

Background: PDZK1 (four PDZ domains) regulates the hepatic HDL receptor SR-BI.
Results: PDZK1's PDZ2 and PDZ3 domains are not required, whereas PDZ4 is, possibly because PDZ4 mediates membrane binding.
Conclusion: Regulation of SR-BI via PDZK1's PDZ domains is complex.
Significance: Combined canonical (target peptide binding) and noncanonical (peptide binding-independent) PDZ domain functions can result in optimal activity of a PDZ domain-containing adaptor protein.
Keywords: Cholesterol Metabolism, High Density Lipoprotein (HDL), Protein-Protein Interactions, Receptors, Transgenic Mice
Abstract
The four PDZ (PDZ1 to PDZ4) domain-containing adaptor protein PDZK1 controls the expression, localization, and function of the HDL receptor scavenger receptor class B, type I (SR-BI), in hepatocytes in vivo. This control depends on both the PDZ4 domain and the binding of SR-BI's cytoplasmic C terminus to the canonical peptide-binding sites of either the PDZ1 or PDZ3 domain (no binding to PDZ2 or PDZ4). Using transgenic mice expressing in the liver domain deletion (ΔPDZ2 or ΔPDZ3), domain replacement (PDZ2→1), or target peptide binding-negative (PDZ4(G389P)) mutants of PDZK1, we found that neither PDZ2 nor PDZ3 nor the canonical target peptide binding activity of PDZ4 were necessary for hepatic SR-BI regulatory activity. Immunohistochemical studies established that the localization of PDZK1 on hepatocyte cell surface membranes in vivo is dependent on its PDZ4 domain and the presence of SR-BI. Analytical ultracentrifugation and hydrogen deuterium exchange mass spectrometry suggested that the requirement of PDZ4 for localization and SR-BI regulation is not due to PDZ4-mediated oligomerization or induction of conformational changes in the PDZ123 portion of PDZK1. However, surface plasmon resonance analysis showed that PDZ4, but not the other PDZ domains, can bind vesicles that mimic the plasma membrane. Thus, PDZ4 may potentiate PDZK1's regulation of SR-BI by promoting its lipid-mediated attachment to the cytoplasmic membrane. Our results show that not all of the PDZ domains of a multi-PDZ domain-containing adaptor protein are required for its biological activities and that both canonical target peptide binding and noncanonical (peptide binding-independent) capacities of PDZ domains may be employed by a single such adaptor for optimal in vivo activity.
Introduction
The expression, localization, and function of membrane-associated proteins, including cell surface receptors, are often regulated by cytoplasmic adaptor proteins (1). One of the largest protein interaction domain families is the PSD-95, Discs-large, ZO-1 (PDZ)3 family. Indeed, human PDZ domain-containing proteins are encoded by 152 genes comprising 267 PDZ domains (2, 3). PDZ domains typically bind to the C termini of their binding partners (1) via a well defined binding pocket (4). PDZ domain-containing proteins play prominent roles in regulating the activity of their targets and signal transduction. A large number of these proteins contain multiple PDZ domains and therefore are able to serve as scaffold proteins to coordinate complex cellular functions. PDZK1 (5), a 519-amino acid cytoplasmic protein, is an example of such a protein as it contains four PDZ domains. It interacts with several membrane-associated proteins such as cell surface receptors and ion channels (2, 6). One of these PDZK1-interacting proteins is the HDL receptor scavenger receptor class B type I (SR-BI)(7–9).
SR-BI is a 509-amino acid cell-surface glycoprotein with a large extracellular loop, two trans-membrane domains, and short cytoplasmic N and C termini (10). It plays a major role in the transfer of cholesterol from HDL particles into cells (10, 11) by a process called selective uptake (12). Inactivation of the SR-BI gene in mice results in a major increase in plasma cholesterol (∼2.2-fold) in the form of abnormally large HDL particles (13). In apoE knock-out (KO) mice (14, 15), the additional inactivation of the SR-BI gene leads to occlusive atherosclerosis, myocardial infarction, and premature death (16, 17). Ikemoto et al. (7) first showed that the C-terminal tail of SR-BI binds to the N-terminal PDZ domain (PDZ1) of PDZK1. We confirmed this high affinity (Kd ∼2.6 μm) binding in vitro, and we also found lower affinity (Kd ∼37.0 μm) binding of the C terminus of SR-BI to the third PDZ domain (PDZ3) of PDZK1 but no binding to PDZ2 or PDZ4 (18, 19). The C terminus of SR-BI binds to the canonical carboxylate binding loops of PDZ1 and PDZ3 (18, 19), the binding site used by most PDZ domains to bind their target peptides (20).
Inactivation of the PDZK1 gene in mice leads to a dramatic decrease in steady state SR-BI protein expression in some tissues (∼95% decrease in hepatocytes and ∼50% in small intestinal epithelial cells), but not others (e.g. steroidogenic organs or endothelial cells) (8, 21, 22). As a result, PDZK1 KO mice show an increase of plasma cholesterol (∼1.7-fold) in the form of abnormally large HDL particles that is similar to, but not as severe as, that seen in SR-BI KO mice (8). Because we have been unable to robustly reproduce the PDZK1 dependence of SR-BI in cultured cells,4 we have analyzed the functional consequences of altering PDZK1's structure on its function using PDZK1 transgenes expressed in hepatocytes of wild-type (WT) or PDZK1 KO mice (18, 19, 23, 24). Analyses of a variety of such transgenic mice established that PDZK1 controls SR-BI's expression, localization, and function. For example, we introduced point mutations in PDZK1 that disrupt the binding of either the PDZ1 or PDZ3 domains to SR-BI's C terminus and showed that only one active SR-BI-binding PDZ domain is necessary for normal PDZK1-dependent regulation of hepatic SR-BI (18, 19). A C-terminally truncated PDZK1 containing only the first three N-terminal PDZ domains (PDZ123) is unable to restore full, normal expression and function of SR-BI in PDZK1 KO mice, whereas a truncated PDZK1 transgene containing all four PDZ domains (PDZ1234), but missing the C-terminal 58 amino acids of the protein, is fully functional (23). These findings suggested that the fourth PDZ domain (PDZ4), which does not bind directly to SR-BI's C terminus (19), performs a critical role in promoting normal SR-BI function.
In this study, we have further explored the in vivo roles of PDZK1's PDZ2, PDZ3, and PDZ4 domains in regulating hepatic SR-BI expression and activity. Using transgenic mice, we have determined that the PDZ2 or PDZ3 domains can be deleted from PDZK1 without compromising PDZK1's ability to mediate normal hepatic SR-BI expression or function. Furthermore, we have determined using immunohistochemistry that the PDZ4 domain is necessary for normal cell surface membrane association of PDZK1. However, this function of PDZ4 is not mediated by classical canonical binding to the C terminus of a target protein but rather by direct binding of this PDZ domain to plasma membrane lipids as demonstrated using surface plasmon resonance (SPR). Biophysical analyses of recombinant PDZK1 in vitro demonstrated that PDZ4 is not responsible for protein oligomerization, and it does not alter the backbone conformation of the PDZ123 domains (hydrogen/deuterium exchange mass spectrometry, HXMS). These studies raise the possibility that PDZ4 may be required for normal PDZK1-mediated regulation of hepatic SR-BI in vivo because of its ability to target PDZK1 to the inner leaflet of the plasma membrane of hepatocytes.
EXPERIMENTAL PROCEDURES
Generation of PDZK1 cDNAs Encoding Proteins with Amino Acid Substitutions or PDZ Domain Substitution/Deletions
Vectors encoding PDZK1 with Y388A, G389P, or G389A single residue substitutions or Y388G/G389P, Y388A/G389A, or Y388A/G389P double residue substitutions within PDZ4's carboxylate binding loop were produced using a previously described wild-type PDZK1/pLiv-LE6 or PDZK1/pCDNA3.1 recombinant plasmids (24) and the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. Oligonucleotide primers containing the corresponding mutations were synthesized and PAGE-purified by Integrated DNA Technologies (IDT). A PDZK1 cDNA gene in which the second PDZ domain (PDZ2, residues 133–212) was replaced by the PDZ1 sequence (residues 7–86, resulting in two PDZ1 domains in tandem (PDZ2→1)) and the PDZK1 cDNA genes in which either the PDZ2 (ΔPDZ2, residues 133–212) or PDZ3 (ΔPDZ3, residues 241–319) domains were deleted were synthesized by IDT. KpnI-XhoI DNA fragments corresponding to each of these mutant PDZK1 constructs were subcloned into the pLiv-LE6 plasmid (24), which was kindly provided by Dr. John M. Taylor (Gladstone Institute of Cardiovascular Disease, University of California, San Francisco), and contains the promoter, first exon, first intron, and part of the second exon of the human apoE gene, the polyadenylation sequence, and a part of the hepatic control region of the apoE/C-I gene locus (25). The sequences of the resulting plasmids were confirmed by DNA sequencing.
Recombinant Protein Production and Purification
We generated cDNAs encoding individual PDZ domains, full-length or truncated PDZK1 proteins, all with either wild-type sequences and single or double point mutations. A cDNA encoding full-length WT PDZK1 was used as template to generate WT PDZ3- and PDZ4-encoding PCR products corresponding to residues 241–348 (108 residues) and 376–484 (109 residues) of PDZK1. The same cDNA was used to generate truncated PDZK1 containing the first three PDZ domains (PDZ123) corresponding to the first 359 amino acids of PDZK1 or containing the four PDZ domains of PDZK1 (458 amino acids), but lacking the 61 C-terminal amino acids of the protein (PDZ1234). cDNAs encoding full-length PDZK1 with single or double residue substitutions in the PDZ4 domain of PDZK1 (see above) were used as templates to generate mutated PDZ4 domains containing the substitutions described above.
All of the cDNAs were cloned into pGEX-4T-3 and expressed in Escherichia coli JM109 cells to produce glutathione S-transferase fusion proteins. The expressed proteins were purified on glutathione-Sepharose 4B (GE Healthcare) and released by digestion with thrombin. All recombinant proteins contained a Gly-Ser dipeptide encoded by the cloning vector at the N terminus that is not normally present in PDZK1 and generated as the N terminus by thrombin cleavage. The recombinant proteins were further purified by fast protein liquid chromatography (FPLC) using Superdex S75 or S200 columns (GE Healthcare) in a buffer containing 150 mm NaCl, 25 mm Tris, pH 8.0, at 4 °C under reducing conditions (0.5 mm tris(2-carboxyethyl)phosphine), and they were shown to be homogeneous using SDS-PAGE.
Isothermal Titration Calorimetry (ITC)
Binding of recombinant proteins to the C-terminal heptapeptide of transient receptor potential cation channel subfamily C member 5 (TRPC5) (969EQVTTRL975, “target peptide”) was measured using a VP-ITC microcalorimeter (GE Healthcare). The TRPC5 peptide was synthesized and purified by HPLC at the Tufts University Core Facility (Boston). Briefly, 1.0 mm of the target peptide was titrated against the recombinant proteins at a concentration of 0.03 mm in a buffer containing 150 mm NaCl, 25 mm Tris, pH 8.0, at 20 °C under reducing conditions (0.5 mm tris(2-carboxyethyl)phosphine). Titration curves were analyzed and Kd values determined using ORIGIN 7.0 software (OriginLab).
