

Background: Cyclin/CDKs play an important role in cell cycle regulation.
Results: MCM7 can be phosphorylated at Serine-121 by cyclin E/Cdk2 and cyclin B/Cdk1. Excess of phosphorylated MCM7 blocks S phase entry, and its phosphorylation is essential for proper mitotic exit.
Conclusion: Cyclin/CDKs can phosphorylate MCM7 to regulate cell cycle progression.
Significance: We revealed the novel function of MCM7 on checkpoint activation and mitotic exit regulated by cyclin/Cdks.
Keywords: Cyclin-dependent kinase (CDK), Cell Cycle, Cell Signaling, Cyclins, Phosphorylation, MCM7
Abstract
MCM7 is one of the subunits of the MCM2–7 complex that plays a critical role in DNA replication initiation and cell proliferation of eukaryotic cells. After forming the pre-replication complex (pre-RC) with other components, the MCM2–7 complex is activated by DDK/cyclin-dependent kinase to initiate DNA replication. Each subunit of the MCM2–7 complex functions differently under regulation of various kinases on the specific site, which needs to be investigated in detail. In this study, we demonstrated that MCM7 is a substrate of cyclin E/Cdk2 and can be phosphorylated on Ser-121. We found that the distribution of MCM7-S121A is different from wild-type MCM7 and that the MCM7-S121A mutant is much less efficient to form a pre-RC complex with MCM3/MCM5/cdc45 compared with wild-type MCM7. By using the Tet-On inducible HeLa cell line, we revealed that overexpression of wild-type MCM7 but not MCM7-S121A can block S phase entry, suggesting that an excess of the pre-RC complex may activate the cell cycle checkpoint. Further analysis indicates that the Chk1 pathway is activated in MCM7-overexpressed cells in a p53-dependent manner. We performed experiments with the human normal cell line HL-7702 and also observed that overexpression of MCM7 can cause S phase block through checkpoint activation. In addition, we found that MCM7 could also be phosphorylated by cyclin B/Cdk1 on Ser-121 both in vitro and in vivo. Furthermore, overexpression of MCM7-S121A causes an obvious M phase exit delay, which suggests that phosphorylation of MCM7 on Ser-121 in M phase is very important for a proper mitotic exit. These data suggest that the phosphorylation of MCM7 on Ser-121 by cyclin/Cdks is involved in preventing DNA rereplication as well as in regulation of the mitotic exit.
Introduction
In eukaryotes, chromosomal DNA replication is a precise event to maintain genome stability that needs to be duplicated only once in each cell cycle. This is accomplished by accurate cell cycle-dependent licensing control (1). During late mitosis and early G1 phase, origin recognition complex, Cdt1, and cdc6 are loaded at replication origins independently and then recruit the minichromosome maintenance (MCM)2 protein MCM2–7 complex onto chromatin sequentially under regulation of mitotic kinases (2, 3). Then, the MCM2–7 complex is triggered into an active helicase at replication forks by concerted activity of cdc7/dbf4 (4) and cyclin E/Cdk2 at the G1-S transition (5), which ultimately leads to recruitment of additional factors to pre-RCs, resulting in the initiation of DNA replication. When cells enter into S phase, the pre-RC is disassociated by high cyclin-dependent kinase (CDK) activity, which will cause destruction of select pre-RC components and the inhibition of pre-RC rebinding onto chromatin and prevent DNA rereplication (5, 6). Therefore, the substrates of each kinase complex are the key subjects to be investigated in detail for a comprehensive study of tight cell cycle regulation.
The most important function of the MCM2–7 complex is to initiate DNA replication through replicative helicase activity at replication forks (7). However, each MCM component functions differently in cell cycle regulation. It has been reported that only the subassembly of the MCM4/6/7 double-trimeric complex possesses DNA helicase activity, whereas MCM2/3/5 does not. The conserved ATPase motifs of MCM7 are essential for ATPase and DNA helicase activities of the MCM4/6/7 complex (8). It has been elucidated that MCM3 phosphorylated by cyclin B/Cdk1 regulates MCM2–7 complex regulation (9), whereas phosphorylation of MCM3 on Thr-722 will cause S phase checkpoint activation under regulation of cyclin E/Cdk2 (10). Meanwhile, there is more and more evidence to show that MCMs have other functions beyond DNA replication initiation (11). Previous studies found that MCM2 and MCM3 can be phosphorylated directly by ataxia telangiectasia-mutated/ATR separately under genotoxic stress (12). MCM5 was confirmed to load onto the centrosome in the association of cyclin E and regulate centrosome duplication (13). It was also found that MCM7 participates in mRNA transcription and DNA damage regulation (12, 14, 15). It has also been reported that MCM7 is a typical marker of various types of cancer and is a potential therapeutic target in human cancers (16–18).
We previously identified MCM7 as one of the Cdk2-associated proteins by tandem affinity-tagged purification (19). In this report, we demonstrated that MCM7 is the substrate of cyclin E/Cdk2 that phosphorylates MCM7 at Ser-121 and regulates its distribution in cells. We also found that an excess of MCM7 but not MCM7-S121A blocks S phase progression. Further analysis indicated that an excess of MCM7 can activate the Chk1-p53 checkpoint pathway through ssDNA accumulation in a p53-dependent manner. In addition, we found that MCM7 could also be phosphorylated by cyclin B/Cdk1 on Ser-121, which is essential for mitotic exit. Collectively, phosphorylations of MCM7 by cyclin/Cdks plays an important role in S phase checkpoint activation as well as in the regulation of proper M phase progression.
EXPERIMENTAL PROCEDURES
Plasmids and Antibodies
MCM7 cDNA was obtained by PCR from human embryonic kidney cDNA and cloned into pENTR-6 (a modified form of pENTR-4, Invitrogen) and then into pDEST-FLAG by Gateway® recombination cloning technology (Invitrogen). MCM7 and its mutants MCM7-S121A, MCM7-S197A, MCM7-S365A, and MCM-T690A were cloned into pCMV FLAG, pET41b, and pGEX6p-1, respectively. The MCM7 single RXL mutants ASA, ATA, and AEA and the triple mutant 3AXA were also cloned into pCMV FLAG. Full-length MCM7, Cdk2, and Cdk1 were cloned into pET41b and pET30a (Novagen). The mammalian expression vectors for myc-tagged cyclins and Cdks were generated as described previously (19).
