

Abstract
Prior to hearing onset, spontaneous action potentials activate voltage-gated Cav1.3 Ca2+ channels in mouse inner hair cells (IHCs), which triggers exocytosis of glutamate and excitation of afferent neurons. In mature IHCs, Cav1.3 channels open in response to evoked receptor potentials, causing graded changes in exocytosis required for accurate sound transmission. Developmental alterations in Cav1.3 properties may support distinct roles of Cav1.3 in IHCs in immature and mature IHCs, and have been reported in various species. It is not known whether such changes in Cav1.3 properties occur in mouse IHCs, but this knowledge is necessary for understanding the roles of Cav1.3 in developing and mature IHCs. Here, we describe age-dependent differences in the biophysical properties of Cav1.3 channels in mouse IHCs. In mature IHCs, Cav1.3 channels activate more rapidly and exhibit greater Ca2+-dependent inactivation (CDI) than in immature IHCs. Consistent with the properties of Cav1.3 channels in heterologous expression systems, CDI in mature IHCs is not affected by increasing intracellular Ca2+ buffering strength. However, CDI in immature IHCs is significantly reduced by strong intracellular Ca2+ buffering, which both slows the onset of, and accelerates recovery from, inactivation. These results signify a developmental decline in the sensitivity of CDI to global elevations in Ca2+, which restricts negative feedback regulation of Cav1.3 channels to incoming Ca2+ ions in mature IHCs. Together with faster Cav1.3 activation kinetics, increased reliance of Cav1.3 CDI on local Ca2+ may sharpen presynaptic Ca2+ signals and improve temporal aspects of sound coding in mature IHCs.
Keywords: inner hair cell, Ca2+ channel, calmodulin
Introduction
Voltage-gated Cav Ca2+ channels conduct inward Ca2+ currents that can depolarize the membrane potential and trigger Ca2+-dependent signal transduction in excitable cells. Multiple classes of Cav channels (Cav1.x-Cav3.x) have been characterized,1 which play distinct roles often within the same cell. In most neurons, somatodendritic Cav1 channels mediate L-type Ca2+ currents that couple neuronal activity to changes in gene transcription,2,3 while presynaptic Cav2 channels generating P/Q- and N-type Ca2+ currents regulate neurotransmitter release from nerve terminals.4-6
In contrast to the diverse complement of Cav channnels in most neuronal cell-types, auditory hair cells express predominantly Cav1.3 channels, which mediate exocytosis of glutamate at ribbon synapses formed with primary afferent neurons.7,8 Genetic inactivation of CACNA1D (Cav1.3 KO), which encodes the pore-forming α1 subunit of Cav1.3, causes ~90% reduction in the whole-cell Ca2+ current in mouse inner hair cells (IHCs).9-11 As a consequence, stimulus-secretion coupling is significantly impaired in Cav1.3 KO IHCs,11 which contributes to deafness in these mice.9,10 Spontaneous Ca2+-dependent action potentials, which support presensory afferent synaptic transmission,12-14 are absent in Cav1.3 KO IHCs. The developmental upregulation of BK K+ channels and pruning of cholinergic efferents from the basal IHC membrane, which normally occurs around the onset of hearing,13,15 are also absent in Cav1.3 KO IHCs.11,16 Thus, Cav1.3 channels play distinct roles in regulating electrical activity and normal developmental programs in immature IHCs, and in transducing stimulus-evoked exocytosis in mature IHCs.
Comparisons of Cav1 currents in mature and immature auditory hair cells suggest differences in the properties of Cav1.3 channels that may tailor Ca2+ signals according to developmental stage. For example, in gerbil and rat IHCs, the extent to which Cav1 channels undergo Ca2+-dependent inactivation (CDI) declines after hearing onset.17,18 CDI is a negative feedback regulation of Cav1 and Cav2 channels by incoming Ca2+ ions, which depends on calmodulin binding to the Cav α1 subunit.19,20 CDI is relatively weak for Cav1 channels in IHCs, which may be due to the antagonistic actions of calmodulin-like Ca2+ binding proteins expressed in IHCs.17,21,22 The rates of Cav1 channel activation and inactivation can significantly influence IHC Ca2+ signals that regulate presensory action potentials or stimulus-secretion coupling. Whether Cav1.3 channels in mouse IHCs undergo developmental changes in their biophysical properties is incompletely characterized. Given the widespread use of mice as experimental models for IHC transmission, filling this void is essential for understanding Ca2+-dependent signaling in IHCs in this species.
In this study, we compared the properties of Ca2+ and Ba2+ currents (ICa and IBa, respectively) in IHCs from mice before and after hearing onset (~P12). We discovered that Cav1.3 channels exhibit significant differences in immature (P6–8) and mature (P16–18) IHCs in terms of activation and inactivation. In particular, CDI differs in magnitude and sensitivity to local rather than global Ca2+ elevations in Ca2+. Our results highlight age-dependent distinctions in the properties of Cav1.3 channels that may fine-tune Ca2+ signals required for the development and mature function of IHCs.