Analytical Ultracentrifugation
Sedimentation velocity analyses were performed on a Beckman Instruments Optima XL-1 Analytical Ultracentrifuge with Rayleigh optics at 50,000 rpm and three loading concentrations (1.803, 0.453, and 0.212 mg/ml). Interference optics were used, and time difference methods were used to correct for optical background systematic noise as described previously (26–29).
Hydrogen/Deuterium Exchange Mass Spectrometry
Deuterium Exchange Reactions
Deuterium labeling was initiated with a 15-fold dilution of an aliquot (60 pmol of PDZ1234 or PDZ123) of each of the purified protein stock solutions into a D2O buffer containing 25 mm Tris, pD 8.03, 150 mm NaCl, 0.5 mm tris(2-carboxyethyl)phosphine (buffer A). After incubation at 21 °C for either 10 s, 10 min, 1 or 4 h, the labeling reaction was quenched with the addition of an equal volume of quench buffer (150 mm potassium phosphate, pH 2.54).
On-line Protein Digestion and Mass Analysis
Immediately following the acid quench, samples were injected into a nanoACQUITY with HDX technology (30) for on-line pepsin digestion with a 2.1 × 30-mm Poroszyme® immobilized pepsin cartridge (Invitrogen). The peptides were trapped and desalted on a VanGuard Pre-Column trap (2.1 × 5 mm, ACQUITY UPLC BEH C18, 1.7 μm) for 3 min. Peptides eluted from the trap using a 5–35% gradient of acetonitrile over 6 min at a flow rate of 60 μl/min and were separated using an ACQUITY UPLC HSS T3, 1.8 μm, 1.0 × 50-mm column. Peptides from an unlabeled protein sample were identified in triplicate using ProteinLynx 2.5 Global Server searches of a protein database, including analyte proteins as well as porcine pepsin A1 and A2.
All mass spectra were acquired using a Waters QTOF Premier mass spectrometer. Each deuterium labeling experiment was performed in duplicate. In this instrumental setup, the error of measuring the deuterium incorporation for each peptide was at or below ±0.2 Da (31). Typically, differences of relative deuterium incorporation less than ∼0.4 Da are considered to be within the error of measurement (31–33). All mass spectra were processed using DynamX software (Waters). The data were not corrected for back exchange and are therefore reported as relative values (34, 35). The relative differences in deuterium incorporation for each common peptide in PDZ1234 and PDZ123 were calculated by subtracting the relative deuterium incorporation for PDZ123 from PDZ1234 (PDZ1234 − PDZ123 = relative difference).
Lipid Vesicle Preparation and SPR Analysis
Plasma membrane (PM) mimetic vesicles (POPC/1 palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine/phosphoinositol/cholesterol/1,2-dipalmitoyl derivatives of phosphatidylinositol-(4,5)-biphosphonate (12:35:22:8:22:1)) and control 100% POPC vesicles were prepared as described previously (36). All SPR measurements were performed in 20 mm Tris-HCl, pH 7.4, containing 160 mm NaCl at 23 °C using a lipid-coated L1 chip in the BIACORE X system as described previously (37). PM mimetic and POPC vesicles were used to coat the active and control surfaces, respectively. Vesicles were injected at 5 μl/min onto the corresponding sensor chip surfaces to yield the identical resonance units, ensuring the equal concentration of the coated lipids (PM mimetic and control POPC). Equilibrium measurements were performed at a flow rate of 5 μl/min, which allowed enough time for the R values of the association phase to reach near equilibrium levels (Req) (36). Each sensorgram was background-corrected by subtracting the control (POPC) surface response from the active (PM mimetic) surface response. A minimum of five different protein concentrations (0.25, 0.5, 1.0, 3.0, and 5 μm) were injected to collect a set of Req values that were plotted against the protein concentrations (Po). An apparent dissociation constant (Kd) was then determined by nonlinear least squares analysis of the binding isotherm using the following equation: Req = Rmax/(1 + Kd/Po), where Rmax indicates the maximal Req value. Because the concentration of lipids coated on the sensor chip cannot be accurately determined, Kd is defined as Po yielding half-maximal binding with a fixed lipid concentration. The measurement was repeated at least three times to determine average and standard deviation values.
Animals
All animal experiments were performed according to IACUC guidelines. All mice (25:75 FVB/N:129SvEV genetic background) were maintained on a normal chow diet (38), and ∼6–8-week-old male mice were used for experiments. All procedures on transgenic and nontransgenic mice were performed in accordance with the guidelines of the Beth Israel Deaconess Medical Center and the Massachusetts Institute of Technology Committee on Animal Care.
PDZK1 KO mice, PDZK1 KO mice expressing a PDZ1234 transgene, founder 1159 (∼10-fold greater PDZ1234 protein than PDZK1 in WT liver), and PDZK1 KO mice expressing a PDZ123 transgene, founder 644 (∼8-fold greater PDZ1234 protein than PDZK1 in WT liver), were generated as described previously (23, 38). Mice on a 50:50 C57Bl/6:129 background with heterozygous or homozygous null mutations of SR-BI were generated as described previously (13). Previously unreported founder transgenic animals in an FVB/N genetic background were generated using the PDZ2→1, ΔPDZ2, ΔPDZ3, and full-length PDZK1 containing a G389P mutation in the PDZ4 domain (PDZK1(G389P)) pLiv-LE6 expression vectors described above (see also Fig. 1, D–G). The pLIV-LE6 plasmid, kindly provided by Dr. John M. Taylor (Gladstone Institute of Cardiovascular Disease, University of California, San Francisco), contains the promoter, first exon, first intron, and part of the second exon of the human apoE gene, the polyadenylation sequence, and a part of the hepatic control region of the apoE/C-I gene locus (25).
FIGURE 1.

Schematic diagrams of wild-type (A) and mutant (B–G) PDZK1 proteins. A, full-length wild-type PDZK1 with the PDZ domains that bind the C terminus (C-term.) of SR-BI (PDZ1 and PDZ3) indicated. B, C-terminally truncated PDZ123. C, C-terminally truncated PDZ1234. D, full-length protein with PDZ2 domain replaced by a duplication of the PDZ1 domain (speckled circle), PDZ2→1. E, PDZK1 with deletion of the PDZ2 domain (dashed line), ΔPDZ2. F, PDZK1 with deletion of the PDZ3 domain (dashed line), ΔPDZ3. G, full-length PDZK1 with an inactivating G389P mutation in the canonical target peptide-binding site of the PDZ4 domain (yellow), PDZK1(G389P).
The transgenic expression vectors were digested with SacII/SpeI and the resulting 6.3–6.5-kb constructs were used to generate transgenic mice using standard procedures (39). Transgenic animals were identified by PCR performed on tail DNA using the following oligonucleotide primers: one within the PDZK1 cDNA and one corresponding to the 3′-end of the human apoE gene sequence included in the cloning vector: for PDZ2→1 and ΔPDZ3, CAATGGTGTCTTTGTCGACAAG and AGCAGATGCGTGAAACTTGGTGA; for ΔPDZ2, GCAGAGGCAGCTGGCTTGAAGAAC and GCCAGCAGATGCGTGAAACTTGGTGAATC; and for PDZK1(G389P), GAGGCAGCTGGCTTGAAGAAC and AGCAGATGCGTGAAACTTGGTGA. Founders expressing PDZK1 transgenic mutants were crossed with PDZK1 KO mice (129SvEv background). Heterozygous pups expressing the transgenes were crossed with WT and PDZK1 KO mice to obtain transgenic and control nontransgenic WT and PDZK1 KO mice, thus ensuring that the mixed genetic backgrounds of experimental and control mice in each founder line were matched. PDZK1 KO mice were genotyped as described previously (38). Two independent founder lines for each construct were generated.
Blood and Tissue Sampling, Processing, and Analysis
Plasma and liver samples were collected and processed, and total plasma cholesterol levels and size-fractionated FPLC cholesterol profiles were obtained as described previously (24). Plasma cholesterol results presented here for transgenic mice are pooled from data collected from animals derived from the different founder transgenic animals because the results obtained were not significantly different between founders expressing the same transgene.
For immunoblotting, protein samples (∼30 μg) from total liver lysates were fractionated by SDS-PAGE, transferred to nitrocellulose membranes, and incubated with either a rabbit polyclonal SR-BI antibody (mSR-BI(495–112)) (1:1000) or a rabbit polyclonal PDZK1 antibody (1:30,000) (24), followed by an anti-rabbit IgG conjugated to horseradish peroxidase (Invitrogen, 1:10,000), and visualized by ECL chemiluminescence (GE Healthcare). Immunoblotting using a polyclonal anti-ϵ-COP antibody (1:5000) (40) was used to control for small variations in loading. The relative amounts of proteins were determined quantitatively using a Kodak Image Station 440 CF and Kodak 1D software.
Immunoperoxidase Analysis
Livers were harvested, fixed, and frozen, and 5-μm sections were stained with an anti-mPDZK1 (23) or anti-mSR-BI(495–112) antibody and biotinylated anti-rabbit IgG, visualized by immunoperoxidase staining, and counterstained with Harris modified hematoxylin, as described previously (8).
Statistical Analysis
Data are shown as the means ± S.D. Statistically significant differences were determined by either pairwise comparisons of values using the unpaired t test, with (if variances differed significantly) or without Welch's correction, or by one-way analysis of variance followed by the Tukey-Kramer multiple comparison post test when comparing three or more variables. Mean values for experimental groups are considered statistically significantly different at p < 0.05 for both types of tests.
RESULTS
Roles of the PDZ2 and PDZ3 Domains on the Regulation of Hepatic SR-BI
To examine the influence of the PDZ2 and PDZ3 domains of PDZK1 on hepatic SR-BI in vivo, we generated transgenic (Tg) mice expressing three types of essentially liver-specific PDZK1 mutant transgenes. In these mutants the following occurred: 1) the PDZ2 domain was replaced by PDZ1 (PDZ2→1, Fig. 1D) resulting in two successive PDZ1 domains as the two first PDZ domains of PDZK1 (PDZ2→1); 2) the PDZ2 domain (residues 133–212) was deleted from the full-length PDZK1 protein (ΔPDZ2, Fig. 1E); and 3) the PDZ3 domain (residues 241–319) was deleted from the full-length PDZK1 protein (ΔPDZ3, Fig. 1F). We generated two founder lines for PDZ2→1 (designated 8418 and 8419), two founder lines for ΔPDZ2 (designated 8421 and 8423), and two founder lines for ΔPDZ3 (designated 8440 and 8442), which were used to generate background-matched wild-type (WT-Tg) and PDZK1 KO (KO-Tg) transgenic mice on a mixed FVB/129SvEv background. Background-matched nontransgenic WT and PDZK1 KO mice were used as controls for each founder.
