

Abstract
Stroke is the fourth leading cause of death in the United States and the leading cause of long-term disability. Ischemic stroke, due to an interruption in blood supply, is particularly prevalent; 87% of all strokes are ischemic. Unfortunately, current options for acute treatment are extremely limited and there is a great need for new treatment strategies. This review will discuss evidence that mild sensory stimulation can completely protect the jeopardized brain from an impending stroke in a rodent model. When delivered within the first 2 hours following ischemic onset, this stimulation results in complete protection, including a full reestablishment of cortical function, sensorimotor capabilities, and blood flow. Identical stimulation, however, initiated 3 hours following ischemic onset, results in an increase in damage compared with untreated animals. The protective effect is not specific to a single sensory modality, anesthesia, or age, and increasing evoked cortical activity by increasing stimulation accelerates recovery. Taken together, these findings demonstrate that cortical activity is a critical factor for protection and suggest a new, exciting potential avenue for the development of acute stroke treatment strategies that may produce a noninvasive, drug-free, equipment-free, and side effect-free means of protecting from ischemic stroke.
Keywords: stroke, ischemia, protection, reperfusion, stimulation treatment, rodent model
Introduction
Despite major advances in prevention and rehabilitation, few neurological injuries are as debilitating as stroke. The disease is currently the fourth leading cause of death and the leading cause of long-term disability in the United States and is similarly devastating in other nations (Roger and others 2011). Current statistics suggest that a new stroke occurs about every 40 seconds and a death by stroke occurs about every 4 minutes on average and only about half of those who survive stroke are able to return to full-time work following the injury (NINDS 2011). Old age is associated with an enhanced susceptibility to stroke, decreased recovery from infarct and reduced effectiveness of neuroprotective treatments (DiNapoli and others 2008; Dirnagl 2010; Popa-Wagner and others 2011; Popa-Wagner and others 2007; Wang-Fischer 2009). With the increasing average age of the population, these already staggering numbers are likely to grow. Indeed, the estimated 2011 cost of stroke was US$73.7 billion and it is projected to exceed US$1.52 trillion by 2050 (Roger and others 2011).
In the United States, as many as 87% of all strokes are ischemic, meaning that they are caused by an interruption in blood supply (NINDS 2011). The majority of ischemic strokes are due to blood clots that become lodged within an artery, and the resulting decrease in blood flow leads to damage in the region of the brain that is fed by the artery. Three major arteries supply blood to the cortex in mammals: middle cerebral artery (MCA), posterior cerebral artery (PCA), and anterior cerebral artery (ACA), with about two thirds of all strokes occurring in MCA (Roger and others 2011); stroke within this vessel's territory can result in extensive dysfunction or death (Caplan 2009; Dirnagl and others 1999; Durukan and Tatlisumak 2007). Given the strong predominance of ischemic stroke and the high likelihood of stroke in MCA, this review will focus on ischemic stroke in the MCA.
There is currently only one Food and Drug Administration-approved treatment for ischemic stroke: Tissue plasminogen activator (tPA; NINDS 1995), which acts by breaking down clots and re-introducing blood into an ischemic brain region (reperfusion). Unfortunately, tPA administration remains extremely limited—only 3% of stroke patients qualify—because current guidelines recommend administration within 3 hours of symptom onset (although some studies suggest this window may be expanded to 4.5 hours; Roger and others 2011). The existence of such critical time windows for treatment of stroke has led to the understanding among clinicians that “time is brain” for stroke victims (Saver 2006). Patients who qualify and receive tPA still often sustain a large degree of tissue damage (Kent and others 2005). In addition, tPA has a number of potential side effects, such as weakening of blood vessel walls, edema, disruption of the blood-brain barrier, conversion to hemorrhagic stroke, and reperfusion injury (Adibhatla and Hatcher 2010).
Although clearly less than ideal, tPA does confer a benefit to some patients, and importantly, it is the first demonstration that it is possible to improve the condition of a stroke victim, given rapid treatment (Wardlaw and others 2009). tPA has been shown to be more effective if additional treatments are administered in parallel or prior to its use (Turner and others 2011; Wahlgren and Ahmed 2004). As a result, there is an intense interest in discovering novel therapies for use in acute ischemic stroke that could provide an alternative to, or be used in conjunction with, tPA treatment.
In response to this growing challenge, an enormous scientific effort has been put forth to identify the underlying causes of stroke, and potential treatment options, including the development of many different pre-clinical animal models of stroke (Wahlgren and Ahmed 2004). Many researchers model ischemic stroke in rodents (Greenberg and Jin 2007; Murphy and Corbett 2009; Wahlgren and Ahmed 2004). Rats, in particular, are well suited for modeling human stroke because in both humans and rats, the MCA delivers a great majority of blood to the lateral surface of the cortex (Blumenfeld 2002), which supports motor, somatosensory, and auditory areas.
Rodent Models of Ischemic Stroke
Although the rat has become a popular model for stroke research, the most clinically relevant means of inducing ischemia within this model is still widely debated (Bederson and others 1986; Carmichael 2005; Chen and others 1986; Graham and others 2004; Hoyte and others 2004; Turner and others 2011). No one model perfectly replicates every aspect of human ischemia—each has its advantages and limitations. In general, an effective model of stroke results in movement deficits contralateral to stroke damage (Cirstea and Levin 2000; Nakayama and others 1994; Nudo and others 2001), loss or disruption of motor and somatosensory representation (Cramer and Riley 2008; Traversa and others 1997), loss or disruption of evoked electrophysiological activity (Chiganos and others 2006; Weber and others 2008), reductions in cerebral blood flow (Dirnagl and others 1989; Wintermark and others 2002), and infarct (Crafton and others 2003; Nudo and Eisner-Janowicz 2006). See Box 1 for a brief synopsis of the primary classifications of rodent models of cerebral ischemia that satisfy the majority (if not all) of these characteristics, along with the primary advantages and disadvantages of each technique (a description of method used in the experiments described in this review can be found under the header “direct surgical occlusion”).
Although much has been learned of the underlying pathophysiology based on these models (Lo and others 2003; Moskowitz and others 2010), rodent studies have yet to produce an effective treatment for acute ischemic stroke (Del Zoppo 1995; Greenberg and Jin 2007; Grotta 1995; Martinez-Vila and Sieira 2001; Wahlgren and Ahmed 2004). More than 1000 candidate compounds have been designated as neuroprotective agents based on positive results in animal models of stroke, and none have ever proven clinically useful in humans (Del Zoppo 1995; Del Zoppo and others 2009; Grotta 1995). Furthermore, more than 160 human clinical trials evaluating candidate pharmacological treatments have been conducted to date, and none have demonstrated efficacy compared to placebo (Ginsberg 2007; Wahlgren and Ahmed 2004). Positive outcomes in stroke models do not guarantee success in clinical trials.
There is a clear need for an alternative approach. Ideally speaking, a truly “protective” treatment for ischemic stroke would completely protect the brain from all impending damage. For the remainder of this review, we will therefore refer to compounds or interventions that demonstrate efficacy in animal models of acute ischemic stroke as “protective” if they result in a reduction of ischemic damage and “completely protective” when they prevent all impending ischemic damage and dysfunction.
The “holy grail” for treatment of an impending stroke should include the following criteria: 1) because “time is brain,” the treatment should be delivered immediately following the first signs of stroke; 2) the treatment should be completely protective; and 3) the treatment should be noninvasive. This review will present a treatment that fits all three criteria, as demonstrated in an adult rat model of ischemic stroke.
