

This Methods paper describes a novel approach for the isolation of ribonucleoprotein (RNP) complexes mediated by microRNAs (miRNAs). The authors developed a cell-free translation system to study interactions between hepatitis C virus (HCV) RNA and miRNA-122, which binds to the 5′-UTR region of HCV and stimulates its replication. This method could potentially be applicable to the characterization of miRNA–mRNA interactions.
Keywords: miRNA, hepatitis C virus, translation, RNA affinity chromatography
Abstract
Micro(mi)RNAs are 21- to 23-nt RNAs that regulate multiple biological processes. In association with Argonaute (Ago) proteins and other factors that form the RNA-induced silencing complex (RISC), miRNAs typically bind mRNA 3′ untranslated regions (UTRs) and repress protein production through antagonizing translation and transcript stability. For a given mRNA–miRNA interaction, cis-acting RNA elements and trans-acting RNA-binding proteins (RBPs) may influence mRNA fate. This is particularly true of the hepatitis C virus (HCV) genome which interacts with miR-122, an abundant liver miRNA. miR-122 binding to HCV RNA considerably stimulates virus replication in cultured cells and primates, but the mechanism(s) and associated host factors required for enhancement of HCV replication have not been fully elucidated. We recapitulated miR-122–HCV RNA interactions in a cell-free translation system derived from cells that express miR-122. Specifically, lysates produced from HEK-293 cells that inducibly transcribe and process pri-miR-122 were characterized alongside those from isogenic cells lacking miR-122 expression. We observed a stimulatory effect of miR-122 on HCV reporter mRNAs in a manner that depended on expression of miR-122 and intact target sites within the HCV 5′ UTR. We took advantage of this system to affinity-purify miR-122-HCV RNP complexes. Similar to functional assays, we found that association of immobilized HCV internal ribosome entry site (IRES) RNA with endogenous Ago2 requires both miR-122 expression and intact miR-122 target sites in cis. This combined approach may be generalizable to affinity purification of miRNP complexes for selected target mRNAs, allowing identification of miRNP components and RBPs that may contribute to regulation.
INTRODUCTION
Considerable effort has been devoted to understanding mechanisms by which microRNAs (miRNAs) regulate target mRNAs and identification of protein factors that modulate or are required for miRNA function (Pillai et al. 2007; Chekulaeva and Filipowicz 2009; Fabian et al. 2010; Fabian and Sonenberg 2012). miRNAs associate directly with Argonaute (Ago) proteins which, along with GW182 proteins, form the core of RNA-induced silencing complexes (RISCs) (Hammond et al. 2001; Liu et al. 2005; Meister et al. 2005; Rehwinkel et al. 2005). In general, RISC-associated miRNAs negatively regulate gene expression by inducing translational repression and decay of targeted transcripts. Multiple model organisms, cultured cell lines, and cell-free extracts have been utilized to dissect miRNA mechanisms. The latter approach has been essential to understanding biochemical mechanisms of mRNA splicing, translation, and turnover. Moreover, cell-free systems offer tight control of multiple experimental parameters and facilitate isolation of functional ribonucleoprotein (RNP) complexes for downstream proteomic analysis.
Hepatitis C virus (HCV) is a pathogenic human RNA virus that replicates in liver hepatocytes of infected individuals (Houghton et al. 1989). The positive-strand RNA genome of HCV lacks a 5′ 7-methylguanosine cap that is typically critical for translation initiation and protection from 5′ to 3′ exonucleases. Instead, HCV translation depends on an internal ribosome entry site (IRES) located within the viral 5′ UTR that drives initiation through direct recruitment of the 40S ribosomal subunit and eIF3 complex (Tsukiyama-Kohara et al. 1992; Pestova et al. 1998; Kieft et al. 2001). An intimate relationship exists between HCV RNA and miR-122, a highly abundant and primarily liver-specific miRNA (Jopling et al. 2005). HCV has evolved binding sites for two copies of miR-122 at the viral genomic 5′ end (Jopling et al. 2008). Antagonizing these interactions by mutation or modified anti-miR-122 oligonucleotides severely restricts HCV replication, and targeting miR-122 may represent a novel therapeutic intervention for treatment of chronic HCV infections (Lanford et al. 2010). Multiple reports have addressed mechanisms by which miR-122 enhances HCV replication. Initially, miR-122 was suggested to enhance viral RNA synthesis without affecting translational efficiency (Jopling et al. 2005). More recently, roles for miR-122 in stimulating translation and stabilizing viral RNA have been reported (Henke et al. 2008; Jangra et al. 2010; Roberts et al. 2011; Li et al. 2012; Shimakami et al. 2012a), suggesting that miR-122 may aid HCV replication at multiple phases of the viral life-cycle.
Not unexpectedly, Ago proteins, and Ago2 in particular, have been implicated as functionally important for miR-122-dependent HCV replication (Roberts et al. 2011; Wilson et al. 2011; Shimakami et al. 2012a). Factors that mediate miRNA processing have also been defined as HCV host factors (Zhang et al. 2012), presumably due to their requirement for generation of mature miR-122. However, the precise composition of the HCV RNP that is formed as a consequence of miR-122 binding has yet to be defined. Given the unusual aspects of miR-122–HCV RNA interactions (binding to the 5′ UTR instead of 3′ UTR; multiple important nonseed base pairs; close proximity of the two miR-122 binding sites; and proximity of the miR-122 binding sites to the HCV IRES), we hypothesize that the miR-122/HCV RNP differs functionally and compositionally from canonical RISC.
In order to begin addressing this hypothesis, we established a cell-free translation system based on stable HEK-293 cell lines that inducibly express wild-type or mutant miR-122. Importantly, HEK-293 cells do not express endogenous miR-122 (Landthaler et al. 2008), allowing simultaneous production of cell lysates with or without miR-122 expression from the same cell line. Using this system, we recapitulated a stimulatory effect of miR-122 on subgenomic, nonreplicating HCV reporter RNAs, confirming a role for miR-122 in stimulating HCV gene expression independently of viral RNA synthesis. We also developed an RNA affinity chromatography approach that allowed purification of HCV IRES RNP complexes, including the miR-122-dependent RNP. This combined approach of using cell lines that inducibly express selected miRNAs, cell-free extracts for reporter RNA translation assays, and RNA affinity chromatography should be applicable to other miRNAs and their selected target transcripts.