For each group of WT-Tg and KO-Tg mice and their nontransgenic controls, we determined the hepatic expression levels of PDZK1 and SR-BI proteins by quantitative immunoblotting (Figs. 2A to 4, and ϵ-COP was used as a loading control). The ratios of band intensities (transgenic/nontransgenic) are shown in Table 1. The relative steady state levels of PDZK1 transgene-encoded proteins varied between 2.5 ± 0.5- and 17.2 ± 4.7-fold greater than that of endogenous PDZK1 in nontransgenic WT mice (most >5-fold). Thus, there was substantial overexpression of the transgene-encoded hepatic PDKZ1 proteins relative to that of the endogenous PDZK1 protein in nontransgenic WT mice. We also measured plasma total cholesterol levels (Figs. 2B to 4B), the size distribution of plasma lipoproteins (FPLC lipoprotein cholesterol profiles, Figs. 2C to 4C), and the distribution of SR-BI protein in hepatocytes in vivo (immunohistochemistry, Fig. 5, A–H).
FIGURE 2.
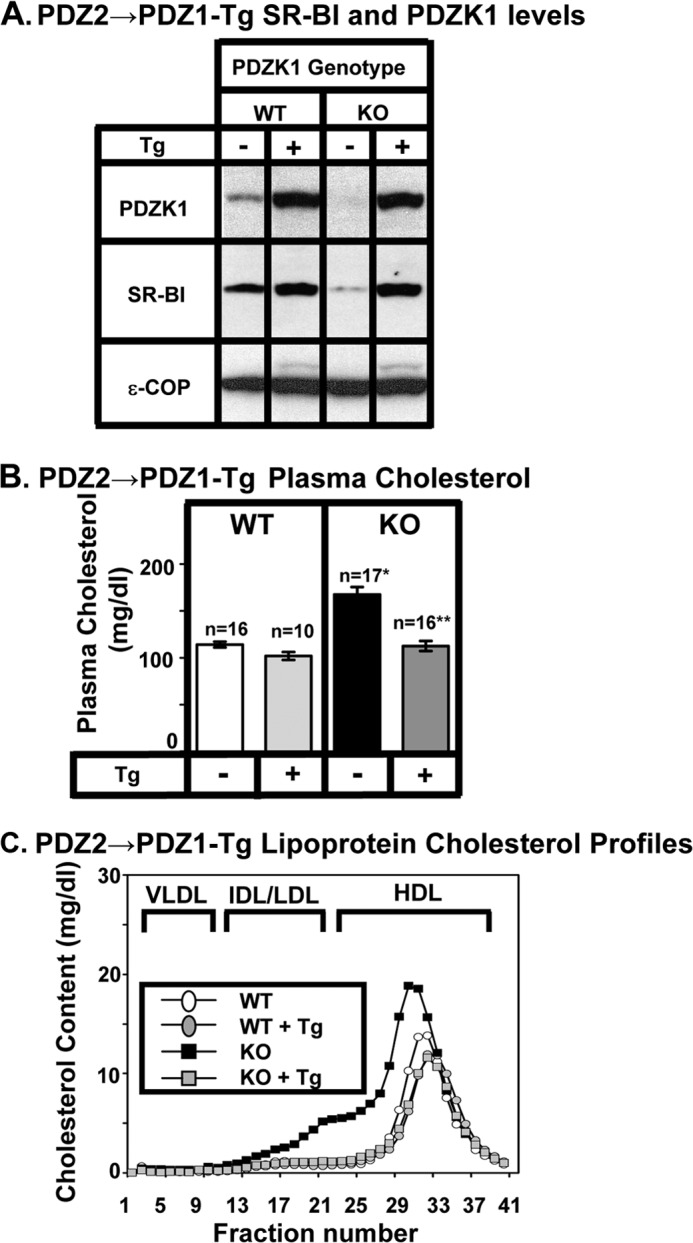
Effects of expression of the PDZ2→1 transgene on hepatic SR-BI protein levels (A) and plasma lipoprotein cholesterol (B and C) in WT and PDZK1 KO mice. A, liver lysates (∼30 μg of protein) from mice with the indicated genotypes, with (+) or without (−) the PDZ2→1 transgene (Tg) (replacement of the PDZ2 domain with a second PDZ1 domain, see Fig. 1D) were analyzed by immunoblotting (data from founder 8419), and bands representing PDZK1 (∼70 kDa) and SR-BI (∼82 kDa) were visualized by chemiluminescence. ϵ-COP (∼34 kDa) was used as a loading control. Note there is a faint SR-BI band in the nontransgenic PDZK1 KO lane. Replicate experiments with multiple exposures and sample loadings were used to determine the relative levels of expression of SR-BI (Table 2). B, plasma samples were harvested from mice with the indicated genotypes and PDZ2→1 transgene. Total plasma cholesterol levels were determined in individual samples by enzymatic assay, and mean values from the indicated numbers of animals (n) are shown for each genotype. Independent WT and KO control animals for each founder line were generated to ensure that the mixed genetic backgrounds for experimental and control mice were matched. Values from the two independent founder lines 8419 and 8418 were not statistically significantly different and thus were averaged together here. * indicates the nontransgenic KO plasma cholesterol levels were statistically significantly different from those plasma cholesterol levels of WT (p < 0.0001). ** indicates PDZK1 KO [PDZ2→1-Tg] plasma cholesterol levels were statistically significantly different from those plasma cholesterol levels of nontransgenic PDZK1 KO mice (p < 0.0001). WT [PDZ2→1-Tg] and KO [PDZ2→1-Tg] plasma cholesterol levels were not statistically significantly different. C, pooled plasma samples (described in B) from four to five animals (data from founder 8419) were size-fractionated by FPLC, and the total cholesterol content of each fraction was determined by an enzymatic assay. The chromatograms are representative of multiple individually determined profiles. Approximate elution positions of native VLDL, IDL/LDL, and HDL particles are indicated by brackets and were determined as described previously (13). The chromatograms for the WT, WT+Tg, and KO+Tg are overlapping.
FIGURE 4.
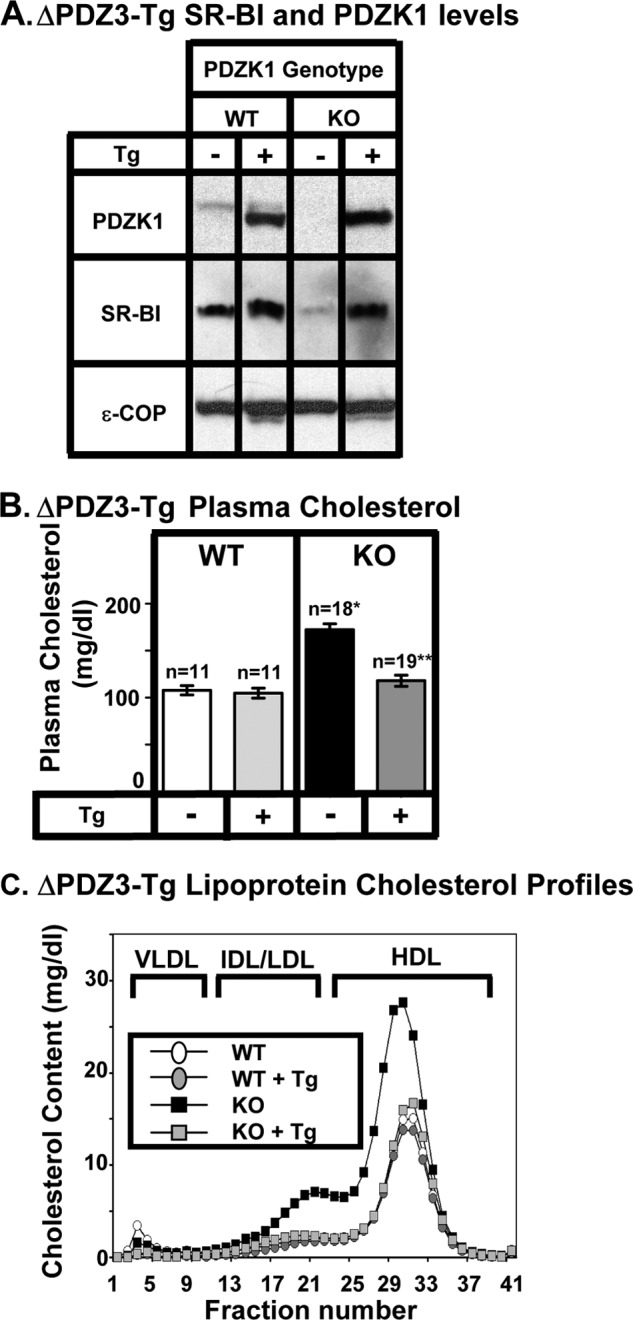
Effects of expression of the ΔPDZ3 transgene on hepatic SR-BI protein levels (A) and plasma lipoprotein cholesterols (B and C) in WT and PDZK1 KO mice. A, liver lysates (∼30 μg of protein) from mice with the indicated genotypes, with (+) or without (-) the ΔPDZ3 Tg (deletion of the PDZ3 domain, see Fig. 1F), were analyzed by immunoblotting (data from founder 8642), and bands representing PDZK1 (∼70 kDa for WT) and SR-BI (∼82 kDa) were visualized by chemiluminescence. ϵ-COP (∼34 kDa) was used as a loading control. Note the faint SR-BI band in the nontransgenic PDZK1 KO lane. Replicate experiments with multiple exposures and sample loadings were used to determine the relative levels of expression of SR-BI (Table 2). B, plasma samples were harvested from mice with the indicated genotypes and ΔPDZ3 transgene. Total plasma cholesterol levels were determined in individual samples by enzymatic assay, and mean values from the indicated numbers of animals (n) are shown for each genotype. Independent WT and KO control animals for each founder line were generated to ensure that the mixed genetic backgrounds for experimental and control mice were matched. Values from the two independent founder lines, 8642 and 8640, were not statistically significantly different and thus were averaged together here. * indicates the nontransgenic KO plasma cholesterol levels were statistically significantly different from those plasma cholesterol levels of WT (p < 0.0001). ** indicates PDZK1 KO (ΔPDZ3-Tg) plasma cholesterol levels were statistically significantly different from those plasma cholesterol levels of nontransgenic PDZK1 KO mice (p < 0.0001). WT (ΔPDZ3-Tg) and KO (ΔPDZ3-Tg) plasma cholesterol levels were not statistically significantly different. C, pooled plasma samples (described in B) from four to five animals (data from founder 8642) were size-fractionated by FPLC, and the total cholesterol content of each fraction was determined by an enzymatic assay. The chromatograms are representative of multiple individually determined profiles. Approximate elution positions of native VLDL, IDL/LDL, and HDL particles are indicated by brackets and were determined as described previously (13). The chromatograms for the WT, WT+Tg, and KO+Tg are overlapping.
TABLE 1.