How Repeated Failures to Induce Ischemic Stroke Led to a Serendipitous and Surprising Finding Regarding Complete Protection from Stroke
As our lab research focus has been on adult plasticity, we initially intended to study how such plasticity helps rats overcome the structural and functional deficits resulting from ischemic stroke. To study this plasticity, a model of adult ischemic stroke was created using a permanent MCA occlusion (pMCAO). Permanent occlusion was achieved by tying two ligatures around what is termed the M1 branch of MCA—at this point, an occlusion blocks all blood supply to MCA cortical branches without affecting any subcortical blood flow through MCA. The artery is then severed between the two ligatures, cutting off MCA blood supply to the cortex.
Regarding the degree of brain injury sustained following pMCAO: Human strokes are most typically small in size. In large clinical studies, strokes range from 28 to 80 cm3. Using volumetric measurements, this translates to 4.5% to 14% of the ipsi-ishemic hemisphere (Carmichael 2005). In contrast, rat pMCAO infarct size ranges from approximately 9 to 35 mm3, with most infarcts between 3% and 12% of the ipsi-ishemic hemisphere. As a result, pMCAO in rats produces infarcts that closely resemble human ischemic stroke infarct volumes.
To assess cortical function before and after pMCAO, we used the functional imaging technique known as intrinsic signal optical imaging. This functional imaging technique has been used extensively to image the functional organization of the cortex and can provide high spatial resolution maps of evoked cortical activity (Frostig and others 1990; Grinvald and others 1986; Ts'o and others 1990; for a recent review, see Frostig and Chen-Bee, 2009). Our lab also has a long history and detailed familiarity with functional imaging of the “barrel cortex” subdivision of the adult rat, an area that contains exquisite anatomical and functional maps of the large facial whiskers (vibrissae). For these reasons, and because MCA supplies this region with blood, the barrel cortex is an ideal venue for the study of ischemia. To evoke activity within the barrel cortex, computer-controlled mechanical rostrocaudal 5-Hz patterned stimulation of a single whisker using an intermittent stimulation protocol (stimulation was given randomly every 15 ± 6 seconds) was used. Functional imaging of a typical evoked response following single whisker stimulation is triphasic, as shown in Figure 1A (top row). It consists of an initial darkening known as the “initial dip” phase that is rapid (beginning <0.5 seconds after stimulus onset) and transient (lasting 1.5–2 seconds) followed by a slower-onset brightening “overshoot” phase that is stronger in magnitude (>2×), longer in duration and typically larger in areal extent. The overshoot phase of the intrinsic signal is followed by a second darkening known as the “undershoot” phase, which is also a slower-onset signal that is stronger in magnitude (~2×) and longer in duration than the initial dip (Chen-Bee and others 2007). The initial dip represents the area of evoked neuronal activity, whereas the overshoot represents blood flow to the activated area (the overshoot is also the phase used by functional MRI). The undershoot is still poorly understood.
Figure 1.

Experimental design and representative cases. (A) Representative cases of a typical alternating triphasic evoked functional response to whisker stimulation (top, black), including the initial dip (dark patch), overshoot (white patch), and undershoot (dark patch). Next, the expected imaging result following permanent middle cerebral artery occlusion (pMCAO; middle, red), and the observed imaging result collected following pMCAO (bottom, blue). Each box represents 500 ms. Step function located to left of imaging examples indicates whisker stimulation. Scale bar, middle row right side, indicates 4 mm. Linear grayscale bar, located bottom right, indicates intrinsic signal strength ×10−4 fractional change (FC). (B) Top, schematic of the within animal experimental design. Each box denotes manipulations performed before and after pMCAO (denoted by red vertical line). Below, representative images from +0h and +3h subjects' initial dip and overshoot phase of the evoked functional response to whisker stimulation, local field potential (LFP), multi-unit activity (MUA), blood flow imaging (step function indicates stimulus delivery), 2,3,5-triphenyltetrazolium chloride (TTC; arrow indicates region of ischemia) histology collected before and 24 hours after pMCAO. Within the TTC images, note that the area devoid of staining (white) within the +3h subject's left cortex indicates ischemic infarct due to an occlusion of the left MCA. (C) The evoked functional response to whisker stimulation, quantified for immediate (+0h) and delayed (+3h) groups. In each plot, +0h (blue) and +3h (red) group data at pre-manipulation baseline is paired with +24-hour data. Means and standard errors are provided for the area (left column) and amplitude (right column) of the initial dip and overshoot of the evoked functional response to whisker stimulation before and 24 hours after pMCAO. A value of zero indicates no response to whisker stimulation. Asterisks indicate significant differences between baseline and 24-hour values for the +0h group (***P = .0086). Data in panels A, B, and C are from Lay and others (2010); used with permission of authors and publisher.
The experimental model used was a “within animal control” design in which assessment of animals was done as a comparison of post-treatment responses to each individual's pre-manipulation baseline evoked responses. Briefly, a pre-pMCAO baseline was established by imaging cortical activation in response to the above-described intermittent whisker stimulation; pMCAO was then induced, followed by delivery of the same whisker stimulation with concurrent assessment over the following 2 hours. Changes induced by pMCAO were then evaluated at different times after pMCAO (Fig. 1B, schematic, top).
At 24 hours post-pMCAO, a lack of functional evoked response to whisker stimulation was expected (Fig. 1A, middle row). Surprisingly, however, a typical triphasic cortical response to whisker stimulation was observed (Fig. 1A, bottom row). This totally unexpected finding was replicated in a large group of rats, clearly demonstrating our failure to induce ischemic stroke by pMCAO. We reasoned that the stimulation of a whisker immediately (+0 hours or +0h group) following pMCAO somehow changes the cortex, a change that leads to this apparent protection. To test this hypothesis, the experimental design was modified to delay whisker stimulation for 1 hour (+1h group) following pMCAO. A normal triphasic response to whisker stimulation was imaged and replicated in this group as well, disproving this hypothesis. This finding represented a second failure to induce stroke despite the 1-hour delay between pMCAO and whisker stimulation. Incredibly, even following a 2-hour delay (+2h group), 70% of the rats showed normal triphasic responses whereas the rest exhibited a present but compromised triphasic response. Only by extending the delay between pMCAO and whisker stimulation to 3 hours (+3h group), finally result in the expected lack of evoked functional response to whisker stimulation—evidence of a stroke (Lay and others 2010). When these findings were quantified in terms of the amplitude and areal extent of each of the phases of the response, another surprising result became apparent. Not only did we observe all phases of a typical functional response to whisker stimulation 24 hours following pMCAO in protected groups, in most cases there was a trend, and in some cases a significant increase in amplitude or area of each phase (for an example, see Fig. 1C, upper right panel). Paradoxically, pMCAO seemed to lead to what appeared to be a better cortical response post-pMCAO and protective treatment. Notably these changes were limited to the ipsi-ischemic cortex as no changes were observed in the contra-ischemic cortex 24 hours following pMCAO (Lay and others 2010). Therefore, by repeatedly failing to induce ischemic stroke, we stumbled upon a way to completely protect the cortex from impending damage using mild sensory stimulation. The findings described so far were, however, based only on functional imaging. It was clear to us that these extraordinary findings needed to be further substantiated by additional independent techniques.
Neuronal Recording: Electrophysiological Assessment of the Endangered Cortex
Although functional imaging is a proxy for assessing neuronal activity, we wanted to confirm healthy neuronal firing in our protected animals. Evoked suprathreshold (multi-unit firing) and evoked subthreshold (local field potential) activity were both assessed and found to be at pre-pMCAO baseline level at 24 hours post-occlusion in animals that received treatment immediately (+0h group) after pMCAO. Therefore, this technique further confirmed our assertion that our treated animals were completely protected (Lay and others 2010; Fig. 1B).