The polyclonal antibodies against human MCM7, MCM5, and MCM3 were generated by immunizing the rabbits with GST-MCM7 (amino acids 10–30), GST-MCM5 (amino acids 644–734), or GST-MCM3 (amino acids 490–807). FLAG antibody (M2) was purchased from Sigma. Myc (9E10), His (H-3), MCM7 (141–2), cdc45 (H-300), PCNA (PC-10), Lamin B (M-20), Cdk2 (d-12), cdc6 (180.2), and β-actin (1–19) antibodies were purchased from Santa Cruz Biotechnology. Phospho-Chk1 (Ser-345) antibody (2341S) were purchased from Cell Signaling Technology. Phospho-Cdk2-Thr-14 antibody (EP2234Y) was purchased from Abcam. PRA70 antibody (04-1521) was purchased from Millipore. Phospho-MCM7-Ser-121 antibody was generated by immunizing the rabbits with synthesized phosphopeptide (GMVRpSPQNQYPAE).
Coimmunoprecipitation and GST Pull-down
myc-tagged cyclin and Cdk together with FLAG-tagged MCM7 or its RXL mutants were coexpressed in 293T cells by transient transfection. The cell lysates were harvested and immunoprecipitated with myc or FLAG antibodies and subjected to immunoblot analysis with the indicated antibodies. For the GST pull-down assay, 1 μg of GST-MCM7 or GST as a control was incubated with the cell lysates from 293T cells overexpressing myc-tagged cyclin and Cdk or with His-Cdk purified from BL21. Glutathione beads were then added and incubated for 2 h. The bound proteins were eluted with sample loading buffer and analyzed by immunoblotting with myc or Cdk antibodies. For endogenous immunoprecipitation, 293T cell lysates were immunoprecipitated with Cdk antibody or normal mouse IgG as a control, followed by incubation with protein A beads. The bound proteins were subjected to immunoblot analysis with MCM7 antibody.
Cell Culture and Synchronization
HEK 293T, HeLa, and HCT116 cells were cultured in DMEM containing 10% fetal calf serum. Human normal liver cells HL-7702 (Cell Resource Center, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences) was cultured in RPMI 1640 medium containing 20% fetal calf serum. For synchronization, cells were synchronized at G1/S phase by double thymidine treatment as described previously (10). In brief, HeLa cells were incubated for 16 h in complete medium with 2 mm thymidine, released for 8 h into fresh medium, and then incubated with 2 mm thymidine for 16 h. At this point, most of the cells were synchronized at G1/S transition. To obtain cells synchronized at mitosis, HeLa cells were incubated for 16 h in the presence of 2 mm thymidine, released for 6 h, and then treated with 100 ng/ml of nocodazole for 6 h. The mitotic cells were obtained by shaking off.
Cell Fractionation
Cells harvested from 3.5-cm dishes were washed with PBS and extracted with CSK buffer (10 mm PIPES, 100 mm NaCl, 300 mm sucrose, 1 mm MgCl2, 1 mm ATP, 0.5% Triton X-100, protease inhibitor tablet (Roche)) for 20 min on ice and then subjected to centrifugation at 3200 × g for 5 min. The supernatant was collected as a CSK-soluble fraction. The pellet was washed once with CSK buffer and then dissolved in SDS loading buffer as a CSK-insoluble fraction.
In Vitro Kinase Assay
GST-fused, full-length MCM7, MCM7-S121A, MCM7-S197A, MCM7-S365A, and MCM7-T690A and truncated forms of MCM7, GST-cyclin E/cyclin A, and GST-Cdk2 proteins were expressed in the BL21 strain of Escherichia coli and then purified by standard procedures. Cyclin B/Cdk1-activated complex was purchased from Millipore. For the in vitro kinase assay, 1 μg of GST-MCM7 protein with 1 μg of GST-cyclin E and Cdk2, GST-cyclin A, and Cdk2 or cyclinB1/Cdk1 was incubated in kinase buffer (50 mm Tris (pH 7.5), 10 mm MgCl2, 0.02% BSA, 0.04 mm ATP) in the presence of 0.5 μCi of [γ32P]ATP for 30 min at 30 °C. Samples were resolved by 10% SDS-PAGE and autoradiographed to x-ray film.
Generation of Tet-On Stable Cell Lines
FLAG-tagged MCM7, MCM7-S121A, and MCM7-S121D were cloned into the HindIII-NotI sites of the pcDNATM/TO vector (Invitrogen) and transfected into T-RExTM-HeLa cells (Invitrogen). 48 h after transfection, cells were selected with 100 μg/ml zeocin and 5 μg/ml blasticidin for 3 weeks. Monoclones were picked, and expression of MCM7 was tested by immunoblotting in the presence of tetracycline for 24 h.
RNAi Treatment
The knockdown of MCM7 was achieved by transfection of HeLa cells with 50 nm siRNA for 72 h. Human MCM7 siRNA target sequences were as follows: #1, GGAGCAGAACATACAGCTA; #2, CTAGTAAGGATGCCACCTA; and #3, GCTCATGAGGCGTTACATA. The control siRNA sequence was obtained from the manufacturer (RiboBio).
Flow Cytometry
For DNA content analysis, cells were fixed in ice-cold 75% ethanol, washed with PBS/1% BSA, and stained with 20 μg/ml of propidium in PBS/1% BSA with 100 μg/ml of RNaseA. All samples were analyzed on a FACSCalibur cytometer (BD Biosciences). The percentage of cells in each phase of the cell cycle was estimated with ModFit.