Relative PDZK1 and SR-BI protein expression levels in Tg and nontransgenic mice
| PDZK1 mutant | Founder | Relative PDZK1 protein level, WT-Tg/WT | Relative SR-BI protein levela |
|
|---|---|---|---|---|
| WT-Tg/WT | KO-Tg/WT | |||
| PDZ2→1 | 8419 | 5.40 ± 0.61b | 2.16 ± 0.32c | 2.18 ± 0.78d |
| 8418 | 4.50 ± 1.12b | |||
| ΔPDZ2 | 8421 | 17.15 ± 4.66b | 2.29 ± 0.73c | 1.80 ± 0.38d |
| 8423 | 16.59 ± 1.57b | |||
| ΔPDZ3 | 8642 | 2.45 ± 0.46b | 1.33 ± 0.21c | 0.85 ± 0.33d |
| 8640 | 15.48 ± 4.17b | |||
| PDZK1(G389P) | 8743 | 14.25 ± 1.02b | 1.70 ± 0.29c | 2.07 ± 0.56d |
| 8744 | 22.39 ± 4.69b | |||
a Averages include both founders.
b p values comparing WT-Tg versus WT ranged from 0.02 to <0.0001.
c p = 0.01 for WT-Tg versus WT.
d p > 0.05 for WT-Tg or KO-Tg versus WT.
FIGURE 5.

Immunohistochemical analysis of hepatic SR-BI in WT and PDZK1 KO nontransgenic (A and B), PDZ2→1 transgenic (C and D), ΔPDZ2 transgenic (E and F), ΔPDZ3 transgenic (G and H), and PDZK1(G389P) transgenic (I and J) mice. Livers from mice of the indicated genotypes and Tg were fixed, frozen, and sectioned (data from the corresponding founders 8419, 8421, 8642, and 8743, respectively). The sections were then stained with a polyclonal anti-SR-BI antibody and a biotinylated anti-rabbit IgG secondary antibody and visualized by immunoperoxidase staining. Magnification, ×600.
Effects of Mutant PDZK1 Constructs in Wild-type (WT) Mice
In transgenic WT (WT-Tg) mice relative to nontransgenic WT mice, expression of the ΔPDZ2 and ΔPDZ3 protein had no significant influence on the levels of hepatic SR-BI protein expression determined by immunoblotting (Figs. 2A to 4A and Table 1). Expression of the PDZ2→1 protein resulted in a small, but significant, increase in hepatic SR-BI protein expression (2.16 ± 0.32-fold, p = 0.01). None of the three transgenes altered the sinusoidal membrane-associated distribution of SR-BI determined by immunohistochemistry (Fig. 5, A, C, E, and G), plasma cholesterol levels (left panels in Figs. 2B to 4B), or lipoprotein cholesterol size distribution profiles determined by FPLC (circles in Figs. 2C to 4C)). Thus, these mutated transgenes did not produce substantial dominant-negative effects when overexpressed in WT mice (an example of a dominant-negative truncated PDZK1 is described in Ref. 24).
Effects of Mutant PDZK1 Constructs in PDZK1 KO Mice
As described previously (8, 23, 24, 41), the absence of PDKZ1 expression in nontransgenic PDZK1 KO mice resulted in a dramatic loss of hepatic SR-BI protein (Figs. 2A to 4A and 5B), increased plasma total cholesterol (black bars in right panels of Figs. 2B to 4B), and an increase in the size of HDL particles (leftward shift in lipoprotein profiles, black squares in Figs. 2C to 4 C). All of these abnormalities are corrected by hepatic expression of a wild-type PDZK1 transgene in otherwise PDKZ1 KO animals (24). Hepatic transgenic expression of the PDZ2→1, ΔPDZ2, and ΔPDZ3 mutants in PDZK1 KO mice also corrected all of these abnormalities (Figs. 2–4, 5, D, F, and H, and Table 1). Thus, a PDZK1 transgene containing a deletion of either the PDZ2 or PDZ3 domain can promote essentially full normalization of hepatic SR-BI steady state expression, localization, and function. PDZK1 mutants with only three PDZ domains (PDZ1 and PDZ4 with either PDZ2 or PDZ3) could restore normal SR-BI activity as can the four PDZ domain containing full-length PDZK1 (Fig. 1A) and the C-terminally truncated PDZ1234 (Fig. 1C). Thus, the spacing between the PDZ1 and PDZ4 domains in the linear sequence (compare PDZ2→1 to ΔPDZ2, Fig. 1, D and E) does not appear to play a critical role in the regulation of hepatic SR-BI.
FIGURE 3.
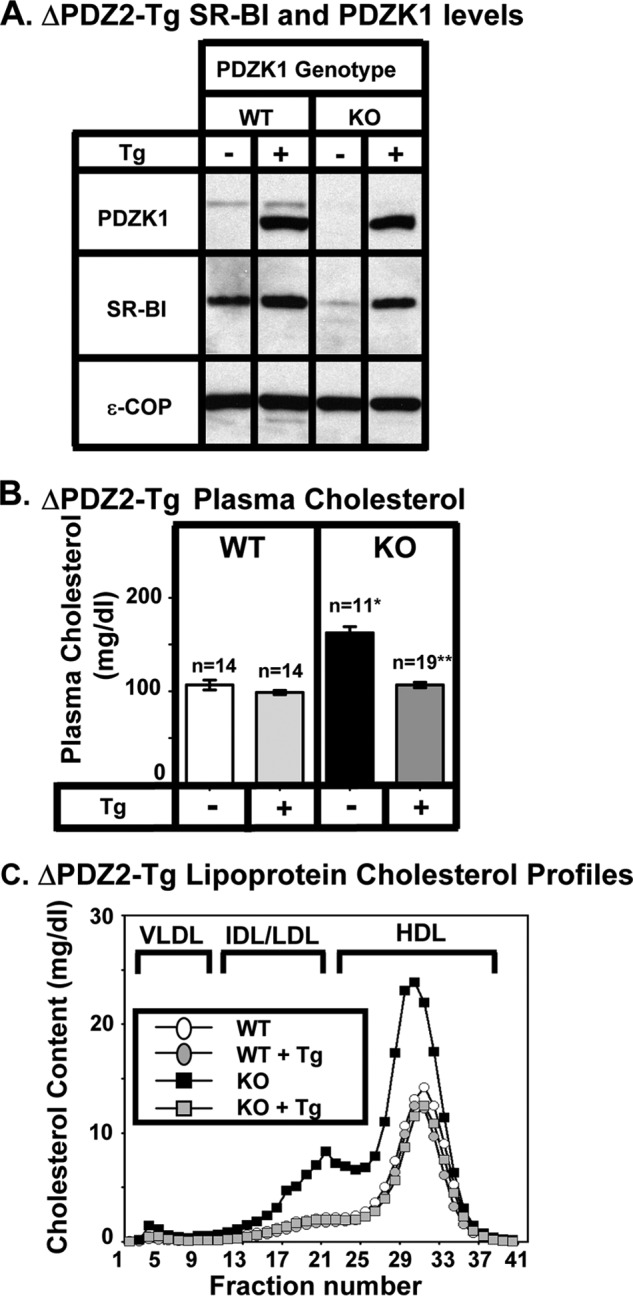
Effects of expression of the ΔPDZ2 transgene on hepatic SR-BI protein levels (A) and plasma lipoprotein cholesterol (B and C) in WT and PDZK1 KO mice. A, liver lysates (∼30 μg of protein) from mice with the indicated genotypes, with (+) or without (−) the ΔPDZ2 Tg (deletion of the PDZ2 domain, see Fig. 1E), were analyzed by immunoblotting (data from founder 8421), and bands representing PDZK1 (∼70 kDa for WT) and SR-BI (∼82 kDa) were visualized by chemiluminescence. ϵ-COP (∼34 kDa) was used as a loading control. Note the faint SR-BI band in the nontransgenic PDZK1 KO lane. Replicate experiments with multiple exposures and sample loadings were used to determine the relative levels of expression of SR-BI (Table 2). B, plasma samples were harvested from mice with the indicated genotypes and ΔPDZ2 transgene. Total plasma cholesterol levels were determined in individual samples by enzymatic assay, and mean values from the indicated numbers of animals (n) are shown for each genotype. Independent WT and KO control animals for the founder line were generated to ensure that the mixed genetic backgrounds for experimental and control mice were matched. Values from the two independent founder lines, 8421 and 8423, were not statistically significantly different and thus were averaged together here. * indicates the nontransgenic KO plasma cholesterol levels were statistically significantly different from those plasma cholesterol levels of WT (p < 0.0001). ** indicates PDZK1 KO (ΔPDZ2-Tg) plasma cholesterol levels were statistically significantly different from those plasma cholesterol levels of nontransgenic PDZK1 KO mice (p < 0.0001). WT (ΔPDZ2-Tg) and KO (ΔPDZ2-Tg) plasma cholesterol levels were not statistically significantly different. C, pooled plasma samples (described in B) from four to five animals (data from founder 8421) were size-fractionated by FPLC, and the total cholesterol content of each fraction was determined by an enzymatic assay. The chromatograms are representative of multiple individually determined profiles. Approximate elution positions of native VLDL, IDL/LDL, and HDL particles are indicated by brackets and were determined as described previously (13). The chromatograms for the WT, WT+Tg, and KO+Tg are overlapping.
Mechanism by Which the PDZ4 Domain Influences Hepatic SR-BI
We previously showed that a C-terminally truncated PDZK1 containing only the first three N-terminal PDZ domains (PDZ123, Fig. 1B) is unable to restore full normal expression and function of SR-BI in PDZK1 KO mice, whereas a truncated PDZK1 transgene containing all four PDZ domains (PDZ1234, Fig. 1C) can do so (23). Thus, even though PDZ4 does not bind directly to SR-BI's C terminus (19), this domain in PDZK1 performs a critical role(s) in promoting normal SR-BI protein expression and activity.
To further explore the role of the PDZ4 domain, we examined by immunohistochemistry using an anti-PDZK1 polyclonal antibody the cellular distribution of PDZK1, PDZ1234, and PDZ123 in hepatocytes in liver sections. Fig. 6A shows that in WT mice immunoperoxidase staining of endogenous PDZK1 exhibited a cell surface membrane-associated distribution similar to that seen for SR-BI (compare with Fig. 5A) (8, 19, 23). This staining is absent in nontransgenic PDZK1 KO liver (Fig. 6B), indicating that the staining was PDZK1-specific. PDZ1234 in the liver of PDZK1 KO mice expressing a PDZ1234 transgene (23) (∼10-fold greater PDZ1234 protein than PDZK1 in WT liver, see under “Experimental Procedures”) exhibited a similar cell surface membrane-associated distribution (Fig. 6D). Strikingly, in the livers of PDZK1 KO mice expressing a PDZ123 transgene (∼8-fold greater PDZ123 protein than PDZK1 in WT liver, see “Experimental Procedures”), we detected little cell surface membrane association (Fig. 6C), although there was clearly a significant level of intracellular staining relative to the negative control (Fig. 6B). Thus, the PDZ4 domain of PDZK1 appears to be necessary for this adaptor to associate robustly with the cell surface membrane of hepatocytes in vivo.
FIGURE 6.

Immunohistochemical analysis of the subcellular localization of PDZK1 in hepatocytes in vivo. Livers were harvested from WT mice (A), nontransgenic PDZK1 KO mice (B), transgenic PDZK1 KO mice expressing in hepatocytes either PDZ123 (C), PDZ1234 (D), PDZK1(G389P) (E), heterozygous (F), or homozygous (G) null SR-BI KO mice. Livers were fixed, frozen, and sectioned. The sections were then stained with a polyclonal anti-PDZK1 antibody, and a biotinylated anti-rabbit IgG secondary antibody, and visualized by immunoperoxidase staining. Magnification, ×600. Arrows indicate examples of cell surface membrane-associated staining.
