 1995, 51A, 855–867. [Google Scholar]
- Dunkers J.; Ishida J. Spectrochim. Acta 1995, 51A, 1061–1074. [Google Scholar]
- Martens H.; Naes T.. Multivariate Calibration; John Wiley and Sons: Chichester, 1998. [Google Scholar]
- Hamerton I.; Herman H.; Mudhar A. K.; Chaplin A.; Shaw S. J. Polymer 2002, 43, 3381–3386. [Google Scholar]
- Hamerton I.; Emsley A. M.; Hay J. N.; Herman H.; Howlin B. J.; Jepson P. J. Mater. Chem. 2006, 16, 255–265. [Google Scholar]
- Patnaik P.Dean’s Analytical Chemistry Handbook; McGraw-Hill: New York, 2004. [Google Scholar]
- Keresztury G.; Billes F.; Kubinyi M.; Sundius T. J. Phys. Chem. A 1998, 102, 2559–2567. [Google Scholar]
- Monni J.; Nemelä P.; Alvila L.; Pakkanen T. T. Polymer 2008, 49, 3865–3874. [Google Scholar]
- Takeichi T.; Kawauchi T.; Agag T. In Handbook of Polybenzoxazine Resins; Ishida H., Agag T., Eds.; Elsevier: New York, 2011; Chapter 20, pp 382–383. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.