

Background: Overproduction of nitric oxide by dimeric inducible nitric-oxide synthase (iNOS) is physiologically harmful.
Results: Pyrimidine imidazole derivative (PID) binds to both the iNOS dimer and monomer causing irreversible monomerization and inhibition of dimerization, respectively.
Conclusion: PID can physiologically inhibit iNOS both during and after its assembly into active enzyme.
Significance: Our study reveals PID's dual ability to inhibit iNOS as well as their kinetic mechanisms.
Keywords: Enzyme Inhibitors, Enzyme Kinetics, Nitric Oxide, Nitric-oxide Synthase, Nitrosative Stress, Dimerization Inhibitors, Pyrimidine Imidazoles
Abstract
Overproduction of nitric oxide (NO) by inducible nitric-oxide synthase (iNOS) has been etiologically linked to several inflammatory, immunological, and neurodegenerative diseases. As dimerization of NOS is required for its activity, several dimerization inhibitors, including pyrimidine imidazoles, are being evaluated for therapeutic inhibition of iNOS. However, the precise mechanism of their action is still unclear. Here, we examined the mechanism of iNOS inhibition by a pyrimidine imidazole core compound and its derivative (PID), having low cellular toxicity and high affinity for iNOS, using rapid stopped-flow kinetic, gel filtration, and spectrophotometric analysis. PID bound to iNOS heme to generate an irreversible PID-iNOS monomer complex that could not be converted to active dimers by tetrahydrobiopterin (H4B) and l-arginine (Arg). We utilized the iNOS oxygenase domain (iNOSoxy) and two monomeric mutants whose dimerization could be induced (K82AiNOSoxy) or not induced (D92AiNOSoxy) with H4B to elucidate the kinetics of PID binding to the iNOS monomer and dimer. We observed that the apparent PID affinity for the monomer was 11 times higher than the dimer. PID binding rate was also sensitive to H4B and Arg site occupancy. PID could also interact with nascent iNOS monomers in iNOS-synthesizing RAW cells, to prevent their post-translational dimerization, and it also caused irreversible monomerization of active iNOS dimers thereby accomplishing complete physiological inhibition of iNOS. Thus, our study establishes PID as a versatile iNOS inhibitor and therefore a potential in vivo tool for examining the causal role of iNOS in diseases associated with its overexpression as well as therapeutic control of such diseases.
Introduction
The mammalian nitric-oxide synthase (NOS)3 produces nitric oxide (NO), which is important for several life-sustaining physiological functions like blood circulation, vision, memory, and immune functions (1–3). The enzyme catalyzes an NADPH and O2-dependent oxidation of its substrate, l-arginine (Arg) to citrulline and NO, through the intermediate formation of N-ω-hydroxy-l-arginine (NOHA).
NOS exists as three different isoforms, inducible NOS (iNOS or type II), neuronal NOS (nNOS or type I), and endothelial NOS (eNOS or type III) as well as their splice variants, which differ in their tissue distribution, catalytic activity, and response to calcium (4–8). However, the three isoforms have similar bi-domain structures with an N-terminal oxygenase domain that is linked to a C-terminal reductase domain through a calmodulin-binding motif. The oxygenase domain contains binding sites for heme, H4B, and Arg, whereas the reductase domain provides binding motifs for NADPH, FAD, and FMN. The enzyme undergoes dimerization through the interaction of two oxygenase domains of two monomeric NOS subunits, although the reductase domains remain suspended as freely hanging tails (9–11).
It is noteworthy that NOS is active only as a homodimer (1–3, 12). This is primarily because the electron transfer between the NOS reductase and oxygenase heme takes place only in trans, where one reductase domain can only donate electrons to the oxygenase domain on the partner subunit to initiate oxidative conversion of Arg to NO (11–15). Because homo-dimerization is an essential post-translational step for creating an active NOS enzyme, it is also a prospective target for controlling NOS activity by potential NOS dimerization inhibitors.
The constitutive NOSs, namely nNOS and eNOS, typically generate low levels of NO (16–18), whereas iNOS can produce large amounts of NO that can be physiologically harmful (18, 19). Although NO is vital for supporting life, its abnormal production, particularly by iNOS, could lead to life-threatening diseases like stroke, arthritis, sepsis, asthma, diabetes, Parkinson disease, Alzheimer disease, and amyotropic lateral sclerosis (2–4, 18, 20–24). This has naturally led to the quest for potent iNOS inhibitors that could be used for therapeutic interventions of such diseases (2, 3). Thus, several iNOS dimerization inhibitors are being explored to accomplish effective control of abnormal iNOS activity and the high levels of NO produced under pathological conditions (25). Various imidazole-based compounds have been tested as dimerization inhibitors largely because of their low toxicity and high selectivity for iNOS, and attempts have also been made to modify them to analogs with high affinity and inhibitory efficacy to bring it close to clinical use (26–29). Original studies by our group on the effect of N1-substituted pyrimidine imidazoles, like clotrimidazole on iNOS-synthesizing RAW cells, revealed that they inhibited iNOS activity by preventing heme insertion (26). Later studies with PID-like compounds showed that they bind to the NOS heme during its expression and produce an irreversible iNOS-monomer-inhibitor complex (28, 30). Indeed, among the imidazoles examined for iNOS dimerization inhibition, pyrimidine imidazoles have shown considerable promise to qualify as effective nontoxic iNOS inhibitors (27, 28). However, the molecular details and mechanism of their action on the enzyme are still not clearly understood, especially with respect to the possible interaction with the active dimeric form of iNOS under physiological conditions, as well as the mechanism of monomerization and the relative kinetics and affinity of binding to the monomeric and dimeric forms of the enzyme.
Indeed, one of the reasons we decided to revisit the mechanism of inhibition of iNOS by pyrimidine imidazoles, amid several previous reports on the same subject (26–28), is because we subsequently discovered that these compounds could also bind and accomplish inhibition of the dimeric iNOS, the active form of iNOS in a physiological setting, an essential prerequisite for an inhibitor to qualify as a potential candidate for therapeutic intervention of iNOS activity both for the effective clinical control of diseases associated with iNOS overexpression and nitrosative stress as well as a research tool for elucidating the role of this physiologically versatile enzyme in vital life functions in vivo. We therefore elucidated the mechanisms of inhibitor action on the iNOS monomer and dimer, which are the two quaternary states of the enzyme that exist in cells during and after its assembly, using two bulky pyrimidine imidazole compounds, PIC and PID. We found that either compound interacted with the iNOS monomer at a significantly faster rate than the dimer in both the purified form and in iNOS-synthesizing cells. Moreover, we found that PID can disrupt the fully assembled, active dimeric form of iNOS in cells, indicating that it has the potential to accomplish complete physiological inhibition of the enzyme and to serve as an in vivo tool for elucidating the role of iNOS in diseases associated with its dysfunctional overexpression as well as a therapeutic inhibitor for clinical management of these diseases.
EXPERIMENTAL PROCEDURES
Reagents and Chemicals
Monoclonal antibody against iNOSfl was obtained from BD Transduction Laboratories, and IFN-γ was procured from Genentech. Resins used for purification of the iNOS proteins and the anti-mouse secondary antibody were procured from GE Healthcare. All other reagents and chemicals used were of analytical grade and were obtained from Sigma.
iNOS Inhibitors (PIC and PID)
The two novel pyrimidine imidazoles used in our study, namely PIC or methyl-3-(((benzo(d)(1,3)dioxol-6-yl)methylcarbamoyl)methyl)-4-(2-(1H-imidazol-1-yl)pyrimidin-4-yl)piperazine-1-carboxylate and PID or 2-(1-(2(1H-imidazol-1-yl)pyrimidin-4-yl)piperazin 2-yl)-N-((benzo(d)(1,3)-dioxol-6-yl)methyl)acetamide were synthesized and purified in the laboratory of Prof. Brindaban C. Ranu (Division of Organic Chemistry, Indian Association for the Cultivation of Science, Kolkata, India), following protocols reported earlier (31), and then characterized and checked for their purity using NMR and mass spectrometry. Fig. 1 shows the chemical structures of PIC and PID as well as their mass spectrometric profiles following purification.
FIGURE 1.

Chemical structures and mass spectral profiles of PIC and PID. Chemical structures of the novel pyrimidine imidazoles, PIC and PID, used in our study indicating the positions of their benzodioxole and the imidazole moieties proposed to be involved in iNOS binding. Mass spectral profiles of the synthesized compounds are also shown indicating their purity and observed molecular weights of 422.19 against theoretical molecular weight of 421.45 for PIC (C21H23N7O3) and 480.18 against theoretical molecular weight of 479.49 for PID (C23H25N7O5). Details on synthesis of the compounds are provided under “Experimental Procedures.”
Molecular Biology
Bacterial transformations and isolation of DNA fragments were performed using standard procedures (32). NOS mutants used in the study, namely D92AiNOSoxy and K82AiNOSoxy, were previously generated in the laboratory of Prof. Dennis J. Stuehr, Cleveland Clinic (32). Escherichia coli strain BL21(DE3) containing pCWori plasmids with iNOSoxy wild type (WT), D92AiNOSoxy, and K82AiNOSoxy mutants as well as iNOSfl (wild type) DNA inserts were used for protein expression and purification.
Expression and Purification of Wild Type and Mutant iNOS Proteins
WT and mutant iNOSoxy proteins (K82AiNOSoxy and D92AiNOSoxy) containing a His6 tag attached to their N termini were overexpressed in E. coli strain BL21(DE3) using a modified pCWori vector in the absence of H4B and Arg as described before (33). The iNOSoxy proteins were purified by affinity chromatography on Ni2+-nitrilotriacetic acid resin followed by chromatography on Q-Sepharose anion exchange resin (34). The proteins were finally eluted from the Q-Sepharose column using a buffer containing 40 mm EPPS, 10% glycerol, 1 mm DTT, and 0.25 m NaCl. The full-length wild type iNOS protein (WT-iNOSfl) was purified by sequential chromatography on Ni2+-nitrilotriacetic acid and then 2′,5′-ADP-Sepharose resins as described previously (35). The proteins were concentrated and dialyzed at 4 °C, and aliquots were stored at a temperature of −85 °C for further use. The ferrous–CO adduct absorbance at 444 nm was used to determine heme protein content as a measure of the enzyme concentration using an extinction coefficient of 74 mm−1 cm−1 (A444–A500) (15).