Results
Cav1.3 currents activate with faster kinetics in mature IHCs than in immature IHCs
Because a goal of this study was to compare CDI before and after hearing onset in mouse IHCs, we first analyzed activation of both ICa and IBa in P6–8 and P16–18 mouse IHCs. Whole-cell patch clamp recordings were performed at room temperature in solutions designed to isolate Cav1.3 currents from other ionic currents, particularly voltage-gated K+ currents (see Materials and Methods). The intracellular solution contained 2 mM EGTA, which is permissive for measuring CDI in IHCs.17,18 Relatively high concentrations of extracellular Ca2+ or Ba2+ were used (5 mM) to amplify current amplitudes, thus increasing resolution of activation and inactivation kinetics. Consistent with previous studies of Cav1 channels in other cell-types,23 ICa underwent a progressive increase in amplitude (run-up) upon attaining whole-cell configuration in immature IHCs (Fig. 1). For this reason, we first monitored the amplitude of ICa evoked by steps to +10 mV every 20 sec until there was no further run-up. Interestingly, run-up was not observed in mature IHCs, which was confirmed by the same protocol. Thus, data were collected ~5 min after whole cell patch rupture in immature IHCs, when steady-state ICa amplitudes were achieved. In mature IHCs, data were collected ~1 min after patch rupture to ensure equilibration with intracellular solution components.
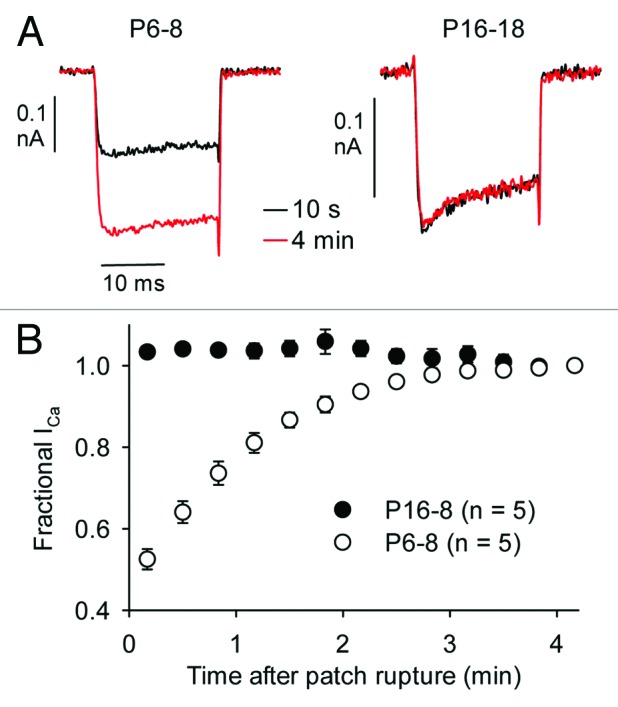
Figure 1. Run-up of ICa in immature mouse IHCs. (A) Representative current traces for ICa evoked by 20-ms test pulses to +10 mV, 10 sec and ~4 min after patch rupture. (B) ICa was normalized to that at 4 min after patch rupture and plotted against time.
As noted previously,11,14 the most dramatic difference in Cav1.3 currents was the smaller amplitude of ICa and IBa in mature IHCs compared with immature IHCs. In current-voltage (I-V) relationships, the maximum current amplitude was obtained with a −10-mV pulse for ICa and was significantly smaller in mature IHCs than in immature IHCs (19.4 ± 15.4%; Figure 2A–D). A similar difference was observed for IBa (32.9 ± 10.2%) except that the peak IBa was obtained between −10 and −0 mV, due to changes in surface charge screening when Ba2+ rather than Ca2+ is used as the permeant ion.24 As has been reported for Cav1.3 in heterologous expression systems,25 there was some rectification in the I-V relationship at positive voltages, probably due to limited permeability of Cav1.3 channels to Cs+ ions. Thus for greater accuracy, we measured parameters for voltage-dependent activation in normalized tail current-voltage relationships fit with the Boltzmann equation. By this analysis, the voltage for half-maximal activation (Vh) was more positive and there was a significant increase in the slope factor (k) with age for ICa and IBa (Table1, Fig. 2E and F). In addition, the activation kinetics of ICa were significantly faster in mature IHCs than in immature IHCs (~5–29% from −40 mV to +40 mV, p < 0.001 by two-way ANOVA; Fig. 3A). The acceleration in Cav1.3 activation depended on Ca2+ as the charge carrier since it was not observed for IBa (p = 0.34 by two-way ANOVA; Fig. 3B). These results indicate that, in addition to a reduction in Cav1.3 current density, the intrinsic biophysical properties of Cav1.3 channels also change with development.
