

Abstract
This paper describes the biological functions of PTEN and the PTEN regulated signaling pathway in pancreatic β-cells. PTEN has been shown to regulate the regeneration of β-cells. We review the pathways that are controlled by PTEN signaling and their functions in β-cell regeneration. In particular, we describe the unique effect of Pten deletion in β-cells. Unlike its effect in other tissues, Pten deletion does not lead to tumor formation but does enhance β-cell proliferation and function. In addition to the literature review, we also report new results exploring PTEN loss in adult β-cells. We demonstrate that inducing PTEN loss in adult cells has the same regenerative effects previously found for prenatal deletion.
Keywords: PTEN, AKT, pancreatic β-cells
PTEN (phosphatase and tensin homologue deleted on chromosome 10) (also named MMAC1/TEP1) was discovered in 1997 independently by three laboratories as a tumor suppressor of which the expression is often lost in tumors [1-3]. Later studies established that PTEN is a negative regulator of a major cell growth and survival signaling pathway, namely the phosphatidylinositol-3-kinase (PI3K)/AKT signaling pathway [4, 5]. In this paper, we describe the canonical signaling regulated by PTEN as well as a variant of PTEN signaling that is localized in the cell nucleus. We also summarized the ways that PTEN can be regulated transcriptionally, post-transcriptionally and through regulation of its subcellular localization. We review the biological functions of PTEN and its downstream target proteins. The role that proteins downstream of PTEN play in β-cells had recently been reviewed [6]. Here, we focus on the specific effects of PTEN on β-cell regeneration and islet tumor development and issues relating to these effects. Toward this end, we review the phenotypes of several AKT transgenic and Pten deletion studies and report new observations that we have made with Pten mutant mice. Lastly, we report new results that illustrate the specific effects of Pten deletion in β-cells and discuss the unique biology of β-cells that requires further research.
PTEN REGULATED SIGNALING PATHWAYS
Canonical Signaling Pathways Regulated by PTEN
PTEN is a dual lipid and protein phosphatase. The protein structure of PTEN shares homology with known protein phosphatases and is capable of dephosphorylating phospho-peptides as well as phospho-lipids in vitro. The biological effects of PTEN, however are dominated by its ability to dephosphorylate the lipid substrate phosphati-dylinositol-3,4,5-triphosphate (PI-3,4,5-P3) whereas protein substrates for PTEN are being discovered [4]. PI-3,4,5-P3 is formed when PI3K is stimulated as a result of growth factors binding to their receptors that are coupled to PI3K (Fig. 1). The lipid phosphatase motif of PTEN dephosphorylates PI-3,4,5-P3 at the 3′ position and converts it into PI-4,5-P2 [4]. This enzymatic function of PTEN thus reduces the cellular concentration of PI-3,4,5-P3 and acts as a negative regulatory signaling for the PI3K mitogenic signaling pathway (Fig. 1). Accumulation of PI-3,4,5-P3 serves as a major signal for growth factor stimulation. PI-3,4,5-P3 binds to the pleckstrin homology (PH) domain of downstream proteins (e.g. AKT) and provides a lipid moiety for these proteins to bind to the lipid membranes. Binding of PI-3,4,5-P3 to the PH domain also changes the confirmation of these proteins so they can later be activated by phosphorylation. By reducing the intracellular levels of PI-3,4,5-P3, PTEN inhibits the activation of downstream proteins of the PI3K pathway, including the serine/threonine kinase AKT and the protein kinase C (PKC).
Fig. (1). The biological function of PTEN.

PTEN is a lipid phosphatase. Its function is to remove the phosphate from the 3′ position of PI-3,4,5-P3 (PIP3) and form PI-4,5-P2 (PIP2). The accumulation of PI-3,4,5-P3 is responsible for the activation of a number of downstream kinases that contain the Pleckstrin Homology (PH) domain. PI-3,4,5-P3 binds to the PH domains of these downstream molecules and initiates their activation events. Since PI-3,4,5-P3 accumulation is caused by the activation of PI3K, a kinase that adds the 3′ phosphate to PI-4,5-P2, the functions of PTEN and PI3K directly oppose each other. PI3K is activated when growth factors bind to their receptor tyrosine kinases (RTKs) to induce mitogenic signals. It is also activated when insulin binds to its receptor. Thus, PTEN, by antagonizing the function of PI3K inhibits mitogenic signals and blocks the signal transduction of insulin receptor activity.
A well known downstream protein of the PTEN signal is AKT, which plays a critical role in regulating a number of cellular activities including cell growth, survival, cell migration and differentiation, cell and organ size control, metabolism, et al. (for detailed review, see [7]). AKT regulates these cellular processes mainly through direct phosphorylation of its downstream targets (Fig. 2). For instance, phosphorylation of the pro-apoptotic factors BAD, caspases 3 and 9 by AKT renders them inactive and thus promotes cell survival [8-10]. Phosphorylation of BAD allows it to bind to 14-3-3 proteins and prevents it from translocating into the nucleus where it normally binds to and inhibits BCL-XL, preventing cell survival. Phosphorylation of caspases by AKT inhibits their protease activities. Cell cycle modulators such as p21, p27 and MDM2 are also directly regulated by AKT. Phosphorylation of p21 on T145 and p27 on T157 leads to their nuclear exclusion and the inability of these cell cycle inhibitors to inhibit cell proliferation [11, 12]. Likewise, AKT also directly phosphorylates MDM2 and MDMX [13, 14]. Such phosphorylation of MDM2 and MDMX leads to their binding to 14-3-3 proteins and stabilization of the MDM2-MDMX complexes. The MDM2-MDMX complex mediates the degradation of p53 and acts as an E3 ubiquitin ligase to keep the level of p53 low in the cells. Stablization of MDM2-MDMX complexes is another way by which AKT activation induces cell survival and proliferation.