Histology: Assessment of Infarct Volumes
Volume of infarct (cell death resulting from ischemia) in the cortex was also assessed using 2,3,5-triphenyltetrazolium chloride (TTC). TTC is reduced by an enzyme in active mitochondria and one of the byproducts of this reduction is a bright red compound that stains healthy tissue. It is therefore commonly used to assess regions of infarct (Fig. 1B, post-pMCAO assessment) in animal models of stroke. In line with our functional imaging results, all animals treated with stimulation within an hour of pMCAO were completely protected from infarct. In addition, the same 30% of animals treated at 2 hours post-pMCAO that had functional deficits also sustained a small degree of infarct, whereas the remaining 70% of these +2h animals were completely protected (Lay and others, 2011b).
Behavioral Assessment: Determination of Protection from Behavioral Deficits
Perhaps the most important concern for a stroke patient is physical capability and a return to pre-stroke lifestyle. Other parameters have little meaning to the patient unless they affect his or her ability to function as previously following stroke. To determine whether animals suffered any behavioral deficits, they were assessed according to several tasks. First, animals were assessed and given a Bederson neurological score, which measures a variety of neurological deficits associated with stroke. Animals were then also assessed according to whisker-based and forepaw-based exploration behavior, where impaired animals will favor exploration with the unaffected whisker pad or forepaw. Once again, animals that received immediate stimulation (+0h group) were protected; they had no sign of behavioral deficit as assessed 1 week post-ischemic onset. Further, animal groups that were not treated in time (+3h group) and suffered damage along previously described parameters were also significantly impaired according to all behavioral tasks at 1 week post-stroke (Lay and others 2010). These findings again supported our hypothesis that early activation of the ischemic cortex can completely protect from impending stroke damage.
Assessment of Other Physiological Parameters
To confirm that changes in other physiological parameters were not underlying the observed protection, a subset of subjects' heart rate, breathing rate, blood pressure, and blood oxygenation were assessed. None of the above-listed parameters were altered by pMCAO or treatment and did not differ between groups. We therefore concluded that none of these physiological parameters were responsible for the observed protection (Lay and others 2011b).
Blood Flow Assessment
Given that the cortex was functioning normally in protected animals despite the complete occlusion of MCA, the assumption was that blood flow must be returning to the endangered cortex as a result of stimulation treatment. Indeed, when blood flow was assessed, using laser speckle imaging (LSI), in the cortical branches of MCA 24 hours post-pMCAO, blood was flowing at pre-pMCAO baseline levels (Fig. 1B, bottom). This was perplexing initially because, as mentioned, MCA was severed prior to its cortical branches during pMCAO, but further investigation using LSI revealed a clear explanation, which will be discussed shortly.
Complete Protection
No sign of damage or functional or behavioral deficits was found in any of the animals receiving stimulation within the first hour after ischemic onset according to any of the above listed series of techniques (and in most cases at 2 hours after ischemic onset). In addition, it was confirmed that protection could not be associated with any changes in physiological parameters such as blood pressure, oxygenation, heart rate, or breathing. The alignment of consistent results using all of the above-described complimentary techniques satisfied our criteria for describing our protected animals as completely protected.
Parameters of Completely Protective Mild Sensory Stimulation
The term mild is used to describe the stimulation delivered because 1) only a single whisker was stimulated, 2) the mechanical stimulation amplitude (~9 degrees) is minor compared with amplitudes of natural whisker movement in rodents, and 3) whisker stimulation was intermittent and delivered for only 3.5% of the 120-minute post-pMCAO treatment period; the whisker was actually stimulated for a total of only 4.2 minutes!
Given the overlap of the area of cortex endangered by ischemia in our model with the area activated by whisker stimulation, it was hypothesized that cortical activation of the ischemic cortex was the critical component of the observed protection and that generating this cortical activity in any fashion would result in protection. To investigate this hypothesis further, a series of experiments was conducted wherein animals still underwent pMCAO and whisker stimulation treatment, but the pattern and duration of the stimulation were altered.
Random whisker stimulation (the same number of whisker deflections as delivered in the previous experiments, but condensed either into a 10-minute delivery period or dispersed randomly as single deflections across 120 minutes) resulted in protection equivalent to previous experiments' patterned stimulation when administered immediately following pMCAO and assessed 24 hours later (Davis and others 2011a). Additionally, increasing stimulation treatment from single whisker to full whisker array also resulted in complete protection.
Collectively, these findings supported the hypothesis that cortical activity in the ischemic brain region, occurring within the critical time window post-pMCAO, irrespective of the exact parameters of peripheral stimulation that initiated it, is the important variable in protection from ischemic stroke.
Underlying Mechanism of Reperfusion of the Ischemic Area
Given the severity of the pMCAO (completely severed MCA), how could blood flow return to MCA cortical branches? It is known that the distal ends of MCA branches can sometimes be connected to other arteries, forming collateral vessels or anastomoses (this occurs in both rodents and humans), and we posited that these collateral vessels could be a source of reperfusion into MCA cortical branches (Coyle 1987; Schaffer and others 2006; Fig. 2A). For more than two decades, it has been widely recognized that collateral vessels could play a critical role in the reperfusion of ischemic brain tissue and that blood flow reversal within arteries is possible (Burnett and others 2006; Cook and others 2012; Coyle 1986, 1987; Liebeskind 2003; Schaffer and others 2006).
Figure 2.

Major cerebral arteries of the human (left) and rat (right), and their potential collateral connection points. (A) Diagram representing the three major cerebral arteries of the human (left) and rat (right): anterior cerebral artery (ACA), middle cerebral artery (MCA), and posterior cerebral artery (PCA). Connection points between these three vessels, known as collateral vessels (examples marked in yellow) are potential locations for the redirection of blood flow from one major artery to another. (B) +0h animals (which received whisker stimulation immediately post-pMCAO) had intact structure and evoked functional response to whisker stimulation at 24 hours post-occlusion (representative functional imaging data and stained coronal brain slice showing normal function and lack of infarct shown at upper right). In addition, baseline level blood flow was observed within the branches of the occluded MCA 24 hours post–permanent MCA occlusion (post-pMCAO; after having dropped significantly in response to occlusion; see graph for quantification of blood flow drop following pMCAO and return following stimulation treatment). The question, based on our knowledge of collateral vessels and the return of blood flow and function in the protected +0h animals, was then, is collateral flow supporting this return and the protection observed? (C) +0h distal occlusion experiments, wherein stimulation was still delivered immediately post-occlusion, but the distal ends of MCA were occluded in addition to the standard base occlusion pMCAO. These animals no longer maintained functional cortical integrity and sustained infarct (arrow). These experiments demonstrate that patent distal branches of MCA are necessary for protective plasticity, suggesting collateral vessel blood flow as the mechanism for the protection observed in +0h animals. Data in panels B and C are from Lay and others (2010); used with permission of authors and publisher.
To determine if blood could be flowing through these collaterals to re-establish blood flow in MCA cortical branches, additional +0h rats were assessed. These animals underwent not only our standard pMCAO but also permanent occlusions at the distal ends of all main MCA cortical branches in a procedure similar to that described by Wei and others (1998). We reasoned that if blood reperfusion were established via collateral flow, this procedure would prevent such re-establishment and therefore prevent the protection previously observed in +0h animals. Indeed, in stark contrast to the protection of functionality and tissue health observed in +0h animals that did not receive distal occlusions in addition to pMCAO (Lay and others 2010; Fig. 2B); the evoked functional response to whisker stimulation disappeared and substantial infarct was observed in all +0h distal subjects despite the fact that whisker stimulation was delivered immediately following occlusion (Fig. 2C).