RESULTS
miR-122 stimulates activity of HCV reporter RNAs in hepatoma and HEK-293 cells
We initially set out to measure effects of point mutations within miR-122 seed-binding sites on two separate HCV IRES-driven subgenomic reporter mRNAs in Huh7.5 hepatoma cells which are known to express miR-122 (Chang et al. 2004; Jopling et al. 2005). Both HCV reporter constructs contain the HCV IRES and 3′ UTR flanking Renilla luciferase (RLuc) but differ in amount of HCV core sequence fused to RLuc: either 16 or 168 codons (Fig. 1A). For each construct, two separate mutations were introduced into the tandem miR-122 seed-binding sites, leading to generation of p3mut (C to G at positions 27 and 42) or p34mut (UC to AG at positions 26–27 and 41–42) variants (Fig. 1B). Cotransfection of these mRNAs with a capped and polyadenylated firefly luciferase (FLuc) mRNA was performed to ascertain the relative potencies of these mRNAs in Huh7.5 cells. Mutation of the miR-122 seed-binding sites led to an approximate fivefold reduction in RLuc synthesis after normalization to FLuc (Fig. 1C), suggesting that loss of functional interactions with miR-122 leads to destabilization and/or translational incompetency of the HCV reporter mRNAs. Moreover, these effects were independent of the presence of core-coding sequence, in agreement with previous findings (Roberts et al. 2011).
FIGURE 1.

(A) Depiction of reporter RNAs used in this study. In descending order: capped and polyadenylated FLuc RNA, HCV16LUC reporter RNA containing viral 5′ and 3′ UTRs plus 16 codons of the core gene fused to RLuc, HCV168LUC transcript with 168 codons from the core gene, and polyadenylated CBV3LUC RNA. (B) The predicted secondary structure of the HCV genomic 5′ end is shown with miR-122 seed-binding sites in boxes. The p3 and p34 mutations are indicated. Below is the sequence of mature miR-122 with seed sequence indicated by a box. (C) Results of HCV reporter RNA cotransfection into Huh7.5 hepatoma cells. Values for the wild-type versions of each RNA (HCV16LUC and HCV168LUC) were normalized to 1.0 for comparison to p3 and p34 mutant variants. Error bars indicate standard deviation (SD) values.
We next set out to establish a system whereby miR-122 expression could be easily manipulated. To this end, two HEK-293 cell lines were created that inducibly overexpress either wild-type (WT) primary (pri) miR-122 transcript or a variant containing a G to C transition mutation at position 3 of the mature miRNA (miR-122 p3). Each of these primary transcripts was readily detectable by standard RT-PCR after treatment of cells with tetracycline for 48 h (Fig. 2A). We next wished to confirm the capacity of miR-122 overexpressed in HEK-293 cells to target a capped and polyadenylated mRNA for post-transcriptional repression. The human CLIC4 chloride ion channel gene is predicted by TargetScan (Lewis et al. 2003) to have two miR-122 binding sites located within an ∼85-nt region of its 3′ UTR. We amplified this segment, along with 100 nt of flanking sequence, and inserted tandem copies into a vector for synthesis of capped and polyadenylated RLuc mRNA, yielding a transcript with four predicted miR-122 binding sites (Fig. 2B). For comparison, we utilized a CLIC4 reporter variant with C to G mutations that would disrupt base-pairing with position 3 of miR-122, as well as an RLuc reporter mRNA lacking CLIC4 3′ UTR sequence. These transcripts were cotransfected with FLuc mRNA into HEK-293 cells that were mock- or tetracycline-treated to induce miR-122 overexpression 48 h prior to transfection. This analysis revealed that only the CLIC4 reporter mRNA with intact miR-122 seed-binding sites was repressed due to miR-122 overexpression (Fig. 2C). The level of repression was modest, ∼40% compared to mock-treatment, but this magnitude of repression appears to be typical of many miRNA-targeted transcripts. These data indicate that CLIC4 mRNA contains at least one functional miR-122 binding site and that pri-miR-122 overexpressed in stable HEK-293 cells is processed into a functional form that can negatively target mRNA with canonical 5′ and 3′ ends.
FIGURE 2.

(A) HEK-293 cell lines that express pri-miR-122 or pri-miR-122 p3 were established. The mutant mature miR-122 sequence is shown above. RT-PCR was used to detect the pri-miRNA transcripts and U1 snRNA from each cell line treated with tetracycline or mock-treated. (B) Depiction of capped and polyadenylated RLuc reporter RNAs with or without tandem fragments derived from the CLIC4 3′ UTR. “X” indicates seed C to G mutation at p3 in predicted miR-122 seed-binding sites. (C) Reporter RNAs shown in B were cotransfected along with FLuc RNA into HEK-293 cells either mock-treated or tetracycline-treated for 48 h prior to transfection. For each RNA, the mock-treated condition was set to 1.0 for comparison to the tetracycline-treated condition. Error bars indicate SD values.
We next tested HCV reporter mRNAs in the context of cell lines that express either WT miR-122 or the miR-122 p3 variant. Specifically, four reporter mRNAs were examined: the fully intact HCV reporter with 16 codons fused to the RLuc sequence (henceforth referred to as HCV16LUC) (Fig. 1A), variant HCV16LUC constructs with p3 or p34 mutations, and an IRES-driven construct containing the unrelated coxsackievirus B3 IRES (CBV3LUC). In the WT miR-122 cell line, only HCV16LUC was substantially affected by tetracycline induction of the miRNA, producing ∼75% more normalized RLuc activity compared to the mock-induced condition (Fig. 3A). Correspondingly, experiments performed with the miR-122 p3 variant-expressing cell line produced a stimulatory effect (∼50%) only for the HCV16LUC mRNA with a p3 mutation that restores base-pairing with the miR-122 p3 mutant (Fig. 3B). These findings are in agreement with previous reports and demonstrate that direct interaction of miR-122 with subgenomic HCV reporter mRNAs in HEK-293 cells leads to enhanced RNA stability and/or translation.
FIGURE 3.

Viral reporter RNAs were separately evaluated in HEK-293 cells expressing either WT miR-122 (A) or the p3 mutant variant (B). WT, p3 mutant, and p34 mutant versions of HCV16LUC (16LUC) were analyzed alongside the CBV3 reporter RNA (B3LUC). As in Figure 2C, the mock-treated condition for each reporter RNA was normalized to 1.0 and error bars indicate SD values.
miR-122 stimulates activity of HCV reporter RNAs in cell-free lysates
Having established cell lines that endogenously transcribe and process active miR-122, we asked whether cytoplasmic lysates produced from these cells would be amenable to cell-free assays for analysis of HCV reporter mRNA translation and decay. In particular, we were motivated by the possibility that cell-free lysates would allow purification of miR-122-dependent HCV RNP complexes. To begin approaching this goal, we simultaneously generated S10 cytoplasmic lysates from batches of HEK-293 cells that were treated identically except for the presence or absence of tetracycline in the culture media for 48 h prior to extract production. A typical extract preparation entailed processing of 20 × 15-cm dishes of stable HEK-293 cells, only half of which were stimulated to express miR-122. Cells were then harvested and S10 extracts generated by mechanical disruption (see Materials and Methods).