Additional analysis suggests that the PDZ4 domain is not sufficient for this membrane association. We examined the localization of endogenous PDZK1 in livers from mice with heterozygous (Fig. 6F) or homozygous (Fig. 6G) null mutations of SR-BI, as well as the amount of PDZK1 in the livers. Immunoblotting analysis (supplemental Fig. S1 and data not shown) indicates that the absence of SR-BI does not alter the levels of endogenous PDZK1. However, unlike the WT-like intracellular distribution of PDZK1 in heterozygous null SR-BI mice (Fig. 6F), there was little, if any, cell surface membrane association of endogenous PDZK1 in homozygous null SR-BI KO mice (Fig. 6G). These immunohistochemical localization results suggest that both SR-BI and the PDZ4 domain of PDZK1 are required for robust cell surface membrane association of PDZK1. They also raise the possibility that the PDZ4 domain may be required for normal regulation of SR-BI by virtue of its ability to influence PDZK1's subcellular distribution.
Given the scaffold function of many multiple PDZ domain-containing proteins (20), it seemed possible that PDZK1 might mediate cell surface membrane localization and regulate SR-BI by linking SR-BI (while bound to PDZ1 or PDZ3) to some other protein whose C terminus binds in the canonical peptide-binding site of PDZ4. To test this possibility, we first generated and tested in vitro the binding activities of a set of mutant recombinant PDZ4 proteins and full-length PDZK1 proteins to identify a mutation that would abrogate classic C-terminal peptide binding. We then generated transgenic mice expressing full-length PDZK1 with a C-terminal peptide-binding negative mutation in the PDZ4 domain (Fig. 1G) and characterized in the transgenic animals its subcellular localization in the liver and its influence on SR-BI expression and activity.
ITC Analysis of the Effects of Amino Acid Substitutions within the Carboxylate-binding Loop of PDZ4 on Classic C-terminal Peptide Binding
The C terminus of the transient receptor potential cation channel subfamily C member 5 (TRPC5) was previously identified as a target of the PDZ4 domain of PDZK1 (42). We generated in E. coli and purified a group of wild-type and mutant recombinant PDZ4 domains and used ITC to measure their binding to a peptide with the sequence of the C terminus of TRPC5, 969EQVTTRL975 (target peptide). The amino acid substitutions in the carboxylate binding loops of these mutants included single residue substitutions (Y388A, G389P, or G389A) or double residue substitutions (Y388G/G389P, Y388A/G389A, or Y388A/G389P). The Y388A substitution was made because the analogous substitution in the PDZ1 and PDZ3 domains prevents binding of a C-terminal peptide from SR-BI to these domains (18, 19). Substitutions at Gly389 were made because this residue is one of the most highly conserved in the PDZ superfamily of proteins, and larger side chains are expected to disrupt target peptide binding (1).
The results of the ITC analyses are shown in Table 2, and selected titration binding curves are shown in Fig. 7. The TRPC5 target peptide bound to wild-type recombinant PDZ4 with a Kd of 10.8 ± 0.6 μm (Fig. 7A). The single Y388A substitution increased the Kd value of target peptide binding by >10-fold, but the residual binding was too high to permit definitive conclusions to be drawn from previously reported in vivo transgenic mouse functional studies that employed this mutation incorporated into the full-length PDZK1 (PDZK1(Y388A)) (19). Transgenic mice overexpressing PDZK1(Y388A) (18–29-fold above that of endogenous PDZK1 in wild-type mice) can complement the defective hepatic SR-BI phenotypes in PDZK1 KO mice (19); however, the residual binding activity of the PDZ4 domain might have been responsible for this activity. There was also residual binding activity for PDZ4 with a single G389A substitution (Table 2). In contrast, the G389P substitution, singly or in combination with Tyr388 substitutions, completely blocked binding (Fig. 7B). Therefore, we incorporated the single G389P substitution into full-length PDZK1 (PDZK1(G389P)) and compared binding of the target peptide to this mutant with that of full-length, wild-type PDZK1 and with the C-terminally truncated PDZ123 protein (Fig. 1B) that does not contain the PDZ4 domain (all recombinant proteins generated in E. coli). Fig. 7, C–E, shows that the binding affinities of TRPC5 target peptide to all three proteins were similar, with Kd values ranging from 7.8 to 12.3 μm; although the approximate number of binding sites for WT PDZK1 (n = 2.17 ± 0.03) was almost twice that of either PDZK1(G389P) (n = 1.37 ± 0.01) or PDZ123 (n = 1.40 ± 0.02). These results suggest that there are two binding sites with similar affinities for the TRPC5 target peptide in WT PDZK1 and that the binding site on the PDZ4 domain is lost when that domain is removed (PDZ123) or when 389Gly is replaced by a Pro. We conclude that expression of the PDZK1(G389P) mutant as a transgene would permit evaluation of the role of classic target peptide binding to PDZ4 in PDZK1's regulation of hepatic SR-BI in vivo.
TABLE 2.
Isothermal titration calorimetric analysis of the binding of a C-terminal peptide from TRPC5 to full-length and truncated PDZK1 recombinant proteins
| Kda | No. of binding sites | |
|---|---|---|
| PDZ4(WT) | 10.8 ± 0.6 μm | 1.07 ± 0.02 |
| PDZ4(Y388A) | 151.1 ± 27.6 μm | 1.80 ± 0.54 |
| PDZ4(G389A) | 37.3 ± 7.25 μm | 0.49 ± 0.20 |
| PDZ4(G389Y) | No binding | NAb |
| PDZ4(G389P) | No binding | NA |
| PDZ4(Y388G/G389P) | No binding | NA |
| PDZ4(Y388A/G389A) | No binding | NA |
| PDZ4(Y388A/G389P) | No binding | NA |
| PDZK1(WT) | 12.3 ± 0.8 μm | 2.17 ± 0.03 |
| PDZK1(G389P) | 7.8 ± 0.3 μm | 1.37 ± 0.01 |
| PDZ123 | 10.2 ± 0.6 μm | 1.40 ± 0.02 |
a The peptide sequence is 969EQVTTRL975.
b NA means not applicable.
FIGURE 7.

Isothermal titration calorimetric analysis of the binding of a C-terminal peptide from TRPC5 to PDZK1-derived recombinant proteins. The indicated individual recombinant wild-type and mutant PDZK1 variants (0.03 mm in 1.8 ml of 150 mm NaCl, 0.5 mm tris(2-carboxyethyl)phosphine, 25 mm Tris, pH 8.0) were placed in a titration cell and equilibrated at 20 °C. These proteins were: A, WT PDZ4 domain; B, PDZ4(G389P) domain; C, full-length WT-PDZK1; D, full-length PDZK1(G389P); and E, PDZ123. A solution containing 1.0 mm of the C-terminal heptapeptide from TRPC5, 969EQVTTRL975, was injected in 10-μl aliquots with an interval of 4 min between each addition to permit re-equilibration. Titration curves were analyzed, and the values of Kd and N (number of binding sites/molecule) were determined using ORIGIN 7.0 software.
In Vivo Activity of the PDZK1(G389P) Transgene
We established two founder lines with the PDZK1(G389P) transgene that expressed the mutant protein at either 14.3 ± 1.0-fold (founder 8743) or 22.4 ± 4.7-fold (founder 8744) above normal endogenous WT PDZK1 levels (Table 1). These founders were used to generate matched wild-type (WT-Tg) and PDZK1 KO (KO-Tg) transgenic mice on a mixed FVB/129SvEv background. Background-matched nontransgenic WT and PDZK1 KO mice were used as controls for each founder. The corresponding WT and KO transgenic mice derived from each these two founders exhibited similar phenotypes. The results are shown in Figs. 5, I and J, and 8. In the WT background the hepatic SR-BI protein levels (Fig. 8A) and distributions (Fig. 5I) and plasma cholesterol levels (Fig. 8B) and lipoprotein size distributions (Fig. 8C) were unchanged by the transgene. Thus, the transgene did not exhibit dominant-negative effects. All of the SR-BI-associated phenotypic abnormalities were corrected by hepatic expression of the PDZK1(G389P) transgene in otherwise PDKZ1 KO animals (Figs. 5J and 8). Furthermore, the subcellular distribution of the PDZK1(G389P) in PDZK1 KO mouse liver (Fig. 6E) was similar to that of WT PDZK1 in WT mice (Fig. 6A). Therefore, we conclude that both PDZ4-mediated cell surface membrane localization of PDZK1 and the mechanism by which PDZ4 contributes to PDZK1's regulation of hepatic SR-BI do not involve canonical binding of a target protein's C terminus to the canonical target peptide-binding site of PDZ4.
FIGURE 8.
