

Abstract
The AMP-activated protein kinase in yeast, Snf1, coordinates expression and activity of numerous intracellular signaling and developmental pathways, including those regulating cellular differentiation, response to stress, meiosis, autophagy, and the diauxic transition. Snf1 phosphorylates metabolic enzymes and transcription factors to change cellular physiology and metabolism. Adr1 and Cat8, transcription factors that activate gene expression after the diauxic transition, are regulated by Snf1; Cat8 through direct phosphorylation and Adr1 by dephosphorylation in a Snf1-dependent manner. Adr1 and Cat8 coordinately regulate numerous genes encoding enzymes of gluconeogenesis, the glyoxylate cycle, β-oxidation of fatty acids, and the utilization of alternative fermentable sugars and nonfermentable substrates. To determine the roles of Adr1, Cat8, and Snf1 in metabolism, two-dimensional gas chromatography coupled to time-of-flight mass spectrometry and liquid chromatography coupled to tandem mass spectrometry were used to identify metabolites whose levels change after the diauxic transition in wild-type-, ADR1-, CAT8-, and SNF1-deficient yeast. A discovery-based approach to data analysis utilized chemometric algorithms to identify, quantify, and compare 63 unique metabolites between wild type, adr1Δ, cat8Δ, adr1Δcat8Δ, and snf1Δ strains. The primary metabolites found to differ were those of gluconeogenesis, the glyoxylate and tricarboxylic acid cycles, and amino acid metabolism. In general, good agreement was observed between the levels of metabolites derived from these pathways and the levels of transcripts from the same strains, suggesting that transcriptional control plays a major role in regulating the levels of metabolites after the diauxic transition.
Keywords: GC×C–OFMS, Chemometrics, PARAFAC, Snf1 protein complex
Introduction
Metabolomics studies are capable of offering a unique, and complementary, perspective on cellular processes [1–3]. The complex chemical nature of cellular metabolite extracts requires powerful instrumentation to isolate and identify specific analytes (i.e., metabolites) [4–6]. Additionally, powerful data analysis techniques are often necessary in order to glean the most useful information from the complex data these instruments generate. A “discovery-based” approach can be particularly useful to draw out specific chemical differences between samples in hypothesis-driven studies, thus providing insight into cellular physiology [7–13].
AMP-activated protein kinase (AMPK) has been referred to as the energy sensor of the cell and can have a large impact on cellular physiology, thus is a good subject for metabolic studies. AMPK responds to a variety of internal and external signals to balance anabolic and catabolic processes and ensure that a constant supply of glucose is available to maintain metabolic homeostasis. Changes in AMPK activity allow the cell to modulate its use and production of energy stores, changing from an energy consuming to an energy producing state as conditions necessitate [14, 15]. AMPK controls the activity of metabolic enzymes at two levels [16]. An immediate consequence of AMPK activation is phosphorylation of metabolic enzymes. A longer-lasting effect on metabolism is brought about by transcriptional changes induced by AMPK, of which there are many examples [17–19].
Snf1 is the AMPK homolog in the budding yeast, Saccharomyces cerevisiae. Its trimeric structure, consisting of an activating γ subunit (Snf4), alternative regulatory scaffolding β-subunits (Sip1, Sip2, and Gal83) and a catalytic α-subunit (Snf1) is similar to that of the mammalian AMPK [15]. Snf1 is regulated in an analogous manner to mammalian AMPK via activation by upstream kinases that phosphorylate the α-subunit and inactivation by dephosphorylation. Snf1 controls the response of many enzymes to glucose starvation, allowing yeast cells to shift from fermentative to aerobic metabolism during the diauxic transition [20]. During fermentation, glucose is metabolized through the glycolytic pathway to produce ATP inefficiently but abundantly as long as a fermentable substrate is present. When glucose is depleted, metabolism is adjusted to utilize previously secreted by-products of fermentation such as ethanol and glycerol, as well as fatty acids stored as membrane lipids. The acetyl CoA produced from these pathways enters the tricarboxylic acid (TCA) cycle in the mitochondrion to be oxidized via the respiratory chain, producing ATP and reduced NAD for energy-requiring processes. At the same time, gluconeogenesis and the glyoxylate cycle are activated to produce intermediates for the biosynthesis of proteins, carbohydrates, and nucleic acids.
Evidence of changes related to the available carbon source can be observed at the metabolite level [7]. The same discovery-based metabolomics approach was also used to illuminate differences between a wild-type strain and a snf1Δ strain. Studying the role of Snf1 was initiated by analyzing the time dependence of Snf1-dependent changes in metabolite levels during the transition from fermentation to respiration by comprehensive two-dimensional gas chromatography coupled to time-of-flight mass spectrometry (GC× GC–TOFMS) [8]. Expanding upon these observations, metabolite data from a separate snf1Δ study was incorporated with transcriptome and proteome data to begin highlighting the regulatory network [21]. Some differences were found between the two studies, likely due to the difference in growth conditions: batch culture [8] as compared with steady-state chemostat cultivation [21].
The influence of Snf1 on metabolism is brought about by both phosphorylation of metabolic enzymes and by transcriptional reprogramming during the diauxic transition. Little is known about the role of enzyme phosphorylation by Snf1 [22, 23] but its control of transcription factors has been studied extensively [24–26]. The best-studied transcription factor target of Snf1 is the DNA binding repressor, Mig1. Snf1 phosphorylates and inactivates Mig1 when glucose is depleted, allowing glucose-repressed genes to be activated. One of these is the transcriptional activator, Cat8, whose expression is repressed by Mig1. Upon glucose depletion, Snf1 phosphorylates and activates Cat8, which in turn binds to and activates genes encoding enzymes in the gluconeogenic and glyoxylate cycle pathways. Snf1 also activates Adr1, a transcription factor necessary for expression of genes encoding enzymes required for the utilization of alternative carbon sources such as ethanol, glycerol, lactate, amino acids, and fatty acids [27], by promoting dephosphorylation of Ser98 in the DNA binding domain [28] and Ser230 in the regulatory domain [29].
Transcriptomics studies are very powerful for providing insight into biochemical processes, especially when augmented with metabolomics studies. A global analysis of the yeast transcriptome in wild-type, snf1Δ, cat8Δ, adr1Δ, and adr1Δcat8Δ mutants allowed the contribution of the two transcription factors to Snf1-dependent gene expression to be distinguished. Many of the affected genes were shown to be direct targets of Adr1 and Cat8 [27, 30]. A major question raised by these transcriptional studies is to what extent mRNA levels accurately reflect the activity of the pathways downstream of the encoded enzymes. This question is particularly important because many enzymes of intermediary metabolism are encoded by multiple isozymes whose individual contributions to metabolite levels are unknown. Investigating downstream effects with proteomics or metabolomics are approaches to answer this question. Integrating metabolomics with other global analyses, such as genomic, transcriptomic, or proteomic studies, presents a broad and ultimately comprehensive picture of cellular physiology [2, 3, 31–35]. When metabolomics is combined with analysis of the transcriptome, specifically, the influence of regulatory pathways on these two mechanistically different aspects of metabolic control can begin to be teased apart.
Discovery-based metabolomics is well suited to address this question because specific metabolite differences in various strains can be determined. A global approach to study metabolism in AMPK-deficient cells requires powerful instrumentation, such as GC×GC–TOFMS [36–40] that is well suited for separating complex samples [7, 8, 11–13, 41– 44]. Chemometric data analysis algorithms are useful in order to draw information out of the large amount of data generated by this instrumentation [7, 8, 11–13, 45]. The discovery-based approach utilizes the separation and the mass spectral information in the raw data cube to find specific analyte differences between sample classes. This often provides the most insight into how those sample classes (i.e., mutant strains) differ overall. The analytes that differ between classes can then be identified and quantified with another software algorithm, parallel factor analysis (PARAFAC), which isolates the pure analyte signal from background noise and any overlapping peaks [46–49]. The combination of these chemometric data analysis tools provides a powerful approach in mutant studies as differences between strains are readily identified and quantified for further interpretation.
Because the metabolome contains molecules having a wide range of chemical properties, these studies can benefit from using more than one complementary instrumentation approach. In fact, there is currently no single chemical analysis method available to study all metabolites simultaneously [4–6]. GC is more amenable to smaller volatile analytes or those that can be made volatile with a chemical derivatization step while liquid chromatography (LC) is better suited for the larger, less volatile, compounds. The combination of these complementary approaches offers greater metabolite coverage than either one independently. If enough metabolites are detected, the compilation of the information can be used to determine how mutations impact functionality and the activity of specific metabolic pathways.
In order to provide a more global investigation of metabolic pathways, in this report, both GC×GC–TOFMS and liquid chromatography coupled to tandem mass spectrometry (LC–MS/MS) were used to determine differences between wild-type yeast and four mutant strains (snf1Δ, adr1Δ, cat8Δ, and adr1Δcat8Δ) grown in the absence of glucose. Metabolites that differed between strains were identified based on information from previous related studies [7, 8, 11] and from a feature selection algorithm, referred to as the S-ratio method [12]. The S-ratio method refers to finding the locations in the 2D separation space for GC×GC, where the signal ratio is significantly different than one (indicating an up- or down-regulation of a particular metabolite). The metabolites located via the S-ratio method were then quantified using PARAFAC. Principal component analysis (PCA) was used as a comparison tool to attain a global view of metabolite information in these strains. Additionally, statistical analyses were performed to determine metabolites that showed significant changes between strains. To gain a better understanding of the different aspects of metabolic control, the metabolite levels from this study were integrated with previously published RNA data [27] for pathways containing statistically significant metabolites (primarily the TCA cycle, glyoxylate cycle, and gluconeogenesis.) Good agreement was found between mRNA and metabolite levels in these pathways, indicating that regulation at the level of transcription is an important determinant of metabolic activity.