Furthermore, to control for possible damage caused by additional surgical procedures inherent in the +0h distal animal experimental design, function was assessed in +0h sham distal control animals. This group underwent the same surgical protocol (including permanent occlusion of MCA's base) as +0h distal animals except that the distal occlusion suture knots were never tightened to occlude the distal ends of MCA branches. Despite having the same additional surgical intrusion as +0h distal animals, these controls maintained normal evoked functional response to whisker stimulation and tissue health according to functional imaging and TTC staining. Taken together, these data confirmed our hypothesis that the distal MCA branches are necessary for stimulation-induced protection of the cortex from ischemic injury (Lay and others 2010).
Directionality of blood flow cannot be determined with LSI, but because pMCAO included a complete transection of the base of MCA (between the two tightened suture knots), blood flow observed in the cortical branches of MCA (distal to the occlusion) at 24 hours post-occlusion, must be flowing in the reverse direction, coming from another arterial source via collateral vessels. In conjunction with the distal experiments discussed above, these findings demonstrate that a mechanism underlying the observed protection from ischemic injury is a redirection of blood from an alternate source, via collaterals to MCA, allowing re-establishment of blood flow to the ischemic cortex (Fig. 3).
Figure 3.
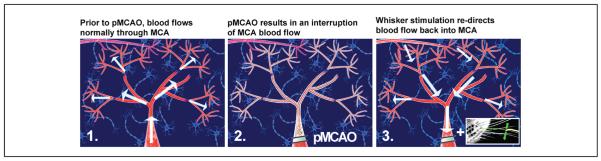
Mild sensory stimulation results in a redirection of blood flow into the ischemic region. Illustration of middle cerebral artery (MCA) blood flow before manipulation, during pMCAO, and following whisker stimulation treatment. MCA is pictured as the large, red, central vessel in each panel. The distal portions of MCA are indicated by the smaller branches extending from MCA. The direction of blood flow is indicated by the arrows. Neuron icons represent the underlying cortical tissue. (1) Prior to manipulation, blood flows up through MCA, into the anterior and posterior branches of the vessel, and into the most distal segments, supplying a large area of the cortex with blood. (2) Following permanent MCA occlusion (pMCAO), blood flow through MCA is interrupted, resulting in ischemia of the surrounding cortical tissue. (3) In response to whisker stimulation treatment, collateral vessels (pictured top left in each panel) form a new supply route, allowing blood to flow in reverse back into the ischemic territory.
Sensory Stimulation Treatment as a Double-Edged Sword
As mentioned, when stimulation was delivered at 3 hours post-ischemic onset, it no longer conferred protection, but when these animals were compared to untreated controls that never received stimulation following pMCAO, another surprising, but important result was revealed. Animals that received the late stimulation at 3 hours post-pMCAO (+3h group) actually sustained significantly more damage (larger infarct) than untreated pMCAO animals (63.4 ± 3.9 mm3 vs. 28.4 ± 2.4 mm3). Therefore stimulation treatment at 3 hours post-occlusion not only failed to protect but actually exacerbated stroke damage.
Furthermore, animals that received multiple whisker stimulation at 3 hours post-ischemic onset were even worse off than single whisker counterparts. Not only did they sustain infarct and lose evoked functional response according to functional imaging, but they also sustained an even larger volume of cortical infarct (~50% larger) than their single whisker counterparts (91.85 ± 9.8 mm3 vs. 63.4 ± 3.9mm3).
Collectively, this evidence thus far suggests that cortical activation is not only a critical factor in the induced complete protection from stroke, but is also critical in the exacerbation of damage when treatment is delivered outside of the protective window (Davis and others 2011a; Fig. 4). This fits well with the previously mentioned human literature concerning recommended time windows for administration of tPA. Not only is the time window (2–3 hours for both tPA and stimulation in our experiments) roughly similar, the concern of an exacerbation of damage with late treatment is also an issue for both.
Figure 4.
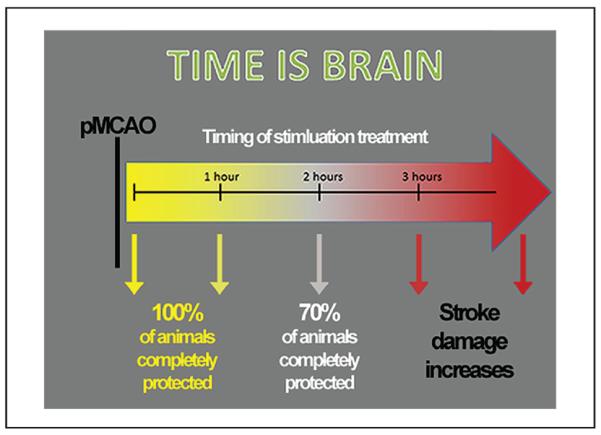
Schematic of the effect of whisker stimulation on outcome depending on timing of administration. Complete protection is observed in all subjects when stimulation is delivered within the first hour following permanent middle cerebral artery occlusion (pMCAO), but only in about 70% of subjects when delivered at 2 hours post-pMCAO. By 3 hours post-pMCAO, whisker stimulation not only ceases to be beneficial, but actually exacerbates impending stroke damage.
Return of Function within 120 Minutes of Post-MCA Treatment
After confirming return of function at 24 hours post-pMCAO, we sought to determine the acute effect of this completely protective sensory stimulation. A gradual return of cortical function was observed over the treatment period itself for all evoked responses measured. Data from functional imaging (Fig. 5A and B), evoked subthreshold (Fig. 6A, top), and suprathreshold recordings (Fig. 6A, bottom) exhibited a “v-shaped” recovery profile.
Figure 5.

Evoked functional response returns gradually on stimulation onset in all protected animals throughout the treatment period. (A) Following permanent middle cerebral artery occlusion (pMCAO), a return of cortical function is evident in +0h, +1h, and +2h groups during protective stimulation treatment, and a loss of all three phases of the evoked function response in animals that receive treatment 3 hours post-occlusion (+3h animals). Representative data from functional imaging of the initial dip for each group (+0h, +1h, +2h, and +3h) were arranged according to minutes of treatment delivered (treatment time is included as white font in the top left corner of each image). Groups +0h, +1h, and +2h all regained evoked functional response comparable to baseline after 90 minutes of whisker stimulation, whereas +3h animals never demonstrated any post-pMCAO cortical activity. (B) In each plot, group baseline is plotted with 120 minutes of post-occlusion stimulation period data. Means and standard errors are provided for the area (left) and amplitude (right) of the ipsi-ischemic C2 initial dip (top) and overshoot (bottom) phases of evoked functional response to whisker stimulation before and after pMCAO. +3h animals had no response to stimulation at any time point post-pMCAO. For all other groups (sham-pMCAO, +0h, +1h, and +2h), asterisks within the gray band above each plot indicate a significant difference from baseline, *P < .05, **P < .01, and ***P < .001. Data in panels A and B are from Lay and others (2011b); used with permission of authors and publisher.
Figure 6.

Evoked neuronal activity returns gradually on stimulation onset following permanent middle cerebral artery occlusion (pMCAO). (A) Representative +0h animals' local field potential (LFP), and multi-unit activity (MUA) responses at baseline and during the stimulus period following pMCAO. Stepping function indicates stimulus delivery. Right, LFP and MUA mean and standard error is plotted prior to and post-occlusion. Asterisks indicated a significant difference from baseline, **P < .01 and ***P < .001. (B) Blood flow imaging (via laser speckle imaging; LSI) experiments demonstrate that post-pMCAO blood flow return in MCA is induced by whisker stimulation treatment. +0h group LSI color scaled images taken at baseline and following pMCAO during treatment at roughly 30-minute intervals. Scale bar indicates 1 mm. The red vessel diagonally traversing the image in each case is a cortical branch of MCA. Right, means and standard errors for +0h animals at baseline, following pMCAO, and during whisker stimulation. Dagger indicates a significant increase in +0h flow compared with value collected immediately following pMCAO, P < .01. Data in panels A and B are from Lay and others (2011b); used with permission of authors and publisher.