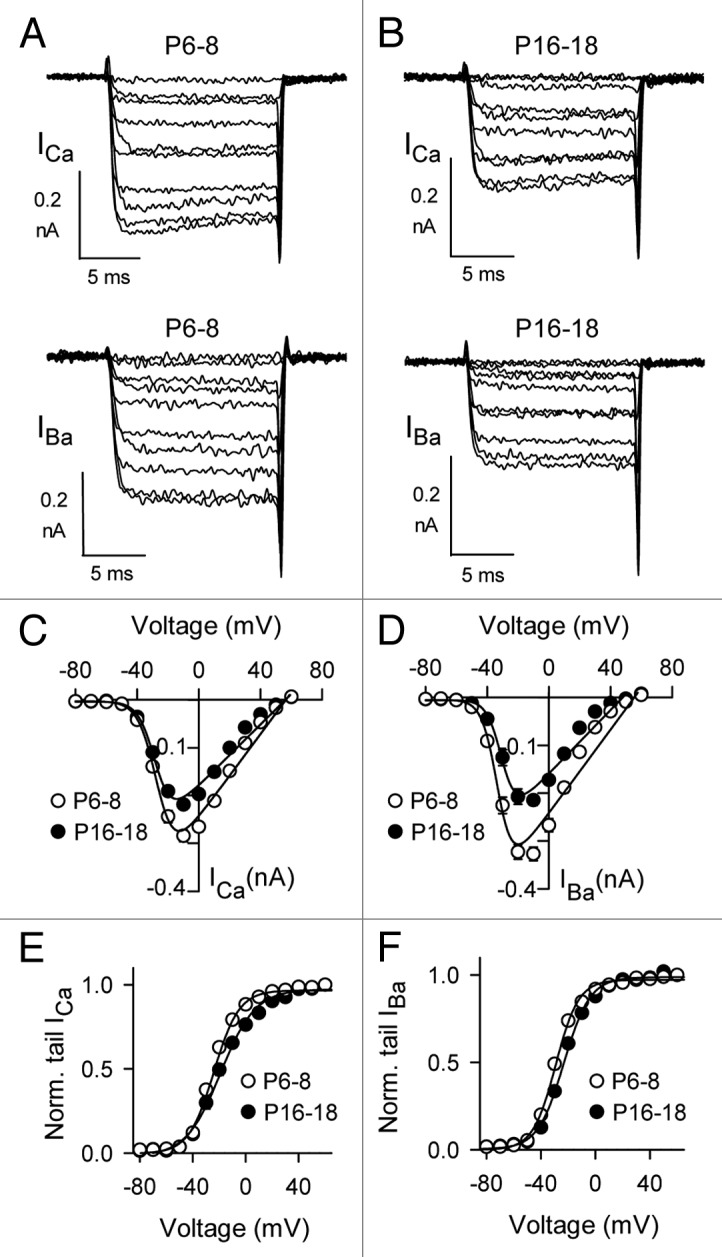
Figure 2. Voltage-dependent properties of ICa and IBa in mature IHCs and in immature IHCs. (A and B) Representative ICa (upper panels) and IBa (lower panels). Currents were evoked by 15– ms depolarizing pulses from a holding voltage of −84 mV. (C and D) I-V relationships for currents in (A). (E and F) G-V relationships for currents in (A and B). Tail current amplitudes were normalized to that obtained with a pulse to +50 mV (Norm. tail) and plotted against test voltage. Smooth lines in (C–F) represent curve fits from the Boltzmann equation. The parameters for the fits shown were: in (C), Vh = −26.6, k = −5.9, Vrev = 56.4 mV, Gmax = 4.3 nS for P6–8 (n = 22), Vh = −27.7, k = −5.4, Vrev = 56.1 mV, Gmax = 3.2 nS for P16–18 (n = 15); in (D), Vh = -32.8, k = −4.9, Vrev = 55.5 mV, Gmax = 4.3 for P6–8 (n = 22), Vh = −30.1 mV, k = −4.7, Vrev = 48.0 mV, Gmax = 3.3 for P16–18 (n = 15); in (E), Vh = −24.1 mV, k = 9.5 for P6–8 (n = 22), Vh = −19.0 mV, k = 13.1 for P16–18 (n = 9); in (F), Vh = −29.2 mV, k = 8.4 for P6–8 (n = 11), Vh = −23.2 mV, k = 9.6 for P16–18 (n = 15).