Materials and methods
Yeast cells and culture conditions
A wild-type yeast strain (CKY19-1) was compared with four mutant yeast strains. The appropriate open reading frames were replaced with a kanMX gene cassette to create the following mutations; adr1Δ (CKY13-1), cat8Δ (CKY15-1), snf1Δ (CKY17-1), and adr1Δcat8Δ (CKY23-1). All strains were grown in fermentable (repressing) synthetic complete (SC) medium containing 5% glucose as the carbon source. At time 0 h, the cell cultures were collected by centrifugation at 4 °C and washed once with cold sterile synthetic medium, (SC lacking amino acids or carbon source). A volume of medium corresponding to 1×107 cells (determined by O.D.) was then collected, centrifuged, and the cell pellet suspended in nonfermentable (derepressing) medium, prewarmed to 30 ° C, containing 3% ethanol and 0.05% glucose as carbon sources. Three biological replicates were analyzed for each strain. Glucose consumption per strain was monitored over time by analyzing aliquots of medium with a PGO enzymes kit (Sigma Aldrich, St. Louis, Missouri, USA).
Extraction of metabolites
Based on our previous time-course study, cells were incubated at 30 °C for 6 h [8]. The metabolites, small polar molecules, were extracted from the cells using a previously described protocol [50]. Briefly, 5 mL of each culture were rapidly diluted into 20 mL of −40 °C quenching buffer (10 mM tricene, pH 7.4, in 60% methanol) and held at −40 °C for 5 min to halt metabolic activity. The cell suspensions were pelleted by centrifugation (Sorvall RC-5B Plus) at 1,000×g for 3 min at −20 °C and washed once with 5 mL of −40 °C quenching buffer. The pellets were resuspended in 1.67 mL of 80 °C extraction buffer (0.5 mM tricine, pH 7.4, in 75% ethanol), held at this temperature for ∼3 min, and then cooled on ice for 5 min. The suspensions were spun twice at high speed in a microcentrifuge to remove large cellular debris. A volume of each ethanol metabolite extract, corresponding to 1×107 cells, was dried in a SpeedVac at room temperature and stored at −80 °C under argon.
Derivatization of metabolites for GC analysis
Any residual water condensate on the cold sample was removed by drying in a SpeedVac at room temperature for 1 h immediately prior to derivatization. The metabolites were derivatized using a previously reported two-step protocol [51]. Methoximation was accomplished by adding 30 µL of a 20 mg/mL methoxyamine solution in pyridine to the extracts and heating at 30 °C for 90 min. The samples were then trimethylsilyated by adding 70 µL of BFTSA/ TMCS (99:1) reagent to the methoximated extracts and heating at 60 °C for 60 min.
GC instrument parameters
GC×GC–TOFMS instrumentation was used to analyze the yeast extracts. An Agilent 6890N gas chromatograph (Agilent Technologies, Palo Alto, CA, USA) coupled to a LECO Pegasus III TOFMS (LECO, St. Joseph, MI, USA) was upgraded with both an Agilent 7683 auto injector and the commercially available 4D thermal modulator (LECO, St. Joseph, MI, USA). A nonpolar to polar column arrangement was used combining a 20m×250 µm i.d.×0.5 µm RTX-5MS (Restek, Bellefonte, PA, USA) as the first column to a 2 m×180 µm i.d.×0.2 µm RTX-200MS (Restek, Bellefonte, PA, USA) for the second. A constant He flow of 1 mL/min was maintained at the head of the first column. The GC inlet and transfer line were both set to 280 °C throughout the chromatographic run. A temperature program was employed starting the first column at 60 °C and the second column at 70 °C for 0.25 min and ramping at a rate of 8 °C/min to 280 °C where the columns were held at constant temperature for 10 min. The effluent from the first column was modulated onto the second column every 1.5 s with the modulator temperature maintained 40 °C higher than the first column. The TOFMS ion source was set to 250 °C. After a 5-min solvent delay, mass channels 40–500m/z were collected at 100 spectra/s. Three injections of 1 µL were made in splitless mode for each sample (five strains×three biological replicates) for a total of 45 chromatographic runs.
GC data analysis
LECO’s ChromaTOF software v 3.32 (St. Joseph, MI, USA) was used to collect the GC×GC–TOFMS data. Previous work performed in our labs, including studies on the snf1Δ yeast strain, was utilized to generate an initial list of metabolites of interest [7, 8, 11]. The discovery-based approach, outlined by the bolded arrows in Fig. 1, was then used to find additional class distinguishing compounds within the GC data. In order to adequately mine the data for the unstudied strains, the data were exported to Matlab v 7.0.4 (Mathworks, Natick, MA, USA). The S-ratio algorithm [12] was used to identify the GC×GC chromatographic locations of additional analytes that exhibited differences in signal intensity between the various strains. These locations were then added to the initial list of metabolite locations. The locations with the largest S-ratio values indicated GC×GC retention times of analytes that differed between samples. These analytes were identified as metabolites using the LECO ChromaTOF software to search the mass spectra at these chromatographic locations against the National Institute of Standards and Technology library and a standard metabolite library created in-house. Identification was determined by match value and, in most cases, retention time confirmation with standards (as noted in Table 2.) Relative quantification information was attained for the metabolites using the in-house developed target-analyte PARAFAC GUI [47]. The algorithm mathematically resolves (deconvolutes) the pure component chromatographic peak profile and the pure mass spectrum of an individual component from overlapping peaks and background noise for quantification and identification confirmation, respectively. This step of analysis accomplishes several important tasks: baseline correction as noise is factored out, chromatographic peak profiles (for both GC columns) and m/z spectra are deconvoluted, and integration as the dot product of the first column peak profile and the second column peak profile provides a signal “volume” that is proportional to analyte concentration. The PARAFAC signal volumes were normalized to the total ion current (TIC).
Fig. 1.

Data analysis flow chart for GC×GC data. The discovery-based data analysis approach utilizes ChromaTOF and Matlab to locate, identify, and quantify class distinguishing compounds. The traditional ChromaTOF approach (thin arrows) follows the path on the left while the discovery-based approach (thick arrows) applied herein follows the path on the right incorporating some elements of ChromaTOF data processing. The discovery-based approach could also be done in a nontargeted way as is demonstrated with the dotted arrow
Table 2.
The 65 detected metabolites (63 unique) are sorted by score on PC1 (as shown in Fig. 2)
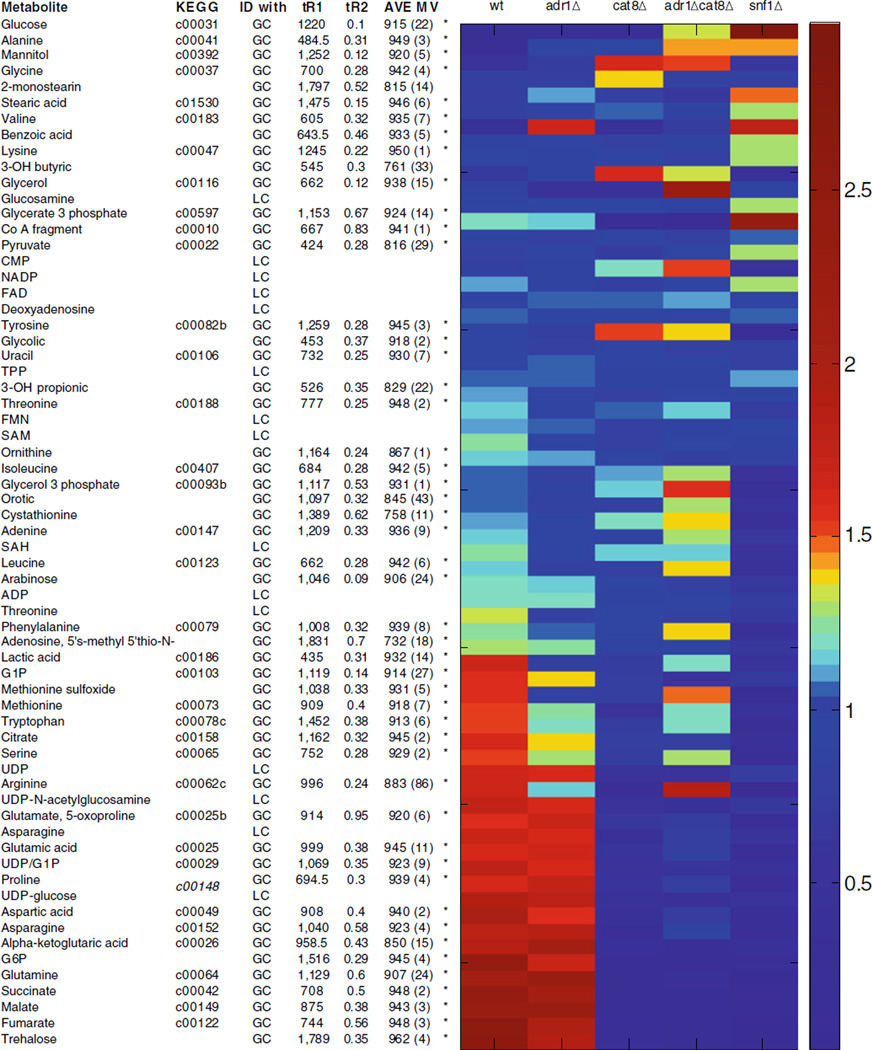 |
The PCA captured patterns are evident in the relative amounts normalized to the mean for each strain. Retention time (tR1 and tR2, noted with an asterisk if matched to standard) and match values (with standard deviation in parenthesis) are provided for analytes identified with GC. ADP adenosine diphosphate, CMP cytidine monophosphate, FAD flavin adenine dinucleotide, FMN flavin mononucleotide, G1P glucose-1-phosphate, G6P glucose-6-phosphate, NADP nicotinamide adenine dinucleotide phosphate, SAH S-adenosyl homocysteine, SAM S-adenosyl methionine, TPP thiamine pyrophosphate, UDP uridine diphosphate
LC analysis
LC data were collected using an Applied Biosystems 3200 Q TRAP hybrid triple quadrupole linear ion trap LC–MS/ MS, as previously referenced [45]. Briefly, metabolite extracts were suspended in ∼30 µL of 0.1% formic acid (or 5 mM NH4OAc). Extracts were separated on a C-18 column with polar embedded groups (Synergi Fusion, 150×2.0 mm×4 µm, Phenomenex). Up to 150 multiple-reaction monitoring could be detected simultaneously throughout the run, so parent/daughter pairs corresponding to metabolites previously run as standards were monitored. Peak areas were quantified with Analyst software and normalized to the TIC, as done in GC analysis.