RESULTS
MCM7 Interacts with Cyclin E/Cdk2 in an RXL-independent Manner
We have previously identified a number of novel Cdk2-associated proteins by tandem affinity tag purification (19). Among them, the MCM7 protein, considered to be a potential substrate that is a subunit of the MCM2–7 complex, functions as replicative DNA helicase in eukaryotes. First, we took the approach of immunoprecipitation to confirm the association between MCM7 and Cdk2. 293T cells were transfected with pCMV FLAG-MCM7 and pCMV myc-cyclin E or Cdk2. The cell lysates were subjected to immunoprecipitate with FLAG antibody and immunoblotted with myc antibody or vice versa. As shown in Fig. 1, A and B, cyclin E and Cdk2 could interact with FLAG-MCM7. We then performed a GST pull-down assay. The data indicated that GST-MCM7 can interact with myc-cyclin E and myc-Cdk2 expressed in 293T cells (Fig. 1C). We then we examined whether endogenous MCM7 and Cdk2 were associated with each other. The cell lysates from 293T cells were immunoprecipitated with Cdk2 antibody and then immunoblotted with MCM7 and cyclin E antibodies. The data showed that MCM7 could interact with Cdk2 and cyclin E (Fig. 1D). These results confirm that MCM7 can interact with Cdk2 in vivo. To examine the direct interaction between MCM7 and Cdk2, GST-MCM7 and His-Cdk2 were purified for the GST pull-down assay. As shown in Fig. 1E, MCM7 can interact with Cdk2 directly.
FIGURE 1.

Interaction between MCM7 and cyclin E/Cdk2. A, 293T cells were transfected with FLAG-MCM7 together with myc-cyclin E and myc-Cdk2. Cell lysates were immunoprecipitated (IP) with myc beads followed by immunoblotting (IB) with FLAG antibody. B, 293T cells were transfected with the indicated plasmids, and the cell lysates were immunoprecipitated with FLAG beads followed by immunoblotting with myc antibody. C, 293T cells were transfected with myc-cyclin E and myc-Cdk2. The cell lysates were subjected to a pull-down assay with GST or GST-MCM7 and immunoblotted with myc antibody. D, 293T cell lysates were immunoprecipitated with Cdk2 antibody or normal mouse IgG and then immunoblotted with MCM7 and cyclin E antibodies. E, GST or GST-MCM7 proteins immobilized on glutathione beads were incubated with His-Cdk2 in lysis buffer for 2 h. The associated protein was eluted with SDS loading buffer and immunoblotted with Cdk2 antibody. F, MCM7 and its RXL mutants were generated as indicated and transfected into 293T cells together with myc-cyclin E/Cdk2. Cell lysates were immunoprecipitated with FLAG beads and eluted for immunoblotting with myc antibody. TCE, total cell extract; NMS, normal mouse serum.
The RXL motif has been shown previously to be the essential cyclin binding motif in a wide range of cyclin/Cdk-interacting proteins (20). Therefore, we asked whether the interaction between MCM7 and cyclin E/Cdk2 is dependent on the RXL motif of MCM7. There are three RXL motifs in MCM7 (Fig. 1F). To find out which one is critical for its binding with cyclin E/Cdk2, we generated a set of MCM7 AXA mutants in which RXL was mutated to AXA for a coimmunoprecipitation assay. Surprisingly, the data in Fig. 1F show that the mutation on neither the single RXL nor all of the RXLs of MCM7 affected the interaction between Cdk2 and MCM7, suggesting that MCM7 interacts with Cdk2 independently of the RXL motif.
MCM7 Can Be Phosphorylated by Cycline/Cdk2
To examine whether MCM7 is the substrate of Cdk2, GST-MCM7, GST-cyclin E, GST-cyclin A, and GST-Cdk2 were prepared for an in vitro kinase assay. As shown in Fig. 2A, MCM7 can be phosphorylated by cyclin E/Cdk2 much stronger than by cyclin A/Cdk2. There are four serine/threonine at MCM7 (Ser-121, Ser-197, Ser-365, and Thr-690) that are potential phosphorylation sites for cyclin E/Cdk2 (Fig. 2B). To identify the phosphorylation site(s) of MCM7 by cyclin E/Cdk2, GST-MCM7-S121A, S197A, S365A, T690A, or 4A, in which all Ser/Thr were mutated to Ala, were generated for an in vitro kinase assay. The data showed that phosphorylation of MCM7-S121A and MCM7–4A has a certain reduction compared with wild-type MCM7 (Fig. 2C). To further confirm that MCM7 Ser-121 is the phosphorylation site by cyclin E/Cdk2, we generated the MCM7 (1–129) and MCM7 (1–129) S121A mutants and performed an in vitro kinase assay. As shown in Fig. 2D, the phosphorylation of MCM7 (1–129) S121A was obviously reduced compared with that of MCM7 (1–129), indicating that MCM7 Ser-121 was phosphorylated by cyclin E/Cdk2 in vitro.
FIGURE 2.

MCM7 can be phosphorylated by cyclin E/Cdk2, not cyclin A/Cdk2, on Ser-121 in vitro. A, in vitro kinase assay. GST or GST MCM7 were incubated with GST-cyclin E and GST-Cdk2 or GST-cyclin A and GST-Cdk2, respectively, in the presence of [γ32P]ATP. Phosphorylation of MCM7 was assessed by SDS-PAGE followed by autoradiography of the gel. B, potential phosphorylation sites on MCM7 by cyclin/CDKs. C, GST and GST-MCM7 and its mutants were incubated with GST-cyclin E and GST-Cdk2 for an in vitro kinase assay in the presence of [γ32P]ATP. Coomassie Blue (CBB) gel shows the amount of each protein. The relative intensities of phosphorylated protein were calculated. D, GST-MCM7 (1–129) and GST-MCM7 (1–129) S121A were purified for an in vitro kinase assay with GST-cyclin E and GST-Cdk2.