Induction and Purification of iNOSfl from RAW Cells
Mouse macrophage RAW 264.7 (RAW) cells were grown (1 × 106 cells per ml) in 90-mm diameter tissue culture plates in DMEM with 10% calf serum. The cells were induced to express iNOSfl under specific treatment conditions based on experimental requirement with 50 μg/ml E. coli LPS and 10 ng/ml IFNγ (36). Cells were either induced for 10 or 14 h before being subjected to relevant experimental treatments. After treatment, the cells were washed twice with 1× PBS before being harvested by centrifugation at 8000 rpm for 10 min in a Beckman J2-HS centrifuge. The harvested cells were then lysed by three cycles of freezing and thawing in a lysis buffer containing 40 mm EPPS (pH 7.6), 10% glycerol, 3 mm DTT, 100 mm NaCl, and 0.1% Nonidet P-40 and again centrifuged at 15,000 rpm for 30 min for their supernatants, which were then used for iNOS immunoblotting or purification of iNOSfl protein through mini-ADP columns as described above.
Binding Assays
UV-visible spectrophotometric analysis of inhibitor binding to iNOS was recorded at 37 °C on a Hitachi U-3110 spectrophotometer. Spectra were either collected against time of incubation using fixed concentrations of the compounds (10 μm) or titrated for a fixed time with different concentrations of the compound for studying the kinetics of inhibitor binding with iNOS. All binding assays were typically done in cuvettes containing 2 μm iNOS protein in 1 ml of assay buffer containing 40 mm EPPS (pH 7.6), 10% glycerol, 250 mm NaCl, and 1 mm DTT in the presence and absence of 1 mm Arg or 10 μm H4B either separately or in combination. Binding rates were determined from the recorded time-dependent spectral perturbation or shift from 393 nm (in the presence of H4B/Arg) or 460 nm (DTT-bound) to 427 nm (indicating imidazole binding to iNOS heme). Inhibitor binding rates were derived from the slopes of the double-reciprocal plots of the absorbance differences (Δ(460–427) nm or Δ(393–427) nm) versus time of incubation or concentration using Origin® 8.0 (OriginLab).
NO Synthesis Assays
NO synthesis was assayed in 96-microwell plates using 2 μm of iNOSfl protein either expressed in bacteria or purified from induced RAW cells in each well, in an assay buffer containing 40 mm EPPS, 3 mm DTT, 4 μm FAD, 4 μm FMN, 10 μm H4B, 10 mm Arg, 0.5 mm EDTA, 1.2 mm CaCl2, 1 μm calmodulin, 0.1 mg/ml bovine serum albumin (BSA), 18 units/ml catalase, and 10 units/ml superoxide dismutase. NADPH (10 mm) was used to trigger NO production at 37 °C for a period of 30 min before being stopped by enzymatic oxidation of NADPH. Griess reagent (0.1 ml) was then added to each well, and the absorbance of the azo dye formed was measured at 550 nm in a VersaMax microplate reader (Molecular Devices) as a measure of NO produced. The nitrite produced as a result of NO solubilization in the aqueous reaction mixture was quantified using NaNO2 standards. The enzyme was either incubated with fixed (10 μm) or different concentrations of the inhibitor at 37 °C for different times or fixed times, respectively, and aliquots of such reactions constituting 2 μm of iNOSfl were subjected to the NO synthesis assay as described. The percentage inhibition of iNOS activity due to PIC or PID treatment was evaluated with respect to the activity of the relevant inhibitor-untreated WT-iNOSfl protein either in its recombinantly expressed, purified form or in RAW cells induced to synthesize the enzyme.
Gel Filtration Chromatography (FPLC)
Gel filtration chromatographic analysis of dimer-monomer content of pure iNOS proteins or iNOS purified from treated RAW cell lysates was done using ÄKTA purifier fast protein liquid chromatography system (GE Healthcare). Equal amounts of protein were loaded into a prepacked Superdex-200 gel filtration column (GE Healthcare) at 4 °C. The column was pre-equilibrated at a flow rate of 0.5 ml/min with 40 mm EPPS buffer (pH 7.6), containing 2 mm DTT, 10% glycerol, and 250 mm NaCl before the iNOS proteins were injected through a 50-μl loading loop. The isocratic flow rate was maintained at 0.5 ml/min at a column pressure of 0.7 MPa. The column effluent was detected at 280 nm using a flow-through UV detector. Molecular weights of the protein peaks were estimated relative to protein molecular weight standards, and the monomer-dimer contents of the proteins were determined using UNICORN software from the instrument manufacturer (GE Healthcare).
Rapid Stopped-flow Kinetic Analysis
Rapid mixing reactions were performed to study the kinetics of inhibitor binding in a stopped-flow spectrophotometer using a diode array detector (SF-61DX2) (HiTech Ltd., UK). All stopped-flow experiments were performed at 22 °C using 3 μm iNOSoxy wild type or mutant proteins in 40 mm EPPS buffer (pH 7.6) containing 10% glycerol and 250 mm NaCl. The kinetics of reaction was studied using Hi-Tech Scientific KinetAssyst software (Hi-Tech Ltd.). Averaged spectra or absorbance traces were compiled from 5–6 reactions, and the traces at wavelength 427 nm were fit to single or double exponential functions using the software provided by the instrument manufacturer to determine the kinetic rate constants for PID binding to WT-iNOSoxy and mutant proteins under different treatment conditions using either fixed (10 μm) or different concentrations of the inhibitor. The fitting did not include any absorbance change that was calculated to take place in the mixing dead time.
Low Temperature SDS-PAGE Immunoblotting of iNOS
Dimer and monomer contents of iNOSfl proteins in the treated RAW cell lysates were also examined using the low temperature SDS-PAGE method of Klatt et al. (13) with modifications. Cell lysates were mixed with 20 μl of sample buffer containing 0.125 m Tris-HCl (pH 6.8), 2% (w/v) SDS, 20% (w/v) glycerol, and 0.02% (w/v) bromphenol blue and subjected to SDS-PAGE in an ice-water bath at a constant current of 10 mA using a Mini Protean III apparatus (Bio-Rad). All gels (8%) and buffers were equilibrated at 4 °C prior to electrophoresis. Following electrophoresis, the proteins were transferred from the gels to PVDF membranes and subjected to immunoblotting for iNOS using anti-mouse iNOS antibody (BD Transduction Laboratories) following standard protocols (37). Protein bands representing the iNOSfl dimer and monomer were detected using mouse secondary antibody and ECL chemiluminescence detection kit (GE Healthcare).
RESULTS
PIC and PID Binding and Consequent Inhibition of iNOS
PIC and its derivative, PID, both showed time-dependent binding to Arg and H4B-free iNOSfl protein as evidenced by the shift of the iNOS heme iron to a low spin state of 427 nm from its DTT- bound Soret maxima at 460 nm, manifesting imidazole binding to the heme iron as the sixth ligand (Fig. 2A) as well as time-dependent inhibition of the activity of the enzyme caused by such binding (Fig. 2B). The inhibition of iNOS activity by PID was twice as fast (0.8 min−1 versus 0.4 min−1) compared with the parent PIC analog (Fig. 2B). This is consistent with their different rates of binding to the enzyme heme measured in terms of change of absorbance Δ(460 to 427) nm versus time of binding, which showed PID binding to iNOSfl was 1.9 min−1 as compared with 1.4 min−1 obtained for PIC (Fig. 2A).
FIGURE 2.
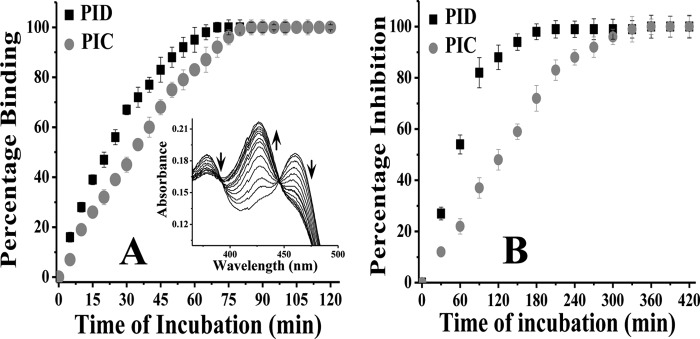
Comparative affinity for iNOSfl (A) and inhibition potency (B) of PIC and PID. A, 10 μm of each compound (PID and PIC) was added to 2 μm of the enzyme in a cuvette, and the spectrum was recorded in situ during such incubation to measure the degree of inhibitor binding at 427 nm in a time-dependent manner at 37 °C. The inset shows typical spectra for time-dependent shift in absorbance at 427 nm against a decrease at 460 nm in the iNOSfl protein free of Arg and H4B due to PID binding. Percentage binding was determined as a ratio of the change in absorbance Δ(460–427) nm versus time of incubation with inhibitor against that recorded for the inhibitor-untreated enzyme. B, aliquots from a similar reaction between the iNOSfl protein and PIC/PID as shown in A were collected at the indicated time points (x axis) after incubation with PIC/PID and the NO synthesis assay performed at 37 °C for 30 min and the generated NO measured as a function of nitrite content by extrapolation of the nitrite standard curve recorded at 550 nm through Griess reaction. The percentage inhibition due to PIC/PID treatment was then determined as a ratio of the drop in activity with respect to the wild type iNOSfl against time of incubation with the inhibitors. For each assay, the corresponding changes obtained for untreated controls, where the iNOS was incubated with the reaction buffer in the absence of the inhibitor under identical conditions and time, as the inhibitor-treated counterparts, were subtracted from the inhibitor-treated values to arrive at the actual values obtained due to the effect of the inhibitors and plotted against time of incubation. Data shown in A and B are mean ± S.D. of three independent experiments.