Fig. (2). Substrates of AKT.

A well characterized target of PI3K and PTEN signaling is AKT. Accumulation of PI-3,4,5-P3 leads to the activation of AKT. AKT is a serine/threonine kinase and has a number of downstream targets. Through these targets, AKT regulates cell survival (through caspase, BAD and FOXO); cell growth (through p21, p27, MDM2, cyclin D (Cyn D) as well as FOXO); AKT also regulates mTOR signaling indirectly by inhibiting TSC2 (tuberous sclerosis complex 2 ). Through mTOR and glycogen synthase kinase (GSK3 β), AKT signaling pathway also controls protein translation. In addition, activation of AKT also leads to upregulation of lipogenic and glycolytic genes such as steroyl-CoA response element binding protein (srebp), fatty acid synthase (FAS) and glucokinase (GK). Together with mobilization of glucose transporters (GLUT), induction of these genes by AKT activation leads to enhanced cellular metabolic processes.
In addition, AKT also phosphorylates forkhead transcriptional factors and induces their binding to 14-3-3 proteins [15]. This process blocks their translocation to the nucleus. Outside the nucleus, the forkhead transcriptional factors are unable to control transcription of genes. Several members of the forkhead transcriptional factor family are targets of AKT, including FOXO1 and FOXO3. The binding elements for these forkhead transcriptional factors are widely spread on promoter regions of genes that regulate cell proliferation, survival and metabolic changes [16]. For example, FOXO3a binds to the promoters of Bim and PUMA and can initiate apoptosis cascades by inducing the transcription of these death genes [17, 18]. FOXO1 transcriptionally activates p21 and p27 and inhibits cell proliferation through these actions [19, 20]. Furthermore, these forkhead transcriptional factors are also responsible for many metabolic effects induced by insulin signaling through the PI3K/AKT signaling pathway [21]. Recent evidence suggests that the forkhead transcriptional factors may play a key role in the feedback regulation of the Insulin/PI3K/AKT signaling pathway [22].
Two substrates of AKT, GSK3β and TSC2, play important roles in mediating cross-talks between PI3K/AKT signaling pathway and other signaling pathways (Fig. 2). GSK3β is phosphorylated by AKT on Serine 21/9 which inhibits its activity [23]. GSK3β is an important regulator in Wnt signaling. It phosphorylates β-catenin, resulting in its ubiquitin-mediated degradation. The crosstalk between PTEN and Wnt signaling may underlie some of the effects of PTEN on the regulation of stem cell maintenance and G0-G1 cell cycle regulation [24-28]. Another substrate of AKT is tuberous sclerosis complex 2 (TSC2). TSC2 plays a key role in incorporating metabolism and cell size controls together with cell growth and proliferation regulation [29]. The heterodimer of TSC1 and TSC2 is essential for suppressing the function of mTOR (mammalian target of rapamycin). TSC2 activity is inhibited when phosphorylated by AKT [30]. Therefore, by acting on TSC2, AKT induces the activity of mTOR and the downstream events of mTOR activation that include metabolic changes, protein translation as well as cell proliferation.
Nuclear PTEN
The majority of PTEN studies focus on the enzymatic function of PTEN and its role in inhibiting the PI3K/AKT signaling pathway. Growing evidence indicates that it may have another role beyond its ability to dephosphorylate PI-3,4,5-P3. The ability of PTEN to directly dephosphorylate protein substrates is being explored although the identities of these substrates remain elusive [31]. Many of the recent works on the protein phosphatase activity have come from the analysis of nuclear PTEN. In earlier studies, PTEN was reported to be a protein that is exclusively localized in the cytoplasm. However, more recent evidence suggested that PTEN can be both cytoplasmic and nuclear [32-34]. PTEN is often found in the nucleus of more differentiated and resting cells even though it was originally identified to be a cytosolic protein (using primarily tumor cells) [32]. PTEN in the nucleus appears to play an important role in inhibiting tumor development. Nuclear PTEN also plays other roles in addition to its lipid phosphatase activity (Fig. 3). PTEN in the nucleus has been shown to promote the stability and transcriptional activity of the tumor suppressor p53 by directly associating with p53 [35-37]. Forced expression of PTEN in the nucleus also led to inhibition of cyclin D1 expression [38]. This inhibition is thought to be dependent on MAP kinase [39]. Nuclear expression of PTEN leads to dephosphorylation of MAP kinase. Whether this is a direct effect of the protein phosphatase activity of PTEN is not clear. In addition, PTEN in the nucleus is found to be associated with the centromere by direct binding to the centromere specific binding protein C (CENP-C) [40]. Specific disruption of this binding, which does not reduce PTEN expression or alter its nuclear localization, led to premature centromere separation. In addition, PTEN is also found to collaborate with E2F to induce the expression of Rad 51 and thus enhance DNA repair [40]. This relationship between PTEN and Rad 51 may explain the observation that double stranded DNA breakage rate is found to be increased when nuclear PTEN function is interrupted.