Data interpretation
Each metabolite was normalized to its mean in order to focus on trends between strains rather than intensity. PCA was then used as a comparison tool for the complete list of metabolites from both GC and LC analyses, as in previous work [8, 12]. Statistical significance of the metabolite differences in the strains analyzed was determined. An analysis of variance (ANOVA) calculation was performed to identify statistically significant differences in metabolite profiles between all five strains in a single test. The metabolites found to have significantly different signals were correlated with previously published RNA data and incorporated into pathways when possible [27]. For the pathway comparisons, metabolite levels for the mutant strains were normalized to the wild-type strain and shown as ratio to wild type, as noted. Additionally, a Student’s t test analysis was performed for all possible pair combinations of the strains (a total of ten) in order to identify potentially interesting metabolite differences between any two strains.
Results and discussion
Discovery-based approach to find metabolite differences in GC×GC–TOFMS data
Applying the discovery-based approach for data analysis allows for focusing on the analytes most responsible for differentiating the sample classes (i.e., the metabolites that differ between strains). As outlined in Fig. 1, this approach combines ChromaTOF, the data analysis software available from LECO for GC×GC–TOFMS data, with further processing of the raw data in Matlab. The standard ChromaTOF approach follows the outline on the left of Fig. 1. The raw data are collected; then, peaks are found, identified, and quantified to generate a peak table. The discovery-based approach applied herein, instead exports the raw GC×GC–TOFMS data into Matlab where peak finding and feature selection are performed to identify which analytes distinguish the classes. These analytes are then initially identified with ChromaTOF software, with refined mass spectral identification and further quantification by PARAFAC in Matlab. Another option is to bypass the initial ChromaTOF identification and perform PARAFAC in a nontargeted way, as shown with the dotted arrow [52]. Using nontargeted PARAFAC, refined mass spectral identification and quantification is provided directly from the raw data. Using these analysis tools, the metabolites that differ between these strains were readily found, identified, and quantified.
Similar metabolites pools in snf1Δ and cat8Δ
Snf1 controls the activity of the transcriptional activators, Adr1 and Cat8, allowing transcriptional activation of dependent genes [53, 54]. Adr1 and Cat8 act both cooperatively and independently to activate numerous genes encoding enzymes of alternative carbon metabolism. However, the relative contribution of Adr1 and Cat8 to metabolite pools during nonfermentative growth is unknown. Snf1 could also impact metabolite levels directly by phosphorylating and influencing specific enzyme activities, in addition to the activation of Adr1 and Cat8. The discovery-based approach found the metabolite differences between the mutant strains to help discern the specific impact of the regulatory factors Snf1, Adr1, and Cat8.
A representative GC×GC chromatogram for the wild-type strain is shown in Fig. 2. From this chromatogram, it is possible to note the complexity of this sample type and the benefit of the second separation dimension. There are many analytes that would be overlapped if only a single dimension were used that can be separated into the 2D space. The additional dimension makes it possible to isolate more metabolites from the complex sample. This wild-type chromatogram could be compared with the chromatograms of other strains by eye, but the chemometric tools in the discovery-based approach are better suited to do so with confidence. The GC data was supplemented with an LC method to allow greater metabolite coverage. Hence, these yeast extracts were analyzed with both LC–MS/MS and GC×GC–TOFMS approaches. In this study, 50 unique metabolites were identified with GC and another 15 with LC. A total of 63 unique metabolites were identified as there were two metabolites, threonine and asparagine, that were observed in both methods. Comparing these two metabolites, the trends in the data are consistent between the two methods. Table 1 shows that the comparative data for these two metabolites are in good agreement. It should be noted that LC analyses do have the potential to provide greater metabolite coverage than is shown here. We have included this representative LC analysis to demonstrate the type of data that can be generated with LC and the complementary nature of these instrumentation approaches.
Fig. 2.

GC × GC–TOFMS chromatographic data for a wild-type strain. Metabolites from five yeast strains (wild type, snf1Δ, adr1Δ, cat8Δ, and adr1Δcat8Δ) were extracted and derivatized for GC analysis as described in “Materials and methods”. The derivatized metabolites were analyzed via GC×GC–TOFMS, with a representative wild-type chromatogram provided here. The complex samples benefit from 2D–GC as the peaks spread out in both GC separation dimensions
Table 1.
Comparison between LC and GC approaches
| Asparagine | Threonine | |||
|---|---|---|---|---|
| LC | GC | LC | GC | |
| adr1Δ | 0.95±0.25 | 1.03±0.61 | 0.76±0.50 | 0.82±0.19 |
| cat8Δ | 0.24±0.21 | 0.19±0.14 | 0.68±0.02 | 0.94±0.10 |
| snf1Δ | 0.29±0.13 | 0.07±0.12 | 0.52±0.48 | 0.64±0.26 |
| WT | 1.00±0.21 | 1.00±0.41 | 1.00±0.13 | 1.00±0.05 |
| adr1Δcat8Δ | 0.45±0.04 | 0.49±0.18 | 0.77±0.11 | 1.02±0.11 |
The signal averages and standard deviations, normalized to wild type, are provided for the two analytes that were detected with both LC and GC
PCA was used to obtain a global picture of how various metabolites and strains relate to each other [8, 12]. PCA is commonly used as a classification tool to determine evidence for class assignment for a set of samples. This application of PCA provides comparison information showing which metabolites behave similarly to each other in the context of yeast strain and which strains are similar to each other in the context of the metabolites analyzed. The 65 metabolites (63 of which are unique) identified in this study are listed in Table 2. In addition to retention time and match value information for each metabolite, the relative amounts normalized to the average for each metabolite are also shown. When these metabolites are treated as samples for PCA analysis, the loadings plot provides information on which strains are most similar to each other, as shown in Fig. 3a. The samples, shown in Fig. 3b, spread out across the PC space based on the patterns between the strains. This global approach shows that the adr1Δ metabolome, as defined by our analysis, is similar to the wild-type metabolome while the cat8Δ, snf1Δ, and adr1Δcat8Δ metabolomes have reduced levels of many metabolites. Previously published gene expression data has also shown that there are fewer Adr1 dependent genes (∼100) than Snf1 (>400) or Cat8 (>200) [27]. The metabolites in Table 2 are listed in the order of their scores on PC1. This facilitates observing the patterns and trends that are responsible for the distribution on PC1 and also determining which metabolites are similar to each other as determined by PCA. From the quantification values, relative amounts normalized to mean for each metabolite, i.e., the columns of Table 2, it can be noted that the metabolites close to each other on PC1 have similar patterns or trends between the strains, as expected.
Fig. 3.

PCA of metabolites and strains. PCA was employed as a data classification tool. When mean-centered metabolites are loaded as samples, the loadings (a) provide information on which strains are most similar to each other, in the context of the analyzed metabolites. The scores (b) provide information on which metabolites are similar to each other, in the context of these strains. The metabolite levels spread out in the PC space based on the various trends, which can be seen in Table 2
To further focus on the metabolites that change the most between strains, the statistical significance of these differences were determined with ANOVA analysis. The ANOVA calculation provides an F value (a statistic) for each metabolite that can be compared with the threshold values. A calculated F value larger than the threshold indicates a metabolite with a statistically significant difference across all five strains. By comparing F values, the metabolites that most significantly differ between strains can be readily determined. ANOVA results for the five strain comparison are listed in Table 3. The number of metabolites with significantly differing levels between the strains was 19 at a 95% confidence level (F> 2.58) and 23 at a 90% confidence level (F>2.09).
Table 3.