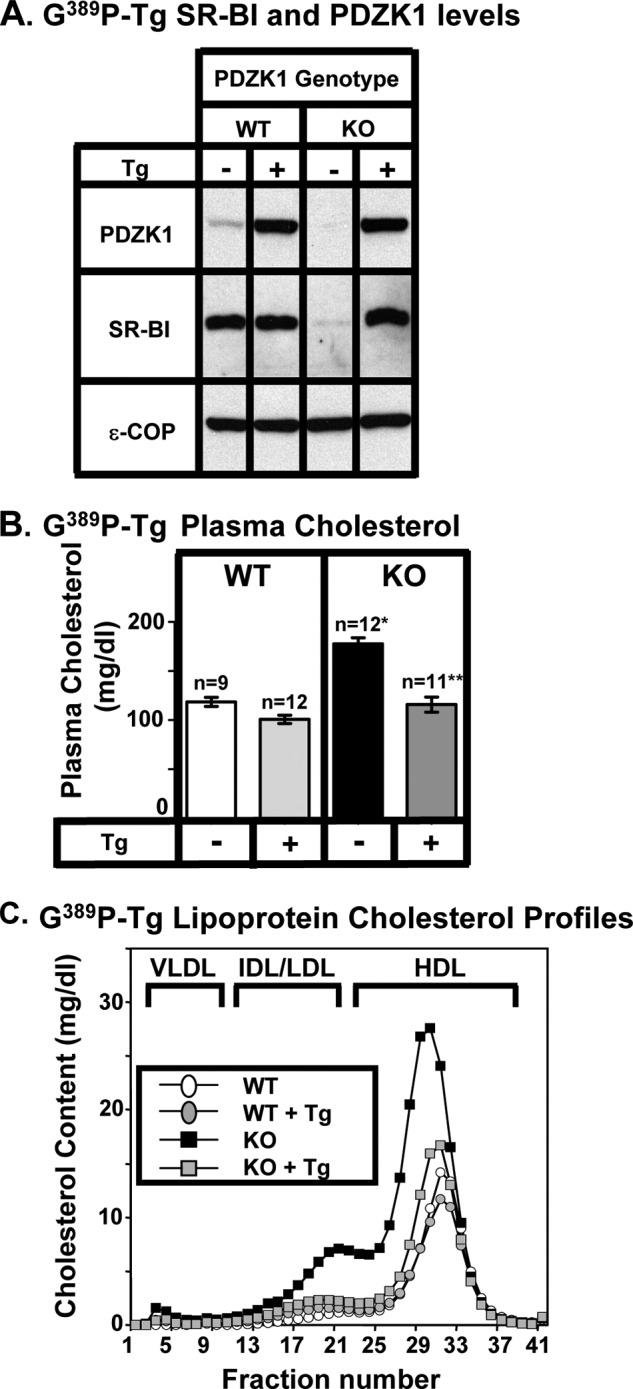
Effects of expression of the PDZK1(G389P) single mutant transgene on hepatic SR-BI protein levels (A) and plasma lipoprotein cholesterol (B and C) in WT and PDZK1 KO mice. A, liver lysates (∼30 μg of protein) from mice with the indicated genotypes, with (+) or without (−) the PDZK1(G389P) transgene (Tg) (inactivating single residue substitution in the PDZ4 domain, see Fig. 1G), were analyzed by immunoblotting (data from founder 8744), and bands representing PDZK1 (∼70 kDa) and SR-BI (∼82 kDa) were visualized by chemiluminescence. ϵ-COP (∼34 kDa) was used as a loading control. Note the faint SR-BI band in the nontransgenic PDZK1 KO lane. Replicate experiments with multiple exposures and sample loadings were used to determine the relative levels of expression of SR-BI (Table 2). B, plasma samples were harvested from mice with the indicated genotypes and PDZK1(G389P) transgene. Total plasma cholesterol levels were determined in individual samples by enzymatic assay, and mean values from the indicated numbers of animals (n) are shown for each genotype. Independent WT and KO control animals for each founder line were generated to ensure that the mixed genetic backgrounds for experimental and control mice were matched. Values from the two independent founder lines, 8744 and 8743, were not statistically significantly different and thus were averaged together here. * indicates the nontransgenic KO plasma cholesterol levels were statistically significantly different from those plasma cholesterol levels of WT (p < 0.0001). ** indicates PDZK1 KO (G389P) plasma cholesterol levels were statistically significantly different from those plasma cholesterol levels of nontransgenic PDZK1 KO mice (p < 0.0001). WT (G389P) and KO (G389P) plasma cholesterol levels were not statistically significantly different. C, pooled plasma samples (described in B) from four to five animals (data from founder 8743) were size-fractionated by FPLC, and the total cholesterol content of each fraction was determined by an enzymatic assay. The chromatograms are representative of multiple individually determined profiles. Approximate elution positions of native VLDL, IDL/LDL, and HDL particles are indicated by brackets and were determined as described previously (13). The chromatograms for the WT, WT+Tg, and KO+Tg are overlapping.
The correlation of the PDZ4-dependent cell surface membrane localization of PDZK1 and its mutants with the regulation of hepatic SR-BI is consistent with the possibility that PDZ4 might mediate this regulation as a consequence of mediating PDZK1's association with the cell surface membrane. We explored this possibility by examining (using biophysical techniques in vitro) three potential mechanisms by which PDZ4 might influence the intracellular distribution of PDZK1 as follows: 1) PDZ4-mediated homo-oligomerization of PDZK1; 2) PDZ4-induced conformational changes in the PDZ123 portion of PDZK1, and 3) PDZ4-mediated binding to the phospholipids in the inner leaflet of the plasma membrane.
Analytical Ultracentrifugation Examination of PDZK1 Oligomerization
Several reports have suggested that PDZ domain-containing proteins, including PDZK1, may be able to dimerize due to intermolecular associations, such as domain swapping or interdomain associations (19, 43–47) (see below for additional discussion). Therefore, we considered the possibility that the PDZ4 domain might mediate homo-oligomerization of PDZK1 and in so doing influence its membrane association and regulation of SR-BI. We used analytical ultracentrifugation to determine the extent of homo-oligomerization of recombinant full-length PDZK1. Fig. 9 shows the results of sedimentation velocity analysis with essentially no observable concentration dependence of the normalized sedimentation patterns indicating that under these conditions there was only very weak (arrow), if any, intermolecular self-association of PDZK1 molecules. Global Lamm equation curve fitting to these data with SEDANAL gave a molar mass of 54,000 g/mol with no indication of self-association under these conditions. Lalonde and Bretscher (47) have also reported weakly interacting, modest homodimerization of PDZK1 mediated by the PDZ3 but not the PDZ4 domain. It is possible that the absence of strong intermolecular self-association seen in vitro does not reflect PDZK1's status in vivo. However, if the lack of significant homodimerization of PDZK1 in vitro reflects its state in vivo, it seems unlikely that PDZ4 mediates homo-oligomerization in vivo and thus influences SR-BI regulation via such oligomerization.
FIGURE 9.

Sedimentation velocity ultracentrifugation analysis of PDZK1. Sedimentation velocity profiles of PDZK1 were determined at three different concentrations as follows: 1.803 (red), 0.453 (blue), and 0.212 (green) mg/ml. No observable concentration dependence of the normalized sedimentation patterns was seen, indicating that under these conditions there was only very weak, if any, intermolecular self-association of PDZK1 molecules. Vertical axis, normalized sedimentation pattern, g(S*)/Co; horizontal axis: apparent sedimentation coefficient, S*. The slight shift (arrow) to higher S* values may reflect a very weak self-association under these conditions. However, global fitting of these data to a single species model did not indicate the presence of significant amount of dimer at 1.8 mg/ml.
Analysis of the Influence of PDZ4 on the Conformation of PDZ123 Using HXMS
We examined another mechanism by which PDZ4 might potentiate the activity of the otherwise not fully active PDZ123 (Fig. 1B) portion of PDZK1. It seemed possible that the presence of the PDZ4 domain at the C terminus of the PDZ1234 protein (Fig. 1C) might alter the conformation of the SR-BI binding PDZ123 portion of the PDZ1234 protein, consequently conferring the ability of PDZ1234 to regulate SR-BI activity normally. To explore this possibility, we used the highly sensitive HXMS technique to assess the effects of the PDZ4 domain on the conformation of the PDZ123 portion of PDZ1234. The conformation of a protein and its interaction with other proteins can substantially influence the ability of backbone amide hydrogens to productively exchange with hydrogen/deuterium atoms in the solvent milieu. Our expectation was that substantial conformational changes in the PDZ123 portion of PDZ1234 relative to PDZ123 alone would be reflected in significant relative differences in deuterium exchange for at least some of the peptides common to the two proteins.
We individually incubated PDZ123 (residues 1–359) and PDZ1234 (1–458) from 10 s to 4 h at 21 °C in buffer A containing deuterated water and then quenched the exchange reactions with low pH (2.5) and temperature (0 °C), digested the proteins into peptides, and used tandem mass spectrometry to identify peptides that correspond to 98.2% of the predicted full-length sequences (supplemental Fig. S2). The relative amounts of deuterium incorporated into each peptide were determined for PDZ123 and PDZ1234 at each incubation time (supplemental Fig. S3). Using these data, we calculated the difference in deuterium incorporation for the common peptides detected in both PDZ123 and PDZ1234, which spanned residues 1–337 (bar graph in Fig. 10). In Fig. 10, bars to the left of the zero base line in the center of the figure represent deuterium incorporation that was greater in PDZ123 than PDZ1234, with the reverse for bars to the right. Notably, the differences obtained for all shared peptides of the two proteins were below 0.4 Da (green vertical dashed lines in Fig. 10), the threshold for meaningful differences in HXMS measurements of this type (31, 32). The data in Fig. 10 clearly show that there were no significant changes in deuterium incorporation between the common regions of PDZ123 and PDZ1234 (residues 1–337) as measured by HXMS. We cannot exclude the possibility that interaction between PDZ4 and the PDZ123 region is governed primarily by side chain-side chain interactions and that those interactions might not influence backbone amide hydrogen exchange. However, it seems more likely that the presence of PDZ4 does not induce substantial conformational changes within the PDZ123 region of PDZ1234. Thus, such PDZ4-induced conformational changes may not be responsible for PDZ4's influence on the localization and in vivo activity of PDZK1.
FIGURE 10.

Comparison of the conformations of the PDZ123 regions of PDZ123 and PDZ1234 using hydrogen/deuterium exchange mass spectrometry. Recombinant PDZ123 (residues 1–359) and PDZ1234 (1–458) proteins that also included two additional N-terminal residues (−2Gly and −1Ser, from the cloning vector) were incubated for 10 s (orange), 10 min (red), 1 h (blue), or 4 h (black) in buffer A containing deuterated water. The proton/deuterium exchange reactions were then quenched with low pH (2.5) and temperature (0 °C), and the proteins were digested into peptides, and the average amounts of deuterium incorporated was determined using mass spectrometry. We identified peptides corresponding to 98.2% of the predicted full-length PDZ1234 sequence (supplemental Fig. S2, created using MSTools (55)). Deuterium incorporation plots for the individual peptides presented here are shown in supplemental Fig. S3. The differences in deuterium incorporation for each of the indicated peptides (PDZ1234 to PDZ123) at each time of incubation are expressed in daltons (Da). The error limit for the determination of deuterium incorporation is ±0.4 Da (indicated with vertical dashed green lines (31)).
Analysis of the Membrane Binding Activity of PDZ4 Using SPR
Because we obtained in vivo or in vitro data suggesting that protein-protein interactions mediated by PDZ4 were not responsible for its influence on PDZK1's regulation of hepatic SR-BI, we examined the ability of PDZ4 to mediate protein-lipid interactions. There are several reports that that PDZ domains can directly bind to membrane lipids (36, 48, 49) and thus mediate protein-membrane interactions. Such interactions may play an important part in the mechanisms underlying the functions of some PDZ domain-containing proteins. Because PDZK1 interacts with proteins located in the plasma membrane, we decided to test the ability of PDZ4 to interact with lipids in the inner plasma membrane. We thus measured the binding of PDZ4 and other PDZ domains from PDZK1 to vesicles whose lipid composition mimics that of the inner leaflet of the plasma membrane of mammalian cells (50). We previously reported that PDZ1, PDZ2, and PDZ3 domains from PDZK1 do not individually bind to such vesicles (36). Here, we measured the binding of individual recombinant PDZ4 and PDZ3 domains to plasma membrane mimetic vesicles. The sensorgrams in Fig. 11A (left) show the time courses of binding to the lipid vesicles at the indicated domain concentrations. The sensorgrams show that there was essentially no significant binding of the PDZ3 domain, as reported previously (36), whereas there was a dose-dependent binding of PDZ4. Fig. 11A (right) shows that the concentration dependence of maximal binding for PDZ4 is well represented by a Langmuir-type saturation binding curve with an apparent Kd of 1.2 ± 0.1 μm. Fig. 11B shows similar results for the binding of recombinant proteins containing either only the first three PDZ domains (PDZ123, left panel, no significant binding) or all four PDZ domains (PDZ1234) (left and right panels, significant binding with an apparent Kd of 1.1 ± 0.1 μm that is virtually identical to that of the PDZ4 domain alone). These data suggest that the in vitro membrane binding activity of PDZ1234 is mediated by PDZ4. Taken together with the in vivo cell surface membrane localization results in Fig. 6, the in vitro membrane binding studies strongly suggest that that PDZ4-mediated binding of PDZK1 to cell surface inner membrane lipids may underlie the ability of PDZ4 to confer on PDZ1234 the ability to fully regulate the expression, localization, and function of SR-BI in hepatocytes in vivo (23).