Fig. (3). The function of PTEN in the nucleus.
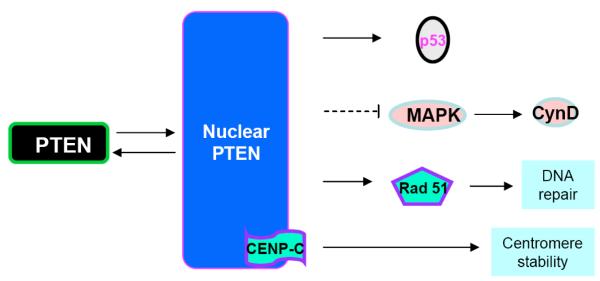
PTEN can shuttle between nucleus and cytosol. In the nucleus, PTEN directly interacts with CENP-C to control Centromere stability. PTEN also interacts with p53 and controls its stability in the nucleus as well. Furthermore, nuclear PTEN control DNA repair and cyclin D through Rad 51 and MAPK, respectively.
Regulation of PTEN
Several non-canonical nuclear localization domains have been found on PTEN [38]. Recent studies suggest that ubiquitination controls the shuttling of PTEN between cytosol and nucleus [41-43]. Monoubiquitination of lysine 289 (K289) is necessary for PTEN to move into the nucleus. Mutation of this site, K289E has been found in familial Cowden’s syndrome that carries multiple mutations of the PTEN gene [41, 44, 45]. A second ubiquitination site K13 and several other sites may also facilitate the nuclear transportation of PTEN. An E3 ubiquitin ligase for PTEN is reported though disagreement exists to whether this is the true E3 ligase for PTEN ubiquintination [42, 45]. NEDD4-1 is thought to be responsible for adding ubiquitin to both K13 and K289 of PTEN molecule, leading to both mono- and poly-ubiquitination of PTEN. Interestingly, poly-ubiquitination of PTEN leads to its degradation whereas mono-ubiquitination leads to its nuclear shuttling. In the nucleus, PTEN is more stable and still capable of inhibiting AKT and inducing cell death.
In addition to cytoplasm-nuclear shuttling, PTEN can also be regulated on the transcriptional and post-transcriptional levels. Several transcriptional factors have been reported to control the transcription of PTEN, including the tumor suppressor p53, an early response gene EGR-1, and a metabolic regulatory gene peroxisomal proliferation activator receptor γ (PPARγ) (for detail, see [46]). Post-translationally, PTEN is modified by acetylation, oxidation and phosphorylation in addition to the ubiquitination discussed above (for detail, see [47]). Phosphorylation of PTEN occurs on several clustered residues in the C-terminal domain of PTEN [48, 49]. Several enzymes are responsible for these phosphorylations including casein kinase 2 (CK2), GSK3β, RhoA kinase and P110δ subunits of PI3K [48, 50, 51]. Phosphorylation of PTEN on these sites generally leads to the stabilization of the molecule but may reduce its activity [48, 49, 51]. A couple of lysine residues at the catalytic domain of PTEN, lysine 125 and 128 are acetylated by PCAF [52]. These acetylations lead to the diminished ability of PTEN to inhibit downstream events such as AKT activation. PTEN is also regulated by the redox status of the cells. Two cysteine residues (124 and 71) form disulfide bonds in response to H2O2 treatment that leads to reduced activity of PTEN [53]. The Cys 124 is one of the “hot spots” that are often found mutated in human cancers. Thus, inhibition of PTEN can provide some molecular basis for the role of oxidative stress in tumor development.
BIOLOGICAL FUNCTIONS REGULATED BY PTEN AND PTEN-CONTROLLED SIGNALS
Insulin/IGF and PI3K Signaling
The impact of PI3K signaling on metabolism has been extensively studied. Most of these studies focus on the insulin and IGF signaling system as insulin and IGF-2 share the same insulin receptor, and IGF-1 also signals through the PI3K pathway. Mice carrying combinations of mutations of the Insulin/IGF and their receptor systems have been used to investigate the role of these ligands and their receptors in cell metabolism. These studies, which have been extensively reviewed [54-57], elucidated the differential functions of insulin, IGF-1, IGF-2 and the various receptors. They also determined how these ligands couple with the various receptors and receptor substrates to control the diverse processes of metabolism.
Divergence in the signaling pathway downstream of the receptors may dictate the responses to insulin signaling. These downstream signaling events are mediated by the multiple isoforms of PI3K [54-57]. Various combinations of these isoforms constitute three classes of PI3K enzymes that are composed of both catalytic and regulatory subunits (There are exceptional cases without regulatory subunits). Studies targeting the PI3K isoforms have obtained conflicting results regarding the role of the regulatory subunit. Gene knockout of all isoforms of the regulatory subunits led to loss of PI3K function and lethal phenotypes, but knockout of individual isoforms led to enhanced function with increased insulin sensitivity and hypoglycemia. The inconsistency may be explained by different isoforms interfering with one another’s functions. The complexity of the PI3K isoforms in metabolism and cell growth needs further investigation.