Statistical comparison by analysis of variance (ANOVA)
| Metabolite | ANOVA |
||
|---|---|---|---|
| 90%> 2.09 |
95%> 2.58 |
F value |
|
| Glucose | x | x | 3.09 |
| Alanine | 0.53 | ||
| Mannitol | 0.79 | ||
| Glycine | 0.30 | ||
| 2-Monostearin | 0.82 | ||
| Stearic acid | 0.48 | ||
| Valine | 0.41 | ||
| Benzoic acid | 0.30 | ||
| Lysine | 0.22 | ||
| 3-OH butyric | x | 2.17 | |
| Glycerol | 1.02 | ||
| Glucosamine | 1.54 | ||
| Glycerate 3 phosphate | x | x | 3.16 |
| CoA fragment | 0.07 | ||
| Pyruvate | 0.40 | ||
| CMP | 0.52 | ||
| NADP | 0.79 | ||
| FAD | 0.20 | ||
| Deoxyadenosine | 0.58 | ||
| Tyrosine | 1.20 | ||
| Glycolic | 0.01 | ||
| Uracil | 0.04 | ||
| TPP | 0.48 | ||
| 3-OH propionic | 0.04 | ||
| Threonine | 0.95 | ||
| FMN | 0.07 | ||
| SAM | 0.17 | ||
| Ornithine | 0.14 | ||
| Isoleucine | 1.25 | ||
| Glycerol 3 phosphate | x | 2.11 | |
| Orotic | 0.20 | ||
| Cystathionine | 0.96 | ||
| Adenine | 0.33 | ||
| SAH | 0.44 | ||
| Leucine | x | x | 2.68 |
| Arabinose | 0.29 | ||
| ADP | x | 2.20 | |
| Threonine | 0.30 | ||
| Phenylalanine | x | x | 4.85 |
| Adenosine, 5′S-methyl 5′thio- | 0.10 | ||
| N- | |||
| Lactic acid | 0.58 | ||
| G1P | x | x | 2.83 |
| Methionine sulfoxide | 1.97 | ||
| Methionine | 1.90 | ||
| Tryptophan | 0.68 | ||
| Citrate | 1.52 | ||
| Serine | x | x | 3.39 |
| UDP | x | x | 4.20 |
| Arginine | 0.43 | ||
| UDP-N-acetylglucosamine | x | x | 7.00 |
| Glutamate, 5-oxoproline | x | x | 13.13 |
| Asparagine | x | x | 3.94 |
| Glutamic acid | x | x | 10.75 |
| UDP/G1P | x | x | 8.25 |
| Proline | x | x | 3.46 |
| UDP-glucose | x | x | 3.63 |
| Aspartic acid | x | x | 2.58 |
| Asparagine | 1.61 | ||
| Alpha-ketoglutaric acid | x | x | 5.91 |
| G6P | x | x | 4.31 |
| Glutamine | 1.17 | ||
| Succinate | x | x | 3.86 |
| Malate | x | 2.18 | |
| Fumarate | x | x | 4.50 |
| Trehalose | 1.97 | ||
ANOVA provides information on which of the 65 metabolites have statistically significant differences in terms of all five strains, identified collectively
Most of the metabolites found to be statistically different between wild-type and mutant strains are in one of several related pathways; gluconeogenesis, TCA cycle, glyoxylate cycle, or pathways using or providing TCA cycle intermediates. However, not all of the significantly changing metabolites are associated with these pathways, and Tables 2, 3, 4, 5 contain complete data for all metabolites detected in this study. A more thorough analysis of these specific metabolic pathways along with the incorporation of gene expression data is instructive for gaining a better understanding of these differences and determining how closely transcriptome data predicts metabolic activity.
Table 4.
Statistical comparison by Student’s t test. Each strain pair was compared to identify statistically significant differences. A t-value is provided for each metabolite that differed at the 99.0% confidence level (>2.58) for that pair combination. When the t-value is greater 3.29, this indicates a significant difference at the 99.9% confidence level. For metabolites where the single standard deviation error bars did not overlap, a < or > sign indicates which strain showed larger levels, relative to their order in the column header. For example, in the top left cell, glucose is significantly higher in wt than adrl Δ with a t-value of 4.07
| Metabolite | wt: adrl Δ |
wt: cat8Δ |
wt: adrlΔcat8Δ |
wt: snflΔ |
adrlΔ: cat8Δ |
adrlΔ: adrlΔcat8Δ |
adrlΔ: snflA |
cat8Δ: adrlΔcat8Δ |
cat8Δ: snflΔ |
adrlΔcat8Δ: snflΔ |
|---|---|---|---|---|---|---|---|---|---|---|
| Glucose | 4.07 > | 3.88 > | 2.99 | 7.19 < | 3.26 < | 7.46 < | 3.24 < | 7.44 < | 3.22 | |
| Alanine | 14.05 < | 27.34 < | 3.37 < | 7.00 < | ||||||
| Mannitol | 3.10 | 5.67 < | 4.05 < | 3.75 | ||||||
| Glycine | 2.61 | |||||||||
| 2-Monostearin | 3.11 | 4.03 > | 2.75 | 2.64 | 4.39 < | |||||
| Stearic acid | 2.71 | |||||||||
| Valine | 4.49 < | 4.19 < | 3.13 | |||||||
| Benzoic acid | ||||||||||
| Lysine | ||||||||||
| 3-OH butyric | 6.66 < | 4.55 < | 2.84 | 3.91 | 3.06 | 8.26 > | 6.16 > | |||
| Glycerol | 3.31 | 3.14 | 2.67 | 3.63 < | 3.59 < | 2.62 | ||||
| Glucosamine | 2.67 | 4.36 < | 3.46 | 4.79 < | 5.61 < | |||||
| Glycerate 3 phosphate | 5.62 > | 5.90 > | 3.21 | 8.45 > | 8.86 > | 3.61 | 6.84 > | 7.00 < | ||
| CoA fragment | ||||||||||
| Pyruvate | 3.41 | |||||||||
| CMP | 2.69 | 3.00 | ||||||||
| NADP | 4.31 > | 5.44 > | 4.31 < | 5.03 < | ||||||
| FAD | ||||||||||
| Deoxyadenosine | 5.02 > | 3.84 | 3.32 | 2.97 | ||||||
| Tyrosine | 2.82 | 5.62 > | 2.99 | 4.13 > | 6.06 > | 4.94 > | ||||
| Glycolic | ||||||||||
| Uracil | ||||||||||
| TPP | 5.99 < | 4.87 < | ||||||||
| 3-OH propionic | ||||||||||
| Threonine | 2.83 | 4.02 > | 2.79 | 3.18 | 3.98 > | |||||
| FMN | ||||||||||
| SAM | 3.13 | |||||||||
| Ornithine | 3.37 | |||||||||
| Isoleucine | 4.51 > | 2.53 | 3.97 | 4.22 > | 5.38 > | |||||
| Glycerol 3 phosphate | 4.36 < | 6.09 > | 4.05 < | 3.12 | 3.45 | 6.80 > | 11.91 > | |||
| Orotic | 3.04 | 2.58 | ||||||||
| Cystathionine | 4.36 > | 3.06 | 4.27 > | 4.56 > | ||||||
| Adenine | 3.02 | 3.23 | ||||||||
| SAH | 3.92 > | 6.39 > | 3.26 | |||||||
| Leucine | 5.19 < | 8.28 > | 3.18 | 4.20 > | 4.41 < | 5.58 > | 9.97 > | |||
| Arabinose | 2.60 | 2.96 | ||||||||
| ADP | 16.03 > | 4.40 > | 5.93 > | 7.14 > | 3.63 | 5.40 > | 2.63 | |||
| Threonine | 7.57 > | 4.08 | 2.92 | 2.61 | ||||||
| Phenylalanine | 4.38 > | 4.24 < | 11.65 > | 3.65 | 6.18 > | 6.36 < | 6.30 > | 12.90 > | ||
| Adenosine, 5’S-methyl 5’thio-N- | ||||||||||
| Lactic acid | 2.77 | 2.65 | 3.01 | 3.11 | 4.18 > | |||||
| G1P | 5.31 > | 6.89 > | 5.10 > | 6.59 > | 9.08 > | 5.50 > | 3.58 | |||
| Methionine sulfoxide | 5.67 > | 15.89 > | 3.67 | 3.97 | 4.21 > | 8.91 > | ||||
| methionine | 8.76 > | 2.58 | 7.25 > | 3.69 | 3.70 | 5.63 < | 4.93 > | |||
| Tryptophan | 2.92 | 4.74 > | 2.66 | 5.08 < | 5.65 > | 11.35 > | ||||
| Citrate | 4.14 | 3.48 | 7.20 > | 3.80 | 2.89 | 9.15 > | ||||
| Serine | 6.90 > | 2.85 | 30.65 > | 2.72 | 6.58 > | 3.83 | 5.08 > | 12.22 > | ||
| UDP | 7.38 > | 7.21 > | 5.65 > | 8.60 > | 8.29 > | 6.36 > | 4.07 | 3.54 | ||
| Arginine | 3.74 | 4.59 > | 2.60 | |||||||
| UDP-N-acetylglucosamine | 11.27 > | 13.76 > | 11.19 > | 7.35 > | 8.63 > | 6.87 > | 5.33 < | |||
| Glutamate, 5-oxoproline | 12.80 > | 9.23 > | 18.53 > | 13.78 > | 10.14 > | 19.35 > | 5.54 > | 5.24 > | ||
| Asparagine | 7.77 > | 7.92 > | 8.72 > | 6.49 > | 5.91 > | 6.96 > | 2.93 | 3.49 | ||
| Glutamic acid | 12.04 > | 7.09 > | 16.44 > | 13.90 > | 7.91 > | 18.82 > | 5.66 > | 5.88 > | ||
| UDP/G1P | 9.97 > | 9.53 > | 10.18 > | 10.81 > | 10.28 > | 11.01 > | ||||
| Proline | 6.41 > | 5.26 > | 8.75 > | 5.65 > | 4.75 > | 7.44 > | 5.65 > | 8.16 > | ||
| UDP-glucose | 5.70 > | 6.13 > | 5.57 > | 8.10 > | 8.70 > | 7.82 > | 4.02 > | 3.45 | ||
| Aspartic acid | 19.52 > | 16.89 > | 18.46 > | 3.57 > | 2.58 | 3.78 > | 7.51 < | 6.96 > | ||
| Asparagine | 5.63 > | 3.39 | 6.48 > | 4.00 > | 4.56 > | 4.05 | 5.79 > | |||
| Alpha-ketoglutaric acid | 6.59 > | 6.79 > | 6.23 > | 13.21 > | 13.19 > | 11.78 > | ||||
| G6P | 22.96 > | 21.75 > | 21.72 > | 4.22 > | 4.08 > | 4.10 > | ||||
| Glutamine | 6.11 > | 5.09 > | 6.37 > | 3.17 | 2.74 | 3.30 > | 3.00 | 3.87 | ||
| Succinate | 28.17 > | 21.65 > | 28.87 > | 4.41 > | 4.36 > | 4.59 > | 4.95 > | 5.23 > | ||
| Malate | 5.97 > | 5.94 > | 6.04 > | 4.53 > | 4.50 > | 4.58 > | ||||
| Fumarate | 23.19 > | 23.78 > | 24.42 > | 4.20 > | 4.39 > | 4.66 > | 5.20 > | 2.91 | ||
| Trehalose | 5.60 > | 5.64 > | 6.07 > | 3.61 > | 3.65 > | 4.04 > | 6.36 > | 5.18 > |
Table 5.