According to functional imaging, the spatial extent and amplitude of evoked functional response to whisker stimulation returned gradually to pre-pMCAO baseline level by 90 minutes of stimulation treatment in +0h animals. Surprisingly, the same time course of recovery was observed in all protected animals irrespective of whether stimulation treatment was initiated—0, 1, or 2 hours post-pMCAO (+0h, +1h, and +2h groups, respectively; Fig. 5B). In addition, multiple whisker stimulation delivered immediately post-pMCAO (+0h group) resulted in an accelerated recovery of evoked function: cortical function was restored 30 minutes faster compared with single whisker treatment. This result was also consistent with our hypothesis that cortical activation of the ischemic region is a key aspect in protecting the region from damage as in this case, increased activation led to accelerated recovery. These results suggest that the cortex remains capable of functional recovery for an extended period of time (2 hours) under ischemic conditions. The current dogma states that irreversible damage occurs within minutes of MCA occlusion (Hossmann 1994; Iadecola and Nedergaard 2007). The fact that stimulation treatment could result in complete recovery even at 2 hours after pMCAO considerably widens the previously envisioned protective “window of opportunity.”
Evoked suprathreshold (MUA) activity was detectable within 17.5 ± 4.5 minutes after the initiation of stimulation treatment and reached baseline levels by 90 minutes (Fig. 6A, bottom right). Evoked subthreshold (LFP) activity also gradually increased throughout treatment, though it remained below baseline levels during treatment (Fig. 6A, top right). Interestingly, spontaneous suprathreshold activity remained constant throughout the experiment, even during periods when evoked suprathreshold activity was completely absent, demonstrating that neurons in the endangered cortex retained their ability to fire action potentials despite the fact that ischemia had rendered the same region of neurons unable to fire evoked responses (Lay and others 2011b).
At what point do collateral vessels begin to supply blood to the ischemic cortex? Given the gradual return of evoked functional and neuronal responses described above, it was expected that blood flow to MCA cortical branches would also gradually increase throughout the treatment period. Reperfusion induced by whisker stimulation treatment was characterized in the ischemic cortex using LSI blood flow before pMCAO and at four time points (spaced approximately 30 minutes apart) during the 120-minute stimulation treatment period in +0h animals. Blood flow in MCA in this group was then compared with MCA flow in a “no stimulus” control group, which underwent pMCAO, but never received whisker stimulation treatment. In both groups, a substantial decrease in blood flow within MCA cortical branches following pMCAO was observed. LSI of MCA demonstrated that perfusion dropped substantially in both groups following occlusion (to 26% ± 4% of pre-pMCAO baseline values). Similar to evoked neuronal recordings, +0h animals' blood flow levels increased gradually from stimulation onset (Fig. 6B, left) and were significantly different from values collected immediately following pMCAO by the conclusion of stimulation treatment (Fig. 6B, right). This reperfusion did not occur in “no stimulus” control animals that underwent pMCAO without stimulation treatment (and sustained infarct)—their flow levels remained at post-pMCAO drop levels (Lay and others 2011b).
Can Mild Sensory Stimulation Completely Protect Impending Ischemic Stroke in Aged Rats?
An important caveat to the findings described above is that all these experiments were conducted in young adult rats (3–4 months old). Old age is associated with an enhanced susceptibility to stroke, poor recovery from infarct (DiNapoli and others 2008; Popa-Wagner and others 2011), and the decreased effectiveness of protective therapy in both humans and rats (Dirnagl 2010; Popa-Wagner and others 2011; Popa-Wagner and others 2007; Wang-Fischer 2009). Could the same mild whisker stimulation treatment protect the aged rat, despite the increased vulnerability to ischemic damage? To answer this question, aged rats (21–24 months old; equivalent to roughly 60–65 years of age in humans) underwent the same pMCAO and whisker stimulation treatment as their young counterparts from the previous studies (Davis and others 2011a; Lay and others 2010). Aged animals were assessed for baseline sensorimotor capacity using the same three behavioral tests used to assess their young counterparts: the Bederson neurological scale, forepaw-guided exploration, and whisker-guided exploration. One week later, all rats underwent pMCAO and were then placed randomly into one of two groups: treated rats that received stimulation treatment immediately following pMCAO and untreated control rats that never received stimulation treatment. One week following pMCAO, both groups underwent behavioral re-assessment and postmortem histological analysis. Aged treated rats were unimpaired on all three sensorimotor tasks and did not sustain infarct. Aged untreated controls, however, demonstrated sensorimotor impairment and sustained infarct (Lay and others 2012).
An additional group of treated aged rats and a group of treated young adult rats underwent functional imaging prior to, during treatment, and 24 hours following pMCAO to assess degree and rate of functional recovery in protected aged animals versus protected young adults (Fig. 7A). Surprisingly, aged rats recovered cortical function at the same rate as young adults (Fig. 7B), demonstrating that the protective effect of single whisker stimulation treatment is not limited to young brains (Lay and others 2012).
Figure 7.

Return of cortical function in aged rats is equivalent to young adult rats. Return of cortical function in aged rats is equivalent to young adult rats. (A) Representative data from functional imaging of the initial dip in aged and young adult animals that underwent permanent middle cerebral artery occlusion (pMCAO) and received whisker stimulation treatment immediately post-occlusion. Functional imaging was conducted prior, during, and 24 hours following pMCAO and whisker stimulation treatment. Both young adult and aged rats regained evoked functional response comparable to baseline after 90 minutes of whisker stimulation treatment. Linear grayscale bar indicates intrinsic signal amplitude (fractional change FC ×10−4). Scale bar indicates 4 mm. (B, C) Group baseline is plotted with 120 minutes of post-occlusion stimulus period data and 24-hour re-assessment data for each graph. Means and standard errors are provided for the area (left) and amplitude (right) of the ipsiischemic C2 initial dip (B) and overshoot (C) phases of evoked functional response to whisker stimulation before and following pMCAO. Data in panels A, B, and C are from Lay and others (2012); used with permission of authors and publisher.
Current Directions
As stated, the central hypothesis driving this line of research has been that cortical activation plays a key role in the observed protection from stroke. If correct, this hypothesis suggests that it is possible to protect the adult rat cortex not only through mechanical whisker stimulus but also through other means of inducing cortical activity in the ischemic area.
Currently, this hypothesis is being further tested by delivering auditory stimulation (5 Hz white noise delivered in the same pattern and over the same period as in the whisker stimulation model) immediately post-pMCAO in a new experimental group. Animals are assessed using functional imaging and histological assessment with TTC. Thus far, auditory stimulation treatment has resulted in complete or near complete cortical protection compared to untreated pMCAO controls. The majority of treated animals had been completely protected (normal evoked functional response to auditory stimulation and no sign of infarct). A minority did suffer minor disruptions of functional responses and very small volumes of infarct (5.4 ± 2.3 mm3), but damage was minimal compared with +3h groups or untreated controls. These results demonstrate that protective cortical activity is not limited to activation in somatosensory cortex. Perhaps even more interestingly, the animals that were not protected were more likely to sustain infarct in regions remote from auditory cortex (in the anterior extent of the ischemic region), suggesting that protection was best conferred to areas within or close to the area activated by auditory stimulation (Davis and others 2011b).