Functions of Different AKT Isoforms
Of the downstream targeting molecules of PI3K, one serine/threonine kinase, AKT, has been extensively studied. In mammals, there are three AKT isoforms encoded by three different genes [58]. Genetic experiments targeting the three AKTs either individually or in combination have revealed the biological functions of these AKT isoforms. These studies demonstrate that AKT1 is not necessary for the regulation of glucose metabolism but is crucial for cell growth and survival [59-62]. Mice lacking AKT2 and AKT3 developed normally without any growth phenotypes as long as AKT1 was present [61]. Mouse embryonic stem cells lacking AKT1 are significantly more susceptible to apoptosis [63]. Together, this genetic evidence suggests that AKT1 is indispensable for the regulation of cell growth and survival. Loss of AKT2 alone or loss of both AKT2 and AKT3 renders mice glucose and insulin intolerant [61, 64, 65]. This genetic evidence suggests that AKT1 has a general role in cell growth and survival while the endogenous function of AKT2 is the regulation of metabolic signals through the PI3K/AKT signaling pathway [58, 66].
THE FUNCTION OF PTEN-REGULATED SIGNALS IN β-CELLS
The functions of various components in the insulin/IGF-PI3K-AKT signaling pathway in β-cells were explored through cell type-specific gene targeting approaches. These studies, collectively suggest that insulin regulates both β-cell proliferation and insulin secretion [56]. This effect is confirmed by studies that target AKT and PTEN. Transgenic expression of constitutively active AKT in β-cells (with membrane targeted forms of either a full-length AKT or an AKT that lacks the PH domain) led to dramatic increases in both the size and numbers of pancreatic islets in the transgenic mice [67, 68]. These mice also displayed more efficient insulin secretion when challenged with glucose. Similarly, deletion of Pten specifically in the β-cells using rat insulin promoter driven Cre (PtenloxP/loxP; Rip-Cre+), which results in increased AKT activity, leads to increased islet mass and resistance to streptozotocin (STZ) induced β-cell death [34]. In response to glucose challenge, the Pten null β-cells secrete insulin much more efficiently than the control cells [69]. The effect of loss of function of the AKT isoforms specifically in β-cells has not been evaluated. Global Akt1 gene deletion studies have not addressed the effects (if any) on β-cells [59, 60, 70]. Mice lacking AKT2 (Akt2−/−) displayed mild hyperglycemia [64]. This development of hyperglycemia in Akt2−/− mice is attributed to the inability of insulin to suppress hepatic glucose output [64]. The β-cell mass actually increased in the Akt2−/− mice compared to the controls, indicating proper β-cell compensation in response to increased blood glucose levels. Haploinsufficiency of Akt1 in Akt2−/− mice pushes the mice to diabetes due partially to the inability of β-cells to compensate for hyperglycemia [71]. Similarly, when we introduced Akt2 deletion into the β-cell-specific Pten null mice (PtenloxP/loxP; Rip-Cre+; Akt2−/−), we observed only very minimum effects on β-cell mass (Fig. 4). The effect of AKT1 on the function of Pten mutation-induced β-cell regeneration remains to be studied. However, introduction of a kinase-dead dominant-negative AKT to β-cells only resulted in decreased insulin secretion but not changes in β-cell mass [72]. Together, these data suggests that the effect of PTEN on β-cell regeneration may not depend on AKT signaling especially given the fact that PTEN is located predominantly in the nucleus in β-cells [69].
Fig. (4). Deletion of Akt2 does not affect islet mass in Pten null islets.

We deleted Akt2 in the PtenloxP/loxP; Rip-Cre+ mice to test whether the PI3K/AKT signaling mediated β-cell regeneration. We found that deletion of Akt2 has little effect on the regenerative islet phenotype we observed in the Pten deletion models. Pm, PtenloxP/loxP; Rip-Cre+; Dm, PtenloxP/loxP; Rip-Cre+;Akt2−/−. Repre-sentative image from 10 animals.
A major consequence of activating PI3K/AKT signaling is the inhibition of cell cycle inhibitors such as p21 and p27, as well as upregulation of cyclins such as cyclin D. The function of cyclins, cyclin dependent kinases (cdks) and other cell cycle regulators have been studied in β-cells [73]. These studies found that the complexes that regulate the G1 stage of the cell cycle including E2F, pRb, cyclin D and cdks are important in the regulation of β-cell proliferation [73]. Loss of cyclin D2 leads to significantly smaller islet mass [74]. Loss of both cyclin D1 and D2 leads to significant loss of insulin secretion in addition to islet mass reduction [75]. Similar phenotypes were observed in cdk4 knockout animals [75, 76]. Other cell cycle regulators including p21, p27, p53 and other cdks and cyclins have also been shown to regulate β-cell proliferation even though multiple events may be required [77, 78]. Collectively, these studies showed that cyclin D, cdk4, and p27 are needed for maintaining β-cell proliferation dynamics while p21 and p-RB are dispensable [73-78].