Signal ratios between each strain pair
| Metabolite | wt.: adr1Δ |
wt.: cat8Δ |
wt.: adr1Δcat8Δ |
wt.: snf1Δ |
adr1Δ: cat8Δ |
adr1Δ: adr1Δcat8Δ |
adr1Δ: snf1Δ |
cat8Δ: adr1Δcat8Δ |
cat8Δ: snf1Δ |
adr1Δcat8Δ: snf1Δ |
|---|---|---|---|---|---|---|---|---|---|---|
| Glucose | 1.50 | 1.44 | 0.22 | 0.10 | 0.96 | 0.14 | 0.06 | 0.15 | 0.07 | 0.45 |
| Alanine | 0.28 | 0.26 | 0.18 | 0.18 | 0.95 | 0.66 | 0.66 | 0.69 | 0.69 | 1.00 |
| Mannitol | 0.71 | 0.32 | 0.34 | 0.82 | 0.45 | 0.47 | 1.15 | 1.06 | 2.55 | 2.42 |
| Glycine | 1.09 | 0.61 | 0.83 | 0.83 | 0.56 | 0.77 | 0.77 | 1.37 | 1.37 | 1.00 |
| 2-Monostearin | 0.68 | 0.76 | 1.22 | 0.52 | 1.10 | 1.78 | 0.76 | 1.61 | 0.68 | 0.42 |
| Stearic acid | 0.90 | 0.80 | 0.95 | 0.66 | 0.90 | 1.06 | 0.73 | 1.18 | 0.82 | 0.69 |
| Valine | 0.21 | 0.78 | 0.49 | 0.20 | 3.73 | 2.33 | 0.93 | 0.62 | 0.25 | 0.40 |
| Benzoic acid | 0.97 | 0.88 | 1.05 | 0.69 | 0.91 | 1.08 | 0.72 | 1.19 | 0.79 | 0.66 |
| Lysine | 1.03 | 0.97 | 0.88 | 0.71 | 0.94 | 0.85 | 0.68 | 0.91 | 0.73 | 0.80 |
| 3-OH butyric | 0.76 | 0.42 | 0.50 | 1.63 | 0.55 | 0.66 | 2.14 | 1.21 | 3.91 | 3.24 |
| Glycerol | 2.05 | 1.95 | 0.38 | 1.02 | 0.95 | 0.19 | 0.50 | 0.20 | 0.52 | 2.65 |
| Glucosamine | 0.98 | 1.03 | 1.10 | 0.74 | 1.04 | 1.12 | 0.75 | 1.08 | 0.72 | 0.67 |
| Glycerate 3 phosphate |
1.05 | 10.59 | 20.53 | 0.49 | 10.12 | 19.62 | 0.47 | 1.94 | 0.05 | 0.02 |
| CoA fragment | 0.91 | 0.95 | 0.89 | 0.85 | 1.04 | 0.97 | 0.93 | 0.93 | 0.90 | 0.96 |
| Pyruvate | 1.03 | 1.36 | 1.06 | 0.78 | 1.32 | 1.02 | 0.75 | 0.78 | 0.57 | 0.74 |
| CMP | 0.97 | 0.69 | 0.56 | 1.56 | 0.71 | 0.57 | 1.60 | 0.81 | 2.25 | 2.79 |
| NADP | 1.28 | 1.39 | 1.23 | 0.88 | 1.09 | 0.96 | 0.69 | 0.88 | 0.63 | 0.72 |
| FAD | 0.84 | 0.84 | 0.81 | 0.98 | 1.01 | 0.97 | 1.17 | 0.96 | 1.16 | 1.21 |
| Deoxyadenosine | 1.13 | 1.08 | 1.14 | 0.99 | 0.95 | 1.00 | 0.88 | 1.06 | 0.92 | 0.87 |
| Tyrosine | 1.09 | 0.60 | 0.66 | 3.09 | 0.55 | 0.61 | 2.85 | 1.10 | 5.18 | 4.70 |
| Glycolic | 0.97 | 1.04 | 0.97 | 0.97 | 1.07 | 1.01 | 1.00 | 0.94 | 0.93 | 0.99 |
| Uracil | 0.89 | 0.99 | 0.99 | 0.94 | 1.12 | 1.12 | 1.06 | 1.00 | 0.95 | 0.95 |
| TPP | 1.01 | 1.24 | 1.23 | 0.96 | 1.23 | 1.22 | 0.95 | 1.00 | 0.77 | 0.78 |
| 3-OH propionic | 1.12 | 1.09 | 1.16 | 1.16 | 0.98 | 1.04 | 1.04 | 1.06 | 1.06 | 1.00 |
| Threonine | 1.22 | 1.06 | 0.98 | 1.55 | 0.87 | 0.80 | 1.27 | 0.92 | 1.46 | 1.58 |
| FMN | 1.04 | 1.09 | 1.28 | 1.22 | 1.05 | 1.23 | 1.18 | 1.18 | 1.12 | 0.95 |
| SAM | 1.32 | 1.19 | 1.36 | 1.39 | 0.90 | 1.03 | 1.05 | 1.14 | 1.16 | 1.02 |
| Ornithine | 1.07 | 1.22 | 1.43 | 1.17 | 1.15 | 1.34 | 1.09 | 1.17 | 0.95 | 0.82 |
| Isoleucine | 1.04 | 0.94 | 0.81 | 1.86 | 0.90 | 0.78 | 1.79 | 0.87 | 1.99 | 2.28 |
| Glycerol 3 phosphate |
1.20 | 0.91 | 0.67 | 2.96 | 0.76 | 0.56 | 2.47 | 0.73 | 3.24 | 4.44 |
| Orotic | 1.05 | 1.06 | 0.84 | 1.76 | 1.01 | 0.80 | 1.70 | 0.79 | 1.68 | 2.12 |
| Cystathionine | 1.18 | 0.94 | 0.79 | 3.04 | 0.79 | 0.67 | 2.57 | 0.85 | 3.25 | 3.83 |
| Adenine | 1.17 | 1.30 | 0.89 | 1.76 | 1.11 | 0.76 | 1.50 | 0.69 | 1.36 | 1.98 |
| SAH | 1.30 | 1.08 | 10.8 | 2.45 | 0.83 | 0.83 | 1.88 | 1.00 | 2.28 | 2.28 |
| Leucine | 1.13 | 1.11 | 0.83 | 2.51 | 0.98 | 0.74 | 2.23 | 0.76 | 2.27 | 3.01 |
| Arabinose | 1.03 | 1.27 | 1.18 | 1.81 | 1.23 | 1.15 | 1.75 | 0.93 | 1.42 | 1.53 |
| ADP | 0.99 | 1.34 | 1.23 | 1.63 | 1.35 | 1.24 | 1.64 | 0.92 | 1.22 | 1.33 |
| Threonine | 1.31 | 1.48 | 1.29 | 1.93 | 1.13 | 0.98 | 1.47 | 0.88 | 1.31 | 1.49 |
| Phenylalanine | 1.16 | 1.29 | 0.88 | 3.32 | 1.11 | 0.76 | 2.86 | 0.69 | 2.58 | 3.76 |
| Adenosine, 5′S methyl 5′thio-N- |
1.02 | 1.47 | 1.37 | 2.01 | 1.45 | 1.35 | 1.97 | 0.93 | 1.36 | 1.46 |
| Lactic acid | 2.01 | 2.22 | 1.35 | 2.96 | 1.10 | 0.67 | 1.47 | 0.61 | 1.33 | 2.19 |
| G1P | 1.12 | 1.94 | 2.72 | 2.21 | 1.74 | 2.43 | 1.98 | 1.40 | 1.14 | 0.18 |
| Methionine sulfoxide |
1.52 | 2.03 | 1.04 | 8.45 | 1.34 | 0.69 | 5.56 | 0.51 | 4.17 | 8.11 |
| Methionine | 1.25 | 2.72 | 1.27 | 3.26 | 2.18 | 1.01 | 2.60 | 0.47 | 1.20 | 2.57 |
| Tryptophan | 1.29 | 2.04 | 1.26 | 5.59 | 1.59 | 0.98 | 4.35 | 0.62 | 2.74 | 4.42 |
| Citrate | 1.18 | 2.04 | 2.03 | 3.71 | 1.73 | 1.72 | 3.14 | 1.00 | 1.82 | 1.83 |
| Serine | 1.18 | 2.02 | 1.19 | 8.89 | 1.72 | 1.01 | 7.55 | 0.59 | 4.39 | 7.44 |
| UDP | 1.05 | 3.42 | 3.43 | 2.25 | 3.27 | 3.27 | 2.15 | 1.00 | 0.66 | 0.66 |
| Arginine | 1.45 | 5.67 | 0.91 | 28.84 | 3.90 | 0.62 | 19.85 | 0.16 | 5.08 | 31.82 |
| UDP-N acetylglucosamine |
1.08 | 3.27 | 4.26 | 2.68 | 3.02 | 3.93 | 2.47 | 1.30 | 0.82 | 0.63 |
| Glutamate, 5-oxoproline |
0.94 | 2.41 | 2.13 | 5.21 | 2.57 | 2.26 | 5.54 | 0.88 | 2.16 | 2.45 |
| Asparagine | 1.05 | 4.16 | 2.24 | 3.47 | 3.95 | 2.12 | 3.29 | 0.54 | 0.83 | 1.55 |
| Glutamic acid |
0.97 | 2.66 | 1.97 | 6.51 | 2.74 | 2.02 | 6.69 | 0.74 | 2.44 | 3.31 |
| UDP/G1P | 1.10 | 3.40 | 3.05 | 3.72 | 3.08 | 2.76 | 3.37 | 0.90 | 1.09 | 1.22 |
| Proline | 0.93 | 2.78 | 2.13 | 6.71 | 2.99 | 2.29 | 7.21 | 0.77 | 2.42 | 3.16 |
| UDP-glucose | 1.07 | 4.10 | 5.50 | 3.89 | 3.82 | 5.12 | 3.63 | 1.34 | 0.95 | 0.71 |
| Aspartic acid | 1.25 | 4.92 | 2.69 | 6.06 | 3.93 | 2.15 | 4.84 | 0.55 | 1.23 | 2.25 |
| Asparagine | 0.97 | 5.38 | 2.03 | 13.54 | 5.52 | 2.08 | 13.90 | 0.38 | 2.52 | 6.68 |
| Alpha-ketoglutaric acid | 0.89 | 5.02 | 6.08 | 4.58 | 5.61 | 6.80 | 5.12 | 1.21 | 0.91 | 0.75 |
| G6P | 1.40 | 8.72 | 7.48 | 7.63 | 6.23 | 5.34 | 5.45 | 0.86 | 0.87 | 1.02 |
| Glutamine | 0.90 | 14.03 | 4.81 | 32.05 | 15.54 | 5.33 | 36.50 | 0.34 | 2.28 | 6.66 |
| Succinate | 1.17 | 12.98 | 11.62 | 21.82 | 11.10 | 9.94 | 18.66 | 0.90 | 1.68 | 1.88 |
| Malate | 1.00 | 15.89 | 14.70 | 19.67 | 15.90 | 14.70 | 19.68 | 0.92 | 1.24 | 1.34 |
| Fumarate | 1.35 | 10.94 | 16.14 | 50.73 | 8.11 | 11.96 | 37.59 | 1.48 | 4.64 | 3.14 |
| Trehalose | 1.39 | 12.09 | 13.31 | 141.29 | 8.66 | 9.54 | 101.28 | 1.10 | 11.69 | 10.62 |
A signal ratio is provided for each pair relative to the column headings (A:B=A/B). For example, in the top left cell, there is 1.50 times more glucose in wild type than in adr1. The statistical significance of these ratios can be determined in Table 4
TCA cycle intermediates