When binding of PID to iNOSfl was studied in the presence and absence of Arg and H4B, it revealed that the two had strikingly different effects on PID binding affinity, level of monomerization, and capacity to inhibit activity of the treated iNOSfl proteins (Fig. 3 and Table 1). As is apparent from Fig. 3, and the corresponding rates reported in Table 1, H4B had a dominant effect in antagonizing PID binding (giving a PID binding rate of 1.27 min−1 to the iNOSfl as compared with 1.86 min−1 in the absence of H4B and Arg) and the resulting monomerization rate (0.36 min−1 as compared with 0.53 min−1 in the absence of H4B and Arg), as well as rate of inhibition of activity of the treated iNOSfl protein (0.65 min−1 as compared with 0.91 min−1 in the absence of H4B and Arg). In contrast, Arg had a comparatively weaker antagonism toward PID binding, giving a binding rate of 1.77 min−1, a rate of monomer formation of 0.47 min−1, and a rate of inhibition of iNOSfl of 0.83 min−1, which were very close to the values obtained in the absence of H4B and Arg. A combination of both Arg and H4B showed the strongest antagonistic effect in all respects with rates of 1.11, 0.30, and 0.51 min−1 for PID binding and the corresponding monomerization and activity inhibition, respectively (Fig. 3, Table 1).
FIGURE 3.
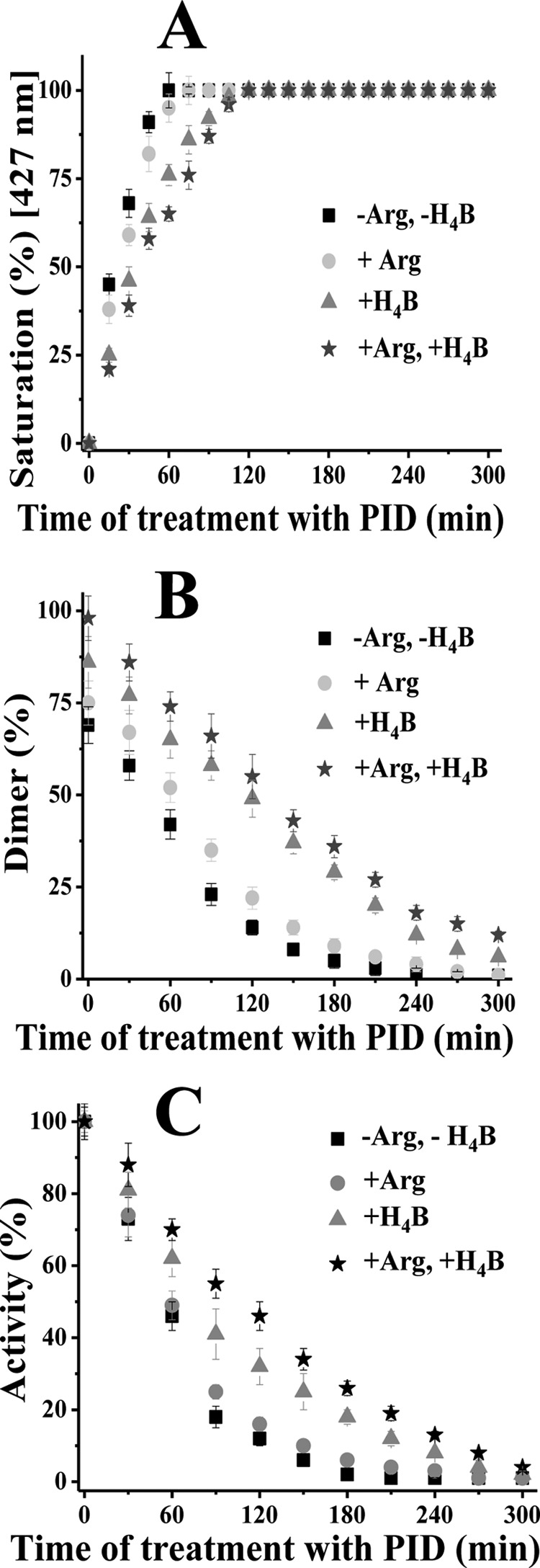
Effect of Arg and H4B on PID binding, monomerization, and inhibition of iNOSfl. Reactions were carried out in a buffer containing 40 mm EPPS (pH 7.4), 10% glycerol, and 250 mm NaCl with addition of 10 μm of PID to 2 μm iNOSfl in the presence or absence of 1 mm Arg and/or 10 μm H4B at 37 °C. A, percentage saturation of the iNOSfl protein by PID was measured in terms of increase in absorbance at 427 nm as a result of PID binding with respect to PID-untreated enzyme and plotted against the time of incubation with PID for all conditions. B, percentage of time-dependent change in NOS dimer content as a function of total dimer-monomer population caused by PID binding was calculated from the corresponding gel filtration profiles of the treated proteins against the PID-untreated iNOSfl. C, percentage activity of iNOSfl was plotted as a function of ratio of activity of treated versus PID-untreated protein against time of incubation with PID. Rates of PID binding, monomerization, and inhibition of iNOSfl were obtained through best linear fit of the corresponding curves obtained with Origin 8.0 software and are reported in Table 1. For each assay, similar inhibitor-untreated control was run along with the inhibitor-treated counterparts under identical conditions as in the experiments depicted in Fig. 2, and the measured changes subtracted from that were recorded for the inhibitor-treated sets for each time point to arrive at the depicted values. Details of the assays are provided under Fig. 2 and “Experimental Procedures.” Data shown in A–C are mean ± S.D. of three independent experiments done under similar conditions.
TABLE 1.
Relative rates of PID binding to purified iNOSfl and consequent iNOS monomerization and inhibition of activity in the presence and absence of Arg and H4B
Rates were derived through linear fitting of the curves depicted in Fig. 3. Details of the experiment are given under “Experimental Procedures.”
| Treatment | Rate of binding | Rate of monomerization | Rate of inhibition |
|---|---|---|---|
| min−1 | min−1 | min−1 | |
| −Arg, −H4B | 1.862 ± 0.071 | 0.533 ± 0.014 | 0.913 ± 0.027 |
| +Arg | 1.774 ± 0.043 | 0.471 ± 0.023 | 0.833 ± 0.014 |
| +H4B | 1.272 ± 0.055 | 0.356 ± 0.011 | 0.654 ± 0.019 |
| +Arg, +H4B | 1.111 ± 0.052 | 0.304 ± 0.005 | 0.511 ± 0.008 |
To study the possible effect of H4B and Arg in recovering active dimers from PID-bound iNOS monomers, we incubated PID-treated, fully monomerized iNOSfl proteins overnight in the separate and conjoint presence of high concentrations of Arg (10 mm) and H4B (100 μm) (almost 10 times that of the physiological concentrations). None of the treatments, including the one carried out with both H4B and Arg, could remotely recover any dimer from the monomers, clearly demonstrating that PID binding induced irreversible monomerization of the iNOSfl protein (supplemental Fig. 1).
Kinetics and Affinity of PID Binding to the iNOS Monomer and Dimer
To elucidate the kinetic mechanism of binding of PID to the iNOS monomer and dimer, we used two monomeric mutants of iNOSoxy, namely K82AiNOSoxy, which is monomeric as purified but can be transformed into a partial dimer by incubation with H4B and Arg, and D92AiNOSoxy, which is also a monomer as purified but cannot be dimerized even with high concentrations of H4B and Arg (33). We studied their PID binding kinetics versus WT-iNOSoxy using rapid stopped-flow spectrophotometry at 22 °C, with the proteins preincubated with or without H4B (10 μm) and Arg (1 mm). All proteins used in such studies were examined for their dimer-monomer content by gel filtration chromatography (Fig. 4, insets) prior to being subjected to such kinetic analysis. The rates of PID binding to the iNOSoxy proteins were measured by change of absorbance at 427 nm. Fig. 4 shows that binding of PID (10 μm) to the WT-iNOSoxy protein (3 μm) in the absence of Arg and H4B followed a biphasic binding kinetics (Fig. 4A and Table 2), as was observed in the case of the H4B and Arg-treated K82AiNOSoxy (Fig. 4D and Table 2), which also yielded a protein with a mixed population of dimers and monomers (approximate percentage ratio of 60:40). In contrast, the completely monomeric iNOSoxy proteins, K82AiNOSoxy (Fig. 4C) and D92AiNOSoxy (Fig. 4E), as well as the completely dimeric WT-iNOSoxy protein (Fig. 4B) showed monophasic kinetics for PID binding (Fig. 4, C, E, and B, and Table 2). The apparent binding rates for PID binding to exclusively monomeric or dimeric proteins closely matched those recorded for its binding to the iNOSoxy proteins consisting of a mixture of dimers and monomers, consistent with there being only two kinetically distinct binding species (Table 2). Fig. 4F shows the gel filtration profiles of the PID-treated dimer-containing proteins represented in Fig. 4, A, B, and D, respectively, after 1 h of incubation with PID. Fig. 5 shows the rates for inhibitor binding to the iNOSoxy dimer (Fig. 5A) and monomer (Fig. 5B) as a function of PID concentration determined by stopped-flow kinetic analysis at 22 °C. The plots indicate a very slow apparent koff for PID binding for both the iNOS dimer (0.0012 s−1) and monomer (0.0053 s−1) as well as a comparatively higher apparent kon for the monomer (0.263 μm−1 s−1) than the dimer (0.0054 μm−1 s−1). Consequently, the apparent Kd (affinity constant) of PID for the iNOS monomer was calculated to be almost 11 times lower (20 nm) than that of the dimeric iNOS (222 nm).