One important cell cycle regulator that received significant attention recently is the INK family cell cycle regulator p16INK4A. p16 was recently found to be correlated with the age related loss of replication observed in β-cells [79, 80]. Accumulation of p16 with age is correlated with a decrease in β-cell regeneration [79, 80]. Transgenic over-expression of p16 leads to age-dependent loss of cell proliferation in multiple organs in vivo [81, 82] including β-cells [81]. The level of p16 increases significantly in mice one year old and older [79, 80]. The timing of the p16 increase in mice coincides with the age at which β-cell proliferation undergoes a steep decline and no longer responds to physical stimuli. We analyzed whether p16 levels are altered in the β-cell specific Pten null mice (PtenloxP/loxP; Rip-Cre+) compared with controls. We found that p16 levels are reduced in islets lacking PTEN (Fig. 5). This reduction occurs in both young and old mice (data not shown). In addition, we also found that deletion of Pten in adult β-cells can still robustly induce the increase of islet mass. We used a model where we induced deletion of Pten in β-cells of 3 month old mice (PtenloxP/loxP; Rip-CreER+). This manipulation resulted in an increased islet mass observed 6 months after induction (Fig. 6). The inhibition of p16 may explain why deletion of Pten at the adult age still leads to increased islet mass. Adult β-cells gradually lose their ability to proliferate with age [83]. p16 is correlated with this age related loss of β-cell proliferation [79, 80]. Thus, our observations linking PTEN signal with p16 represent significant progress in unveiling how β-cell regeneration may be controlled. A previous transgenic approach has demonstrated that over-expressing p16 inhibits the ability of β-cells to proliferate [81]. However, whether inhibiting p16 can allow aged β-cells to regain the regenerating potential has not been previously tested. Our observation shows that inhibition of p16 (through PTEN loss) can lead to enhanced ability of β-cells to regenerate in adult mice. This investigation of how p16 expression can be controlled represents future research directions that are likely to be significant for regeneration of adult β-cells. Since β-cells losing their ability to compensate for hyperglycemia in old age is one of the likely mechanisms for diabetes development, such research is needed for any therapeutic development that aims at either preventing or treating diabetes.
Fig. (5). Reduced p16 expression in Pten mutant β-cells.

Deletion of Pten in the β-cells (PtenloxP/loxP; Rip-Cre+, Mut) led to reduction of cell cycle inhibitor p16 compared to control mice (PtenloxP/loxP; Rip-Cre−, Con). Immunohistochemistry staining of p16 (green) in β-cells (red) show that p16 levels are reduced in the Pten mutant islets vs. control islets. Images are from pancreatic sections of 8 months old mice. Representative of 4 animals.
Fig. (6). Induced Pten loss in adult β-cells leads to increased islet mass.

PTEN loss is induced in 3 month old PtenloxP/loxP; Rip-CreER+ mice with treatment of tamoxifen. Pancreata are sectioned 3 months later. Mice treated with tamoxifen (Pten deleted) displayed larger and more islets. Representative image from 6 animals.
PTEN REGULATED SIGNALS AND INSULINOMA
Functional dissections of various AKT isoforms are a beginning in understanding how signals downstream of insulin/IGF receptor events diverge in their control of cell growth and metabolism. These differences in signaling, especially in light of the mitogenic role that IGF plays in promoting tumor growth in various tissues, is important for any future therapeutic development that hopes to enhance the overall function of β-cells without producing adverse effects such as tumor development. Insulinoma, cancer of the β-cells, is a rare form of tumor in humans. It has been shown that PI3K signaling plays a role in the development of human insulinoma [84]. In rodent models, over-expression of the constitutively active form of AKT in β-cells also leads to tumor development [85]. However, loss of Pten in β-cells (which enhances AKT activity) resulted in increased islet mass without tumor development (Fig. 7 and Ref [86]). This inconsistency warrants further investigation into distinguishing between the endogenous signals that regulate β-cell regeneration and those that induce tumor development. One possible reason for the different observations is the dosage of activated AKT. AKT activity may be substantially higher in the AKT transgenic mice than in the Pten mutant mice. As a result, there may be a “Goldilocks phenomenon” where too little AKT enhancement cannot regenerate β-cells, too much activity leads to tumor development, but a middle level can induce β-cell regeneration without adverse effects. An alternative explanation is that the subcellular localization of PTEN differs significantly between insulinomas and normal islet cells [34, 87]. In normal islet cells, PTEN is predominantly found in the nucleus [34], whereas 82.5% of islet tumors exhibit cytoplasmic labeling patterns for PTEN. The same cytoplasmic staining pattern for PTEN is also observed in adenomas developed in transgenic mice expressing constitutively active AKT in the β-cells [85]. These analyses suggest that the nuclear function of PTEN is associated with suppression of tumor development in the islets. PTEN in the nucleus may not rely on PI3K/AKT signaling to function, even though PI3K and AKT as well as PI-3,4,5-P3 have all been observed in the nucleus [32, 88]. The lack of tumor development in β-cells lacking PTEN is also in striking difference comparing to other epithelial tissues in which PTEN is lost. In the prostate, colon, mammary tissue and liver, loss of PTEN leads to tumor development [5, 33]. Even in the pancreas, deletion of Pten leads to the development of pancreatic ductal carcinomas (PDAs) when targeted to the progenitor cell population using Pdx-1 Cre (PtenloxP/loxP; Pdx-1-Cre+) [89]. This difference suggests that the cell proliferation biology in β-cells may be unique. Further experimentation is needed to discover this unique mechanism that governs the proliferation of β-cells. Such experiments could be highly significant for diabetes research because they may identify a target for therapeutic intervention that can stimulate β-cell regeneration without inducing insulinoma.
Fig. (7). Pten loss does not lead to development of insulinomas.

We analyzed the Pten mutant (PtenloxP/loxP; Rip-Cre+) mice 14.5 months of age (right panels) and found that the islets in these older mice are even larger compared with the controls (left panels). They maintain the normal cellular structures and still produce insulin (red, bottom panels) and other endocrine hormones (green, stained with a cocktail of somatostatin, pancreatic polypeptide and glucagon). n=3.