Because these strains were grown in a nonfermentable carbon source, the TCA cycle is expected to be active in the cells showing wild-type behavior. As expected, several TCA cycle intermediates were identified in extracts from wild-type cells (Table 2). Under these growth conditions, the Snf1 protein kinase is required for maximal expression of a number of genes involved in the TCA cycle [27]. Five of the TCA intermediates were detected and are shown in Fig. 4 along with stearic acid that has previously been observed to change very little between strains and conditions [8]. It should be noted that the error bars (average standard deviation=0.19 for these six metabolites) incorporate the variance from both injection replicates and from biological replicates of each strain. As three different cultures were prepared for each strain, there is good agreement in the data. All of the TCA intermediates fall at the most positive end of PC1 on the scores plot while stearic acid falls at the least positive end. At the 90% confidence level, only citrate was not statistically different, by ANOVA, due to the larger error bars and relatively smaller differences between strains. The trend of adr1Δ showing wild-type levels for many metabolites is consistent with what was observed with PCA (and can be observed in Table 2). The metabolite and RNA levels (normalized to wild type) for snf1Δ are shown in Fig. S1a in the Electronic supplementary material. The same information is shown for adr1Δ, cat8Δ, and adr1Δcat8Δ, respectively, in Fig. S1b–d in the Electronic supplementary material. As already demonstrated by PCA, adr1Δ behaves like wild type for these metabolites while the levels in the other three mutant strains are severely reduced.
Fig. 4.

TCA cycle intermediates. Five of the TCA cycle intermediates were detected with this methodology and were quantified by PARAFAC analysis. The signal value for each metabolite was averaged for each strain, incorporating injection variation and biological variation, and then normalized to wild type. Stearic acid is also provided as a reference as it has been observed to be relatively constant in different growth conditions and strains in a previous study [8]. By ANOVA, α-ketoglutarate, succinate, fumarate, and malate all have statistically significant differences
The RNA expression levels also roughly correlate with the metabolite levels for all four strains, demonstrating that in this system transcript data was a good predictor of metabolic activity. For the three strains showing a reduction in metabolite levels, the citrate to succinyl-CoA portion of the TCA cycle appears to be less affected with an approximate 4-fold decrease while the succinate to malate portion of the TCA cycle shows a 10- to 50-fold decrease. These data suggest that the major defect in the TCA cycle in the snf1Δ and cat8Δ mutants may be attributed to MLS1 and ICL1 expression, which is also more severely reduced in the transcriptome data than IDH, KGD, and LSC in the same mutant strains [27]. This suggests that in the wild-type strain more carbon flows through the glyoxylate shunt (catalyzed by isocitrate lyase and malate synthase, encoded by ICL and MLS isozymes, respectively) than the portion of the TCA cycle catalyzed by isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and succinate dehydrogenase, encoded by IDH, KGD, and LDH, respectively. Indeed, growth on ethanol has been shown to activate the enzymes of the glyoxylate cycle over those in the TCA cycle [55]. The glyoxylate shunt bypasses the TCA cycle steps in which CO2 is lost by going directly from isocitrate to malate through glyoxylate. Sending carbon along this route generates extra TCA intermediates to be shunted off to gluconeogenesis rather than continuing on to regenerate intermediates, particularly oxaloacetate for the TCA pathway. While further investigation of this observation is beyond the scope of this study, the results reported herein suggest both that the glyoxylate shunt is an essential part of switching between glucose and ethanol and that the absence of Adr1 has little effect on this mechanism while a defect in Cat8 or Snf1 prevents carbon from moving into gluconeogenesis. Consistent with these trends, as carbon levels are decreased in these strains there are fewer intermediates to be shunted off to the related pathways. In particular, metabolites in glutamate metabolism (produced from α-ketoglutarate) and aspartate metabolism (produced from oxaloacetate) were found to have statistically significant differences and also show decreased levels in snf1Δ, cat8Δ, and adr1Δcat8Δ.
Gluconeogenesis
An overview of the metabolites and RNA in the gluconeogenesis pathway is shown in Fig. S2a–d in the Electronic supplementary material. Again, pathway metabolites in the adr1Δ strain are present approximately at the wild-type levels. The TCA cycle intermediates that feed into gluconeogenesis are less affected in this strain and levels comparable to wild type are observed through the rest of the cycle as well. Most of the metabolites detected in the three remaining strains (snf1Δ, cat8Δ, and adr1Δcat8Δ) are detected at lower relative levels compared with the wild type, similar to those observed for fumarate and malate. This may suggest that the starting amount of oxaloacetate limits the carbon that can travel through gluconeogenesis, but that there is not a further defect in this pathway. The major exceptions to the “TCA trend” are glucose and glycerate-3-phosphate in the snf1Δ strain where much higher levels are observed as compared with the wild type. This may be related to the availability of glucose after 6 h of growth. The glucose consumption from the medium is shown in Fig. 5 and the snf1Δ strain has very low levels of glucose remaining after 6 h. The other strains all consume the small amounts of glucose (0.05%) within the first 2 h. The glucose uptake mechanism is likely altered in the snf1Δ strain but not in the other strains. There is evidence in the literature that Snf1 is important for glucose sensing and uptake [56–58]. The upregulated expression of glycolytic enzymes [35] concurrent with down-regulation of pathways exiting glycolysis in the snf1Δ strain may account for the elevated glucose and glycerate-3-phosphate. Upregulated RNA levels through the glycolytic pathway in the snf1Δ strain [27] may serve as a compensatory mechanism for the disruption in glucose uptake.
Fig. 5.