FIGURE 4.

Rapid stopped-flow kinetic studies of PID binding to iNOSoxy monomer and dimer. Stopped-flow traces were collected at 427 nm after rapid mixing of PID (10 μm) to the depicted iNOSoxy proteins (3 μm) pretreated with or without H4B and Arg to generate the desired dimer-monomer populations (shown as insets). A, iNOSoxy (−Arg, −H4B), representing dimer-monomer mixture; B, iNOSoxy (+Arg,+H4B), representing 100% dimer; C, K82AiNOSoxy (−Arg, −H4B) representing 100% monomer; D, K82AiNOSoxy (+Arg, +H4B) representing dimer-monomer mixture. E, D92AiNOSoxy (−Arg, −H4B) representing 100% monomer. F shows the final gel filtration profiles and dimer-monomer content of the dimer containing iNOSoxy proteins shown in A, B, and D, respectively, after treatment with 10 μm PID for 1 h. Details of the experiment are described under “Experimental Procedures.” Rate constants obtained by fitting the increase in absorbance to single or double exponential functions are reported in Table 2. The calculated best fit curves are plotted as solid lines. mAU, milli-absorbance unit.
TABLE 2.
Apparent rate constants for PID binding to iNOSoxy proteins with different dimer-monomer contents as depicted in Fig. 4
Scheme and protocol of experiment are described under Fig. 4 and “Experimental Procedures.”
| Protein | Apparent rate constant k1 | Apparent rate constant k2 |
|---|---|---|
| s−1 | s−1 | |
| A: iNOSoxy (−Arg, −H4B) | 2.50 ± 0.18 | 0.055 ± 0.004 |
| B: iNOSoxy (+Arg, +H4B) | 0.06 ± 0.004 | |
| C: K82AiNOSoxy (−Arg, −H4B) | 2.44 ± 0.14 | |
| D: K82AiNOSoxy (+Arg, +H4B) | 2.20 ± 0.11 | 0.058 ± 0.005 |
| E: D92AiNOSoxy (−Arg, −H4B) | 2.62 ± 0.18 |
FIGURE 5.

Affinity of PID for the iNOS dimer and monomer. A, wild type iNOSoxy incubated overnight with Arg (1 mm) and H4B (10 μm), produced 100% dimeric iNOS, and B, K82A iNOSoxy mutant (−Arg, H4B) was used as a 100% monomeric iNOS control. PID binding to both the proteins was analyzed using rapid stopped-flow kinetic measurements at different concentrations of PID (0–10 μm) using 3 μm of the iNOSoxy protein. Rates of binding of PID to both dimeric (A) and monomeric (B) iNOSoxy proteins were plotted against concentrations of PID used. The line of best fit was determined and used for the calculation of kon (slope), koff (y intercept), and Kd (koff/kon) of PID for the iNOS dimer and monomer. Concentration-dependent effect of PID on the inhibition (C) as well as monomerization (D) of the WT-iNOSfl protein at 37 °C was also studied through spectrophotometric analysis of NO synthesis and gel filtration chromatographic analysis, respectively. Data represent mean of three distinct experiments. The S.E. for each data point was less than 10%.
We also carried out a study on the effect of PID concentration on the activity (Fig. 5C) of the WT-iNOSfl (2 μm) and the corresponding level of monomerization (Fig. 5D) over a period of 3 h at 37 °C carried under similar conditions as in Fig. 3. A steady drop in both activity and dimer content was observed up to ∼7 μm PID, after which the changes were less drastic up to ∼10 μm of PID when almost complete inhibition and monomerization of the enzyme were achieved. Here, too, the rate of inhibition of the enzyme was slightly higher than the rate of monomerization as observed in the corresponding time-dependent studies (Fig. 3)
PID-induced Inhibition of iNOS during Its Synthesis in RAW Cells
To study the functional effect of PID binding to iNOS during its physiological synthesis, we studied the effect of PID on the iNOSfl protein during its de novo synthesis in RAW cells under induction with LPS and IFNγ for 10 h. Because NO can block dimer formation in NOS (38), we used 4 mm l-NAME-treated cells to prevent NO formation from the synthesized iNOSfl during the experiment along with l-NAME-untreated counterparts (Fig. 6). We found that the total level of iNOSfl protein did not change due to PID treatment in the l-NAME-treated and -untreated cells (Fig. 6, A and B), although there was complete inhibition of the iNOS activity when the cells were treated with PID (10 μm) for 10 h during induction of the iNOS expression (Fig. 6C, samples 2 and 3), which was associated with an absence of dimeric iNOSfl (Fig. 6, A and B, lanes 2 and 3, and supplemental Fig. 2). There was a noticeable amount of dimer detected by low temperature SDS-PAGE immunoblots (Fig. 6A) of the iNOSfl purified from PID-untreated RAW cells that were induced for iNOS synthesis for 10 h. Expectedly, such dimer levels were considerably lower than that assessed through native gel filtration analysis (Fig. 6B, samples 4 and 5, and supplemental Fig. 2, sample 5). The activity of the iNOSfl purified from such inhibitor-untreated cells both in the presence and absence of l-NAME was significantly high (Fig. 6C, samples 4 and 5). That the iNOSfl proteins in the PID-treated cells were indeed PID-bound was confirmed by spectrophotometric analysis of the purified iNOSfl from such cells, which showed the typical low spin 427 nm absorbance maxima, for the heme-ligated imidazole group of PID (data not shown).
FIGURE 6.
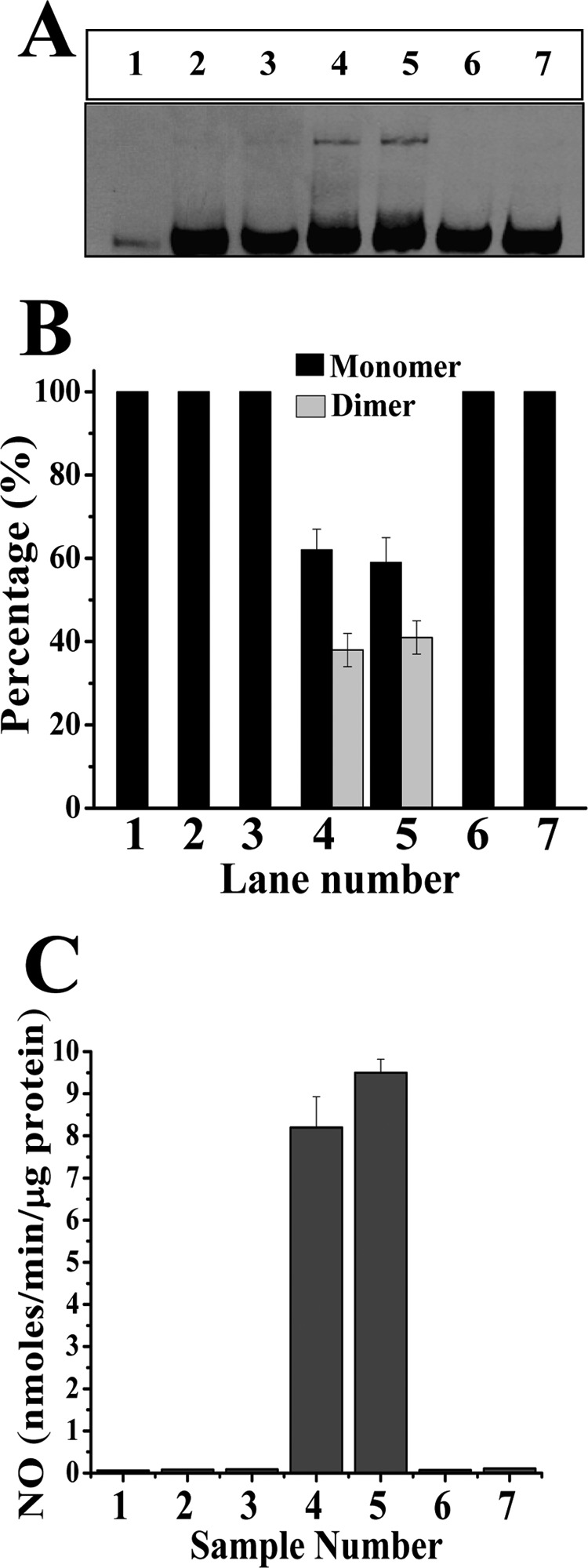
Effect of PID on iNOS during its de novo synthesis as well as after complete assembly into dimeric iNOS in induced RAW 264.7 cells. A shows the expression levels of iNOSfl and its relative dimer-monomer content in the extracts of PID-treated and -untreated cells as determined through low temperature SDS-PAGE immunoblotting of iNOSfl. RAW 264.7 cells were induced with LPS (50 μg/ml) and IFN-γ (10 ng/ml) to express iNOSfl in the presence (A, lanes 2, 4, and 6) or absence (A, lanes 3, 5, and 7) of l-NAME. Cells induced for 10 h without PID showed discernible amounts of dimer both in the presence (A, lane 4) or absence (A, lane 5) of l-NAME. PID (10 μm) was added either along with initiation of induction and harvested after 10 h (A, lanes 2 and 3) or after 10 h of induction following which cells were incubated for 6 h with PID before being harvested (A, lanes 6 and 7). The iNOS immunoblot of the uninduced control is shown in A, lane 1. B graphically depicts the percentage dimer-monomer content as evidenced through gel filtration chromatographic analysis of the purified iNOSfl from the treated cells. C shows the corresponding NOS activities in terms of the nitrite produced in a Griess reaction assay by the iNOSfl proteins purified from the above-treated cells.