FIGURE 11.

Determination of the binding of recombinant PDZ3, PDZ4, PDZ123, and PDZ1234 to plasma membrane mimetic vesicles by equilibrium SPR analysis. A, left panel, sensorgrams of the time courses of binding of PDZ3 (2 μm, broken line) and PDZ4 (0.25, 0.5, 1, 2, 3, and 5 μm, lowest to highest sensorgram responses, respectively). Equilibrium SPR measurements were performed in 20 mm Tris-HCl, pH 7.4, containing 0.16 m NaCl. Proteins were injected over an L1 chip coated with PM mimetic vesicles at 5 μl/min. For PDZ4, the membrane association was too slow for the sensorgrams to reach saturation within the injection period, and thus the final resonance units (RU) values were arbitrarily taken as Req. Right panel, concentration dependence of binding of PDZ4 generated from Req values (averages of triplicate measurements). The solid line represents the curve determined by nonlinear least squares fit of the data to the equation Req = Rmax/(1 + Kd/P0), where P0 is the concentration of PDZ4, Rmax = 34 ± 2, and Kd = 1.2 ± 0.1 μm. B, left panel, sensorgrams of the time courses of binding of PDZ123 (2 μm, broken line) and PDZ1234 (0.25, 0.5, 1, 2, 3, and 5 μm, lowest to highest sensorgram responses, respectively) as described above. Right panel, concentration dependence of binding of PDZ1234 with the solid line representing the nonlinear least squares fit of the data (Rmax = 38 ± 1 and Kd = 1.1 ± 0.1 μm).
DISCUSSION
PDZK1 is a four PDZ domain-containing adaptor protein that controls the expression, localization, and function of several membrane-associated proteins, among them the HDL receptor SR-BI in the liver (8). PDZK1 is normally concentrated at cell surface membranes in hepatocytes in vivo, and this concentration depends on the presence of SR-BI (this study). Using transgenic mice, we have shown that the PDZ2 or PDZ3 domains can be deleted without abrogating PDZK1-mediated in vivo regulation of hepatic SR-BI protein expression, intracellular distribution, and function as an HDL receptor. The results for the ΔPDZ3 construct are consistent with our previous study that showed inactivation of the canonical target peptide-binding site on PDZ3 did not prevent essentially normal PDZK1-dependent regulation of hepatic SR-BI (19). The results also suggest that neither the PDZ2 nor PDZ3 domains play functionally important roles as spacers, maintaining appropriate separation of the other portions of the protein. Because PDZK1 is a multifunctional adaptor protein that interacts with many membrane proteins (2, 6), it is not surprising that two of its four PDZ domains would not be essential for one of its functions, i.e. regulation of hepatic SR-BI.
Although the fourth PDZ domain (PDZ4) of PDZK1 does not bind to the C terminus of SR-BI (19), it appears to be essential for normal PDZK1-mediated regulation of hepatic SR-BI in mice; hepatic expression of PDZ1234 fully corrects all hepatic SR-BI-related abnormal phenotypes in PDZK1 KO mice, whereas hepatic expression of PDZ123 only partially corrects these abnormalities (24). The PDZ4 domain also appears to be necessary for the normal cell surface membrane localization of PDZK1 (this study). We have used a combination of in vivo transgenic mice and in vitro biochemical and biophysical approaches to explore the role of PDZ4. For example, we examined the in vivo localization and function of PDZK1 with a point mutation in PDZ4 that prevents its binding to the C terminus of target proteins via its canonical peptide-binding site. This analysis established that PDZK1 localization and function are independent of target protein binding via this site (protein scaffold mechanism). Our in vitro studies also appear to exclude a role for PDZ4 in mediating robust oligomerization of PDZK1 and thus its bound SR-BI (PDZ4-mediated oligomerization) or altering substantially the conformation of the PDZ123 portion of PDZK1 (conformational activation). However, we have discovered a new activity of PDZ4 that provides an attractive explanation for the mechanism by which this domain mediates the cell surface membrane localization of PDZK1 and PDZK1's regulation of the activity of SR-BI (23). PDZ4 binds directly to phospholipids membranes that mimic the composition of the inner leaflet of the plasma membrane. The PDZ4-mediated binding of PDZK1 to lipids in the inner leaflet of the plasma membrane would increase the juxtamembrane concentration of PDZK1 and thus facilitate its interactions with SR-BI (PDZ4-mediated membrane proximity activation).
LaLonde and Bretscher (47) previously reported studies of PDZK1 that are directly relevant to the work reported here. They described detailed analyses of intramolecular and intermolecular (homodimerization) PDZK1 interactions (47). They observed modest (weak) homodimerization of PDZK1 detected in cultured cells by co-immunoprecipitation of overexpressed epitope-tagged forms of PDZK1 and in vitro by chemical cross-linking (0.1% glutaraldehyde) of purified recombinant PDZK1. The modest homodimerization they observed might correspond to the very weak intermolecular self-association we observed using analytical ultracentrifugation, although it is difficult to directly compare the results of these very different methods. The oligomerization they detected was mediated by the PDZ3 domain. Our observation of essentially full in vivo activity of a ΔPDZ3 protein suggests that any modest PDZ3-dependent homodimerization of PDZK1 in vivo is not required for effective PDZK1-mediated regulation of hepatic SR-BI, although it may contribute to other in vivo functions of PDZK1. LaLonde and Bretscher (47) also reported that a recombinant, N-terminally truncated portion of PDZK1 comprising only the PDZ4 domain and the remaining C-terminal residues does not dimerize in vitro (chemical cross-linking assay). These findings provide additional support for the proposal that intermolecular PDZ4-PDZ4 interactions are unlikely to mediate oligomerization that is functionally relevant to the hepatic regulation of SR-BI. Our conclusion that mechanism PDZ4-mediated oligomerization is unlikely appears to be consistent with the results of LaLonde and Bretscher (47).
LaLonde and Bretscher (47) also reported that an intramolecular interaction between the C terminus of PDZK1 and its PDZ1 domain can induce “the protein to adopt a more compact conformation.” The formation of the compact conformation that they detected using native PAGE depended on the presence of both the C terminus of PDZK1 and an intact carboxylate binding loop in the PDZ1 domain. We have previously reported that deletion of the C terminus of PDZK1 or inactivating the carboxylate binding loop in the PDZ1 domain does not prevent full PDZK1-mediated regulation of hepatic SR-BI (23). Thus, formation of the compact conformation of PDZK1 reported by LaLonde and Bretscher (47) apparently is not required for PDZK1-mediated regulation of hepatic SR-BI, although it may contribute to other in vivo functions of PDZK1.
Future studies will be required to define in greater detail the PDZ4-mediated membrane proximity activation mechanism for the in vivo subcellular localization of PDZK1 and its promotion of normal expression, localization, and function of hepatic SR-BI. In future work, the reagents and methods used in this study may help establish if PDZK1's PDZ2, PDZ3, or PDZ4 domains influence the other activities of this adaptor, such as mediating SR-BI signaling in endothelial cells (22, 51) or the activities of other membrane proteins (e.g. cystic fibrosis transmembrane regulator, chloride-formate exchanger, or Oatp1a1 (52–54)). Our results suggest that not all of the PDZ domains of a multifunctional, multi-PDZ domain-containing adaptor protein are required for each of its biological activities and that both the canonical target peptide binding and noncanonical (lipid binding) capacities of PDZ domains may be employed by a single such adaptor for optimal in vivo activity.
Supplementary Material
Acknowledgments
We thank Joel Lawitts from the Beth Israel Deaconess Medical Center Transgenic Facility and Debby Pheasant from the Massachusetts Institute of Technology Biophysical Instrumentation Facility for helping with the ITC experiments and Verna Frasca from Microcal for helping with analysis and interpretation of the ITC data.
This work was supported, in whole or in part, by National Institutes of Health Grants HL077780 (to O. K.), HL52212 and HL66105 (to M. K.), GM68849 (to W. C.), and GM086507 and GM101135 (to J. R. E.). This work was also supported by a research collaboration with the Waters Corp. (to J. R. E.).

This article contains supplemental Figs. S1–S3.
K. Tsukamoto, L. Buck, W. Inman, L. Griffith, O. Kocher, and M. Krieger, unpublished results.
- PDZ
- PSD-95, Discs-large, ZO-1
- SR-BI
- scavenger receptor class B, type I
- HXMS
- hydrogen/deuterium exchange mass spectrometry
- ITC
- isothermal titration calorimetry
- POPC
- 1 palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- SPR
- surface plasmon resonance
- Tg
- transgene
- PM
- plasma membrane
- IDL
- intermediate density lipoprotein.