FUTURE CONSIDERATIONS
In a number of tissue types, PTEN is a critical tumor suppressor. When PTEN is lost or its expression is reduced, tumors often develop. In β-cells, loss of PTEN does not lead to tumor formation but instead leads to enhanced β-cell regeneration. This enhancement is not limited to effects during organogenesis but can also be obtained when Pten is deleted in adult mice. Even in adult mice, Pten deletion does not cause insulinoma formation, despite the fact that over-expression of activated AKT, the downstream target of PTEN, does induce insulinoma development. These observations suggest that either the signaling molecules downstream of PTEN are different in β-cells than other cells or they respond differently due to something unique in the β-cell environment. The unique mechanism in β-cells that allows them to respond to growth signals requires detailed studies to identify the mechanism responsible. Such studies could be advantageous for both the cancer development and diabetes treatment fields.
ACKNOWLEDGEMENTS
We thank Dr. Bryan Stiles for editing the manuscript. Bangyan Stiles acknowledges fundings from NIDDK and USC TREC center. Ni Zeng acknowledges funding from CBM training grants. Jennifer-Ann Bayan acknowledges funding from ARRA minority student dissertation awards.
REFERENCES
- [1].Li JYC, Liaw D, Podsypanina K, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- [2].Liaw DMD, Li J, Dahia PL, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–7. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- [3].Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–9. [PubMed] [Google Scholar]
- [4].Downes CP, Ross S, Maccario H, et al. Stimulation of PI 3-kinase signaling via inhibition of the tumor suppressor phosphatase, PTEN. Adv Enzyme Regul. 2007;47:184–94. doi: 10.1016/j.advenzreg.2006.12.018. [DOI] [PubMed] [Google Scholar]
- [5].Stiles B, Groszer M, Wang S, Jiao J, Wu H. PTENless means more. Dev Biol. 2004;273:175–84. doi: 10.1016/j.ydbio.2004.06.008. [DOI] [PubMed] [Google Scholar]
- [6].Elghazi L, Bernal-Mizrachi E. Akt and PTEN: beta-cell mass and pancreas plasticity. Trends Endocrinol Metab. 2009;20:243–51. doi: 10.1016/j.tem.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kermer PKN, Labes M, Bahr M. Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent akt phosphorylation and inhibition of caspase-3 In vivo. J Neurosci. 2000;20:722–8. [PubMed] [Google Scholar]
- [9].Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- [10].Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- [11].Zhou BP, Liao Y, Xia W, et al. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–52. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- [12].Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2002;277:28706–13. doi: 10.1074/jbc.M203668200. [DOI] [PubMed] [Google Scholar]
- [13].Feng J, Tamaskovic R, Yang Z, et al. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J Biol Chem. 2004;279:35510–7. doi: 10.1074/jbc.M404936200. [DOI] [PubMed] [Google Scholar]
- [14].Lopez-Pajares V, Kim MM, Yuan ZM. Phosphorylation of MDMX mediated by Akt leads to stabilization and induces 14-3-3 binding. J Biol Chem. 2008;283:13707–13. doi: 10.1074/jbc.M710030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- [16].Hedrick SM. The cunning little vixen: Foxo and the cycle of life and death. Nat Immunol. 2009;10:1057–63. doi: 10.1038/ni.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].You H, Pellegrini M, Tsuchihara K, et al. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203:1657–63. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–4. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- [19].Liu R, Wang L, Chen G, et al. FOXP3 up-regulates p21 expression by site-specific inhibition of histone deacetylase 2/histone deacetylase 4 association to the locus. Cancer Res. 2009;69:2252–9. doi: 10.1158/0008-5472.CAN-08-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stahl M, Dijkers PF, Kops GJ, et al. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–31. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- [21].Gross DN, Wan M, Birnbaum MJ. The role of FOXO in the regulation of metabolism. Curr Diab Rep. 2009;9:208–14. doi: 10.1007/s11892-009-0034-5. [DOI] [PubMed] [Google Scholar]
- [22].Marr MT, 2nd, D’Alessio JA, Puig O, Tjian R. IRES-mediated functional coupling of transcription and translation amplifies insulin receptor feedback. Genes Dev. 2007;21:175–83. doi: 10.1101/gad.1506407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- [24].Bachman ES, Dhillon H, Zhang CY, et al. betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science. 2002;297:843–5. doi: 10.1126/science.1073160. [DOI] [PubMed] [Google Scholar]
- [25].Groszer M, Erickson R, Scripture-Adams DD, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–9. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- [26].Kwon CH, Zhu X, Zhang J, et al. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–11. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- [27].Yilmaz OH, Valdez R, Theisen BK, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- [28].Zhang J, Grindley JC, Yin T, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–22. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- [29].Leung AK, Robson WL. Tuberous sclerosis complex: a review. J Pediatr Health Care. 2007;21:108–14. doi: 10.1016/j.pedhc.2006.05.004. [DOI] [PubMed] [Google Scholar]
- [30].Li Y, Corradetti MN, Inoki K, Guan KL. TSC2: filling the GAP in the mTOR signaling pathway. Trends Biochem Sci. 2004;29:32–8. doi: 10.1016/j.tibs.2003.11.007. [DOI] [PubMed] [Google Scholar]