Glucose consumption per strain. Glucose levels in the media were measured using a PGO Enzymes kit for all five strains over the course of the experiment. Glucose was essentially depleted in all strains by 2 h, except for snf1Δ in which glucose remained at low levels throughout the experiment
Individual strain comparisons
While it is interesting to compare all of the strains simultaneously, it can also be informative to compare individual strain pairs. For example, the identification of differences between snf1Δ and cat8Δ or snf1Δ and adr1Δ helps to illuminate when Snf1 works directly through the phosphorylation of enzymes and indirectly by regulating the activity of Adr1 and Cat8. A comparison between adr1Δ and cat8Δ indicates the relative importance of these transcription factors in different pathways. In this regard, the data indicate that deletion of Cat8 is quite detrimental while a deletion of Adr1 is less so for the metabolites evaluated in this data set. A comparison of adr1Δ and adr1Δcat8Δ identifies instances in which Adr1 plays a more major role in pathways that are coordinately regulated by both transcription factors. In order to more easily make these observations from the large data set, a Student’s t test analysis was performed between each strain pair (a total of ten pairs) with results provided in Table 4. Within each strain pair, the calculated t value is provided when a statistically significant difference was identified at the 99.0% confidence level (t>2.58). A t-value greater than 3.29 indicates significance at the 99.9% confidence level. A qualitative screen was also done in conjunction with the t test and a > or < symbol indicates which strain has higher levels if the single standard deviation error bars did not overlap between the two strains in comparison. A complete set of strain pair ratios is also provided in Table 5. For this table, the ratio of the average PARAFAC signals between each strain pair is calculated and provided. The statistical significance of these differences can be determined with Table 4. Thus, these tables, together, provide a wealth of information from which comparisons, such as those that have been highlighted, can be made.
The analysis of metabolite data with gene expression data provides useful information on the impact of gene expression defects on metabolism. Overall, these results suggest that a defect in Snf1 or Cat8 has broader effects on the metabolism of yeast cells grown in nonfermentable carbon sources than a defect in Adr1. The metabolite isolation procedure used in this study extracts primarily small polar analytes, thus it is possible that a nonpolar extraction would illuminate other pathways, such as fatty acid synthesis and breakdown, in which a defect in Adr1 would have a greater impact on metabolite levels.
It is noteworthy that the adr1Δ mutation had little effect on the level of most amino acids and related metabolites. This is surprising because loss of Adr1 is associated with defects in expression of many amino acid transporters [27]. In contrast, loss of Cat8 and Snf1 caused large reductions of many amino acids and related products. Since Cat8 and Snf1 defects would reduce amino acid precursors generated through the glyoxylate cycle, the abundance of intracellular amino acids after the diauxic transition may be regulated primarily by synthesis rather than uptake.
Conclusions
Correlating the RNA data with the metabolite data, provided via a state-of-the-art global analysis approach, helped to discern the following trends related to the diauxic shift. For the metabolites of gluconeogenesis and the TCA and glyoxylate cycles, the RNA data were a good predictor of the metabolite levels measured in many cases. Furthermore, metabolic pathways that were impacted by Snf1 but not by Cat8 were not detected. This could be because the changes observed were due to long-term effects of Snf1 on metabolite levels. More rapid, short-term responses to flux in carbon source may be required to observe rapid responses that are due to direct Snf1 effects on the metabolic enzymes themselves rather than transcriptional regulation. The selection of the 6-h time point was based on previous observations [8], but it is possible that further insight could be gained from monitoring early time points. More generally, it has been demonstrated that these data collection and discovery-based data analysis approaches are useful for determining differences between mutant strains and in pinpointing specific metabolic pathways influenced by regulatory proteins.
Supplementary Material
Acknowledgement
This research was supported by NIH grant no. GM26079 to ETY.
Abbreviations
- AMPK
AMP-activated protein kinase
- ANOVA
Analysis of variance
- GC×GC–
Two-dimensional (2D) gas chromatography
- TOFMS
coupled to time-of-flight mass spectrometry
- GUI
Graphical user interface
- LC–MS/MS
Liquid chromatography coupled to two-dimensional mass spectrometry
- NIST
National Institute of Standards and Technology
- PARAFAC
Parallel factor analysis
- PCA
Principal component analysis
- TCAcycle
Tricarboxylic acid cycle
- TIC
Total ion current
Footnotes
Electronic supplementary material The online version of this article doi:10.1007/s00216-011-4800-2) contains supplementary material, which is available to authorized users.
Contributor Information
Elizabeth M. Humston, Department of Chemistry, University of Washington, Box 351700, Seattle, WA 98195-7350, USA
Kenneth M. Dombek, Department of Biochemistry, University of Washington, Box 357350, Seattle, WA 98195-7350, USA
Benjamin P. Tu, Department of Biochemistry, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75390-9038, USA
Elton T. Young, Department of Biochemistry, University of Washington, Box 357350, Seattle, WA 98195-7350, USA
Robert E. Synovec, Email: synovec@chem.washington.edu, Department of Chemistry, University of Washington, Box 351700, Seattle, WA 98195-7350, USA.
References
- 1.Goodacre R, Vaidyanathan S, Dunn WB, Harrigan GG, Kell DB. Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol. 2004;22(5):245–252. doi: 10.1016/j.tibtech.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Hollywood K, Brison DR, Goodacre R. Metabolomics: current technologies and future trends. Proteomics. 2006;6:4716–4723. doi: 10.1002/pmic.200600106. [DOI] [PubMed] [Google Scholar]
- 3.Weckwerth W, Morgenthal K. Metabolomics: from pattern recognition to biological interpretation. Drug Discov Today. 2005;10(22):1551–1558. doi: 10.1016/S1359-6446(05)03609-3. [DOI] [PubMed] [Google Scholar]
- 4.Dunn WB, Bailey NJ, Johnson HE. Measuring the metabolome: current analytical technologies. Analyst. 2005;130:606–625. doi: 10.1039/b418288j. [DOI] [PubMed] [Google Scholar]
- 5.Dunn WB, Ellis DI. Metabolomics: current analytical platforms and methodologies. Trends Anal Chem. 2005;24(4):285–294. [Google Scholar]
- 6.Guttman A, Varoglu M, Khandurina J. Multidimensional separations in the pharmaceutical arena. Drug Discov Today. 2004;9(3):136–144. doi: 10.1016/S1359-6446(03)02972-6. [DOI] [PubMed] [Google Scholar]
- 7.Mohler RE, Dombek KM, Hoggard JC, Young ET, Synovec RE. Comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry analysis of metabolites in fermenting and respiring yeast cells. Anal Chem. 2006;78(8):2700–2709. doi: 10.1021/ac052106o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Humston EM, Dombek KM, Hoggard JC, Young ET, Synovec RE. Time-dependent profiling of metabolites from Snf1 mutant and wild type yeast cells. Anal Chem. 2008;80(21):8002–8011. doi: 10.1021/ac800998j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humston EM, Zhang Y, Brabeck GF, McShea A, Synovec RE. Development of a GC×GC-TOFMS method using SPME to determine volatile compounds in cacao beans. J Sep Sci. 2009;32(13):2289–2295. doi: 10.1002/jssc.200900143. [DOI] [PubMed] [Google Scholar]
- 10.Humston EM, Knowles JK, McShea A, Synovec RE. Quantitative assessment of moisture damage for cacao bean quality using two-dimensional gas chromatography combined with time-of-flight mass spectrometry and chemometrics. J Chromatogr A. 2010;1217(12):1963–1970. doi: 10.1016/j.chroma.2010.01.069. [DOI] [PubMed] [Google Scholar]
- 11.Mohler RE, Dombek KM, Hoggard JC, Pierce KM, Young ET, Synovec RE. Comprehensive analysis of yeast metabolite GC×GC–TOFMS data: combining discovery-mode and De convolution Chemometric Software. Analyst. 2007;132:756–767. doi: 10.1039/b700061h. [DOI] [PubMed] [Google Scholar]
- 12.Mohler RE, Tu BP, Dombek KM, Hoggard JC, Young ET, Synovec RE. Identification and evaluation of cycling yeast metabolites in two-dimensional comprehensive gas chromatography-time-of-flight-mass spectrometry data. J Chromatogr A. 2008;1186(1–2):401–411. doi: 10.1016/j.chroma.2007.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pierce KM, Hoggard JC, Hope JL, Rainey PM, Hoofnagle AN, Jack RM, Wright BW, Synovec RE. Fisher ratio method applied to third-order separation data to identify significant chemical components of metabolite extracts. Anal Chem. 2006;78(14):5068–5075. doi: 10.1021/ac0602625. [DOI] [PubMed] [Google Scholar]
- 14.Carling D. Branching out on AMPK regulation. Cell Metab. 2009;9(1):7–8. doi: 10.1016/j.cmet.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Sanz P. AMP-activated protein kinase: structure and regulation. Curr Protein Pept Sci. 2008;9(5):478–492. doi: 10.2174/138920308785915254. [DOI] [PubMed] [Google Scholar]