We subsequently assembled cell-free translation reactions using a design similar to that described previously (Bergamini et al. 2000). These reactions contained 50% S10 extract and 0.3 nM HCV reporter RNA (150 pg/μL) as well as supplemented MgOAc2, KOAc2, ATP, GTP, creatine phosphate, creatine phosphokinase, spermidine, HEPES buffer, and amino acids. For each lysate, reactions were assembled with either HCV16LUC reporter RNA or the p3 variant and incubated for 150 min at 30°C. Reaction aliquots were removed at 30-min intervals for analysis of RLuc activity. In lysate from mock-treated cells, RLuc activity was generated at constant and similar rates for each reporter RNA over the course of 120 min and then plateaued during the final 30 min of incubation (Fig. 4A). Importantly, expression of RLuc from the same RNAs yielded a different profile in extract from cells expressing miR-122, with HCV16LUC generating significantly more reporter protein between the 60- and 150-min time points than the p3 mutant RNA (Fig. 4B). More precisely, the WT RNA produced nearly twofold more RLuc after 120 min of incubation than the corresponding p3 mutant (Fig. 4D). In summary, we observed that relatively efficient RLuc output is evident only when both miR-122 is abundant in the lysate and the reporter RNA harbors intact miR-122 binding sites (Fig. 4C). A similar analysis was also performed using the miR-122 p3 cell line. As expected, the p3 variant reporter RNA was specifically transactivated by the p3 mutant miR-122 (Fig. 4E). Together with results shown in Figure 3, these observations further suggest that the p3 mutation in miR-122 does not affect production of the mature miRNA.
FIGURE 4.

Cell-free translation reactions using WT and p3 HCV16LUC reporter RNAs were conducted using lysates derived from mock-treated (A) or tetracycline-treated (B) HEK-293 cells that harbor an inducible pri-miR-122 transgene. At the indicated time points, aliquots of reaction mixtures were removed for measurements of RLuc activity. The data shown are representative of multiple independent experiments. Data from A and B are shown together in panel C. A separate experiment performed in triplicate with a single 120-min time point is shown in panel D. For each lysate (mock or tet), the p3 reporter RNA was normalized to 1.0 for comparison to the WT reporter RNA. A similar experiment was performed with the same reporter RNAs in lysates from cells harboring the p3 pri-miR-122 mutant transgene (E). In panel E, the WT reporter RNA was normalized to 1.0 for each lysate (mock or tet) for comparison to the p3 mutant. All error bars indicate SD values.
Cell-free translation systems allow direct temporal assessment of RNA integrity. We, therefore, asked whether the differential production of RLuc by WT and mutant HCV reporter RNAs (Fig. 4) could be explained, at least in part, by effects on RNA turnover. Internally radiolabeled HCV16LUC and p3 mutant reporter RNAs were used to program reactions with lysate derived from miR-122-expressing cells, and levels of each RNA were monitored every 10 min over the course of a 70-min reaction time by denaturing PAGE. Surprisingly, a substantial amount of each transcript (∼50%) was rapidly degraded within the first 10 min of incubation (Fig. 5). The remaining RNAs were relatively stable, with no apparent difference in decay rates between the WT and mutant RNAs. This observation suggests that miR-122 enhances translation of HCV reporter RNA in this system.
FIGURE 5.

Cell-free translation reactions were assembled with either WT or p3 mutant HCV16LUC reporter RNAs using lysate from miR-122-expressing cells as in Figure 4B, except that transcripts were internally radiolabeled. At the indicated time points, reaction aliquots were removed for RNA extraction and analysis by denaturing PAGE (A). Levels of rRNA in each corresponding sample were separately evaluated by agarose gel electrophoresis. Phosphorimager analysis of radiolabeled RNA signals is shown in panel B. Levels of RNA before incubation of reactions (time point 0) were calibrated to 100%.
Ago2 associates with HCV IRES RNA in a miR-122-dependent manner
We next investigated whether the HEK-293 cell-free translation system might be amenable to affinity purification of RNP complexes that form on HCV reporter RNA as a consequence of miR-122 binding. In order to perform RNA affinity chromatography reactions, RNA transcripts that contain the 342-nt HCV IRES plus 48 nt of core-coding sequence and 65 nt of the RLuc open reading frame (ORF) were synthesized. The latter served as the site for hybridization to a 2′-O-methylated (me) oligonucleotide modified at the 5′ end with a biotin moiety and triethlyene glycol (TEG) spacer. The biotin moiety allows coupling of RNAs to streptavidin-sepharose, while the 2′-O-me modification protects against RNase H activity present in cytoplasmic lysates. For each binding reaction, RNA transcripts (1 μg per reaction) (Fig. 6A) were first hybridized to the modified oligonucleotide and then coupled to streptavidin-sepharose in annealing buffer. We routinely observed between 60% and 80% efficiency in RNA immobilization. After washing away unbound RNA with reaction buffer, 0.5-mL binding reactions containing 50% S10 lysate and all components of cell-free translation reactions (MgOAc2, KOAc2, ATP, GTP, creatine phosphate, creatine phosphokinase, spermidine, HEPES buffer, and amino acids) were assembled and used to resuspend RNA-coupled sepharose. These reactions were then incubated at 30°C for 20 min before extensive washing to clear away unbound proteins. After the last wash, sepharose pellets were resuspended in sample buffer for SDS-PAGE analysis. In contrast to the RNA decay analysis shown in Figure 5, RNAs immobilized on sepharose were highly stable (<10% loss) during the course of binding reactions and subsequent washes. We speculate that blocking the 3′ end of the RNA with the modified DNA oligonucleotide may protect from potent 3′ to 5′ exonuclease activity present in the lysates.
FIGURE 6.

(A) RNA transcripts used for affinity chromatography are depicted. RNAs with intact (WT), point mutated (p34mut), or deleted (Δmut) miR-122 binding sites were immobilized on streptavidin sepharose by hybridization to a biotinylated 2′-O-me-modified oligonucleotide. A triethylene glycol (TEG) spacer separates oligonucleotide bases from the biotin moiety. (B) RNA affinity chromatography reactions were conducted using the RNAs shown in A and lysates from mock-treated or tetracycline-treated cells harboring the WT pri-miR-122 transgene. Analysis of the indicated proteins by Western blot of input and eluate fractions is shown.