The effect of PID on the fully assembled, dimer-containing iNOSfl protein was also studied in the iNOS-synthesizing RAW cells, by addition of the inhibitor after the completion of the 10-h induction period followed by incubation for an additional 6 h at 37 °C. This resulted in the generation of solely monomers (Fig. 6, A and B, lanes 6 and 7, and supplemental Fig. 2, sample 7) without catalytic activity, as judged from iNOSfl proteins purified from both the l-NAME-treated and -untreated cells under such conditions (Fig. 6C, samples 6 and 7).
Effect of PID on Fully Assembled Active iNOSfl in RAW Cells
To examine the precise physiological effect of PID on fully assembled (dimeric) iNOSfl protein in the RAW cells, and the time kinetics of its inhibition, we conducted a separate experiment in which RAW cells were induced with LPS and IFNγ for 14 h to ensure substantial formation of the fully formed dimeric iNOSfl protein, and thereafter we treated such cells with PID (10 μm) in fresh media to wash the cells free of LPS and IFNγ and to stop further iNOSfl synthesis (Fig. 7). The supplemental Fig. 3 depicts the gel filtration profiles of the iNOSfl protein in the PID-treated cells as a function of time of incubation, following 14 h of induction of iNOS synthesis. As is also apparent from the low temperature SDS-PAGE immunoblots of the iNOSfl proteins (Fig. 7A), the 14-h induction resulted in considerable accumulation of dimeric iNOSfl in all the PID-untreated controls (Fig. 7, lanes 1, 3, 5, 7, and 9), and there was no discernible monomerization or loss in dimer content (Fig. 7, A and B) nor loss of NO synthesis activity (Fig. 7C) in these cells that were further incubated for 2–8 h in fresh media without PID. However, incubating the cells with PID after a 14-h induction of iNOSfl synthesis caused a time-dependent monomerization of the iNOSfl dimer, prominently observable after 4 h of PID incubation and almost reaching completion at later time points (Fig. 7, A and B, and supplemental Fig. 3, samples 3, 5, and 7). The rate of monomerization as estimated by gel filtration chromatographic analysis of the iNOSfl proteins purified from the cell lysates of the PID-treated cells was estimated to be 0.38 min−1 (Fig. 7B and supplemental Fig. 3). This rate was very close to the loss of NO synthesis activity we observed in the same cell lysates (0.45 min−1) (Fig. 7C), confirming the effective PID-induced shift from active iNOSfl dimer to the inactive monomer.
FIGURE 7.
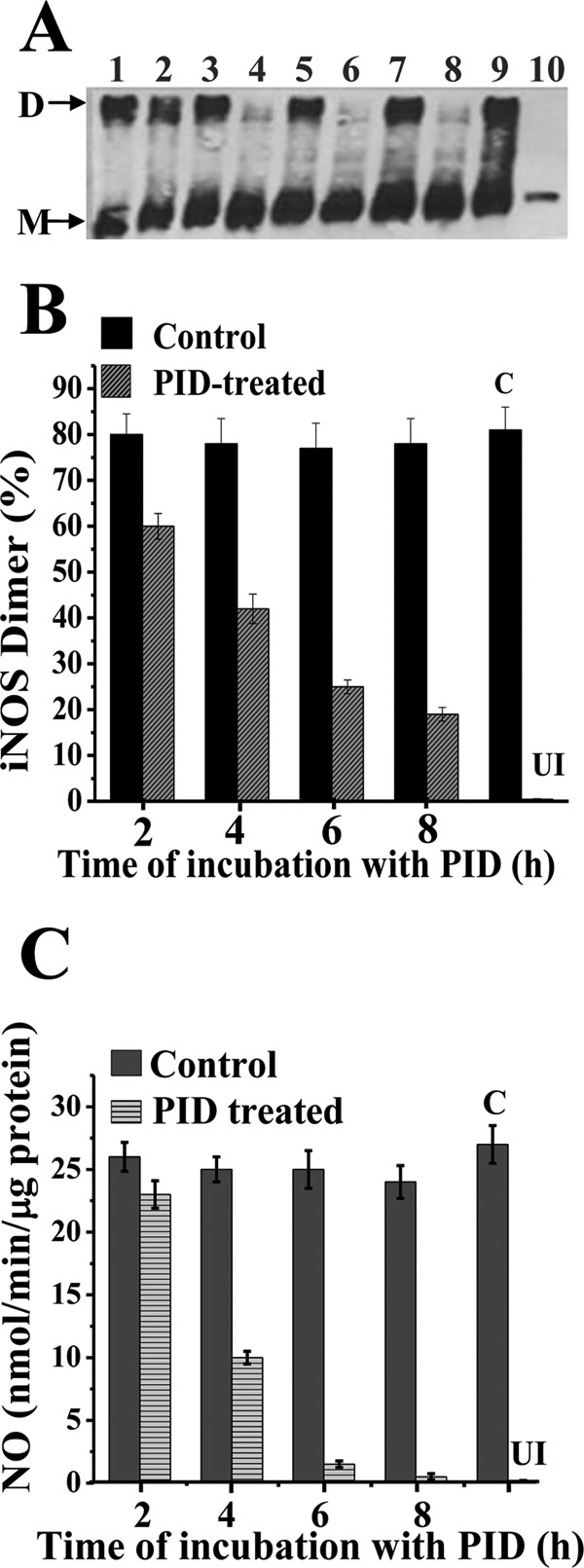
Kinetics of PID-induced monomerization and inhibition of mature dimeric iNOSfl protein in RAW 264.7 cells. RAW cells were induced with LPS (50 μg/ml) and IFN-γ (10 ng/ml) for 14 h, following which they were washed free of LPS and IFN-γ and transferred into fresh culture media to which either PID (10 μm) or an equivalent volume of 1× PBS was added and incubated for 2, 4, 6, and 8 h. Cells were either harvested after 14 h of induction (A, lane 9) or after incubation with or without PID for 2, 4, 6, and 8 h. A shows relative dimer-monomer profiles of the iNOSfl proteins as manifested through low temperature SDS-PAGE and immunoblotting of iNOSfl (D indicates dimer and M indicates monomer), and B and C show the corresponding dimer content from gel filtration chromatography analysis of the purified iNOSfl from the treated cells (supplemental Fig. 3) and their NO synthesis activities, respectively. Lanes 1, 3, 5, and 7 show the respective PID-untreated controls, and lanes 2, 4, 6, and 8 show the corresponding PID-treated iNOSfl after 2, 4, 6, and 8 h of incubation, respectively. Bold bars indicate PID-untreated (Control) and crossed bars represent the PID-treated samples (B and C). Results are representative of three independent experiments done under similar conditions. C and UI, indicate control and uninduced samples, respectively.
DISCUSSION
In this study, we demonstrate the unique capability of a bulky pyrimidine imidazole derivative, PID, to interact with both the functional entities of the iNOS enzyme, namely the iNOS dimer and monomer to either cause monomerization of already assembled active dimers or prevent dimerization of nascent monomers, through a near-irreversible binding of PID to the iNOS heme moiety. Our work also reveals the kinetic mechanism of iNOS inhibition by novel pyrimidine imidazoles like PID, especially with respect to its different rates of interaction with the iNOS heme iron when iNOS is in its dimeric and monomeric forms. Such novel findings bear considerable clinical significance given that an ideal candidate for therapeutic intervention of iNOS should be able to target and inhibit the fully assembled, active dimeric iNOSfl protein as well as the freshly developing monomeric form of the enzyme before it undergoes dimerization to render it functionally mature and active. This study for the first time demonstrates that novel pyrimidine imidazole compounds can accomplish both these functions.
The core compound, PIC, had significantly less iNOS inhibition potential and affinity than its modified analog PID. This suggests that the NOS binding as well as inhibitory properties of these compounds apparently resides in their core structure and that with appropriate modifications both the affinity of binding as well as potency of iNOS inhibition could be tuned to develop an ideal therapeutic iNOS inhibitor. In fact, modification of PIC to PID increased its binding affinity by 1.4 times and almost doubled its iNOS inhibition potential. Expectedly, H4B antagonized the binding of PID to iNOSfl protein when compared with Arg (by almost 1.5 times). This was consistent with the stronger iNOS dimer promoting and stabilizing capability of H4B compared with Arg (39). Such observation was also in agreement with crystal structure inputs, which revealed that in iNOS, H4B mobilizes dimer interaction through its binding with the heme propionate and the helix α7a to further reinforce the dimer through it dihydroxyprolyl side chain interaction with the enzyme's N-terminal hook. This apparently helps to create an iNOS “tight” or strong dimer (40). Interestingly, deletion of this N-terminal hook region renders only iNOS monomeric and nonfunctional, unlike other NOS isoforms (40). Thus, H4B's role in dimer reinforcement in iNOS is significantly stronger than Arg. H4B and Arg together were capable of decreasing the binding affinity of PID to the iNOSfl to the maximum extent even when used close to physiological concentrations in our study, much in agreement with crystal structure data that show H4B binding actually facilitates Arg interaction and its dimer reinforcement capacity by aligning the active center channel and hydrogen bonding to two elements involved in Arg binding, the heme propionate and the helix α7a (40). This perhaps helps to explain why Arg demonstrates better dimer reinforcing capacity when used in combination with H4B than alone. However, despite their combined dimer reinforcing effect, H4B and Arg could not prevent PID from causing monomerization of the iNOS tight dimer (Fig. 4F, B-PID).
In the absence of H4B in particular, iNOSoxy subunits have been found to interact through their helical lariat and helical T regions to form “loose” or weak dimers (39), which usually consist of a dynamic mix of monomers and dimers (Fig. 4A). Although the heme moiety is less stringently sequestered in such loose dimers compared with the tight dimer (41), the heme apparently is not openly accessible for direct PID binding, as the observed rate of PID binding to the heme in such a loose dimer (in the absence of H4B and Arg) is very similar to that observed for the tight dimer (in the presence of H4B and Arg) obtained through our rapid stopped-flow spectroscopic analysis (Table 2). In fact, Arg alone is not capable of forming a tight dimer like H4B, which as mentioned earlier is capable of mobilizing additional structural elements besides the helix α7a to form the tight dimer (40) even in the absence of Arg (39). This also explains why WT-iNOSoxy pretreated with both H4B and Arg showed exclusive dimeric behavior with respect to PID binding kinetics in our study (Fig. 4B and Table 2) as well as the fact that the behavior of Arg-bound iNOS was almost the same as Arg and H4B-free enzyme in response to PID treatment (Fig. 3).