Table 1. Parameters for voltage-dependent activation of Cav1.3 currents in mouse IHCs.
| P6–8 | n | P16–18 | n | p-value1 | ||
|---|---|---|---|---|---|---|
| ICa (2 mM EGTA) |
Peak current density2 (pA/pF) |
−35.6 ±1.8 (at −10 mV) |
22 |
−23.0 ±1.2 (at −10 mV) |
15 |
< 0.001 |
|
V1/2 (mV)3 |
−24.1 ± 0.9 |
−19.0 ± 1.8 |
9 |
0.009 |
||
|
k3 |
9.5 ± 0.3 |
13.1 ± 0.7 |
< 0.001 |
|||
| |
|
|
|
|||
| IBa (2 mM EGTA) |
Peak current density2 (pA/pF) |
−36.2 ± 1.8 (at −10mV) |
11 |
−21.7 ± 1.2 (at −10mV) |
15 |
< 0.001 |
|
V1/2 (mV)3 |
−29.2 ± 1.1 |
−23.2 ± 0.9 |
< 0.001 |
|||
|
k3 |
8.4 ± 0.3 |
9. ± 0.3 |
0.011 |
|||
| |
|
|
|
|||
| ICa (10 mM BAPTA) |
Peak current density2 (pA/pF) |
−34.5 ± 1.3 (at −10 mV) |
25 | −20.0 ± 0.8 (at −10 mV) |
8 | < 0.001 |
|
V1/2 (mV)3 |
−24.4 ± 0.6 |
−18.6 ± 0.9 |
< 0.001 |
|||
|
k3 |
8.3 ± 0.3 |
12.7 ± 0.8 |
< 0.001 |
|||

Figure 3. ICa activation kinetics are faster in mature IHCs than immature IHCs. (A and B) Upper panels, representative traces for ICa and IBa for P6–8 (gray) or P16–18 (black) IHCs. Currents were evoked by a 15-ms step to -10 mV from a holding potential of −84 mV. Dashed lines represent exponential fits of the current trace. The time constants (τ, ms) for the displayed current traces were: for ICa (A) 0.62 (P6–8) and 0.28 (P16–18); for IBa (B) 0.50 (P6–8) and 0.38 (P16–18). Lower panels, time constants for ICa (A) and IBa (B) were plotted against test voltage. For P6–8, n = 22 for ICa, 15 for IBa. For P16–18, n = 11 for ICa, 15 for IBa. *p < 0.001 by two-way ANOVA.
Ca2+-dependent inactivation is greater in mature IHCs than immature IHCs
To determine if Cav1.3 inactivation was also subject to developmental regulation in mouse IHCs, we compared the extent to which ICa and IBa inactivated in response to sustained depolarizations. Like other Cav1 channels, Cav1.3 undergoes inactivation due to Ca2+- or voltage-dependent mechanisms (CDI and VDI, respectively). CDI manifests as stronger inactivation of ICa compared with IBa, due to Ca2+ binding to the calmodulin that is associated with the channel protein.20 Unlike ICa, which undergoes both CDI and VDI, IBa exhibits purely VDI since Ba2+ substitutes poorly for Ca2+ in binding to calmodulin.26 We measured CDI and VDI with a triple-pulse voltage-protocol in which ICa or IBa was evoked by test pulses before (p1) and after (p2) a 200-ms prepulse to varying voltages. Inactivation was measured as the ratio of the p2/p1 current amplitudes; this ratio was less than 1 for prepulses inducing inactivation (Fig. 4A and B). As expected for CDI, the prepulse-voltage dependence of ICa inactivation was U-shaped and was greatest for prepulses evoking the maximal inward ICa. In contrast, inactivation of IBa with this protocol was not evident across the full prepulse voltage range. In mature IHCs, IBa underwent facilitation (p2/p1 > 1) at positive prepulse voltages, likely due to voltage-dependent facilitation (VDF).27 Since ICa is thought to undergo VDI as well as VDF, we isolated the effects of CDI on ICa as the difference in p2/p1 for ICa and IBa (FCDI, Fig. 4C). By this metric, CDI was significantly greater in mature IHCs than in immature IHCs (p < 0.001 by two-way ANOVA).

Figure 4. CDI is greater in mature IHCs than immature IHCs. (A) Top panel, Voltage protocol in which test currents were evoked by pulses (p1, p2) from −84 mV to −20 mV (for ICa) or −30 mV (for IBa) before and after a 200-ms prepulse to various voltages. Bottom panels, representative traces for ICa or IBa recorded in P6–8 or P16–18 mouse IHCs. Currents were evoked by p1 (black) and p2 (red) overlaid for comparison. Scale bars, 0.2 nA (vertical), 2.5 ms (horizontal). (B) Inactivation was measured as the ratio of P2/P1 current amplitude and plotted against prepulse voltage. Results are from mouse IHCs from P6–8 (n = 20 for ICa, n = 6 for IBa) or P16–18 (n = 18 for ICa, n = 8 for IBa). (C) CDI was measured as the difference in p2/p1 for ICa and IBa (FCDI) and is plotted against prepulse voltage for P6–8 and P16–18 IHCs. *p < 0.05 by two-way ANOVA and post-hoc Bonferroni t-test.