Thus far, all experiments had been run in anesthetized animals. Given that the typical human stroke patient is not anesthetized, determining whether similar protection could be conferred in an awake behaving animal seemed a critical next step. Currently, we are testing the hypothesis that sensory induced cortical activity, independent of anesthesia, protects the brain from stroke. To test this hypothesis, natural whisker use is promoted during what would be the mechanical whisker stimulation treatment period in anesthetized counterparts by allowing awake rats to freely explore an enriched environment post-pMCAO. Rats were revived from anesthesia either immediately (+0h group) or 3 hours after pMCAO (+3h group)—at which point both groups were allowed to freely explore an enriched environment. Similar to mechanical stimulation in anesthetized rats, rats that explore (which involves frequent whisker use) immediately (+0h group) after ischemic onset maintained normal cortical function and were histologically equivalent to sham-pMCAO controls (rats that underwent surgery without occlusion of MCA). Rats that were revived 3 hours post-pMCAO (+3h group) exhibited eliminated cortical activity and sustained ischemic infarct (Lay and others 2011a).
Finally, because of the presence of multimodal stimulation (e.g., auditory, olfactory and motor stimuli) during active exploration, another set of experiments was undertaken to determine whether or not the protection conferred to awake behaving rats was specifically dependent on whisker use or might be attributed to the other sensorimotor and experiential stimuli inherent in the exploration of the enriched environment. To address this question, we clipped off the entire whisker array (all large vibrissae) of a group of rats immediately following pMCAO. These rats were then revived and allowed to explore the same novel environment. Thus, these animals underwent the same experimental protocol as the above described un-anesthetized +0h group. Clipped +0h rats that actively explored immediately post-pMCAO, albeit without the use of whiskers, were also completely protected from infarct. Taken together, stimulation-induced protection from ischemic stroke was shown to be effective in actively exploring, whisker-less rats, and thus not specific to whisker stimulation alone (Lay and others 2011a). This further supported our hypothesis that cortical activation in the ischemic area was the important factor in the observed protection, rather than a specific sensory modality or means of activation.
Summary and Conclusions
This review describes the initial discovery and development of a new drug-free, noninvasive method for protecting the cortex against ischemic stroke that could be implemented without the use of equipment. A mild form of sensory stimulation, that when administered within 1 hour, and in most cases, within 2 hours of ischemic onset results in complete protection of structure and function. Complete protection is achieved irrespective of the age of the animal, despite the expected increased vulnerability of aged subject to ischemic damage. Complete protection is also achieved irrespective of the pattern of stimulation or sensory modality and depends on the activation of the endangered ischemic cortex within the protective time window. In fact, pattern and rate of recovery of function are identical and equally complete irrespective of when the stimulation treatment is delivered as long as it is delivered within 2 hours of ischemic onset. It was also demonstrated that the rate of recovery could be augmented by increasing the amount of sensory stimulation from single whisker deflection to simultaneous deflection of the entire whisker array. If given 3 hours after ischemic onset, however, single whisker stimulation exacerbates stroke damage and multiple whisker stimulation worsens this exacerbation. The underlying mechanism for the observed protection is cortical activity–induced redirection of blood flow within the permanently occluded MCA, via collateral connections with other cerebral arteries, which leads to pre-pMCAO level of reperfusion of the ischemic area supporting the complete protection. Both anesthetized (mechanically stimulated) rats and awake, behaving rats that were compelled to induce cortical activity naturally by exploring a novel environment, were completely protected, making this sensory based treatment potentially even more relevant to humans.
Translation to Humans?
One critical question when addressing a human disease with animal studies is, how do the results relate to the current clinical characteristics and understanding of the human disease? In human stroke, there is a widely recognized classification of ischemic tissue that describes regions as either being a “core”—a region for which impending damage is irreversible after a given period of time at a given decrease in blood flow. The development of this region is thought to depend on the severity and duration of ischemia (Jones and others 1981; Kaufmann and others 1999). This “core” region of severely ischemic tissue has been shown to develop over the first 1 to 6 hours of ischemia and is correlated with future damaged tissue (Kaufmann and others 1999). The other is a surrounding area subjected to less severe ischemia and termed penumbra—this region is considered functionally disrupted, but salvageable with early treatment (Baron 2001; Zhang and others 2012). The “core” region is thought to expand with time, taking over the penumbra and making protection of this region no longer possible. Given that the defining characteristic of these two regions is whether or not damage can be reversed or prevented—perhaps mild sensory stimulation treatment renders the entire ischemic brain region “penumbra” and therefore amenable to sensory stimulation induced protection. Because of the disappointing history of translation from animal models to humans in stroke research, one has to be very careful when discussing translation. In the case of sensory stimulation, however, there is a reduced chance of some important issues that have plagued previous studies of neuroprotection in animal models. Unlike many of the agents that have proven unsuccessful in human studies, matters of dosage and dangerous side effects are not an issue for stimulation. Additionally, stimulation treatment was shown to be equally effective in aged animals. Even if sensory stimulation treatment were not completely protective when translated to human stroke, however, perhaps it could provide a means of augmenting the protective ability of other agents that might not have otherwise been effective.
If sensory-based stimulation is indeed relevant to humans, the following potential limitations should be kept in mind: 1) 12% of strokes are hemorrhagic (leakage) and it is not yet clear whether and how sensory based treatment affects this type of stroke, 2) some ischemic strokes occur in white matter or subcortical parts of the brain and it is not yet clear whether and how sensory based treatment affects this type of ischemic stroke, and 3) If given too late, this treatment exacerbates the damage.
However, if applicable to humans this treatment could be a very important breakthrough in protection from stroke for two main reasons: 1) This is a drug-free, equipment-free, and side effects-free treatment that could save lives of stroke victims and 2) because “time is brain,” it may be possible, for the first time, to develop a stroke treatment strategy that could be easily initiated anywhere by anyone, including informed family, friends, or first responder when the first signs of stroke appear, long before the ambulance arrives.
Box 1: Modeling Ischemic Stroke.
Endothelin-1
Endothelin-1 is a powerful vasoconstrictor that is directly injected into brain tissue, diffuses across a short distance, and causes long-lasting and powerful constrictions of the local blood vessel network. The primary advantage to this technique is that researchers dictate the location of ischemia with a high degree of certainty. The primary drawback is the limited control of intensity and duration of ischemia.
Photothrombosis
After intravenous injection of a photosensitive dye (such as `rose-bengal'), the brain is irradiated through the intact skull using laser illumination. Illumination of blood vessels results in chemically induced coagulation of red blood cells, leading to blockage of the vessel, and ischemia within the surrounding tissue. While photothrombosis does not require removal of the skull or invasive surgical procedures and allows for a well-defined ischemic region, this technique does not model the basic phenomena underlying the onset and development of ischemia (i.e. the sudden blockage of an artery due to a clot).
Direct surgical occlusion
In this model, MCA is surgically dissected and occluded, most typically via permanent cauterization or surgical ligature. Temporary occlusion of MCA can also be performed using a vascular clip. Surgical occlusion can be performed on the proximal or distal portion of MCA (Distal occlusion results in ischemic damage restricted to the cerebral cortex), and can be combined with temporal or permanent common carotid artery occlusion. The primary advantages of this model are: the exact site of occlusion is well defined, infarct size is consistent, and the high rate of survival allows for long-term study. The primary disadvantage is that the surgical procedure is relatively invasive as a craniotomy is required in order to access MCA.
Intraluminal monofilament
A surgical filament is introduced into the external carotid artery and forwarded until the tip occludes the base of MCA. This model allows for either transient or permanent ischemia, does not require craniotomy, and results in behavior and pathology similar to profoundly disabled stroke patients. The primary drawbacks of this method are variable degree of MCA occlusion, subarachnoid hemorrhage, hyperthermia, and invasive surgical procedures including: temporary hemispheric ischemia while the carotid artery is blocked during monofilament insertion.