- [31].Leslie NR, Maccario H, Spinelli L, Davidson L. The significance of PTEN’s protein phosphatase activity. Adv Enzyme Regul. 2009;49:190–6. doi: 10.1016/j.advenzreg.2008.12.002. [DOI] [PubMed] [Google Scholar]
- [32].Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci. 2008;121:249–53. doi: 10.1242/jcs.022459. [DOI] [PubMed] [Google Scholar]
- [33].Stiles B, Wang Y, Stahl A, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected] Proc Natl Acad Sci USA. 2004;101:2082–7. doi: 10.1073/pnas.0308617100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stiles BL, Kuralwalla-Martinez C, Guo W, et al. Selective deletion of Pten in pancreatic beta cells leads to increased islet mass and resistance to STZ-induced diabetes. Mol Cell Biol. 2006;26:2772–81. doi: 10.1128/MCB.26.7.2772-2781.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Freeman DJ, Li AG, Wei G, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–30. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- [36].Tang Y, Eng C. p53 down-regulates phosphatase and tensin homologue deleted on chromosome 10 protein stability partially through caspase-mediated degradation in cells with proteasome dysfunction. Cancer Res. 2006;66:6139–48. doi: 10.1158/0008-5472.CAN-06-0772. [DOI] [PubMed] [Google Scholar]
- [37].Tang Y, Eng C. PTEN autoregulates its expression by stabilization of p53 in a phosphatase-independent manner. Cancer Res. 2006;66:736–42. doi: 10.1158/0008-5472.CAN-05-1557. [DOI] [PubMed] [Google Scholar]
- [38].Chung JH, Eng C. Nuclear-cytoplasmic partitioning of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) differentially regulates the cell cycle and apoptosis. Cancer Res. 2005;65:8096–100. doi: 10.1158/0008-5472.CAN-05-1888. [DOI] [PubMed] [Google Scholar]
- [39].Chung JH, Ostrowski MC, Romigh T, et al. The ERK1/2 pathway modulates nuclear PTEN-mediated cell cycle arrest by cyclin D1 transcriptional regulation. Hum Mol Genet. 2006;15:2553–9. doi: 10.1093/hmg/ddl177. [DOI] [PubMed] [Google Scholar]
- [40].Shen WH, Balajee AS, Wang J, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–70. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- [41].Trotman LC, Wang X, Alimonti A, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–56. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang X, Trotman LC, Koppie T, et al. NEDD4-1 is a protooncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–39. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Drinjakovic J, Jung H, Campbell DS, et al. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron. 2010;65:341–57. doi: 10.1016/j.neuron.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chi SG, Kim HJ, Park BJ, et al. Mutational abrogation of the PTEN/MMAC1 gene in gastrointestinal polyps in patients with Cowden disease. Gastroenterology. 1998;115:1084–9. doi: 10.1016/s0016-5085(98)70078-2. [DOI] [PubMed] [Google Scholar]
- [45].Fouladkou F, Landry T, Kawabe H, et al. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc Natl Acad Sci USA. 2008;105:8585–90. doi: 10.1073/pnas.0803233105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Stiles BL. Phosphatase and tensin homologue deleted on chromosome 10: extending its PTENtacles. Int J Biochem Cell Biol. 2009;41:757–61. doi: 10.1016/j.biocel.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene. 2008;27:5464–76. doi: 10.1038/onc.2008.243. [DOI] [PubMed] [Google Scholar]
- [48].Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem. 2005;280:35195–202. doi: 10.1074/jbc.M503045200. [DOI] [PubMed] [Google Scholar]
- [49].Torres J, Rodriguez J, Myers MP, et al. Phosphorylation-regulated cleavage of the tumor suppressor PTEN by caspase-3: implications for the control of protein stability and PTEN-protein interactions. J Biol Chem. 2003;278:30652–60. doi: 10.1074/jbc.M212610200. [DOI] [PubMed] [Google Scholar]
- [50].Papakonstanti EA, Ridley AJ, Vanhaesebroeck B. The p110delta isoform of PI 3-kinase negatively controls RhoA and PTEN. EMBO J. 2007;26:3050–61. doi: 10.1038/sj.emboj.7601763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–8. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- [52].Okumura K, Mendoza M, Bachoo RM, et al. PCAF modulates PTEN activity. J Biol Chem. 2006;281:26562–8. doi: 10.1074/jbc.M605391200. [DOI] [PubMed] [Google Scholar]
- [53].Lee SR, Yang KS, Kwon J, et al. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–42. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- [54].Hribal ML, Oriente F, Accili D. Mouse models of insulin resistance. Am J Physiol Endocrinol Metab. 2002;282:E977–81. doi: 10.1152/ajpendo.00561.2001. [DOI] [PubMed] [Google Scholar]
- [55].Nakae J, Kido Y, Accili D. Tissue-specific insulin resistance in type 2 diabetes: lessons from gene-targeted mice. Ann Med. 2001;33:22–7. doi: 10.3109/07853890109002056. [DOI] [PubMed] [Google Scholar]
- [56].Rother KI, Accili D. Role of insulin receptors and IGF receptors in growth and development. Pediatr Nephrol. 2000;14:558–61. doi: 10.1007/s004670000351. [DOI] [PubMed] [Google Scholar]
- [57].Nandi A, Kitamura Y, Kahn CR, Accili D. Mouse models of insulin resistance. Physiol Rev. 2004;84:623–47. doi: 10.1152/physrev.00032.2003. [DOI] [PubMed] [Google Scholar]
- [58].Whiteman EL, Cho H, Birnbaum MJ. Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab. 2002;13:444–51. doi: 10.1016/s1043-2760(02)00662-8. [DOI] [PubMed] [Google Scholar]