We initially performed RNA affinity chromatography with three distinct transcripts (Fig. 6A): one with intact WT miR-122-binding sites, a second with UC to AG mutations at nucleotide bases that interact with positions 3 and 4 of miR-122 (p34mut), and a third with a 5′ end deletion to the first base of stem–loop II (Δmut) that eliminates both miR-122-binding sites. Binding reactions were simultaneously assembled for each RNA using HEK-293 cell lysates derived from cells either induced to express miR-122 or mock-treated. Protein eluates were then used for SDS-PAGE and Western blotting. Multiple antibodies were used to detect proteins in input and eluate samples, some of which have previously been implicated as factors that interact with the HCV RNA: La autoantigen (Ali and Siddiqui 1997), polypyrimidine tract-binding protein (PTB) (Ali and Siddiqui 1995), a subunit (B) of the eukaryotic initiation factor (eIF) 3 complex (Buratti et al. 1998; Sizova et al. 1998), the 5′ to 3′ exonuclease Xrn1 (Li et al. 2012), and Ago2 (Shimakami et al. 2012a). Levels of each of these proteins in input samples were unchanged by miR-122 overexpression (Fig. 6B). Except for PTB, which was barely detectable in eluates despite high signal in input samples, each of the other proteins interrogated was readily detectable in eluate samples and did not appear to differ significantly between lysates or RNA utilized. Importantly, however, Ago2 pull-down was relatively robust only for the WT RNA in lysate produced from cells overexpressing miR-122. This observation is well correlated with cell-free assays where efficient HCV reporter RNA activity depended on intact cis-acting miR-122 binding sites and induced expression of trans-acting miR-122 (Fig. 4). In lysate from uninduced cells, Ago2 was noticeably more abundant for the WT RNA compared to p34mut and Δmut transcripts, perhaps due to leaky expression of miR-122. Taken together with data presented in Figure 4, we conclude that the RNA affinity chromatography strategy employed here allowed purification of a functional HCV RNP complex containing Ago2 and miR-122.
We next evaluated the complexity of affinity-purified protein mixtures that bound IRES RNAs with differential capability to interact with miR-122. To this end, RNA affinity chromatography reactions were performed with miR-122-expressing HEK-293 lysate and streptavidin-sepharose linked to either WT RNA, Δmut RNA, or biotinylated 2′-O-me oligonucleotide (oligo) alone, which served as a negative control. Eluates were evaluated by Coomassie stain and Western blot using antibodies against Ago2 and PTB. Consistent with data shown in Figure 6, Ago2 was significantly more abundant in WT RNA eluate than in other samples, while PTB appeared to be equally present in each eluate (Fig. 7A). Compared to the oligo control, multiple protein bands were detected by Coomassie stain that bound specifically to both WT and Δmut RNAs (Fig. 7B). The patterns of proteins binding the two RNAs were highly similar, with the exception of a band migrating slightly below the 66-kDa molecular weight marker, which was relatively abundant in the WT eluate compared to that of Δmut. We also noted in several independent experiments that intensities of multiple protein bands in Δmut eluates exceeded those of corresponding bands in WT eluates. Lastly, although Ago2 clearly bound to WT RNA more efficiently than Δmut RNA by Western blot (Fig. 7A), there was no observable Coomassie-stained protein band at the corresponding molecular weight (∼95 kDa). Along with data from Figure 6, the results of this analysis suggest that Ago2 and any associated factors can be purified in a manner that depends on miRNA expression and integrity of cognate target sites using the approach described here. However, it is apparent from Coomassie stain analysis that Ago2 is in relatively low abundance compared to other HCV IRES RNP complexes that form independently of miR-122.
FIGURE 7.

RNA affinity chromatography reactions were conducted using lysate from miR-122-expressing cells and either WT or Δmut transcripts, or biotinylated oligonucleotide without hybridized RNA (oligo). Eluates were probed for PTB and Ago2 (A), or analyzed by SDS-PAGE and Coomassie blue staining (B). The arrow points to a protein that is enriched in WT eluate. Separate RNA affinity chromatography reactions were performed with lysate from miR-122-expressing cells to evaluate interactions between IGF2BP1 and the indicated RNAs or oligonucleotide alone (C,D).
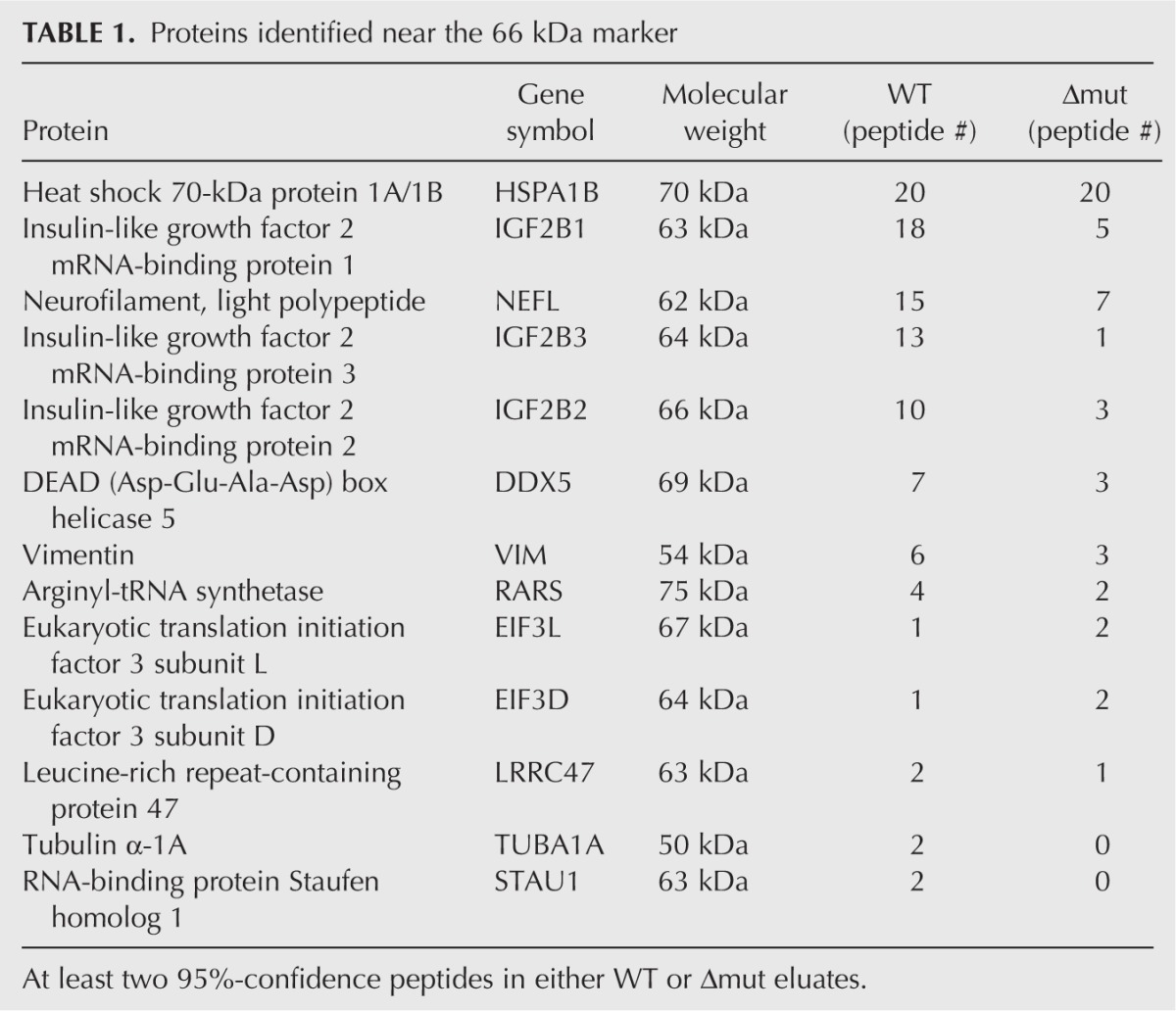