Further deployment of two unique mutants, D92AiNOSoxy, a mutant existing as an irreversible monomer, that cannot be induced to form dimer with H4B, and K82AiNOSoxy, a mutant that can be induced to form partial dimer with H4B (33), helped us to mechanistically elucidate the precise kinetics of PID interaction with iNOS through rapid stopped-flow kinetic studies. Such studies revealed that PID interacted with the iNOS monomer with a profoundly lower Kd (almost 11 times lower) than compared with the iNOS dimer. Because the monophasic kinetic rates of PID binding to the pure iNOS monomer and dimer closely matched the biphasic kinetic rates observed for PID binding to iNOS proteins having mixed dimer-monomer populations (Table 2), it is clear that PID is capable of separately binding to the iNOS monomer and the dimer.
The apparent Kd value for PID binding to the iNOS monomer obtained in our study (20 nm) through rapid stopped-flow kinetic assessment of the absorbance change at 427 nm (spectral perturbation due to PID binding to the heme iron in the sixth axial position) is higher than that determined before through competitive radioligand binding assay (2–3 nm) (28). This discrepancy may be attributed to three major differences in the assay protocols. First, the concentration of enzyme required for stopped-flow kinetic evaluation is almost 1000-fold higher (3 μm) compared with the average concentration of enzyme (3 nm) used for the rapid filtration-based radioligand binding assay. Second, our stopped-flow binding analysis was performed at 22 °C, whereas the ligand binding assay was done at 37 °C (28). Finally, our spectral perturbation method is dependent on the PID imidazole binding to the heme iron (a consequential event following actual PID binding to the enzyme), whereas the radioligand-binding assay is based on competitive ligand interaction that is independent of heme ligation.
Our stopped-flow analysis revealed a kon of 0.263 μm−1 s−1 and a koff of 5.3 × 10−3 s−1 for the PID binding to the iNOS monomer heme and a kon of 0.0054 μm−1 s−1 and a koff of 1.2 × 10−3 s−1 for the PID binding to the iNOS dimer heme through the spectral perturbation measurement at 427 nm (recorded at 22 °C) giving an apparent Kd of 20 nm for PID binding to the iNOS monomer and 222 nm for the iNOS dimer. The extremely low koff values obtained for PID binding to both the monomer and dimer indicated that its binding to the iNOS enzyme as a whole was virtually irreversible as was also manifested by the failure of high concentrations of H4B and Arg to displace the inhibitor from the PID-iNOS complex in our study (supplemental Fig. 1).
Why are the kon values for PID binding to iNOS dimer and monomer so different? Crystal structures show that heme is tightly sequestered in the iNOS dimer and is perhaps not directly accessible for inhibitor binding, unless the heme distal pocket opens up to a sufficient extent to provide access to the imidazole moiety of PID, so that it can bind to the heme in the sixth axial position (27). Previous crystallographic studies revealed that two independent imidazoles can bind to two distinct sites in the iNOSoxy monomer, one directly coordinated to the heme and the other hydrogen-bonded to the Glu-371 residue (42). In comparison, the crystal structure of the PID-iNOS-monomer revealed that although the imidazole moiety of PID is bound to the heme, it is its benzodioxole moiety that actually binds to the Glu-371 residue, which is also the closest hydrophilic residue to the heme iron (27). It is thus likely that dimer binding by PID is initiated by its interaction with this hydrophilic and solvent-exposed glutamic acid residue that is not concealed within the dimer interface (42). Thus, in dimeric iNOS, PID may bind directly to the Glu-371 residue through its benzodioxole group, which may help to open up the heme distal pocket through the displacement of helix α7a and consequent perturbation of the helix 8a and residues 460–462 that form a part of the dimer interface as well as the H4B-binding site (27). Gradual opening of the heme distal pocket potentially serves as the first step for monomerization of the dimer as well as provides subsequent access of the PID imidazole to the heme iron's sixth axial position for accomplishing complete inhibitor binding to iNOS and creation of an irreversible iNOS-PID complex. This consequently forms an irreversible iNOS-monomer-inhibitor complex that is not recoverable even with high concentrations of H4B (100 μm) and Arg (10 mm) (supplemental Fig. 1). Thus, the heme distal pocket in the iNOS dimer is expected to gradually open up following PID binding to make the heme gradually accessible to inhibitor binding, in contrast to the open monomer where the heme is directly accessible for rapid binding by the inhibitor almost immediately. This may explain why the apparent kon value obtained for PID binding to the iNOS dimer was substantially lower than that of the iNOS monomer. Thus, the kon values for the iNOS monomer and dimer are perhaps more reflective of the rate of heme access by the imidazole of PID rather than the actual rate of its surface interaction to the two forms of the enzyme, and this may underlie the observed mismatch with the competitive ligand binding-based affinity constants determined in the previous study (28). In any case, our stopped-flow-based kinetic findings establish, for the first time, the unique capability of PID to interact with both the dimer and monomer for accomplishing catalytic inhibition of the two physiological forms of the iNOS enzyme.
Interestingly, although two free imidazole molecules could simultaneously bind to the iNOS monomer (42), only one bulky pyrimidine imidazole molecule was found to do so (27) during crystal structure studies. Thus, with the increase in bulkiness of PID-like pyrimidine imidazoles, the chances of dual inhibitor binding to the NOS monomer is probably reduced due to the steric inaccessibility of the second inhibitor molecule for binding when one inhibitor molecule is already bound. This would predictably allow a binding ratio of 1:1 between the iNOS monomer and PID and 1:2 between the inhibitor and the iNOS dimer (composed of two monomeric subunits).
Indeed, the observed rate of inhibitor binding as measured through its binding to the iNOS heme is comparatively faster than the rate of enzyme inhibition, and furthermore, the observed rate of such inhibition is somewhat higher than that of the corresponding rate of monomerization. As dimer binding by PID precedes binding to the iNOS heme (causing spectral shift at 427 nm), it is unlikely that the proposed binding of the inhibitor to the dimer through the initial interaction between its benzodioxole moiety and the Glu-371 residue (27) elicits immediate inhibition of the dimeric iNOS, as evidenced by the results obtained in our study (Fig. 3 and Table 1). This can be possibly because such interaction does not substantially disturb the effective interaction between the reductase and oxygenase domains in the dimeric iNOS for productive electron transfer and the consequent heme reduction and NO generation. Thus, the immediate inhibitor-bound iNOS dimer is probably not inactive. However, when such binding paves the way for subsequent disruption of helix α7a, it partially opens up the dimer interface for making access for the binding of the imidazole moiety of the inhibitor to the heme, which potentially initiates the active dimer ripping into monomers. In fact, such dimer disruption into monomers is likely to undergo a transition through a phase in which the effective orientation of the specific electron-exchange motifs between the reductase and the oxygenase domains would no longer support interdomain electron transfer. This would in turn prevent NO formation and render the enzyme inactive even when the dimer has not completely parted into monomers. This would imply that the dimeric enzyme can start losing its activity even before it undergoes substantial monomerization due to PID binding. This may also explain why the rate of inhibition is observed to be higher than the rate of monomerization in our study (Fig. 3 and Table 1). Thus, it appears that the event of inhibition initiates soon after the inhibitor's imidazole moiety binds to the NOS heme and initiates “dimer separation” subsequently leading to irreversible inhibition of the enzyme with the complete monomerization. This possibly explains why the rate of inhibition does not strictly match but is slightly higher than the rate of monomerization of iNOS in our study (Fig. 3 and Table 1). Fig. 8 delineates such a possible mechanism underlying PID interaction with iNOS for achieving holistic inhibition of the enzyme based on our current data.
FIGURE 8.

Scheme for PID-induced inhibition of iNOS. The scheme pictorially describes the mode of inhibition of iNOS by PID as well as the fate of the iNOS monomer and dimer after PID binding. In the proposed model, PID binds to the Glu-371 (E-371) residue of the heme containing iNOS monomer (v) through its benzodioxole moiety (B) as well as the free sixth axial position of the heme iron through its imidazole moiety (I) almost simultaneously and forms an irreversible iNOS-monomer-PID complex (iv). However, in the iNOS dimer (i), the benzodioxole moiety of PID initiates binding to the open Glu-371 residue and consequently disturbs helix α7a and thereafter helix 8 that form the critical part of the dimer interface (ii). This apparently helps to slowly open up the heme distal pocket to provide direct access for the unbound imidazole moiety of the already iNOS-Glu-371-bound PID to the iNOS heme (that was previously sequestered), which on coordinating with the sixth axial position of the heme iron initiates dimer separation (iii) and apparently causes loss of effective contact between the reductase and oxygenase domains and abrogation of the functional inter-domain electron transfer and deactivation of the enzyme, even when the dimers are not fully parted. This subsequently leads to the irreversible monomerization of the enzyme through complete steric separation of the two monomeric subunits constituting the iNOS dimer and formation of an irreversible iNOS-monomer-PID complex (iv) that is not recoverable to the active dimer even with high concentrations of H4B and Arg.
To tie our observations obtained with pure iNOS proteins to the physiological system, we treated RAW cells (that were induced with LPS and IFNγ for synthesis of iNOSfl) with PID under different conditions. We found that when the induced RAW cells were treated with PID from the beginning of the induction, only inactive monomeric iNOSfl proteins showing 427 nm absorption maxima were produced. This indicated that PID was capable of binding newly synthesizing iNOS proteins to form PID-iNOS monomers that were incapable of forming dimers. When PID was added to the cells after 10 h of induction, during which discernible amounts of active iNOS dimers were detectable in the cells, and subsequently incubated for 6 h, it resulted in the accumulation of monomeric iNOSfl, establishing that the inhibitor could also physiologically interact and monomerize the mature iNOS dimer, as seen in our in vitro studies.