To confirm the phosphorylation of MCM7 on Ser-121 in vivo, we generated antibodies against phosphor-MCM7 Ser-121 by immunizing rabbit with a specific phosphopeptide. Then, 293T cells were cotransfected with pCMV FLAG-MCM7 or pCMV FLAG-MCM7-S121A together with myc-cyclin E/Cdk2 expression plasmids. Cell lysates were immunoprecipitated with FLAG antibody followed by immunoblotting with MCM7-p-Ser121 antibody. As shown in Fig. 3A, the phosphorylation of the MCM7-S121A mutant is reduced dramatically compared with that of wild-type MCM7, indicating that Ser-121 of MCM7 can be phosphorylated by cyclin E/Cdk2 in vivo. To further verify that cyclin E/Cdk2 is the main kinase to phosphorylate MCM7 at Ser-121, FLAG-MCM7 and constitutively active Cdk2 (Cdk2-AF) or dominant negative Cdk2 (Cdk2-DN) were coexpressed in 293T cells. The cell lysates were immunoprecipitated with FLAG antibody and immunoblotted with MCM7-p-Ser121 antibody. As shown in Fig. 3B, MCM7-p-Ser-121 is detected in Cdk2-AF- but not Cdk2-DN-coexpressed cells, indicating that MCM7 Ser-121 is phosphorylated by Cdk2 in vivo.
FIGURE 3.

Phosphorylation of MCM7 on Ser-121 in vivo. A, 293T cells were cotransfected with pCMV FLAG-MCM7 or pCMV FLAG-MCM7-S121A together with myc-cyclin E/Cdk2 expression plasmids. The cell lysates were immunoprecipitated (IP) with FLAG antibody and immunoblotted (IB) with MCM7-p-Ser-121 antibody. B, 293T cells were transfected with pCMV FLAG-MCM7 together with pCMV myc-Cdk2AF or myc-Cdk2DN. The cell lysates were immunoprecipitated with FLAG antibody and immunoblotted with MCM7-p-Ser-121 antibody. C, HeLa-6TR/FLAG-MCM7 cells were treated with tetracycline and then with 20 mm roscovitine, 1 mm flavopiridol, or 1 mm BI2536 for 16 h. The level of MCM7-p-Ser-121 was detected as described in A. DMSO, dimethyl sulfoxide. D, 293T cells were transfected with pCMV myc-Cdk2 AF and myc-Cdk2 DN together with myc-cyclin E or empty vector as control. The cell lysates were immunoprecipitated with MCM7 antibody and immunoblotted with MCM7-p-Ser-121 antibody. The total cell lysates were immunoblotted with the indicated antibodies. TCE, total cell extract.
To further assure the phosphorylation of MCM7 on Ser-121 by cyclin/Cdk in vivo, we treated the cells with roscovitine or flavopiridol as Cdk inhibitors or BI2536 as Plk1 inhibitor and then analyzed the status of MCM7 phosphorylation on Ser-121. As shown in Fig. 3C, the phosphorylation of MCM7 on Ser-121 was abolished completely in roscovitine- or flavopiridol-treated cells but was not changed in BI2536-treated cells.
To detect the phosphorylation of endogenous MCM7, we transfected 293T cells with constitutively active Cdk2 (Cdk2 AF) or dominant negative Cdk2 (Cdk2 DN) together with cyclin E. Cell lysates were immunoprecipitated with MCM7 antibody and immunoblotted with MCM7-p-Ser-121 antibody. As shown in Fig. 3D, the phosphorylation of endogenous MCM7 on Ser-121 is much higher in cyclin E/Cdk2 AF-overexpressed cells than in control and cyclin E/Cdk2 DN-overexpressed cells, which indicates that MCM7 could be phosphorylated on Ser-121 in vivo. Taken together, these data suggest that MCM7 Ser-121 is the specific phosphorylation site of cyclin/Cdks.
Phosphorylation of MCM7 at Ser-121 Affects MCM2–7 Complex Formation
Before DNA replication was initiated, the MCM complex was loaded onto chromatin during late M and G1 phase and then disassociated from chromatin when cells entered S phase. We wondered whether phosphorylation of MCM7 would affect its loading onto chromatin. 293T cells were transfected with FLAG-tagged MCM7, MCM7-S121A, or MCM7-S365A expression plasmids, respectively. The cells were then treated with CSK lysis buffer. The amount of MCM7 in total cell lysates and CSK-soluble and CSK-insoluble fractions was analyzed by immunoblotting with FLAG antibody. As shown in Fig. 4A, there is a significant reduction in the CSK-soluble fraction but more in the CSK-insoluble fraction of MCM7-S121A compared with that of MCM7 and MCM7-S365A. This result suggests that the phosphorylation of MCM7 at Ser-121 regulates its loading onto chromatin.
FIGURE 4.

Phosphorylation of MCM7 at Ser-121 affects MCM complex formation. A, 293T cells transfected with pCMV FLAG-MCM7, MCM7-S121A, or MCM7-S365A were treated with CSK buffer to obtain a CSK-insoluble fraction (chromatin-bound fraction) and CSK-soluble fraction (cytoplasm and nucleoplasm). The lysates were immunoblotted with the indicated antibodies. β-actin and Lamin B were used as the internal controls. B, 293T cells were transfected with pCMV FLAG-MCM7 or pCMV MCM7-S121A. The cell lysates were immunoprecipitated (IP) with FLAG beads followed by immunoblotting with MCM3 and MCM5 antibodies. C, Tet-On inducible HeLa cell lines for expressing FLAG-MCM7 and its mutants (MCM7-S121A and MCM7-S121D) were generated. The cell lysates were harvested for immunoprecipitation with FLAG antibody and immunoblotting with the indicated antibodies. D, pCMV FLAG-MCM7 and pCMV myc-Cdk2-AF or Cdk2-DN were cotransfected into 293T cells. The cell lysates were then immunoprecipitated with FLAG beads and immunoblotted with MCM3 and MCM5 antibodies. The total cell lysates were immunoblotted with the indicated antibodies. TCE, total cell extract.