We conducted a proteomic characterization of each eluate shown in Figure 7B by fractionating samples by limited SDS-PAGE and mass spectrometric analysis of six equivalent gel slices per eluate. A total of 65 proteins were positively identified in either the WT or Δmut samples (Supplemental Table S1), the most abundant of which participate in the eIF3 complex. This is consistent with the established direct interaction between the HCV IRES and eIF3. However, Ago2 was not detected in this preliminary analysis, confirming its relatively low abundance in the protein mixture. We subsequently identified proteins in the vicinity of 66 kDa for both WT and Δmut eluates produced from a separate experiment (Table 1). This analysis revealed the presence of insulin-like growth factor 2 mRNA binding proteins (IGF2BPs) 1–3, known regulators of cellular mRNA metabolism. Each of these proteins had more identified peptides in the WT than in the Δmut sample, suggesting that the two RNAs differentially interact with IGF2BPs. To validate the proteomic analysis, we conducted additional RNA affinity assays and checked for the presence of IGF2BP1 by Western blot. Interestingly, IGF2BP1 purification was compromised, but not eliminated, by deletion of miR-122 binding sites (Fig. 7C), while point mutations that disrupt miR-122 binding had no apparent effect on IGF2BP1 pull-down (Fig. 7D). These data suggest that IGF2BPs interact with the discrete region of the HCV 5′ UTR that contains miR-122 binding sites, but these interactions do not depend on miR-122 binding.
TABLE 1.
Proteins identified near the 66 kDa marker
DISCUSSION
We employed a combined approach to isolate RNP complexes that form between HCV RNA and miR-122. Our strategy entailed (1) confirming functional interactions between miR-122 and HCV reporter RNAs in Huh7.5 hepatoma cells and stable HEK-293 cell lines where expression of miR-122 could be easily manipulated, (2) establishing HEK-293 cell-free translation assays that recapitulated positive effects of miR-122 on HCV reporter RNAs observed in cultured cells, and (3) affinity purification of HCV RNP complexes from cell lysates under conditions that depend on both miR-122 expression and intact miR-122-binding sites in HCV IRES RNA. To our knowledge, this is the first report describing miRNP affinity purification using a known authentic RNA target as “bait.” Previous approaches in mammalian systems have used immunoprecipitation (IP) of endogenous or tagged Ago proteins to characterize components of RISC (Mourelatos et al. 2002; Meister et al. 2005; Hock et al. 2007; Landthaler et al. 2008). These studies identified a variety of RNA-binding proteins (RBPs) and helicases as well as components of the translational machinery as specific proteins that co-IP with Ago proteins. Many of these interactions were found to be sensitive to RNase treatment, indicating that Ago IP pulls down a complex mixture of miRNAs, mRNA transcripts, and associated RNP components. A more targeted approach involved RISC purification using biotinylated 2′-O-me oligonucleotides to deplete RISC populations containing a specific miRNA or siRNA (Hutvagner et al. 2004; Flores-Jasso et al. 2013), but this method does not assay for RNA–protein interactions involving a specific transcript targeted by a miRNA of interest.
Although we focused on the unique case of miR-122 and HCV, which is atypical given the location of miRNA binding sites (5′ UTR instead of 3′ UTR) and functional outcome (stimulation instead of repression), we anticipate that a similar combined approach would be applicable to miRNA interactions with cellular mRNA. There is considerable interest in characterizing miRNA–mRNA interactions and their functional outcomes for specific examples of gene regulation. Moreover, there is reason to suspect that not all miRNA–mRNA interactions lead to identical outcomes. For example, the RBP HuR has been described to reverse miR-122 repression of the CAT-1 gene under conditions of cellular stress (Bhattacharyya et al. 2006) and may be a general antagonist of miRNA function (Mukherjee et al. 2011). Unexpectedly, HuR has also been implicated as an RBP that facilitates let-7-mediated repression of c-Myc mRNA translation and stability (Kim et al. 2009). Global analyses of miRNA function in several model systems also suggests nonuniform regulation of mRNA targets that may be due to factors such as mRNA abundance, localization, number and strength of seed sites, and identity of neighboring cis-acting RNA elements (Baek et al. 2008; Guo et al. 2010; Bazzini et al. 2012). RBPs play a significant role in cytoplasmic mRNA regulation and may modify miRNA activity depending on the specific transcript target (Walters et al. 2010). Thus, deciphering the molecular composition of mRNPs targeted by miRNAs and corresponding functional consequences for specific transcripts should enhance our understanding of cytoplasmic gene regulation.