REFERENCES
- 1. Pawson T., Nash P. (2003) Assembly of cell regulatory systems through protein interaction domains. Science 300, 445–452 [DOI] [PubMed] [Google Scholar]
- 2. Kocher O., Krieger M. (2009) Role of the adaptor protein PDZK1 in controlling the HDL receptor SR-BI. Curr. Opin. Lipidol. 20, 236–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Ham M., Hendriks W. (2003) PDZ domains–glue and guide. Mol. Biol. Rep. 30, 69–82 [DOI] [PubMed] [Google Scholar]
- 4. Doyle D. A., Lee A., Lewis J., Kim E., Sheng M., MacKinnon R. (1996) Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell 85, 1067–1076 [DOI] [PubMed] [Google Scholar]
- 5. Kocher O., Comella N., Tognazzi K., Brown L. F. (1998) Identification and partial characterization of PDZK1: a novel protein containing PDZ interaction domains. Lab. Invest. 78, 117–125 [PubMed] [Google Scholar]
- 6. Yesilaltay A., Kocher O., Rigotti A., Krieger M. (2005) Regulation of SR-BI-mediated high-density lipoprotein metabolism by the tissue-specific adaptor protein PDZK1. Curr. Opin. Lipidol. 16, 147–152 [DOI] [PubMed] [Google Scholar]
- 7. Ikemoto M., Arai H., Feng D., Tanaka K., Aoki J., Dohmae N., Takio K., Adachi H., Tsujimoto M., Inoue K. (2000) Identification of a PDZ-domain-containing protein that interacts with the scavenger receptor class B type I. Proc. Natl. Acad. Sci. U.S.A. 97, 6538–6543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kocher O., Yesilaltay A., Cirovic C., Pal R., Rigotti A., Krieger M. (2003) Targeted disruption of the PDZK1 gene in mice causes tissue-specific depletion of the high density lipoprotein receptor scavenger receptor class B type I and altered lipoprotein metabolism. J. Biol. Chem. 278, 52820–52825 [DOI] [PubMed] [Google Scholar]
- 9. Nakamura T., Shibata N., Nishimoto-Shibata T., Feng D., Ikemoto M., Motojima K., Iso-O N., Tsukamoto K., Tsujimoto M., Arai H. (2005) Regulation of SR-BI protein levels by phosphorylation of its associated protein, PDZK1. Proc. Natl. Acad. Sci. U.S.A. 102, 13404–13409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rigotti A., Miettinen H. E., Krieger M. (2003) The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr. Rev. 24, 357–387 [DOI] [PubMed] [Google Scholar]
- 11. Krieger M. (1999) Charting the fate of the “good cholesterol”: identification and characterization of the high-density lipoprotein receptor SR-BI. Annu. Rev. Biochem. 68, 523–558 [DOI] [PubMed] [Google Scholar]
- 12. Morrison J. R., Silvestre M. J., Pittman R. C. (1994) Cholesteryl ester transfer between high density lipoprotein and phospholipid bilayers. J. Biol. Chem. 269, 13911–13918 [PubMed] [Google Scholar]
- 13. Rigotti A., Trigatti B. L., Penman M., Rayburn H., Herz J., Krieger M. (1997) A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc. Natl. Acad. Sci. U.S.A. 94, 12610–12615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Plump A. S., Smith J. D., Hayek T., Aalto-Setälä K., Walsh A., Verstuyft J. G., Rubin E. M., Breslow J. L. (1992) Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 71, 343–353 [DOI] [PubMed] [Google Scholar]
- 15. Zhang S. H., Reddick R. L., Piedrahita J. A., Maeda N. (1992) Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 258, 468–471 [DOI] [PubMed] [Google Scholar]
- 16. Braun A., Trigatti B. L., Post M. J., Sato K., Simons M., Edelberg J. M., Rosenberg R. D., Schrenzel M., Krieger M. (2002) Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ. Res. 90, 270–276 [DOI] [PubMed] [Google Scholar]
- 17. Trigatti B., Rayburn H., Viñals M., Braun A., Miettinen H., Penman M., Hertz M., Schrenzel M., Amigo L., Rigotti A., Krieger M. (1999) Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc. Natl. Acad. Sci. U.S.A. 96, 9322–9327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kocher O., Birrane G., Tsukamoto K., Fenske S., Yesilaltay A., Pal R., Daniels K., Ladias J. A., Krieger M. (2010) In vitro and in vivo analysis of the binding of the C terminus of the HDL receptor scavenger receptor class B, type I (SR-BI), to the PDZ1 domain of its adaptor protein PDZK1. J. Biol. Chem. 285, 34999–35010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kocher O., Birrane G., Yesilaltay A., Shechter S., Pal R., Daniels K., Krieger M. (2011) Identification of the PDZ3 domain of the adaptor protein PDZK1 as a second, physiologically functional binding site for the C terminus of the high density lipoprotein receptor scavenger receptor class B type I. J. Biol. Chem. 286, 25171–25186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pawson T., Scott J. D. (1997) Signaling through scaffold, anchoring, and adaptor proteins. Science 278, 2075–2080 [DOI] [PubMed] [Google Scholar]
- 21. Kocher O., Yesilaltay A., Shen C. H., Zhang S., Daniels K., Pal R., Chen J., Krieger M. (2008) Influence of PDZK1 on lipoprotein metabolism and atherosclerosis. Biochim. Biophys. Acta 1782, 310–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu W., Saddar S., Seetharam D., Chambliss K. L., Longoria C., Silver D. L., Yuhanna I. S., Shaul P. W., Mineo C. (2008) The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity. Circ. Res. 102, 480–487 [DOI] [PubMed] [Google Scholar]
- 23. Fenske S. A., Yesilaltay A., Pal R., Daniels K., Barker C., Quiñones V., Rigotti A., Krieger M., Kocher O. (2009) Normal hepatic cell surface localization of the high density lipoprotein receptor, scavenger receptor class B, type I, depends on all four PDZ domains of PDZK1. J. Biol. Chem. 284, 5797–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fenske S. A., Yesilaltay A., Pal R., Daniels K., Rigotti A., Krieger M., Kocher O. (2008) Overexpression of the PDZ1 domain of PDZK1 blocks the activity of hepatic scavenger receptor, class B, type I by altering its abundance and cellular localization. J. Biol. Chem. 283, 22097–22104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simonet W. S., Bucay N., Lauer S. J., Taylor J. M. (1993) A far-downstream hepatocyte-specific control region directs expression of the linked human apolipoprotein E and C-I genes in transgenic mice. J. Biol. Chem. 268, 8221–8229 [PubMed] [Google Scholar]
- 26. Stafford W. (1994) Methods in Enzymology, Numerical computer methods, Part B, pp. 478–501, Academic Press, Orlando: [DOI] [PubMed] [Google Scholar]
- 27. Stafford W. (2000) Analysis of reversibly interacting macromolecular systems by time derivative sedimentation velocity. Methods Enzymol. 323, 302–325 [DOI] [PubMed] [Google Scholar]
- 28. Stafford W. F., Sherwood P. J. (2004) Analysis of heterologous interacting systems by sedimentation velocity: curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys. Chem. 108, 231–243 [DOI] [PubMed] [Google Scholar]
- 29. Stafford W. F. (1992) Boundary analysis in sedimentation transport experiments: a procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal. Biochem. 203, 295–301 [DOI] [PubMed] [Google Scholar]
- 30. Wales T. E., Fadgen K. E., Gerhardt G. C., Engen J. R. (2008) High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal Chem. 80, 6815–6820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Houde D., Berkowitz S. A., Engen J. R. (2011) The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 100, 2071–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iacob R. E., Engen J. R. (2012) Hydrogen exchange mass spectrometry: are we out of the quicksand? J. Am. Soc. Mass Spectrom. 23, 1003–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iacob R. E., Pene-Dumitrescu T., Zhang J., Gray N. S., Smithgall T. E., Engen J. R. (2009) Conformational disturbance in Abl kinase upon mutation and deregulation. Proc. Natl. Acad. Sci. U.S.A. 106, 1386–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wales T. E., Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 [DOI] [PubMed] [Google Scholar]
- 35. Zhang Z., Smith D. (1993) Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen Y., Sheng R., Källberg M., Silkov A., Tun M. P., Bhardwaj N., Kurilova S., Hall R. A., Honig B., Lu H., Cho W. (2012) Genome-wide functional annotation of dual-specificity protein- and lipid-binding modules that regulate protein interactions. Mol. Cell 46, 226–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stahelin R. V., Cho W. (2001) Differential roles of ionic, aliphatic, and aromatic residues in membrane-protein interactions: a surface plasmon resonance study on phospholipases A2. Biochemistry 40, 4672–4678 [DOI] [PubMed] [Google Scholar]
- 38. Kocher O., Pal R., Roberts M., Cirovic C., Gilchrist A. (2003) Targeted disruption of the PDZK1 gene by homologous recombination. Mol. Cell. Biol. 23, 1175–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Palmiter R. D., Brinster R. L. (1986) Germ-line transformation of mice. Annu. Rev. Genet. 20, 465–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guo Q., Penman M., Trigatti B. L., Krieger M. (1996) A single point mutation in epsilon-COP results in temperature-sensitive, lethal defects in membrane transport in a Chinese hamster ovary cell mutant. J. Biol. Chem. 271, 11191–11196 [DOI] [PubMed] [Google Scholar]
- 41. Yesilaltay A., Kocher O., Pal R., Leiva A., Quiñones V., Rigotti A., Krieger M. (2006) PDZK1 is required for maintaining hepatic scavenger receptor, class B, type I (SR-BI) steady state levels but not its surface localization or function. J. Biol. Chem. 281, 28975–28980 [DOI] [PubMed] [Google Scholar]
- 42. Stiffler M. A., Chen J. R., Grantcharova V. P., Lei Y., Fuchs D., Allen J. E., Zaslavskaia L. A., MacBeath G. (2007) PDZ domain binding selectivity is optimized across the mouse proteome. Science 317, 364–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fanning A. S., Lye M. F., Anderson J. M., Lavie A. (2007) Domain swapping within PDZ2 is responsible for dimerization of ZO proteins. J. Biol. Chem. 282, 37710–37716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Im Y. J., Lee J. H., Park S. H., Park S. J., Rho S. H., Kang G. B., Kim E., Eom S. H. (2003) Crystal structure of the Shank PDZ-ligand complex reveals a class I PDZ interaction and a novel PDZ-PDZ dimerization. J. Biol. Chem. 278, 48099–48104 [DOI] [PubMed] [Google Scholar]
- 45. Im Y. J., Park S. H., Rho S. H., Lee J. H., Kang G. B., Sheng M., Kim E., Eom S. H. (2003) Crystal structure of GRIP1 PDZ6-peptide complex reveals the structural basis for class II PDZ target recognition and PDZ domain-mediated multimerization. J. Biol. Chem. 278, 8501–8507 [DOI] [PubMed] [Google Scholar]
- 46. Kocher O., Comella N., Gilchrist A., Pal R., Tognazzi K., Brown L. F., Knoll J. H. (1999) PDZK1, a novel PDZ domain-containing protein up-regulated in carcinomas and mapped to chromosome 1q21, interacts with cMOAT (MRP2), the multidrug resistance-associated protein. Lab. Invest. 79, 1161–1170 [PubMed] [Google Scholar]
- 47. LaLonde D. P., Bretscher A. (2009) The scaffold protein PDZK1 undergoes a head-to-tail intramolecular association that negatively regulates its interaction with EBP50. Biochemistry 48, 2261–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zimmermann P. (2006) The prevalence and significance of PDZ domain-phosphoinositide interactions. Biochim. Biophys. Acta 1761, 947–956 [DOI] [PubMed] [Google Scholar]
- 49. Feng W., Zhang M. (2009) Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat. Rev. Neurosci. 10, 87–99 [DOI] [PubMed] [Google Scholar]
- 50. Stahelin R. V., Long F., Peter B. J., Murray D., De Camilli P., McMahon H. T., Cho W. (2003) Contrasting membrane interaction mechanisms of AP180 N-terminal homology (ANTH) and epsin N-terminal homology (ENTH) domains. J. Biol. Chem. 278, 28993–28999 [DOI] [PubMed] [Google Scholar]
- 51. Mineo C., Shaul P. (2012) Functions of scavenger receptor class B, type I in atherosclerosis. Curr. Opin. Lipidol. 23, 487–493 [DOI] [PubMed] [Google Scholar]
- 52. Thomson R. B., Wang T., Thomson B. R., Tarrats L., Girardi A., Mentone S., Soleimani M., Kocher O., Aronson P. S. (2005) Role of PDZK1 in membrane expression of renal brush border ion exchangers. Proc. Natl. Acad. Sci. U.S.A. 102, 13331–13336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang P., Wang J. J., Xiao Y., Murray J. W., Novikoff P. M., Angeletti R. H., Orr G. A., Lan D., Silver D. L., Wolkoff A. W. (2005) Interaction with PDZK1 is required for expression of organic anion transporting protein 1A1 on the hepatocyte surface. J. Biol. Chem. 280, 30143–30149 [DOI] [PubMed] [Google Scholar]
- 54. Wang S., Yue H., Derin R. B., Guggino W. B., Li M. (2000) Accessory protein facilitated CFTR-CFTR interaction, a molecular mechanism to potentiate the chloride channel activity. Cell 103, 169–179 [DOI] [PubMed] [Google Scholar]
- 55. Kavan D., Man P. (2011) MSTools–Web-based application for visualization and presentation of HXMS data. Int. J. Mass Spectrom. 302, 53–58 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.