During M to G1 phase, MCM2–7 forms as a complex and functions as DNA helicase at G1/S phase to initiate DNA replication (7). Therefore, we asked whether the phosphorylation of MCM7 at Ser-121 affected the formation of the MCM2–7 complex. To address this question, we transfected 293T cells with FLAG-MCM7 or its mutants expression plasmids. Then the cell lysates were harvested for an immunoprecipitation assay. As shown in Fig. 4B, the association of MCM7-S121A with MCM3 and MCM5 was reduced significantly compared with that of WT MCM7. Then we generated Tet-On inducible HeLa cell lines for expressing MCM7, MCM7-S121A, and the phosphormimic mutant MCM7 S121D and performed a similar experiment. Consistently, MCM7-S121A was less efficient when binding with MCM3 and MCM5, whereas MCM7 S121D could partially rescue the phenotype (Fig. 4C). We also found that MCM7-S121A was deficient in binding with cdc45, which was loaded onto chromatin after the MCM2–7 complex formed (Fig. 4C). We then transfected 293T cells with pCMV FLAG-MCM7 together with Cdk2 AF or Cdk2 DN. The cell lysates were harvested for immunoprecipitation with FLAG antibody and immunoblotted with MCM3 and MCM5 antibodies. As shown in Fig. 4D, FLAG-MCM7 can bind with MCM3 and MCM5 much better in Cdk2 AF-transfected cells than in Cdk2 DN-transfected cells. Collectively, these results suggest that phosphorylation of MCM7 at Ser-121 regulates its incorporation into the hexameric MCM2–7 complex.
Overexpression of MCM7 but not MCM7 Ser-121A Causes an S Phase Delay by Activating the Chk1 Checkpoint Pathway in a p53-dependent Manner
To further analyze whether phosphorylation of MCM7 at Ser-121 affected DNA replication, Tet-On-inducible HeLa cells expressing MCM7-WT or MCM7-S121A were generated and synchronized to the G1/S boundary by double thymidine treatment, released for 3 h, and then subjected to flow cytometry analysis. As shown in Fig. 5A, the percentage of cells that entered S phase in MCM7-overexpressed cells was much lower (nearly 15%) than that in the control cells or MCM7-S121A-overexpressed cells. We then performed similar experiment in HCT116 cells and also observed that overexpression of MCM7 but not MCM7-S121A inhibited S phase entry after double thymidine release (Fig. 5B). However, overexpression of MCM7 in HCT116 p53−/− cells did not inhibit cell cycle progression and even promoted S phase entry in some degree (Fig. 5C), which suggests that overexpression of MCM7 inhibits S phase entry in a p53-dependent manner. Meanwhile, the immunoblotting data in Fig. 5D show that Ser-121 phosphorylation of MCM7 was detected in MCM7-WT-overexpressed cells.
FIGURE 5.
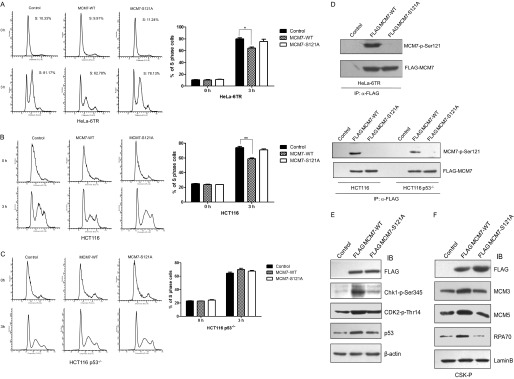
An excess of MCM7 decelerates S phase entry. A, HeLa-6TR Tet-On inducible cell lines for overexpressing MCM7-WT or MCM7-S121A were generated. The cells were synchronized at G1 phase by double thymidine treatment in the presence of tetracycline, released for 3 h, and subjected to flow cytometry analysis (left panel). The percentage of S phase cells was calculated (right panel). B and C, HCT116 and HCT116 p53−/− cells were transfected with pCMV FLAG-MCM7-WT or pCMV FLAG-MCM7-S121A together with pCMV GFP-H2B as the sorting marker. The cells were synchronized at G1 phase, released for 3 h, and subjected to flow cytometry analysis. D, cell lysates from the indicated samples were immunoprecipitated with FLAG antibody and immunoblotted with MCM7-p-Ser-121 antibody. E, HeLa-6TR/MCM7-WT and HeLa-6TR/MCM7-S121A cells were treated with tetracycline for 24 h. The total cell lysates were harvested for immunoblotting (IB) with Chk1-p-Ser-345, Cdk2-p-Thr-14, and p53 antibodies. F, the chromatin-bound fractions (CSK-P) from HeLa-6TR/MCM7-WT, HeLa-6TR/MCM7-S121A, and control cells were immunoblotted with FLAG, MCM3, MCM5, RPA70, and Lamin B. *, p < 0.05; **, p < 0.01.
It has been reported that knockdown of MCM7 does not block S phase entry, suggesting that only a small fraction of MCM7 is enough to form a pre-RC complex with other subunits to initiate DNA replication (21). Consistently, our data shown in supplemental Fig. S1 also confirms that knockdown of MCM7 in HeLa cells by siRNA does not affect S phase progression. Therefore, we propose that the excess of MCM7 may activate the checkpoint pathway to prevent S phase entry. Therefore, we examined the level of active Chk1 in MCM7-overexpressed cells. As shown in Fig. 5E, the level of Chk1-p-Ser-345 and Cdk2-p-Thr-14 were increased greatly in MCM7-WT-overexpressed cells compared with control and MCM7-S121A-overexpressed cells, which indicates that the ataxia telangiectasia-mutated/ATR-Chk1-Cdk checkpoint pathway was activated. We also observed that chromatin-bound RPA70 as an ssDNA accumulation marker was obviously increased in MCM7-WT-overexpressed cells compared with control or MCM7-S121A cells (Fig. 5F). However, the increase of Chk1-p-S345 was not detected in MCM7-WT-overexpressed HCT116 p53−/− cells (data not shown). In the HeLa-6TR stable cell line, there were also moderate increases of p53 levels when excess of MCM7-WT was induced by tetracycline (Fig. 5E), which implied that the ATR-Chk1 pathway was activated in MCM7-overexpressed cells in a p53-dependent manner.