- 16.Hardie DG, Carling D, Carlson M. The AMP-activated/ SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67(1):821. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 17.Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans . Curr Biol. 2007;17(19):1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint Molecular. Cell. 2008;30(2):214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaeger S, Handschin C, St.-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1a. Proc Natl Acad Sc. 2007;104(29):12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hedbacker K, Carlson M. Snf1/AMPK pathways in yeast. Front Biosci. 2008;13:2408–2420. doi: 10.2741/2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Usaite R, Jewett MC, Oliveira AP, Yates JRI, Olsson L, Nielsen J. Reconstruction of the yeast Snf1 kinase regulatory network reveals its role as a global energy regulator. Mol Syst Biol. 2009;5(319):319. doi: 10.1038/msb.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8(10):774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 23.Hardie DG. AMPK and SNF1: snuffing out stress. Cell Metab. 2007;6(5):339–340. doi: 10.1016/j.cmet.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gancedo JM. The early steps of glucose signalling in yeast. FEMS Microbiol Rev. 2008;32(4):673–704. doi: 10.1111/j.1574-6976.2008.00117.x. [DOI] [PubMed] [Google Scholar]
- 25.Sarma NJ, Haley TM, Barbara KE, Buford TD, Willis KA, Santangelo GM. Glucose-responsive regulators of gene expression in Saccharomyces cerevisiae function at the nuclear periphery via a reverse recruitment mechanism. Genetics. 2007;175(3):1127–1135. doi: 10.1534/genetics.106.068932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuller H-J. Transcriptional control of nonfermentative metabolism in the yeast Saccharomyces cerevisiae . Curr Genet. 2003;43(3):139–160. doi: 10.1007/s00294-003-0381-8. [DOI] [PubMed] [Google Scholar]
- 27.Young ET, Dombek KM, Tachnibana C, Ideker T. Multiple pathways are co-regulated by the protein kinase Snf1 and the transcription factors Adr1 and Cat8. J Biol Chem. 2003;278(28):26146–26158. doi: 10.1074/jbc.M301981200. [DOI] [PubMed] [Google Scholar]
- 28.Kacherovsky N, Tachibana C, Amos E, Fox D, III, Young ET. Promoter binding by the Adr1 transcriptional activator may be regulated by phosphorylation in the DNA-binding region. PLoS ONE. 2008;3(9):e3213. doi: 10.1371/journal.pone.0003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ratnakumar S, Kacherovsky N, Arms E, Young ET. Snf1 controls the activity of Adr1 through dephosphorylation of Ser230. Genetics. 2009;182(3):735–745. doi: 10.1534/genetics.109.103432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tachibana C, Yoo JY, Tagne J-B, Kacherovsky N, Lee TI, Young ET. Combined global localization analysis and transcriptome data identify genes that are directly coregulated by Adr1 and Cat8. Mol Cell Biol. 2005;25(6):2138–2146. doi: 10.1128/MCB.25.6.2138-2146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fiehn O. Metabolomics—the link between genotypes and phenotypes. Plant Mol Biol. 2002;48:155–171. [PubMed] [Google Scholar]
- 32.Raamsdonk LM, Teusink B, Broadhurst D, Zhang N, Hayes A, Walsh MC, Berden JA, Brindle KM, Kell DB, Rowland JJ, Westerhoff HV, van Dam K, Oliver SG. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutation. Nat Biotechnol. 2001;19:45–50. doi: 10.1038/83496. [DOI] [PubMed] [Google Scholar]
- 33.Sweetlove LJ, Last RL, Fernie AR. Predictive metabolic engineering: a goal for systems biology. Plant Physiol. 2003;132(2):420–425. doi: 10.1104/pp.103.022004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.ter Kuile BH, Westerhoff HV. Transcriptome meets metabolome: hierarchical and metabolic regulation of the glycolytic pathway. Federation of European Biochemical Societies Letters. 2001;500(3):169–171. doi: 10.1016/s0014-5793(01)02613-8. [DOI] [PubMed] [Google Scholar]
- 35.Haurie V, Sagliocco F, Boucherie H. Proteomics. 2004;4(13):364–373. doi: 10.1002/pmic.200300564. [DOI] [PubMed] [Google Scholar]
- 36.Dalluge J, van Rijn M, Beens J, Vreuls RJ, Brinkman UATh. Comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometric detection applied to the determination of pesticides in food. J Chromatogr A. 2002;965:207–217. doi: 10.1016/s0021-9673(01)01324-3. [DOI] [PubMed] [Google Scholar]
- 37.Dalluge J, Vreuls RJ, Beens J, Brinkman UATh. Optimization and characterization of comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometric detection (GC×GC/TOFMS) J Sep Sci. 2002;25:201–214. [Google Scholar]
- 38.Shellie R, Marriott P, Morrison P. Concepts and preliminary observations on the triple-dimensional analysis of complex volatile samples by using GC×GC/TOFMS. Anal Chem. 2001;73:1336–1344. [Google Scholar]
- 39.Sinha AE, Prazen BJ, Fraga CG, Synovec RE. Valve-based comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometric detection: instrumentation and figures of merit. J Chromatogr A. 2003;1019:79–87. doi: 10.1016/j.chroma.2003.08.047. [DOI] [PubMed] [Google Scholar]
- 40.van Deursen M, Beens J, Reijenga J, Lipman P, Cramers C, Blomberg J. Group-type identification of oil samples using comprehensive two-dimensional gas chromatography coupled to a time-of-flight mass spectrometer (GC×GC-TOF) J High Resolut Chromatogr. 2000;23(7/8):507–510. [Google Scholar]
- 41.Hope JL, Prazen BJ, Nilsson EJ, Lidstrom ME, Synovec RE. Comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometry detection: analysis of amino acid and organic acid trimethylsilyl derivatives, with applications to the analysis of metabolites in rye grass samples. Talanta. 2005;65:380–388. doi: 10.1016/j.talanta.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 42.Shellie R, Welthagen W, Zrostlikova J, Spranger J, Ristow M, Fiehn O, Zimmermann R. Statistical methods for comparing comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry results: metabolomic analysis of mouse tissue extracts. J Chromatogr A. 2005;1086:83–90. doi: 10.1016/j.chroma.2005.05.088. [DOI] [PubMed] [Google Scholar]
- 43.Sinha AE, Hope JL, Prazen BJ, Nilsson EJ, Jack RM, Synovec RE. Algorithm for locating analytes of interest based on mass spectral similarity in GC×GC-TOF-MS data: analysis of metabolites in human infant urine. J Chromatogr A. 2004;1058(1–2):209–215. [PubMed] [Google Scholar]
- 44.Welthagen W, Shellie R, Spranger J, Ristow M, Zimmermann R, Fiehn O. Comprehensive two-dimensional gas chromatography–time-of-flight mass spectrometry (GC×GC-TOF) for high resolution metabolomics: biomarker discovery on spleen tissue extracts of obese NZO compared to lean C57BL/6 mice. Metabolomics. 2005;1(1):65. [Google Scholar]
- 45.Tu BP, Mohler RE, Liu JC, Dombek KM, Young ET, Synovec RE, McKnight SL. Cyclic changes in metabolic state during the life cycle of yeast. Proc Natl Acad Sci. 2007;104(43):16886–16891. doi: 10.1073/pnas.0708365104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bro R. Tutorial PARAFAC. Tutorial and applications. Chemom Intell Lab Syst. 1997;38:149–171. [Google Scholar]
- 47.Hoggard JC, Synovec RE. Parallel factor analysis (PARAFAC) of target analytes in GC×GC–TOFMS data: automated selection of model with an appropriate number of factors. Anal Chem. 2007;79(4):1611–1619. doi: 10.1021/ac061710b. [DOI] [PubMed] [Google Scholar]
- 48.Sinha AE, Fraga CG, Prazen BJ, Synovec RE. Trilinear chemometric analysis of two-dimensional comprehensive time of flight mass spectrometry data. J Chromatogr A. 2004;1027:269–277. doi: 10.1016/j.chroma.2003.08.081. [DOI] [PubMed] [Google Scholar]
- 49.Sinha AE, Hope JL, Prazen BJ, Fraga CG, Nilsson EJ, Synovec RE. Multivariate selectivity as a metric for evaluating comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry subjected to chemometric peak deconvolution. J Chromatogr A. 2004;1056(1–2):145–154. [PubMed] [Google Scholar]
- 50.Castrillo JI, Hayes A, Mohammed S, Gaskell SJ, Oliver SG. An optimized protocol for metabolome analysis in yeast using direct infusion electrospray mass spectrometry. Phytochemistry. 2003;62:929–937. doi: 10.1016/s0031-9422(02)00713-6. [DOI] [PubMed] [Google Scholar]
- 51.Fiehn O, Kopka J, Tretheway RN, Willmitzer L. Identification of uncommon plant metabolites based on calculation of elemental compositions using gas chromatography and quadrupole mass spectrometry. Anal Chem. 2000;72:3573–3580. doi: 10.1021/ac991142i. [DOI] [PubMed] [Google Scholar]
- 52.Hoggard JC, Synovec RE. Automated resolution of non-target analyte signals in GC×GC–TOFMS data using parallel factor analysis (PARAFAC) Anal Chem. 2008;80:6677–6688. doi: 10.1021/ac800624e. [DOI] [PubMed] [Google Scholar]
- 53.Biddick RK, Law GL, Chin KKB, Young ET. The transcriptional coactivators SAGA, SWI/SNF, and mediator make distinct contributions to activation of glucose-repressed genes. J Biol Chem. 33101–33109;283(48) doi: 10.1074/jbc.M805258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biddick RK, Law GL, Young ET. Adr1 and Cat8 mediate coactivator recruitment and chromatin remodeling at glucose-regulated genes. PLoS ONE. 2008;3(1):e1436. doi: 10.1371/journal.pone.0001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Samokhvalov V, Ignatov V, Kondrashova M. Inhibition of Krebs cycle and activation of glyoxylate cycle in the course of chronological aging of Saccharomyces cerevisiae. Compensatory role of succinate oxidation. Biochimie. 2004;86(1):39–46. doi: 10.1016/j.biochi.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 56.Bisson LF, Neigeborn L, Carlson M, Fraenkel DG. The SNF3 gene is required for high-affinity glucose transport in Saccaromyces cerevisiae . J Bacteriol. 1987;169(4):1656–1662. doi: 10.1128/jb.169.4.1656-1662.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasula S, Jouandot Ii D, Kim J-H. Biochemical evidence for glucose-independent induction of HXT expression in Saccharomyces cerevisiae . FEBS Lett. 2007;581(17):3230–3234. doi: 10.1016/j.febslet.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rolland F, Winderickx J, Thevelein JM. Glucose-sensing and -signalling mechanisms in yeast. FEMS Yeast Res. 2002;2(2):183–201. doi: 10.1111/j.1567-1364.2002.tb00084.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.