To address the underlying mechanism(s) for the age-dependent increase in CDI, we analyzed the onset of ICa inactivation by obtaining p2/p1 ratios in voltage protocols with prepulses of varying duration (Fig. 5A). The prepulse was set to −20 mV for ICa and −30 mV for IBa, to partially compensate for the negative shift in activation voltages for IBa compared with ICa (Table 1). The duration of the p1 and p2 test pulse was increased to 5 ms in these experiments to ensure that test currents reached steady-state levels, as long prepulses tended to slow the activation kinetics of the p2 current. The onset of inactivation was obtained by plotting p2/p1 ratios for ICa against prepulse duration. With this protocol, ICa inactivated with a double exponential time course in both immature and mature IHCs. Both fast and slow inactivation were likely influenced by CDI: the fast component was not observed for IBa, and the slow component of ICa inactivation was significantly faster than that for IBa (τslow 2.0 ± 0.4 sec for ICa vs. 3.1 ± 0.6 sec for IBa, P6–8, p = 0.15; τslow = 0.8 ± 0.13 sec for ICa vs. 2.3 ± 0.4 sec for IBa, P16–18, p < 0.001; both by t-test). While there was no age-dependent difference in the time constants for IBa inactivation (p = 0.3), both the slow and fast time constants were significantly faster for ICa inactivation in mature IHCs than in immature IHCs (Fig. 5C, p < 0.05). However, the amplitudes of the slow and fast components of ICa inactivation did not vary with age (Afast = −0.25 ± 0.01 for P6–8 vs. −0.23 ± 0.02 for P16–18, p = 0.20; Aslow = −0.40 ± 0.03 for P6–8 vs. −0.34 ± 0.01 for P16–18, p = 0.31; both by t-test). These results indicate that CDI is governed by fast and slow processes, both of which occur more rapidly in mature IHCs compared with in immature IHCs.

Figure 5. Onset of CDI is faster in mature IHCs than in immature IHCs. (A) Top panel, Voltage protocol in which test currents were evoked by pulses (p1, p2) from −84 mV to −20 mV (for ICa) or −30 mV (for IBa) before and after a prepulse to −10 mV of varying durations. Bottom panels, representative traces for ICa or IBa recorded in P6–8 or P16–18 mouse IHCs with protocols using the indicated prepulse durations. Currents evoked by p1 (black) and p2 (red) are overlaid for comparison. Scale bars, 0.8 nA (vertical), 2.5 ms (horizontal). (B) Onset of inactivation was measured by plotting p2/p1 values against prepulse duration. Smooth line represents fit with a double (ICa) or single (IBa) exponential function. For P6–8, curve fit parameters for ICa were y0 = 0.34, Afast = 0.25, τfast = 32.9 ms, Aslow = 0.40, τslow = 1990 ms (n = 22); for IBa, y0 = 0.91, A = 56.5, τ = 3108 ms (n = 8). For P16–18, curve fit parameters for ICa were y0 = 0.37, Afast = 0.28, τfast = 20.4 ms, Aslow = 0.34, τslow = 795 ms (n = 12); for IBa, y0 = 0.79, A = 30.1, τ = 2313 ms (n = 11). (C) Average values from exponential fits of ICa data in (B). *p < 0.02 by Mann–Whitney rank sum test.
To analyze recovery from inactivation, p2 test currents were measured at varying intervals after the inactivating prepulse and the ratio of p2/p1 plotted against the recovery interval (Fig. 6A). The duration of the prepulse was limited to 200 ms since longer prepulses induced VDI, particularly in mature IHCs (Fig. 5A); this would complicate analyses of recovery from CDI. In single exponential fits of these data, there was no difference in the amplitude (A = 0.32 ± 0.02 for P6–8 vs. 0.37 ± 0.03 for P16–18, p = 0.40, by t-test) or time constant (τ = 425.9 ± 28.3 ms for P6–8 vs. 486.4 ± 87.7 ms for P16–18, p = 0.08, by Mann-Whitney rank sum test; Fig. 6B). These results indicate that greater CDI in mature IHCs results primarily from faster onset, rather than slower recovery, of ICa from inactivation.