To further understand the significance of MCM7 phosphorylation of MCM7 by cyclin E/Cdk2, we performed similar experiments with the human normal liver cell line HL-7702. The coimmunoprecipitation data and in vitro kinase assay indicate that endogenous MCM7 can interact with Cdk2 (Fig. 6A) and that the phosphorylation of MCM7 on Ser-121 by cyclin E/Cdk2 occurred (B) in HL-7702 cells. To examine whether phosphorylation of MCM7 at Ser-121 affects S phase entry, we transfected MCM7-WT, MCM7-Ser121A, or empty vector into HL-7702 cells. After being synchronized at G1/S with double thymidine treatment, cells were released for 3 h and harvested for immunoblotting or flow cytometry analysis. As shown in Fig. 6, C and D, MCM7 was phosphorylated at Ser-121, and the overexpression of MCM7-WT caused the increase of Chk1-p-Ser-345 and Cdk2-p-Thr-14, indicating that the checkpoint pathway was activated. The flow cytometry data show that overexpression of MCM7-WT causes a significant S phase block compared with that of MCM7-Ser121A or control cells, which is consistent with the results observed in HeLa and HCT116 cells.
FIGURE 6.

An excess of phosphorylated MCM7 blocked S phase entry in human normal cells. A, MCM7 interacts with Cdk2 in HL-7702 cells. The lysates from the human normal liver cell line HL-7702 were immunoprecipitated (IP) with Cdk2 antibody or normal mouse IgG and then immunoblotted (IB) with MCM7 antibody. NMS, normal mouse serum. B, MCM7 Ser-121 is phosphorylated by cyclin E/Cdk2. pCMV myc-Cdk2 AF, pCMV and myc-Cdk2 DN together with pCMV myc-cyclin E were transfected into HL-7702 cells. The cell lysates were immunoprecipitated with MCM7 antibody and then immunoblotted with MCM7-p-Ser-121 antibody. C, pCMV FLAG-MCM7-WT or pCMV FLAG-MCM7-S121A was transfected into HL-7702 cells. The total cell lysates were harvested for immunoblotting with Chk1-p-Ser-345 and Cdk2-p-Thr-14 antibodies. D, HL-7702 cells were transfected with pCMV FLAG-MCM7-WT or pCMV FLAG-MCM7-S121A, respectively. Cell lysates were immunoprecipitated with FLAG antibody and immunoblotted with MCM7-p-Ser-121 antibody. E, HL-7702 cells were transfected with pCMV FLAG-MCM7-WT or pCMV FLAG-MCM7-S121A together with pCMV GFP-H2B as the sorting marker. The cells were synchronized at G1 phase, released for 3 h, and subjected to flow cytometry analysis. **, p < 0.01.
Phosphorylation of MCM7 on Ser-121 by Cyclin B/Cdk1 promotes Mitotic Exit
Because Cdk1 is highly homologous to Cdk2, we wondered whether MCM7 is also the substrate of cyclin B/Cdk1. Therefore, we performed an in vitro kinase assay to test this. As shown in Fig. 7A, the phosphorylation of MCM7-S121A is lower than that of MCM7-WT and other MCM7 SA/TA mutants. Furthermore, we found that MCM7 (1–129) can be phosphorylated by cyclin B/Cdk1, whereas the phosphorylation of MCM7 (1–129) S121A was greatly reduced compared with the wild type of MCM7 (Fig. 7B). These data indicate that cyclin B/Cdk1 can phosphorylate MCM7 at Ser-121. We then tried to detect the phosphorylation of MCM7 at M phase in vivo. As shown in Fig. 7C, phosphorylation of MCM7 on Ser-121 was detected in MCM7-overexpressed cells after being synchronized at M phase. Considering that inhibition of Plk1 by BI2536 did not affect the phosphorylation of MCM7 on Ser-121, whereas inhibition of Cdk activity by roscovitine and flavopiridol almost abolished the MCM7 phosphorylation on Ser-121 (Fig. 3C), cyclin B/Cdk1 should be the main kinase for the phosphorylation of MCM7 during M phase.
FIGURE 7.

Overexpression of MCM7-S121A slows down mitotic exit. A, GST and GST-MCM7 and its mutants were incubated with GST-cyclin B1 and GST-Cdk1 for an in vitro kinase assay in the presence of [γ32P]ATP. Coomassie Blue (CBB) gel shows the amount of each protein. B, GST, GST-MCM7 (1–129), or GST-MCM7 (1–129) S121A was incubated with GST-cyclin B/Cdk1 in the presence of [γ32P]ATP for an in vitro kinase assay. C, HeLa-6TR/MCM7, MCM7-S121A, and control cells were synchronized at M phase by nocodazole. The cell lysates were immunoprecipitated (IP) with FLAG antibody and immunoblotted (IB) with MCM7-p-Ser-121 antibody. D, HeLa-6TR/MCM7-WT, MCM7-S121A, and control cells were treated with tetracycline and then synchronized at M phase by nocodazole. The cells were then shaken off to be released in fresh medium for 75 min or 100 min and subjected to flow cytometry analysis (left panel). The percentage of cells entering into G1 phase was calculated (right panel). *, p < 0.05; ***, p < 0.001.
It has been reported that the MCM2–7 complex loaded onto chromatin during mitotic exit and early G1 phase. To analyze whether phosphorylation of MCM7 on Ser-121 by Cdk1 in M phase affects M-to-G1 phase progression, Tet-On-inducible HeLa cells overexpressing MCM7-WT or MCM7-S121A were synchronized at M phase, released for different time periods, and subjected to flow cytometry analysis. Interestingly, we found that the percentage of cells that entered G1 phase in MCM7-S121A-overexpressed cells was significantly lower than that in control and MCM7-overexpressed cells (nearly 20%), whereas MCM7-overexpressed cells that progressed into G1 phase were a little quicker than control cells (Fig. 7D). Because the data shown in Fig. 4C indicate that MCM7-S121A is less efficient to form a complex with other MCM members, we propose that the phosphorylation of MCM7 at Ser-121 may play an important role in regulating the formation of MCM2–7 complexes for a proper mitotic exit to prepare for pre-RC formation in G1.