We also treated RAW cells with PID after induction of iNOS expression for 14 h for different time periods, before harvesting the cells for measurement of iNOSfl monomer-dimer levels. We observed a slow but steady monomerization and deactivation of the dimeric iNOSfl protein over a period of 8 h with attainment of almost complete inhibition in 6 h of incubation with PID. This indicated that PID may take up to 6 h to completely inhibit the active dimeric iNOS to accomplish total inhibition of the available pool of the enzyme in a physiological setting.
We also carried out native gel filtration chromatographic analysis of iNOS proteins for assessing their dimer-monomer content and validated them with corresponding low temperature gel electrophoresis immunoblots in our study. In fact, such a gel electrophoresis technique has been used effectively in several studies to detect the NOS dimer, particularly the tight dimer and monomer (15, 39), although this method is not so effective in depicting the NOS loose dimer as it usually scores such dimers as monomers in contrast to the NOS tight dimer, which it can effectively portray as dimeric. This is in sharp contrast to the gel filtration analysis where both the loose and tight dimers are depicted as dimeric with appreciable reproducibility (37). This difference possibly serves as a clue for the existence of the NOS loose and tight dimers. It may also explain why the dimer content of iNOS measured through gel filtration in our study is comparatively higher than that assessed through corresponding low temperature gel electrophoresis, which is comparatively harsher to the loose or weak dimers when compared with the sensitive gel filtration technique. Thus, dimer analysis through gel filtration measures the total dimer (loose as well as tight), whereas the low temperature gel electrophoresis specifically evaluates the tight dimer in the total dimer population.
PID has almost 1000 times more affinity than free imidazole for the iNOS monomer (43). Notably, the three isoforms of NOS, in particular iNOS, utilize different structural elements as well as dependence on H4B and Arg for reinforcing their dimer interaction, and they show a rank order in their dimer strengths of eNOS > nNOS > iNOS (39). In particular, a 49-residue motif (66–114) in the N-terminal region of iNOS, containing an additional H4B-binding motif and a β-hairpin hook, was found to be indispensable for dimerization in our previous studies (33, 34). Deletion of analogous residues from the N-terminal regions in nNOS and eNOS did not completely abrogate their capability for dimer formation (44, 45). Such differences may explain the different affinities and dimerization inhibitory potential of pyrimidine imidazoles for the three NOS isoforms (27, 30). In fact, the inhibitory potency of bulky pyrimidine imidazoles was found to be in the order of eNOS/nNOS/iNOS as 1:200:1000 (27). Indeed, based on such substantial differences, it has been possible to use them within thresholds of a defined dose window to make them act as specific iNOS inhibitors.
This work transcends all previous reports on the mechanism of iNOS inhibition by pyrimidine imidazoles (26–28) primarily as it unveils critical aspects of interaction of such inhibitors with the active dimeric form of the enzyme for the first time and thereby reveals the true potential of such compounds to act as candidates for clinical control of this physiologically important enzyme. Indeed, our observation of the interaction of PID with the iNOS dimer and demonstration of its capability to accomplish irreversible dissociation and consequent inhibition of the active dimeric form of the enzyme is seminal. Second, this is also the first kinetic study on the relative affinity and binding of pyrimidine imidazoles separately with the two physiological forms of iNOS, namely its dimer and monomer, providing clinically fundamental information on kon and koff of the compound and revealing a kinetic difference of 11-fold on the interaction of such compounds with the monomeric (inactive) versus dimeric (active) forms of the iNOS enzyme, again a vital information that has not been reported before. This work also establishes an unprecedented functional link between the kinetic mechanism of interaction of pyrimidine imidazoles with the iNOS monomer and the dimer in vitro and the actual physiological mechanism of their action on these two functional entities of the enzyme when expressed in induced cells. Finally, our work is the first demonstration of the efficacy of these compounds in a physiological setting, showing its ability to both target and inhibit iNOS during its synthesis or formative stage as well as in its post-translational matured state in cells engaged in dynamic expression of the enzyme. Overall, our findings clearly reveal that these compounds can act as the much required holistic inhibitors of iNOS under a clinical setting, and thereby opens up a renewed scope for evaluating these compounds as therapeutically applicable drugs for combating the numerous diseases causally associated with iNOS overexpression as well as a powerful research tool for studying the role of this enzyme in vivo.
This report is thus of significant importance amid the quest for a nontoxic and versatile iNOS inhibitor for elucidating the role of this enzyme in important physiological and pathophysiological life processes as well as clinically achieving the desired level of inhibition of this enzyme when its overexpression is physiologically harmful. In addition, the open prospects of improving both the affinity as well as inhibition potency of such compounds through appropriate chemical modifications of the core molecule, PIC may also encourage further exploration of such compounds, particularly for their unique capability to physiologically target both the iNOS monomer and dimer and achieve holistic inhibition of the enzyme. Thus, such a novel inhibitor like PID certainly deserves more attention on its development and functional exploration as a candidate for therapeutic intervention of iNOS activity as well as an in vivo research tool for understanding the possible causal role of this enzyme in vital life processes and diseases associated with them. Our present work would hopefully create the required premises for such future endeavors.
Supplementary Material
Acknowledgment
We thank Deborrah Durra for excellent technical support in the Stuehr laboratory.
This work was supported, in whole or in part, by UGC UPE (Modern Biology) research grant (to K.P.) and by National Institute of Health Grants GM51491, CA53914, and HL58883 (to D. J. S.) and INSPIRE-DST Fellowship, Government of India, and Fulbright-Nehru Doctoral and Professional Research Fellowship (to L. N.).

This article contains supplemental Figs. 1–3.
- NOS
- nitric-oxide synthase
- eNOS
- endothelial NOS
- iNOS
- inducible NOS
- nNOS
- neuronal NOS
- iNOSoxy
- inducible NOS oxygenase domain
- iNOSfl
- inducible NOS full-length protein
- PIC
- pyrimidine imidazole core compound
- PID
- pyrimidine imidazole derivative
- H4B
- (6R)-5,6,7,8-tetrahydro-l-biopterin
- EPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid.
REFERENCES
- 1. Daff S. (2010) NO synthase: structures and mechanisms. Nitric Oxide 23, 1–11 [DOI] [PubMed] [Google Scholar]
- 2. Li H., Poulos T. L. (2005) Structure-function studies on nitric oxide synthases. J. Inorg. Biochem. 99, 293–305 [DOI] [PubMed] [Google Scholar]
- 3. Alderton W. K., Cooper C. E., Knowles R. G. (2001) Nitric oxide synthases: structure, function, and inhibition. Biochem. J. 357, 593–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bredt D. S., Hwang P. M., Snyder S. H. (1990) Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 347, 768–770 [DOI] [PubMed] [Google Scholar]
- 5. Nakane M., Schmidt H. H., Pollock J. S., Förstermann U., Murad F. (1993) Cloned human brain nitric oxide synthase is highly expressed in skeletal muscle. FEBS Lett. 316, 175–180 [DOI] [PubMed] [Google Scholar]
- 6. Martin W., Furchgott R. F., Villani G. M., Jothianandan D. (1986) Depression of contractile responses in rat aorta by spontaneously released endothelium-derived relaxing factor. J. Pharmacol. Exp. Ther. 237, 529–538 [PubMed] [Google Scholar]
- 7. O'Dell T. J., Huang P. L., Dawson T. M., Dinerman J. L., Snyder S. H., Kandel E. R., Fishman M. C. (1994) Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science 265, 542–546 [DOI] [PubMed] [Google Scholar]
- 8. Cho H. J., Xie Q. W., Calaycay J., Mumford R. A., Swiderek K. M., Lee T. D., Nathan C. (1992) Calmodulin is a subunit of nitric oxide synthase from macrophages. J. Exp. Med. 176, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michel T., Feron O. (1997) Nitric oxide synthases: which, where, how, and why? J. Clin. Invest. 100, 2146–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schmidt H. H., Walter U. (1994) NO at work. Cell 78, 919–925 [DOI] [PubMed] [Google Scholar]
- 11. Stuehr D. J. (1999) Mammalian nitric oxide synthases. Biochim. Biophys. Acta 1411, 217–230 [DOI] [PubMed] [Google Scholar]
- 12. Baek K. J., Thiel B. A., Lucas S., Stuehr D. J. (1993) Macrophage nitric oxide synthase subunits: Purification, characterization, and role of prosthetic groups and substrate in regulating their association into a dimeric enzyme. J. Biol. Chem. 268, 21120–21129 [PubMed] [Google Scholar]