In-situ thromboembolic
In situ thromboembolic ischemia is achieved by injecting a blood clot (autologous blood taken directly from the experimental subject) into MCA via surgical tube in a method similar to the intraluminal filament technique. Although this technique requires dissection of the carotid artery, the in situ thromboembolic model perhaps most closely mimics the onset of ischemic stroke in humans. A disadvantage of this model is that the blood clot occluding MCA is often spontaneously lysed or dislodged via blood flow. The volume of brain infarct, therefore, is highly variable.
Acknowledgments
We thank Cynthia Bee for her contribution and Aneeka Hancock for her comments on the article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article:
This work was supported by the American Heart Association Predoctoral Fellowship 788808-41910, the NIH-NINDS NS-066001 and NS-055832, and The Center for Hearing Research NIH Training Grant 1T32DC010775-01.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Adibhatla RM, Hatcher JF. Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2010;12(1):125–69. doi: 10.1089/ars.2009.2668. [DOI] [PubMed] [Google Scholar]
- Baron JC. Mapping the ischaemic penumbra with PET: a new approach. Brain. 2001;124(Pt 1):2–4. doi: 10.1093/brain/124.1.2. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17(3):472–6. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H. Neuroanatomy through clinical cases. Sinauer Associates; Sunderland, MA: 2002. [Google Scholar]
- Burnett MG, Shimazu T, Szabados T, Muramatsu H, Detre JA, Greenberg JH. Electrical forepaw stimulation during reversible forebrain ischemia decreases infarct volume. Stroke. 2006;37(5):1327–31. doi: 10.1161/01.STR.0000217305.82123.d8. [DOI] [PubMed] [Google Scholar]
- Caplan LR. Caplan's stroke: a clinical approach. 4 ed. Saunders/Elsevier; Phildelphia: 2009. [Google Scholar]
- Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx. 2005;2(3):396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ST, Hsu CY, Hogan EL, Maricq H, Balentine JD. A model of focal ischemic stroke in the rat: reproducible extensive cortical infarction. Stroke. 1986;17(4):738–43. doi: 10.1161/01.str.17.4.738. [DOI] [PubMed] [Google Scholar]
- Chen-Bee CH, Agoncillo T, Xiong Y, Frostig RD. The triphasic intrinsic signal: implications for functional imaging. J Neurosci. 2007;27(17):4572–86. doi: 10.1523/JNEUROSCI.0326-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiganos TC, Jr., Jensen W, Rousche PJ. Electrophysiological response dynamics during focal cortical infarction. J Neural Eng. 2006;3(4):L15–22. doi: 10.1088/1741-2560/3/4/L01. [DOI] [PubMed] [Google Scholar]
- Cirstea MC, Levin MF. Compensatory strategies for reaching in stroke. Brain. 2000;123(Pt 5):940–53. doi: 10.1093/brain/123.5.940. [DOI] [PubMed] [Google Scholar]
- Cook DJ, Teves L, Tymianski M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature. 2012;483(7388):213–7. doi: 10.1038/nature10841. [DOI] [PubMed] [Google Scholar]
- Coyle P. Different susceptibilities to cerebral infarction in spontaneously hypertensive (SHR) and normotensive Sprague-Dawley rats. Stroke. 1986;17(3):520–5. doi: 10.1161/01.str.17.3.520. [DOI] [PubMed] [Google Scholar]
- Coyle P. Spatial relations of dorsal anastomoses and lesion border after middle cerebral artery occlusion. Stroke. 1987;18(6):1133–40. doi: 10.1161/01.str.18.6.1133. [DOI] [PubMed] [Google Scholar]
- Crafton KR, Mark AN, Cramer SC. Improved understanding of cortical injury by incorporating measures of functional anatomy. Brain. 2003;126(Pt 7):1650–9. doi: 10.1093/brain/awg159. [DOI] [PubMed] [Google Scholar]
- Cramer SC, Riley JD. Neuroplasticity and brain repair after stroke. Curr Opin Neurol. 2008;21(1):76–82. doi: 10.1097/WCO.0b013e3282f36cb6. [DOI] [PubMed] [Google Scholar]
- Davis MF, Lay CC, Chen-Bee CH, Frostig RD. Amount but not pattern of protective sensory stimulation alters recovery after permanent middle cerebral artery occlusion. Stroke. 2011a;42(3):792–8. doi: 10.1161/STROKEAHA.110.607135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MF, Lay CC, Chen-Bee CH, Frostig RD. Neuroscience 2011. Society for Neuroscience; Washington, DC: 2011b. Stimulation of auditory cortex following permanent MCA occlusion confers complete or near complete protection from ischemic stroke in the adult rat. [Google Scholar]
- Del Zoppo GJ. Why do all drugs work in animals but none in stroke patients? 1. Drugs promoting cerebral blood flow. J Intern Med. 1995;237(1):79–88. doi: 10.1111/j.1365-2796.1995.tb01144.x. [DOI] [PubMed] [Google Scholar]
- Del Zoppo GJ, Saver JL, Jauch EC, Adams HP., Jr. Expansion of the time window for treatment of acute ischemic stroke with intravenous tissue plasminogen activator: a science advisory from the American Heart Association/American Stroke Association. Stroke. 2009;40(8):2945–8. doi: 10.1161/STROKEAHA.109.192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNapoli VA, Huber JD, Houser K, Li X, Rosen CL. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging. 2008;29(5):753–64. doi: 10.1016/j.neurobiolaging.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, editor. Rodent models of stroke. Spring Science+Business Media; New York: 2010. [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22(9):391–7. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Kaplan B, Jacewicz M, Pulsinelli W. Continuous measurement of cerebral cortical blood flow by laser-Doppler flowmetry in a rat stroke model. J Cereb Blood Flow Metab. 1989;9(5):589–96. doi: 10.1038/jcbfm.1989.84. [DOI] [PubMed] [Google Scholar]
- Durukan A, Tatlisumak T. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav. 2007;87(1):179–97. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Frostig RD, Chen-Bee CH. Visualizing adult cortical plasticity using intrinsic signal optical imaging. In: Frostig RD, editor. In vivo optical imaging of brain function. 2 ed. CRC Press; Boca Raton, FL: 2009. pp. 255–87. [PubMed] [Google Scholar]
- Frostig RD, Lieke EE, Ts'o DY, Grinvald A. Cortical functional architecture and local coupling between neuronal activity and the microcirculation revealed by in vivo high-resolution optical imaging of intrinsic signals. Proc Natl Acad Sci U S A. 1990;87(16):6082–6. doi: 10.1073/pnas.87.16.6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg MD. Life after cerovive: a personal perspective on ischemic neuroprotection in the post-NXY-059 era. Stroke. 2007;38(6):1967–72. doi: 10.1161/STROKEAHA.106.479170. [DOI] [PubMed] [Google Scholar]
- Graham SM, McCullough LD, Murphy SJ. Animal models of ischemic stroke: balancing experimental aims and animal care. Comp Med. 2004;54(5):486–96. [PubMed] [Google Scholar]
- Greenberg DA, Jin K. Regenerating the brain. Int Rev Neurobiol. 2007;77:1–29. doi: 10.1016/S0074-7742(06)77001-5. [DOI] [PubMed] [Google Scholar]
- Grinvald A, Lieke E, Frostig RD, Gilbert CD, Wiesel TN. Functional architecture of cortex revealed by optical imaging of intrinsic signals. Nature. 1986;324(6095):361–4. doi: 10.1038/324361a0. [DOI] [PubMed] [Google Scholar]
- Grotta J. Why do all drugs work in animals but none in stroke patients? 2. Neuroprotective therapy. J Intern Med. 1995;237(1):89–94. doi: 10.1111/j.1365-2796.1995.tb01145.x. [DOI] [PubMed] [Google Scholar]
- Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36(4):557–65. doi: 10.1002/ana.410360404. [DOI] [PubMed] [Google Scholar]
- Hoyte L, Kaur J, Buchan AM. Lost in translation: taking neuroprotection from animal models to clinical trials. Exp Neurol. 2004;188(2):200–4. doi: 10.1016/j.expneurol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10(11):1369–76. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Jones TH, Morawetz RB, Crowell RM, Marcoux FW, FitzGibbon SJ, DeGirolami U, et al. Thresholds of focal cerebral ischemia in awake monkeys. J Neurosurg. 1981;54(6):773–82. doi: 10.3171/jns.1981.54.6.0773. [DOI] [PubMed] [Google Scholar]
- Kaufmann AM, Firlik AD, Fukui MB, Wechsler LR, Jungries CA, Yonas H. Ischemic core and penumbra in human stroke. Stroke. 1999;30(1):93–9. doi: 10.1161/01.str.30.1.93. [DOI] [PubMed] [Google Scholar]
- Kent DM, Price LL, Ringleb P, Hill MD, Selker HP. Sex-based differences in response to recombinant tissue plasminogen activator in acute ischemic stroke: a pooled analysis of randomized clinical trials. Stroke. 2005;36(1):62–5. doi: 10.1161/01.STR.0000150515.15576.29. [DOI] [PubMed] [Google Scholar]
- Lay CC, Davis MF, Chen-Bee CH, Frostig RD. Mild sensory stimulation completely protects the adult rodent cortex from ischemic stroke. PLoS One. 2010;5(6):e11270. doi: 10.1371/journal.pone.0011270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay CC, Davis MF, Chen-Bee CH, Frostig RD. Neuroscience 2011. Society for Neuroscience; Washington, DC: 2011a. Active exploration completely protects rodent cortex from ischemic stroke following permanent MCA occlusion. [Google Scholar]
- Lay CC, Davis MF, Chen-Bee CH, Frostig RD. Mild sensory stimulation reestablishes cortical function during the acute phase of ischemia. J Neurosci. 2011b;31(32):11495–504. doi: 10.1523/JNEUROSCI.1741-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay CC, Davis MF, Chen-Bee CH, Frostig RD. Mild sensory stimulation protects the aged rodent from cortical ischemic stroke following permanent middle cerebral artery occlusion. J Am Heart Assoc. 2012;1(4):e001255. doi: 10.1161/JAHA.112.001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebeskind DS. Collateral circulation. Stroke. 2003;34(9):2279–84. doi: 10.1161/01.STR.0000086465.41263.06. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4(5):399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Martinez-Vila E, Sieira PI. Current status and perspectives of neuroprotection in ischemic stroke treatment. Cerebrovasc Dis. 2001;11(Suppl 1):60–70. doi: 10.1159/000049127. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67(2):181–98. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci. 2009;10(12):861–72. doi: 10.1038/nrn2735. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Jorgensen HS, Raaschou HO, Olsen TS. The influence of age on stroke outcome. The Copenhagen Stroke Study. Stroke. 1994;25(4):808–13. doi: 10.1161/01.str.25.4.808. [DOI] [PubMed] [Google Scholar]
- NINDS Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333(24):1581–7. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- NINDS NINDS stops trial enrollment in SAMMPRIS trial due to higher risk of stroke and death in stented group. American Academy of Neurology. 2011 [Google Scholar]
- Nudo RJ, Eisner-Janowicz I. Neuronal plasticity after stroke. In: Lomber S, Eggermont J, editors. Reprogramming the cerebral cortex: plasticity following central and peripheral lesions. Oxford University Press; Oxford: 2006. pp. 231–56. [Google Scholar]
- Nudo RJ, Plautz EJ, Frost SB. Role of adaptive plasticity in recovery of function after damage to motor cortex. Muscle Nerve. 2001;24(8):1000–19. doi: 10.1002/mus.1104. [DOI] [PubMed] [Google Scholar]
- Popa-Wagner A, Buga AM, Kokaia Z. Perturbed cellular response to brain injury during aging. Ageing Res Rev. 2011;10(1):71–9. doi: 10.1016/j.arr.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Popa-Wagner A, Carmichael ST, Kokaia Z, Kessler C, Walker LC. The response of the aged brain to stroke: too much, too soon? Curr Neurovasc Res. 2007;4(3):216–27. doi: 10.2174/156720207781387213. [DOI] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation. 2011;123(4):e18–209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saver JL. Time is brain—quantified. Stroke. 2006;37(1):263–6. doi: 10.1161/01.STR.0000196957.55928.ab. [DOI] [PubMed] [Google Scholar]
- Schaffer CB, Friedman B, Nishimura N, Schroeder LF, Tsai PS, Ebner FF, et al. Two-photon imaging of cortical surface microvessels reveals a robust redistribution in blood flow after vascular occlusion. PLoS Biol. 2006;4(2):e22. doi: 10.1371/journal.pbio.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traversa R, Cicinelli P, Bassi A, Rossini PM, Bernardi G. Mapping of motor cortical reorganization after stroke. A brain stimulation study with focal magnetic pulses. Stroke. 1997;28(1):110–7. doi: 10.1161/01.str.28.1.110. [DOI] [PubMed] [Google Scholar]
- Ts'o DY, Frostig RD, Lieke EE, Grinvald A. Functional organization of primate visual cortex revealed by high resolution optical imaging. Science. 1990;249(4967):417–20. doi: 10.1126/science.2165630. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Jickling GC, Sharp FR. Are underlying assumptions of current animal models of human stroke correct: from STAIRs to high hurdles? Transl Stroke Res. 2011;2(2):138–43. doi: 10.1007/s12975-011-0067-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlgren NG, Ahmed N. Neuroprotection in cerebral ischaemia: facts and fancies—the need for new approaches. Cerebrovasc Dis. 2004;17(Suppl 1):153–66. doi: 10.1159/000074808. [DOI] [PubMed] [Google Scholar]
- Wang-Fischer Y. Manual of stroke models in rats. CRC Press; Boca Raton, FL: 2009. pp. 17–30.pp. 239–78. [Google Scholar]
- Wardlaw JM, Murray V, Berge E, Del Zoppo GJ. Thrombolysis for acute ischaemic stroke. Cochrane Database Syst Rev. 2009;(4):CD000213. doi: 10.1002/14651858.CD000213.pub2. [DOI] [PubMed] [Google Scholar]
- Weber R, Ramos-Cabrer P, Justicia C, Wiedermann D, Strecker C, Sprenger C, et al. Early prediction of functional recovery after experimental stroke: functional magnetic resonance imaging, electrophysiology, and behavioral testing in rats. J Neurosci. 2008;28(5):1022–9. doi: 10.1523/JNEUROSCI.4147-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Craven K, Erinjeri J, Liang GE, Bereczki D, Rovainen CM, et al. Local cerebral blood flow during the first hour following acute ligation of multiple arterioles in rat whisker barrel cortex. Neurobiol Dis. 1998;5(3):142–50. doi: 10.1006/nbdi.1998.0199. [DOI] [PubMed] [Google Scholar]
- Wintermark M, Reichhart M, Cuisenaire O, Maeder P, Thiran JP, Schnyder P, et al. Comparison of admission perfusion computed tomography and qualitative diffusion-and perfusion-weighted magnetic resonance imaging in acute stroke patients. Stroke. 2002;33(8):2025–31. doi: 10.1161/01.str.0000023579.61630.ac. [DOI] [PubMed] [Google Scholar]
- Zhang F, Graham SH, Chen J. Blood flow reduction: laser Doppler, 14C-IAP. In: Chen J, Xu X-M, Xu ZC, Zhang JH, editors. Animal models of acute neurological injuries II: injury and mechanistic assessments. volume 2. Springer; New York: 2012. pp. 107–14. [Google Scholar]