DISCUSSION
We previously identified MCM7 as a new interacting protein of Cdk2 using tandem affinity purification. Here we demonstrated that MCM7 Ser-121 can be phosphorylated by cyclin E/Cdk2. MCM7 is a subunit of the putative replicative DNA helicase MCM2–7 complex. The MCM complex, at replication origins, is activated by the concerted actions of cyclin-dependent kinases and the cdc7/dbf4 protein kinase (DDK), which leads to initiation of DNA synthesis. It was found that phosphorylation of MCM3 by cyclin E/Cdk2 causes an S phase delay through checkpoint activation (10). It has also been reported that phosphorylation of MCM4 by cyclin A/Cdk2 inhibits DNA replication through loss of MCM4/6/7 helicase activity (22–23). These data make us speculate whether MCM7 phosphorylation by cyclin E/Cdk2 functions in origin firing and replication fork progression. As shown in this study, overexpression of MCM7-WT but not MCM7-S121A will cause an obviously S phase block. However, when overexpressed in HCT116 p53−/−, MCM7-WT did not inhibit S phase entry, which implied that MCM7 activated the p53-dependent checkpoint pathway. There were already some hints to imply that MCMs could function in the p53-dependent pathway during apoptosis and DNA damage regulation (24–25), which could explain our findings, but the detailed mechanism needs to be elucidated in further studies. It was also reported that overexpression of cdt1 and cdc6, the essential part of pre-RC, will induce ataxia telangiectasia-mutated/ATR checkpoint activation in a p53-dependent pathway (26). We found that the levels of Chk1-p-Ser-345 and p53 increased significantly in MCM7-WT-overexpressed cells. It has been reported that accumulation of RPA70 on chromatin will cause ATR activation. Consistently, we found that chromatin-bound RPA70 increased 3- to 4-fold in MCM7-overexpressed cells, which implied that an excess of MCM7 may turn on the ssDNA checkpoint pathway.
It has been reported that cells with a 90% reduction of MCM7 by using RNA interference display normal S phase progression, which is in agreement with the observation in our study, suggesting that the MCM2–7 complex is overabundant and may have other functions beyond replication initiation over the minimum amount compatible with cell survival in human cells. More and more evidence supports the idea that MCM7 plays important roles in cell cycle regulation beyond DNA replication initiation. It has been found that MCM7 could interact with Rad17 for replication checkpoint regulation when cells were undergoing DNA damage (15). It was also validated that MCM7 was a direct binding partner for the ATR-associated protein ATR-interacting protein and that partial depletion of MCM7 interfered with UV-induced Chk1 activation in U2OS cells (12, 27). Previous reports also determined that the subassembly of the MCM4/6/7 double-trimeric complex possesses DNA helicase activity, and our findings suggest that MCM7-WT could form a proper complex with other MCM subunits, whereas MCM7-S121A is much less efficient, which implied that phosphorylation of MCM7 on Ser-121 may contribute to its helicase activity through binding with other pre-RC family members. Therefore, as MCM7 is in excess and phosphorylated by cyclin E/Cdk2 at G1/S transition, there will be more activated MCM complex on the chromatin that could recruit other pre-RC components to give rise to an improper accumulation of ssDNA. Consequently, it will activate the checkpoint pathway, which is in accordance with a previous report that the engagement of the ATR pathway occurs in response to ssDNA breaks, resulting in checkpoint activation designed to arrest cells for DNA repair.
The MCM2–7 complex is loaded onto chromatin in late M phase and early G1 phase, but the regulation is not quite clear. It has been reported in the database that phosphorylation of MCM7 on Ser-365 peaks in M phase but with unknown function (Phosida), which implies that MCM7 could play an important role during mitotic exit. Meanwhile, it was determined that the loading of MCM onto chromatin started at 60 min after nocodazole release (19). In this report, we found that MCM7 Ser-121 is also phosphorylated by cyclin B/Cdk1. We then further examined whether the Ser-121 phosphorylation of MCM7 by cyclin B/Cdk1 could regulate M phase progression. Surprisingly, we found that MCM7-S121A-overexpressed cells are less deficient for normal mitotic exit compared with control or MCM7-WT-overexpressed cells. In accordance with our findings that MCM7-WT binds with other MCM subunits better than MCM7-S121A, we proposed that cyclin B1/Cdk1 phosphorylated MCM7 at Ser-121 to regulate the formation of the MCM2–7 complex to promote mitotic exit.
In conclusion, we found that both cyclin E/Cdk2 and cyclin B/Cdk1 can phosphorylate MCM7 at Ser-121 to regulate cell cycle progression through either activating the S phase checkpoint pathway or controlling MCM2–7 complex formation for proper mitotic exit.
Supplementary Material
Acknowledgment
We thank Dr. Y Li (Institute of Psychology, Chinese Academy of Sciences) for help with flow cytometry.
This work was supported by National Natural Science Foundation of China Grant 81272272; by Ministry of Science and Technology of China Grants 2013ZX10004611, 2012CB519003, and 2011CB504705; by Chinese Academy of Sciences Innovation Project Grant KSCX2-EW-J-6; and by Innovative Research Group of the National Natural Science Foundation of China Grant 81021003 (to X. Y.).

This article contains supplemental Fig. S1.
- MCM
- minichromosome maintenance
- pre-RC
- pre-replication complex
- CDK
- cyclin-dependent kinase
- ATR
- ATM-Rad3-related
- ssDNA
- single-stranded DNA
- DDK
- Dbf4-dependent kinase
- PCNA
- proliferating cell nuclear antigen.