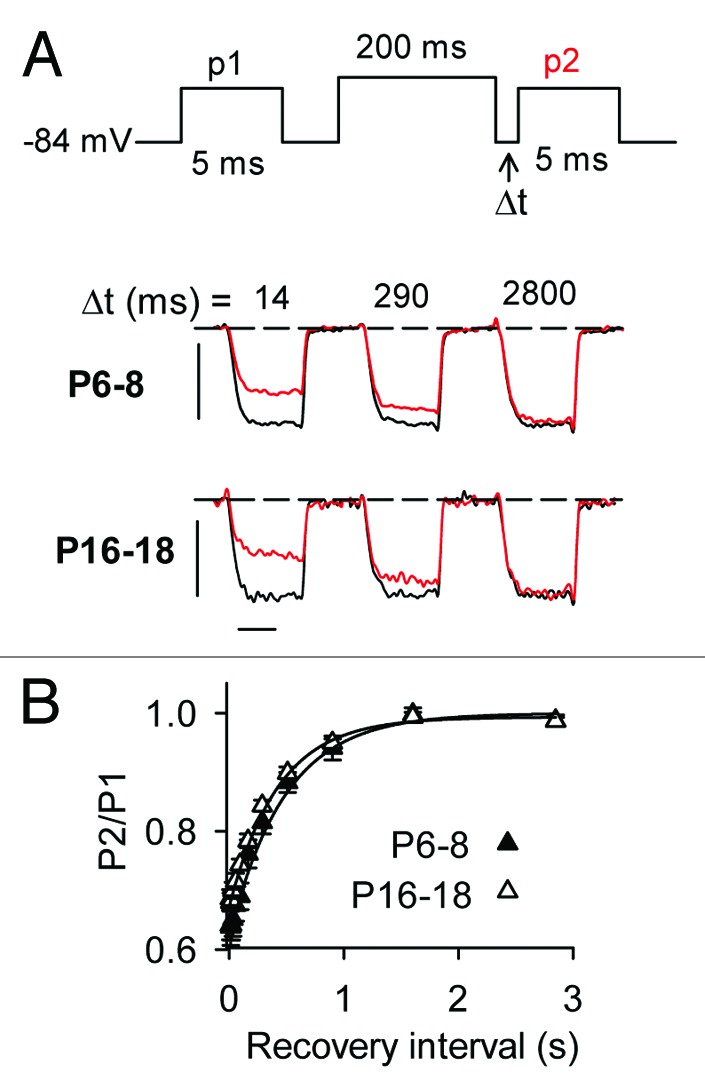
Figure 6. Recovery from CDI does not differ in mature and immature IHCs. (A) Top panel, Voltage protocol in which ICa was evoked by pulses (p1, p2) from −84 mV to −20 mV before and at variable recovery intervals after a 200-ms prepulse to −10 mV. Bottom panel, representative traces for ICa recorded in P6–8 or P16–18 mouse IHCs. Currents evoked by p1 (black) and p2 (red) at the indicated recovery intervals are overlaid for comparison. Scale bars, 0.8 nA (vertical), 2.5 ms (horizontal). (B) Recovery of ICa from inactivation was measured by plotting p2/p1 for data obtained as in (A) against recovery interval. Data were fit with an exponential function (smooth line). Curve fit parameters were: for P6–8 (n = 19), y0 = 0.99, A; −0.321, τ = 425 ms; for P16–18 (n = 8), y0 = 1.0, A = −37.8, τ = 486 ms.
Ca2+-dependent inactivation depends on local Ca2+ in mature IHCs but not immature IHCs
In heterologous expression systems, CDI of Cav1 channels is not affected by relatively high concentrations of intracellular Ca2+ buffers (i.e., 10 mM BAPTA), due to a reliance on rapid, local Ca2+ influx through individual channels.28 However, CDI of Cav1 currents in immature rat IHCs but not in mature gerbil IHCs is suppressed by high intracellular concentrations (10 mM) of EGTA or BAPTA.17,18 These results suggest that the Ca2+ sensitivity of Cav1 CDI may be subject to developmental regulation. To test this, we analyzed the impact of increasing Ca2+ buffering strength on CDI in mature and immature mouse IHCs using triple-pulse protocols with varying prepulse voltages. To rapidly buffer incoming Ca2+ ions, we used relatively high concentrations (10 mM) of BAPTA in the intracellular recording solution. Since BAPTA (10 mM) does not influence inactivation of IBa mediated by Cav1.3,29 we restricted analysis to ICa in these experiments. Compared with our standard solution with EGTA (2 mM), the BAPTA-containing solution significantly blunted CDI in immature IHCs across a range of prepulse voltages (up to ~30% between −30 and +20 mV, p < 0.05 by two-way ANOVA; Fig. 7A and B). Remarkably, this effect of BAPTA was not observed in mature IHCs (p = 0.16 by two-way ANOVA; Fig. 7A–C). These effects of BAPTA were not due to altered voltage-dependence of activation, since G-V parameters were not significantly different with BAPTA or low intracellular EGTA (Table 1, Fig. S1).

Figure 7. CDI is inhibited by BAPTA in immature IHCs but not mature IHCs. (A) Top, Voltage protocol in which test currents were evoked by pulses (p1, p2) from −84 mV to −20 mV before and after a 200-ms prepulse to various voltages. Bottom, Representative traces for ICa recorded in P6–8 or P16–18 mouse IHCs with protocols using the indicated prepulse voltages. Intracellular recording solution contains 2 mM EGTA or 10 mM BAPTA. Currents evoked by p1 (black) and p2 (red) are overlaid for comparison. Scale bars, 0.2 nA (vertical), 2.5 ms (horizontal). (B) p2/p1 was plotted against prepulse voltage for P6–8 (n = 20 for 2 EGTA, n = 25 for 10 BAPTA) and P16–18 (n = 18 for 2 EGTA, n = 8 for BAPTA). *p < 0.05 for EGTA vs. BAPTA, by two-way ANOVA and Bonferroni t-test. (C) Representative normalized current traces obtained with 2 EGTA or 10 BAPTA. ICa was evoked by a 200-ms test pulse to −10 mV from −84 mV. (D) For P6–8 IHCs, p2/p1 ratios were plotted against ICa amplitude for 2 EGTA or 10 BAPTA. Lines represent fits by linear regression.