Multiple cell-free systems for dissecting miRNA mechanisms have been described (Wang et al. 2006; Mathonnet et al. 2007; Thermann and Hentze 2007; Wakiyama et al. 2007), though none have yet been employed for RNA affinity chromatography to purify miRNPs. A system based on rabbit reticulocyte lysate (RRL) was initially utilized to investigate miR-122–HCV interactions (Henke et al. 2008). This approach suggested that miR-122 stimulates IRES-dependent translation by enhancing formation of ribosomal 48S complexes on HCV reporter RNAs without affecting RNA decay rate. More recently, a HeLa cell-free system that was used to evaluate miR-122–HCV interactions revealed that miR-122 acts to stabilize HCV RNA, in agreement with corresponding RNA electroporation-based assays in hepatoma cells (Shimakami et al. 2012a). The latter system relies on exogenous addition of synthetic miR-122 duplex to lysate which is assumed to efficiently incorporate into RISC, while the RRL system requires pre-annealing of mature miR-122 to the target reporter RNA. In contrast, our strategy takes advantage of endogenous expression of a pri-miR-122 transgene that is processed into mature miR-122 at levels similar to that found in Huh7.5 hepatoma cells (data not shown). It has been documented that pre-miRNA processing is functionally coupled to RISC assembly (Gregory et al. 2005; Maniataki and Mourelatos 2005) and that HEK-293 cells express and process multiple miRNAs that incorporate into RISC (Landthaler et al. 2008). Therefore, we are confident that pri-miR-122 expressed in HEK-293 cells is authentically processed and assembled into a RISC that positively transactivates HCV reporter RNAs in living cells and cell-free lysates. This is further evidenced by the specific association of Ago2 with immobilized HCV RNA in a miR-122-dependent manner.
Our RNA affinity approach purified multiple specific proteins for both the WT and Δmut transcripts compared to the biotinylated 2′-O-me oligonucleotide alone, and these included several proteins previously reported to interact with the HCV IRES. Of note, IGF2BP1 has previously been reported to bind and transactivate the HCV IRES (Weinlich et al. 2009). Banding patterns of the most abundant proteins binding these RNAs were highly similar by Coomassie stain, suggesting that Ago2 and any associated proteins were present in relatively low abundance despite reproducible detection by Western blotting. Indeed, the lack of Ago2 detection by our initial proteomic analysis indicates that mass spectrometry must be performed with as much depth as possible to detect minority protein species that depend on miR-122 in the complex mixture. Recent biochemical analysis of miRNA-loaded RISC complexes has revealed unexpectedly rapid rates of binding and dissociation to RNA targets via the seed sequence (Wee et al. 2012). Whether this is also true for interactions between miR-122 and HCV RNA, which involve both seed pairing and extensive 3′ nonseed pairing (Machlin et al. 2011; Shimakami et al. 2012b) and potential competition between adjacent binding sites for miR-122 interaction (Mortimer and Doudna 2013), is presently unknown. However, this phenomenon could explain the relatively low levels of Ago2 present in our RNA affinity eluates. Thus, implementation of a cross-linking step may improve pull-down of Ago2 and associated factors.
In summary, we have sequentially combined several methods to investigate HCV–miR-122 interactions that may be applicable to other examples of miRNA-target mRNA regulation. Previous studies have implicated miR-122 as a translational enhancer and stabilizer of HCV RNA by protecting viral genomes from the Xrn1 5′ to 3′ exonuclease. However, these effects cannot fully account for the massive decline in HCV replication that occurs when miR-122–HCV RNA interactions are disrupted (Jangra et al. 2010; Li et al. 2012; Mortimer and Doudna 2013). It is, therefore, of significant interest to characterize in detail the viral RNP that depends on miR-122, as this may lead to identification of potentially novel host factors that regulate HCV replication.
MATERIALS AND METHODS
Cell culture and transfections
Establishment of a Flp-In 293 T-REx cell line (Invitrogen) containing inducible pri-miR-122 has been described previously (Chang et al. 2008). A miR-122 p3 mutant Flp-In 293 T-REx cell line was established as directed by the manufacturer. These cell lines were maintained in high glucose DMEM with nonessential amino acids, 10% tetracycline-reduced fetal bovine serum (FBS) (Hyclone), 15 μg/mL blasticidin, and 100 μg/mL hygromycin. Huh7.5 human hepatoma cells (Blight et al. 2002) were maintained in high glucose DMEM supplemented with nonessential amino acids and 10% FBS (Gemcell). For transfection of reporter RNAs, cells were grown on 24-well plates and transfected at a confluency of 90% with 100 ng of HCV, CBV3 or CLIC4 reporter RNA, and 100 ng of capped/polyadenylated FLuc RNA using 0.6 μL of lipofectamine 2000 (Invitrogen) per transfection. Cell lysates were harvested 8 h post-transfection for dual luciferase assays (Promega). All transfection experiments were performed at least three separate times.
Plasmids and in vitro transcription
Construction of HCV, CBV3, and FLUC reporter plasmids has been described previously (Dobrikova et al. 2003; Bradrick et al. 2006, 2007). For establishing CLIC4 reporter constructs, a 189-bp region of the 3′ UTR was synthesized (Integrated DNA Technologies) with or without p3 mutations in predicted miR-122-binding sites and used for PCR amplification to generate fragments for ligation into the pTNT vector (Promega) containing the RLuc ORF. For establishment of the miR-122-p3 mutant expression construct and cell line, the 160-bp miR-122 cassette was PCR-amplified from genomic DNA purified from the miR-122 HEK-293 cell line and inserted into pcDNA5/FRT/TO (Invitrogen). This was used for PCR-based site-directed mutagenesis to generate the p3 mutant construct. HCV transcription templates were generated by PCR as previously described (Bradrick et al. 2006), while CBV3 and CLIC4 templates were produced by plasmid linearization with BamHI. Uncapped and capped RNAs were synthesized using Ambion T7 Megascript and T7 mMessage machine kits, respectively. RNAs used for affinity chromatography were produced from templates generated by PCR using a forward primer containing a T7 promoter and a reverse primer encompassing the first 65 nt of the RLuc ORF. For synthesis of radiolabeled RNAs, reactions were performed in the presence of 10 μCi [α -32P]-UTP (PerkinElmer).
S10 extract preparation and cell-free translation
We modeled our strategy for cell-free translation on the approach described by Bergamini and colleagues (Bergamini et al. 2000). Extract preparation was conducted with 20 × 15-cm dishes of confluent HEK-293 cells that were either treated with 1 μg/mL tetracycline for 48 h to induce miR-122 expression or mock-treated. At the time of harvest, cells were gently washed with phosphate-buffered saline (PBS) and then scraped in 3 mL of PBS per plate and transferred to a prechilled tube. Cells were pelleted at 1500g, resuspended in a volume of hypotonic buffer (10 mM HEPES-KOH [pH 7.4], 10 mM KOAc2, 0.5 mM MgOAc2, 5 mM dithiothreitol, and proteinase inhibitors [EDTA-free; Roche]) equal to that of the cell pellet, and transferred to a 15-mL dounce homogenizer. A tight-fitting pestle was used to disrupt cells with 16 firm strokes, and then homogenate was subjected to centrifugation for 5 min at 12,000g. Supernatants were collected and flash-frozen before storage at −80°C. Cell-free translation reactions were assembled with 50% cytoplasmic extract, 150 ng/mL (∼0.3 nM) reporter RNA, 60 μM amino acids, 0.8 mM ATP, 0.1 mM GTP, 16 mM HEPES-KOH (pH 7.4), 20 mM creatine phosphate, 40 μg/mL creatine phosphokinase, 50 μM spermidine, 3 mM MgOAc2, and 160 mM KOAc2. Reactions were incubated at 30°C and stopped at indicated time points by addition of EDTA to 10 mM and chilling on ice. RLuc was measured (Promega) using a Berthold luminometer. Cell-free translation reactions were performed multiple times with independently prepared cell lysates. For analysis of radiolabeled RNAs, Trizol (Invitrogen) was used for RNA extraction, and samples were subjected to 4% denaturing PAGE. Radioactive RNA levels were measured on a phosphorimager (Molecular Dynamics).