- [59].Chen W, Xu P-Z, Gottlob K, et al. Growth retardation and increased apoptosis in mouse homozygous disruption of the akt1 gene. Genes Dev. 2001;15:2203–8. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum M. Akt1/PKBa is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–52. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- [61].Dummler B, Tschopp O, Hynx D, et al. Life with a single isoform of Akt: mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol. 2006;26:8042–51. doi: 10.1128/MCB.00722-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yang ZZ, Tschopp O, Hemmings-Mieszczak M, et al. Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J Biol Chem. 2003;278:32124–31. doi: 10.1074/jbc.M302847200. [DOI] [PubMed] [Google Scholar]
- [63].Stiles B, Gilman V, Khanzenzon N, et al. Essential role of AKT-1/protein kinase B alpha in PTEN-controlled tumorigenesis. Mol Cell Biol. 2002;22:3842–51. doi: 10.1128/MCB.22.11.3842-3851.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cho H, Mu J, Kim JK, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–31. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- [65].Garofalo RS, Orena SJ, Rafidi K, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bae SS, Cho H, Mu J, Birnbaum MJ. Isoform-specific regulation of insulin-dependent glucose uptake by Akt/protein kinase B. J Biol Chem. 2003;278:49530–6. doi: 10.1074/jbc.M306782200. [DOI] [PubMed] [Google Scholar]
- [67].Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest. 2001;108:1631–8. doi: 10.1172/JCI13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tuttle RL, Gill NS, Pugh W, et al. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med. 2001;7:1133–7. doi: 10.1038/nm1001-1133. [DOI] [PubMed] [Google Scholar]
- [69].Nguyen KT, Tajmir P, Lin CH, et al. Essential role of Pten in body size determination and pancreatic beta-cell homeostasis in vivo. Mol Cell Biol. 2006;26:4511–8. doi: 10.1128/MCB.00238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yang ZZ, Tschopp O, Di-Poi N, et al. Dosage-dependent effects of Akt1/protein kinase Balpha (PKBalpha) and Akt3/PKBgamma on thymus, skin, and cardiovascular and nervous system development in mice. Mol Cell Biol. 2005;25:10407–18. doi: 10.1128/MCB.25.23.10407-10418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Chen WS, Peng XD, Wang Y, et al. Leptin deficiency and beta-cell dysfunction underlie type 2 diabetes in compound Akt knockout mice. Mol Cell Biol. 2009;29:3151–62. doi: 10.1128/MCB.01792-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bernal-Mizrachi E, Fatrai S, Johnson JD, et al. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest. 2004;114:928–36. doi: 10.1172/JCI20016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, et al. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev. 2006;27:356–70. doi: 10.1210/er.2006-0004. [DOI] [PubMed] [Google Scholar]
- [74].Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963–8. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kushner JA, Ciemerych MA, Sicinska E, et al. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25:3752–62. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Rane SG, Dubus P, Mettus RV, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- [77].Cozar-Castellano I, Weinstock M, Haught M, et al. Evaluation of beta-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21cip. Diabetes. 2006;55:70–7. [PubMed] [Google Scholar]
- [78].Vasavada RC, Cozar-Castellano I, Sipula D, Stewart AF. Tissue-specific deletion of the retinoblastoma protein in the pancreatic beta-cell has limited effects on beta-cell replication, mass, and function. Diabetes. 2007;56:57–64. doi: 10.2337/db06-0517. [DOI] [PubMed] [Google Scholar]
- [79].Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes. 2009;58:1365–72. doi: 10.2337/db08-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes. 2009;58:1312–20. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–7. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- [82].Canepa ET, Scassa ME, Ceruti JM, et al. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life. 2007;59:419–26. doi: 10.1080/15216540701488358. [DOI] [PubMed] [Google Scholar]
- [83].Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–67. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- [84].Burtscher I, Compagni A, Lamm GM, Christofori G. An insulin-like growth factor-mediated, phosphatidylinositol 3′ kinase-independent survival signaling pathway in beta tumor cells. Cancer Res. 1999;59:3923–6. [PubMed] [Google Scholar]
- [85].Alliouachene S, Tuttle RL, Boumard S, et al. Constitutively active Akt1 expression in mouse pancreas requires S6 kinase 1 for insulinoma formation. J Clin Invest. 2008;118:3629–38. doi: 10.1172/JCI35237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Radziszewska A, Choi D, Nguyen KT, et al. PTEN deletion and concomitant c-Myc activation do not lead to tumor formation in pancreatic beta cells. J Biol Chem. 2009;284:2917–22. doi: 10.1074/jbc.M805183200. [DOI] [PubMed] [Google Scholar]
- [87].Perren A, Komminoth P, Saremaslani P, et al. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol. 2000;157:1097–103. doi: 10.1016/S0002-9440(10)64624-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Neri LM, Borgatti P, Capitani S, Martelli AM. The nuclear phosphoinositide 3-kinase/AKT pathway: a new second messenger system. Biochim Biophys Acta. 2002;1584:73–80. doi: 10.1016/s1388-1981(02)00300-1. [DOI] [PubMed] [Google Scholar]
- [89].Stanger BZ, Stiles B, Lauwers GY, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8:185–95. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]