The BAPTA-sensitivity of CDI in immature but not mature IHCs suggests that Ca2+ elevations that support CDI are not restricted to local Ca2+ influx through single channels but rather global Ca2+ signals fueled by multiple neighboring channels. In this respect, Cav1.3 CDI in immature IHCs may be similar mechanistically to CDI of Cav2.1 channels, which is strongly suppressed by high concentrations of BAPTA or EGTA.30-34 If so, CDI should increase with channel density in immature IHCs. Consistent with this prediction, p2/p1 ratios decreased (i.e., inactivation increased) with increasing ICa amplitude (Fig. 7D). BAPTA caused a significant increase in the slope of the p2/p1 vs. ICa relationship (4 × 10−4 for EGTA vs. 9.5 × 10−4 for BAPTA, p < 0.03 by ANCOVA; Fig. 7D). These results show that BAPTA increases the current-amplitude dependence of CDI, such that small-amplitude ICa showed weaker CDI (and BAPTA-sensitivity) than large-amplitude ICa.
To determine if the sensitivity of CDI to BAPTA in immature IHCs was due to effects of BAPTA on the onset and/or recovery kinetics of ICa inactivation, we compared these parameters using either 2 mM EGTA or 10 mM BAPTA in the intracellular recording solution. With BAPTA, the onset of inactivation exhibited a double exponential time course as was found for EGTA (Figs. 5 and 8A–C). In these experiments, BAPTA did not affect either the slow or fast time constants (τfast = 48.1 ± 9.2 ms for BAPTA vs. 32.9 ± 3.9 ms for EGTA, p = 0.06; τslow = 1.2 ± 0.3 ms for BAPTA vs. 2.0 ± 0.5 ms for EGTA, p = 0.07, both by Mann–Whitney rank sum test; Fig. 8C). However, BAPTA significantly decreased the corresponding amplitudes (Afast = 0.15 ± 0.02 for BAPTA vs. 0.25 ± 0.01 for EGTA, p < 0.001; Aslow = 0.20 ± 0.01 for BAPTA vs. 0.40 ± 0.03 for EGTA, p < 0.001, both by Mann-Whitney rank sum test) and increased the available current (Y0 = 0.66 ± 0.02 for BAPTA vs. 0.34 ± 0.04 for EGTA, p < 0.001, by Mann–Whitney rank sum test; Fig. 8C). In addition, BAPTA accelerated recovery of ICa from inactivation (τ = 271.9 ± 28.3 ms for BAPTA vs. 425.9 ± 28.3 ms for EGTA, p < 0.001 by t-test; Fig. 8D–F). Thus, BAPTA increases Cav1.3 channel availability in immature IHCs by inhibiting the amount of ICa inactivation as well as by speeding recovery from CDI. Our results highlight age-dependent differences in the intrinsic properties and modulation of Cav1.3 channels in mouse IHCs that may be important for the normal function and/or maturation of these cells as sound-transducers in the inner ear.

Figure 8. BAPTA slows the onset and accelerates recovery of CDI in immature IHCs. (A) Voltage protocol and representative p1 and p2 current traces for ICa with the indicated prepulse durations. Scale bars, 0.2 nA (vertical), 2.5 ms (horizontal). (B) Graph shows data plotted as in Figure 4B for ICa recorded with 2 EGTA (n = 22) or 10 BAPTA (n = 14). Smooth line represents double exponential curve fit (for 10 BAPTA, y0 = 0.66, Afast = 0.14, τfast = 48.1 ms, Aslow = 0.20, τslow = 1251 ms; see Figure 4B for parameters for 2 EGTA). (C) Average parameters for exponential fits of data in (B).*p < 0.001 by Mann-Whitney rank sum test. (D) Voltage protocol and representative p1 and p2 current traces for ICa with the indicated recovery intervals. Scale bars, 0.2 nA (vertical), 2.5 ms (horizontal). (E) Graph shows data plotted as in Figure 5B for ICa recorded with 2 EGTA (n = 8) or 10 BAPTA (n = 14). Smooth line represents exponential curve fit (y0 = 1.0, A = −0.25, τ = 271 ms see Fig. 5B for parameters for 2 EGTA). (F) Average parameters for exponential fits of data in (E).*p < 0.001 by t- test.
Discussion
The aim of the present study was to characterize the biophysical properties of Cav1.3 channels in mouse IHCs before and after the onset of hearing. While comprehensive analyses of Cav1 currents have been performed in auditory hair cells from other species,17,18,35 details regarding the function and modulation of Cav1.3 channels in immature and mature mouse IHCs is necessary to understand the sequelae associated with dysregulated Ca2+ signaling in mouse models of deafness. Our results indicate that Cav1.3 channels undergo significant changes in activation and inactivation that may support distinct roles of these channels at different stages of development.