RNA affinity chromatography, mass spectrometry, and Western blotting
Unless otherwise indicated, all steps were performed at 0°C–4°C. Streptavidin sepharose 4B (20 μL packed volume per binding reaction) was blocked overnight in annealing buffer (10 mM Tris-HCl [pH 7.4], 50 mM NaCl, 1 mM EDTA) containing 0.5 mg/mL BSA (Roche) and 0.1 mg/mL yeast tRNA (Sigma). The next day, sepharose beads were washed twice with 1 mL of annealing buffer without BSA or tRNA. All centrifugation steps to pellet beads were performed for 1 min at 1700g. For each binding reaction, 1 μg (7.5 pmol) of RNA was mixed with 15 pmol of biotinylated 2′-O-me oligonucleotide in 100 μL of annealing buffer and placed into a 70°C heat block which was allowed to cool slowly to 30°C on the laboratory bench. Annealed RNA-oligonucleotide duplexes were adjusted to 0.6 mL with annealing buffer and used to resuspend preblocked sepharose beads. These were placed at 4°C and agitated by rotation for 1.5 h. While coupling RNAs to sepharose, extract aliquots were removed from frozen storage and quickly thawed in a room temperature block before placing on ice. For each binding reaction, 250 μL of lysate was mixed with amino acids, ATP, GTP, HEPES-KOH (pH 7.4), creatine phosphate, creatine phosphokinase, spermidine, MgOAc2, and KOAc2 at the concentrations indicated above and brought to 0.5 mL total volume with H2O. The former seven components were added as a 5× master mix. The lysate mixtures were subsequently precleared for 60 min by rotation with 100 μL of streptavidin sepharose beads that had been washed three times with RNP buffer (16 mM HEPES-KOH [pH 7.4], 3 mM MgOAc2, and 160 mM KOAc2).
After coupling RNA to sepharose, the beads were washed three times with RNP buffer. Binding reactions were then assembled by resuspending immobilized RNA-sepharose with precleared lysate mixtures and incubation at 30°C for 20 min with periodic gentle agitation. Sepharose beads were then washed four times (1 mL each) with room temperature RNP buffer containing 1% Triton X-100 (Sigma). After the last wash, beads were pelleted and resuspended in 25 μL LDS sample buffer to elute proteins (Invitrogen). Eluate samples were analyzed on NuPAGE 4%–12% gradient Bis-Tris gels with MOPS running buffer (Invitrogen). RNA affinity chromatography reactions were performed multiple times from independently prepared cell lysates. Gels were either stained with Coomassie blue R-250 (Bio-rad) or blotted onto nitrocellulose for probing with antibodies. Antibodies used in this study were directed against Ago2 (Cell Signaling Technology), Xrn1 (Novus Biologicals), eIF3 (B subunit; Santa Cruz Biotech.), PTB (kindly provided by M.A. Garcia-Blanco), IGF2BP1 (Sigma), and La protein (kindly provided by J.D. Keene). Liquid chromatography–tandem mass spectrometry was conducted on Coomassie-stained gels by the Duke University Proteomics Core Facility. Analysis of proteomic data sets was performed with Scaffold 4 (Proteome software).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Stacia Phillips, Daneen Schaeffer, and Jason Somarelli for critical reading of this manuscript. We also thank Mariano Garcia-Blanco (Duke University) for mentorship and the PTB antibody and Jack Keene (Duke University) for the antibody to La protein. S.S.B. acknowledges funding from NIH grant 5-K01-DK082613.
REFERENCES
- Ali N, Siddiqui A 1995. Interaction of polypyrimidine tract-binding protein with the 5′ noncoding region of the hepatitis C virus RNA genome and its functional requirement in internal initiation of translation. J Virol 69: 6367–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali N, Siddiqui A 1997. The La antigen binds 5′ noncoding region of the hepatitis C virus RNA in the context of the initiator AUG codon and stimulates internal ribosome entry site-mediated translation. Proc Natl Acad Sci 94: 2249–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP 2008. The impact of microRNAs on protein output. Nature 455: 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Lee MT, Giraldez AJ 2012. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 336: 233–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamini G, Preiss T, Hentze MW 2000. Picornavirus IRESes and the poly(A) tail jointly promote cap-independent translation in a mammalian cell-free system. RNA 6: 1781–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W 2006. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125: 1111–1124 [DOI] [PubMed] [Google Scholar]
- Blight KJ, McKeating JA, Rice CM 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76: 13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradrick SS, Walters RW, Gromeier M 2006. The hepatitis C virus 3′-untranslated region or a poly(A) tract promote efficient translation subsequent to the initiation phase. Nucleic Acids Res 34: 1293–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradrick SS, Dobrikova EY, Kaiser C, Shveygert M, Gromeier M 2007. Poly(A)-binding protein is differentially required for translation mediated by viral internal ribosome entry sites. RNA 13: 1582–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Tisminetzky S, Zotti M, Baralle FE 1998. Functional analysis of the interaction between HCV 5′UTR and putative subunits of eukaryotic translation initiation factor eIF3. Nucleic Acids Res 26: 3179–3187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA, Xu C, Mason WS, Moloshok T, Bort R, et al. 2004. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol 1: 106–113 [DOI] [PubMed] [Google Scholar]
- Chang J, Guo JT, Jiang D, Guo H, Taylor JM, Block TM 2008. Liver-specific microRNA miR-122 enhances the replication of hepatitis C virus in nonhepatic cells. J Virol 82: 8215–8223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekulaeva M, Filipowicz W 2009. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol 21: 452–460 [DOI] [PubMed] [Google Scholar]
- Dobrikova E, Florez P, Bradrick S, Gromeier M 2003. Activity of a type 1 picornavirus internal ribosomal entry site is determined by sequences within the 3′ nontranslated region. Proc Natl Acad Sci 100: 15125–15130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian MR, Sonenberg N 2012. The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nat Struct Mol Biol 19: 586–593 [DOI] [PubMed] [Google Scholar]
- Fabian MR, Sonenberg N, Filipowicz W 2010. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 79: 351–379 [DOI] [PubMed] [Google Scholar]
- Flores-Jasso CF, Salomon WE, Zamore PD 2013. Rapid and specific purification of Argonaute-small RNA complexes from crude cell lysates. RNA 19: 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R 2005. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 123: 631–640 [DOI] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ 2001. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 293: 1146–1150 [DOI] [PubMed] [Google Scholar]