- 13. Klatt P., Pfeiffer S., List B. M., Lehner D., Glatter O., Bächinger H. P., Werner E. R., Schmidt K., Mayer B. (1996) Characterization of heme-deficient neuronal nitric-oxide synthase reveals a role for heme in subunit dimerization and binding of the amino acid substrate and tetrahydrobiopterin. J. Biol. Chem. 271, 7336–7342 [DOI] [PubMed] [Google Scholar]
- 14. Rodríguez-Crespo I., Gerber N. C., Ortiz de Montellano P. R. (1996) Endothelial nitric-oxide synthase: Expression in Escherichia coli, spectroscopic characterization, and role of tetrahydrobiopterin in dimer formation. J. Biol. Chem. 271, 11462–11467 [DOI] [PubMed] [Google Scholar]
- 15. Panda K., Ghosh S., Stuehr D. J. (2001) Calmodulin activates intersubunit electron transfer in the neuronal nitric-oxide synthase dimer. J. Biol. Chem. 276, 23349–23356 [DOI] [PubMed] [Google Scholar]
- 16. Ichinose F., Hataishi R., Wu J. C., Kawai N., Rodrigues A. C., Mallari C., Post J. M., Parkinson J. F., Picard M. H., Bloch K. D., Zapol W. M. (2003) A selective inducible NOS dimerization inhibitor prevents systemic, cardiac, and pulmonary hemodynamic dysfunction in endotoxemic mice. Am. J. Physiol. Heart Circ. Physiol. 285, H2524–H2530 [DOI] [PubMed] [Google Scholar]
- 17. Enkhbaatar P., Murakami K., Shimoda K., Mizutani A., Traber L., Phillips G., Parkinson J., Salsbury J. R., Biondo N., Schmalstieg F., Burke A., Cox R., Hawkins H., Herndon D., Traber D. (2003) Inducible nitric oxide synthase dimerization inhibitor prevents cardiovascular and renal morbidity in sheep with combined burn and smoke inhalation injury. Am. J. Physiol. Heart Circ. Physiol. 285, H2430–H2436 [DOI] [PubMed] [Google Scholar]
- 18. Lincoln J., Hoyle C. H., Burnstock G. (1997) Nitric Oxide in Health and Diseases. pp. 1–356, Cambridge University Press, Cambridge, UK [Google Scholar]
- 19. Stuehr D. J. (1997) Structure-function aspects in the nitric oxide synthases. Annu. Rev. Pharmacol. Toxicol. 37, 339–359 [DOI] [PubMed] [Google Scholar]
- 20. Kröncke K. D., Fehsel K., Kolb-Bachofen V. (1995) The action of NO and its role in autoimmune diabetes mellitus. Biol. Chem. Hoppe-Seyler 376, 327–343 [DOI] [PubMed] [Google Scholar]
- 21. Milstien S., Sakai N., Brew B. J., Krieger C., Vickers J. H., Saito K., Heyes M. P. (1994) Cerebrospinal fluid nitrite/nitrate levels in neurologic diseases. J. Neurochem. 63, 1178–1180 [DOI] [PubMed] [Google Scholar]
- 22. Bieńkowska-Haba M., Liebhart J., Cembrzyńska-Nowak M. (2006) Nitric oxide production by pulmonary leukocytes from induced sputum in patients with asthma and its effect on epithelial cell viability. Arch. Immunol. Ther. Exp. 54, 201–207 [DOI] [PubMed] [Google Scholar]
- 23. Radomski M. W., Palmer R. M., Moncada S. (1990) Glucocorticoids inhibit the expression of an inducible, but not the constitutive, nitric oxide synthase in vascular endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 87, 10043–10047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boullerne A. I., Petry K. G., Meynard M., Geffard M. (1995) Indirect evidence for nitric oxide involvement in multiple sclerosis by characterization of circulating antibodies directed against conjugated S-nitrosocysteine. J. Neuroimmunol. 60, 117–124 [DOI] [PubMed] [Google Scholar]
- 25. Griffith O. W., Stuehr D. J. (1995) Nitric oxide synthases: properties and catalytic mechanism. Annu. Rev. Physiol. 57, 707–736 [DOI] [PubMed] [Google Scholar]
- 26. Sennequier N., Wolan D., Stuehr D. J. (1999) Antifungal imidazoles block assembly of inducible NO synthase into an active dimer. J. Biol. Chem. 274, 930–938 [DOI] [PubMed] [Google Scholar]
- 27. McMillan K., Adler M., Auld D. S., Baldwin J. J., Blasko E., Browne L. J., Chelsky D., Davey D., Dolle R. E., Eagen K. A., Erickson S., Feldman R. I., Glaser C. B., Mallari C., Morrissey M. M., Ohlmeyer M. H., Pan G, Parkinson J. F., Phillips G. B., Polokoff M. A., Sigal N. H., Vergona R., Whitlow M., Young T. A., Devlin J. J. (2000) Allosteric inhibitors of inducible nitric oxide synthase dimerization discovered via combinatorial chemistry. Proc. Natl. Acad. Sci. U.S.A. 97, 1506–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blasko E., Glaser C. B., Devlin J. J., Xia W., Feldman R. I., Polokoff M. A., Phillips G. B., Whitlow M., Auld D. S., McMillan K., Ghosh S., Stuehr D. J., Parkinson J. F. (2002) Mechanistic studies with potent and selective inducible nitric-oxide synthase dimerization inhibitors. J. Biol. Chem. 277, 295–302 [DOI] [PubMed] [Google Scholar]
- 29. Gahman T. C., Herbert M. R., Lang H., Thayer A., Symons K. T., Nguyen P. M., Massari M. E., Dozier S., Zhang Y., Sablad M., Rao T. S., Noble S. A., Shiau A. K., Hassig C. A. (2011) Identification and SAR of selective inducible nitric oxide synthase (iNOS) dimerization inhibitors. Bioorg. Med. Chem. Lett. 21, 6888–6894 [DOI] [PubMed] [Google Scholar]
- 30. Panda K., Chawla-Sarkar M., Santos C., Koeck T., Erzurum S. C., Parkinson J. F., Stuehr D. J. (2005) Visualizing inducible nitric-oxide synthase in living cells with a heme-binding fluorescent inhibitor. Proc. Natl. Acad. Sci. U.S.A. 102, 10117–10122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davey D. D., Adler M., Arnaiz D., Eagen K., Erickson S., Guilford W., Kenrick M., Morrissey M. M., Ohlmeyer M., Pan G., Paradkar V. M., Parkinson J., Polokoff M., Saionz K., Santos C., Subramanyam B., Vergona R., Wei R. G., Whitlow M., Ye B., Zhao Z. S., Devlin J. J., Phillips G. (2007) Design, synthesis, and activity of 2-imidazol-1-ylpyrimidine-derived inducible nitric oxide synthase dimerization inhibitors. J. Med. Chem. 50, 1146–1157 [DOI] [PubMed] [Google Scholar]
- 32. Wu C., Zhang J., Abu-Soud H., Ghosh D. K., Stuehr D. J. (1996) High-level expression of mouse inducible nitric oxide synthase in Escherichia coli requires co-expression with calmodulin. Biochem. Biophys. Res. Commun. 222, 439–444 [DOI] [PubMed] [Google Scholar]
- 33. Ghosh D. K., Crane B. R., Ghosh S., Wolan D., Gachhui R., Crooks C., Presta A., Tainer J. A., Getzoff E. D., Stuehr D. J. (1999) Inducible nitric oxide synthase: role of the N-terminal β-hairpin hook and pterin-binding segment in dimerization and tetrahydrobiopterin interaction. EMBO J. 18, 6260–6270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ghosh D. K., Wu C., Pitters E., Moloney M., Werner E. R., Mayer B., Stuehr D. J. (1997) Characterization of the inducible nitric oxide synthase oxygenase domain identifies a 49-amino acid segment required for subunit dimerization and tetrahydrobiopterin interaction. Biochemistry 36, 10609–10619 [DOI] [PubMed] [Google Scholar]
- 35. Adak S., Ghosh S., Abu-Soud H. M., Stuehr D. J. (1999) Role of reductase domain cluster 1 acidic residues in neuronal nitric-oxide synthase. Characterization of the FMN-free enzyme. J. Biol. Chem. 274, 22313–22320 [DOI] [PubMed] [Google Scholar]
- 36. Stuehr D. J., Marletta M. A. (1987) Synthesis of nitrite and nitrate in murine macrophage cell lines. Cancer Res. 47, 5590–5594 [PubMed] [Google Scholar]
- 37. Klatt P., Schmidt K., Lehner D., Glatter O., Bächinger H. P., Mayer B. (1995) Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and l-arginine in the formation of an SDS-resistant dimer. EMBO J. 14, 3687–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Albakri Q. A., Stuehr D. J. (1996) Intracellular assembly of inducible NO synthase is limited by nitric oxide-mediated changes in heme insertion and availability. J. Biol. Chem. 271, 5414–5421 [DOI] [PubMed] [Google Scholar]
- 39. Panda K., Rosenfeld R. J., Ghosh S., Meade A. L., Getzoff E. D., Stuehr D. J. (2002) Distinct dimer interaction and regulation in nitric-oxide synthase types I, II, and III. J. Biol. Chem. 277, 31020–31030 [DOI] [PubMed] [Google Scholar]
- 40. Crane B. R., Arvai A. S., Ghosh D. K., Wu C., Getzoff E. D., Stuehr D. J., Tainer J. A. (1998) Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science 279, 2121–2126 [DOI] [PubMed] [Google Scholar]
- 41. Pant K., Bilwes A. M., Adak S., Stuehr D. J., Crane B. R. (2002) Structure of a nitric oxide synthase heme protein from Bacillus subtilis. Biochemistry 41, 11071–11079 [DOI] [PubMed] [Google Scholar]
- 42. Crane B. R., Arvai A. S., Gachhui R., Wu C., Ghosh D. K., Getzoff E. D., Stuehr D. J., Tainer J. A. (1997) The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science 278, 425–431 [DOI] [PubMed] [Google Scholar]
- 43. Fedorov R., Vasan R., Ghosh D. K., Schlichting I. (2004) Structures of nitric oxide synthase isoforms complexed with the inhibitor AR-R17477 suggest a rational basis for specificity and inhibitor design. Proc. Natl. Acad. Sci. U.S.A. 101, 5892–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Panda K., Adak S., Aulak K. S, Santolini J., McDonald J. F, Stuehr D. J. (2003) Distinct influence of N-terminal elements on neuronal nitric-oxide synthase structure and catalysis. J. Biol. Chem. 278, 37122–37131 [DOI] [PubMed] [Google Scholar]
- 45. Rodríguez-Crespo I., Moënne-Loccoz P., Loehr T. M., Ortiz de Montellano P. R. (1997) Endothelial nitric oxide synthase: modulations of the distal heme site produced by progressive N-terminal deletions. Biochemistry 36, 8530–8538 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.