REFERENCES
- 1. Sclafani R. A., Holzen T. M. (2007) Cell cycle regulation of DNA replication. Annu Rev. Genet 41, 237–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lei M., Tye B. K. (2001) Initiating DNA synthesis. From recruiting to activating the MCM complex. J. Cell Sci. 114, 1447–1454 [DOI] [PubMed] [Google Scholar]
- 3. Tye B. K. (1999) MCM proteins in DNA replication. Annu. Rev. Biochem. 68, 649–686 [DOI] [PubMed] [Google Scholar]
- 4. Tsuji T., Ficarro S. B., Jiang W. (2006) Essential role of phosphorylation of MCM2 by Cdc7/Dbf4 in the initiation of DNA replication in mammalian cells. Mol. Biol. Cell 17, 4459–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tanaka S., Tak Y. S., Araki H. (2007) The role of CDK in the initiation step of DNA replication in eukaryotes. Cell Div. 2, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Porter A. C. (2008) Preventing DNA over-replication. A Cdk perspective. Cell Div 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maiorano D., Lutzmann M., Méchali M. (2006) MCM proteins and DNA replication. Curr. Opin. Cell Biol. 18, 130–136 [DOI] [PubMed] [Google Scholar]
- 8. You Z., Ishimi Y., Masai H., Hanaoka F. (2002) Roles of Mcm7 and Mcm4 subunits in the DNA helicase activity of the mouse Mcm4/6/7 complex. J. Biol. Chem. 277, 42471–42479 [DOI] [PubMed] [Google Scholar]
- 9. Lin D. I., Aggarwal P., Diehl J. A. (2008) Phosphorylation of MCM3 on Ser-112 regulates its incorporation into the MCM2–7 complex. Proc. Natl. Acad. Sci. U.S.A. 105, 8079–8084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li J., Deng M., Wei Q., Liu T., Tong X., Ye X. (2011) Phosphorylation of MCM3 protein by cyclin E/cyclin-dependent kinase 2 (Cdk2) regulates its function in cell cycle. J. Biol. Chem. 286, 39776–39785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Forsburg S. L. (2004) Eukaryotic MCM proteins. Beyond replication initiation. Microbiol. Mol. Biol. Rev. 68, 109–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cortez D., Glick G., Elledge S. J. (2004) Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc. Natl. Acad. Sci. U.S.A. 101, 10078–10083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferguson R. L., Maller J. L. (2008) Cyclin E-dependent localization of MCM5 regulates centrosome duplication. J. Cell Sci. 121, 3224–3232 [DOI] [PubMed] [Google Scholar]
- 14. Shohet J. M., Hicks M. J., Plon S. E., Burlingame S. M., Stuart S., Chen S. Y., Brenner M. K., Nuchtern J. G. (2002) Minichromosome maintenance protein MCM7 is a direct target of the MYCN transcription factor in neuroblastoma. Cancer Res. 62, 1123–1128 [PubMed] [Google Scholar]
- 15. Tsao C. C., Geisen C., Abraham R. T. (2004) Interaction between human MCM7 and Rad17 proteins is required for replication checkpoint signaling. EMBO J. 23, 4660–4669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y. Z., Jiang Y. Y., Hao J. J., Lu S. S., Zhang T. T., Shang L., Cao J., Song X., Wang B. S., Cai Y., Zhan Q. M., Wang M. R. (2012) Prognostic significance of MCM7 expression in the bronchial brushings of patients with non-small cell lung cancer (NSCLC). Lung Cancer 77, 176–182 [DOI] [PubMed] [Google Scholar]
- 17. Ren B., Yu G., Tseng G. C., Cieply K., Gavel T., Nelson J., Michalopoulos G., Yu Y. P., Luo J. H. (2006) MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene 25, 1090–1098 [DOI] [PubMed] [Google Scholar]
- 18. Padmanabhan V., Callas P., Philips G., Trainer T. D., Beatty B. G. (2004) DNA replication regulation protein Mcm7 as a marker of proliferation in prostate cancer. J. Clin. Pathol. 57, 1057–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deng M., Li F., Ballif B. A., Li S., Chen X., Guo L., Ye X. (2009) Identification and functional analysis of a novel cyclin e/cdk2 substrate ankrd17. J. Biol. Chem. 284, 7875–7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sørensen C. S., Lukas C., Kramer E. R., Peters J. M., Bartek J., Lukas J. (2001) A conserved cyclin-binding domain determines functional interplay between anaphase-promoting complex-Cdh1 and cyclin A-Cdk2 during cell cycle progression. Mol. Cell. Biol. 21, 3692–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ibarra A., Schwob E., Méndez J. (2008) Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. U.S.A. 105, 8956–8961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ishimi Y., Komamura-Kohno Y. (2001) Phosphorylation of Mcm4 at specific sites by cyclin-dependent kinase leads to loss of Mcm4,6,7 helicase activity. J. Biol. Chem. 276, 34428–34433 [DOI] [PubMed] [Google Scholar]
- 23. Ishimi Y., Komamura-Kohno Y., You Z., Omori A., Kitagawa M. (2000) Inhibition of Mcm4,6,7 helicase activity by phosphorylation with cyclin A/Cdk2. J. Biol. Chem. 275, 16235–16241 [DOI] [PubMed] [Google Scholar]
- 24. Abe S., Kurata M., Suzuki S., Yamamoto K., Aisaki K., Kanno J., Kitagawa M. (2012) Minichromosome maintenance 2 bound with retroviral Gp70 is localized to cytoplasm and enhances DNA-damage-induced apoptosis. PLoS ONE 7, e40129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yun H. J., Hyun S. K., Park J. H., Kim B. W., Kwon H. J. (2012) Widdrol activates DNA damage checkpoint through the signaling Chk2-p53-Cdc25A-p21-MCM4 pathway in HT29 cells. Mol. Cell Biochem. 363, 281–289 [DOI] [PubMed] [Google Scholar]
- 26. Vaziri C., Saxena S., Jeon Y., Lee C., Murata K., Machida Y., Wagle N., Hwang D. S., Dutta A. (2003) A p53-dependent checkpoint pathway prevents rereplication. Mol. Cell 11, 997–1008 [DOI] [PubMed] [Google Scholar]
- 27. Nitani N., Nakamura K., Nakagawa C., Masukata H., Nakagawa T. (2006) Regulation of DNA replication machinery by Mrc1 in fission yeast. Genetics 174, 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.