- Henke JI, Goergen D, Zheng J, Song Y, Schuttler CG, Fehr C, Junemann C, Niepmann M 2008. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 27: 3300–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock J, Weinmann L, Ender C, Rudel S, Kremmer E, Raabe M, Urlaub H, Meister G 2007. Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells. EMBO Rep 8: 1052–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton MJ, Carey JC, Seegmiller RE 1989. Pulmonary hypoplasia in mice homozygous for the cartilage matrix deficiency (cmd) gene: A model for human congenital disorder. Pediatr Pathol 9: 501–512 [DOI] [PubMed] [Google Scholar]
- Hutvagner G, Simard MJ, Mello CC, Zamore PD 2004. Sequence-specific inhibition of small RNA function. PLoS Biol 2: E98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangra RK, Yi M, Lemon SM 2010. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J Virol 84: 6615–6625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309: 1577–1581 [DOI] [PubMed] [Google Scholar]
- Jopling CL, Schutz S, Sarnow P 2008. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 4: 77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft JS, Zhou K, Jubin R, Doudna JA 2001. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA 7: 194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M 2009. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev 23: 1743–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landthaler M, Gaidatzis D, Rothballer A, Chen PY, Soll SJ, Dinic L, Ojo T, Hafner M, Zavolan M, Tuschl T 2008. Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. RNA 14: 2580–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H 2010. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327: 198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB 2003. Prediction of mammalian microRNA targets. Cell 115: 787–798 [DOI] [PubMed] [Google Scholar]
- Li Y, Masaki T, Yamane D, McGivern DR, Lemon SM 2012. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc Natl Acad Sci 110: 1881–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Rivas FV, Wohlschlegel J, Yates JR III, Parker R, Hannon GJ 2005. A role for the P-body component GW182 in microRNA function. Nat Cell Biol 7: 1261–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machlin ES, Sarnow P, Sagan SM 2011. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci 108: 3193–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniataki E, Mourelatos Z 2005. A human, ATP-independent, RISC assembly machine fueled by pre-miRNA. Genes Dev 19: 2979–2990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathonnet G, Fabian MR, Svitkin YV, Parsyan A, Huck L, Murata T, Biffo S, Merrick WC, Darzynkiewicz E, Pillai RS, et al. 2007. MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science 317: 1764–1767 [DOI] [PubMed] [Google Scholar]
- Meister G, Landthaler M, Peters L, Chen PY, Urlaub H, Luhrmann R, Tuschl T 2005. Identification of novel argonaute-associated proteins. Curr Biol 15: 2149–2155 [DOI] [PubMed] [Google Scholar]
- Mortimer SA, Doudna JA 2013. Unconventional miR-122 binding stabilizes the HCV genome by forming a trimolecular RNA structure. Nucleic Acids Res 41: 4230–4240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G 2002. miRNPs: A novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev 16: 720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M Jr, Tuschl T, Ohler U, Keene JD 2011. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell 43: 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova TV, Shatsky IN, Fletcher SP, Jackson RJ, Hellen CU 1998. A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev 12: 67–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai RS, Bhattacharyya SN, Filipowicz W 2007. Repression of protein synthesis by miRNAs: How many mechanisms? Trends Cell Biol 17: 118–126 [DOI] [PubMed] [Google Scholar]
- Rehwinkel J, Behm-Ansmant I, Gatfield D, Izaurralde E 2005. A crucial role for GW182 and the DCP1:DCP2 decapping complex in miRNA-mediated gene silencing. RNA 11: 1640–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AP, Lewis AP, Jopling CL 2011. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res 39: 7716–7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, Lemon SM 2012a. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc Natl Acad Sci 109: 941–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimakami T, Yamane D, Welsch C, Hensley L, Jangra RK, Lemon SM 2012b. Base pairing between hepatitis C virus RNA and microRNA 122 3′ of its seed sequence is essential for genome stabilization and production of infectious virus. J Virol 86: 7372–7383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizova DV, Kolupaeva VG, Pestova TV, Shatsky IN, Hellen CU 1998. Specific interaction of eukaryotic translation initiation factor 3 with the 5′ nontranslated regions of hepatitis C virus and classical swine fever virus RNAs. J Virol 72: 4775–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermann R, Hentze MW 2007. Drosophila miR2 induces pseudo-polysomes and inhibits translation initiation. Nature 447: 875–878 [DOI] [PubMed] [Google Scholar]
- Tsukiyama-Kohara K, Iizuka N, Kohara M, Nomoto A 1992. Internal ribosome entry site within hepatitis C virus RNA. J Virol 66: 1476–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakiyama M, Takimoto K, Ohara O, Yokoyama S 2007. Let-7 microRNA-mediated mRNA deadenylation and translational repression in a mammalian cell-free system. Genes Dev 21: 1857–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RW, Bradrick SS, Gromeier M 2010. Poly(A)-binding protein modulates mRNA susceptibility to cap-dependent miRNA-mediated repression. RNA 16: 239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Love TM, Call ME, Doench JG, Novina CD 2006. Recapitulation of short RNA-directed translational gene silencing in vitro. Mol Cell 22: 553–560 [DOI] [PubMed] [Google Scholar]
- Wee LM, Flores-Jasso CF, Salomon WE, Zamore PD 2012. Argonaute divides its RNA guide into domains with distinct functions and RNA-binding properties. Cell 151: 1055–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich S, Huttelmaier S, Schierhorn A, Behrens SE, Ostareck-Lederer A, Ostareck DH 2009. IGF2BP1 enhances HCV IRES-mediated translation initiation via the 3′UTR. RNA 15: 1528–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JA, Zhang C, Huys A, Richardson CD 2011. Human Ago2 is required for efficient microRNA 122 regulation of hepatitis C virus RNA accumulation and translation. J Virol 85: 2342–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Huys A, Thibault PA, Wilson JA 2012. Requirements for human Dicer and TRBP in microRNA-122 regulation of HCV translation and RNA abundance. Virology 433: 479–488 [DOI] [PubMed] [Google Scholar]