Technical considerations and comparisons with other species
Due to the concentration of Ca2+ or Ba2+ (5 mM) in our extracellular recording solution and the fact that our recordings were performed at room temperature (~25°C), parameters for ICa and IBa activation and inactivation in the present study differ from those expected under physiological conditions (~1.3 mM extracellular Ca2+, ~37°C). In addition to increasing the amplitudes of ICa and IBa, the higher than physiological concentration of extracellular permeant ions would cause a positive shift in the half-maximal activation voltage (Vh) due to increased neutralization of cell surface charges.36 Subsequently, the voltage-dependence of time constants for activation and inactivation should be positively shifted with 5 mM compared with 1.3 mM extracellular Ca2+ or Ba2+. However, charge-screening effects of higher divalent concentrations should not influence the voltage-sensitivity of Cav1.3 activation, which is reflected by the steepness of the G-V curves (k from Boltzmann fits).37,38 Assuming that Cav1.3 properties in mature and immature IHCs would be equally affected by the increased Ca2+ or Ba2+ concentration, we would expect our results to accurately report the age-dependent difference, if not the absolute values, pertaining to Cav1.3 properties.
As shown previously,17,18 recordings at room temperature (~25°C) would cause smaller Cav1 current amplitudes and slower activation and inactivation kinetics than at physiological temperature (~37°C). At the same time, signaling processes that post-translationally alter the intrinsic biophysical properties of Cav1.3 channels would proceed more rapidly at 37°C than at 25°C. This could be a concern given that Cav1 amplitudes in immature IHCs show greater temperature sensitivity (larger Q10) than in mature IHCs in the gerbil.18 Notably, we observed greater run-up of ICa upon initiation of whole-cell recordings in immature compared with mature IHCs (Fig. 1). Cav1 channel run-up has been reported in turtle and frog hair cells35,39 and in cardiac myocytes (see for example ref. 40). While the underlying mechanisms are not entirely clear, run-up of cardiac Cav1 currents is associated with patch rupture in whole-cell recordings and exhibits a rapid (< 5 min) and late (> 10 min) phase, the latter of which is not observed at lower than physiological temperatures.40 Therefore, our recordings at room temperature may minimize alterations in channel properties resulting from intracellular dialysis.
Our results in mouse IHCs may not be entirely consistent with those reported for gerbil and rat IHCs due to methodological and/or species-related differences. For example, the age-dependent acceleration in ICa activation kinetics in our study (Fig. 3) agrees with results from gerbil IHCs18 but not rat IHCs, in which ICa activation was slower after hearing onset than before.17 In addition, while we found CDI to be greater in mature IHCs than in immature IHCs (Fig. 4), there was no such age-related difference in gerbil IHCs,18 and CDI was actually greater in immature IHCs than in mature IHCs in the rat.17 Our results regarding the Ca2+ buffer-sensitivity of CDI in immature but not in mature mouse IHCs (Figs. 7 and 8) are consistent with these previous studies: high concentrations of BAPTA (5 mM) did not affect CDI in mature gerbil IHCs18 but did significantly suppress CDI in immature rat IHCs.17 Therefore, the shift in CDI dependence on global Ca2+ signals in immature IHCs, to local Ca2+ signals in mature IHCs, may be a fundamental feature of IHC development.
Possible mechanisms for developmental alterations in Cav1.3 function
The properties of Cav1 channels are subject to multiple forms of regulation, which could account for the difference in Cav1.3 properties we noted during mouse IHC development. We showed previously that the PDZ-domain containing protein harmonin interacts with Cav1.3 channels in mouse IHCs after hearing onset and inhibits IBa.27 In mature mouse IHCs, harmonin enhances proteosomal degradation of Cav1.3, which could contribute to the age-dependent decrease in ICa or IBa current density (Fig. 2C and D). A second possibility is related to alternative splicing of the pore-forming Cav1.3 α1 subunit (α11.3), which produces variants that differ in their biophysical properties and modulation.25,41-47 After hearing onset, there may be an upregulation of α11.3 variants lacking portions of a C-terminal modulatory domain (CTM), which exhibit decreased current density relative to variants with the intact CTM.41,48 However, it is also possible that factors regulating the transcription/translation of Cav1.3 channels could also vary in mouse IHCs with age.
Some CTM-lacking variants exhibit enhanced voltage-dependence of activation and stronger CDI than variants containing the entire CTM.44 Such variants could account for increased CDI in mature IHCs (Fig. 4), but their hyperpolarized activation properties relative to the CTM-containing variants are inconsistent with our findings of weaker voltage-dependent activation of Cav1.3 currents in mature than in immature mouse IHCs (Table 1). Alternative splicing and RNA editing of the calmodulin-binding IQ domain α11.3 also produce Cav1.3 variants with limited CDI,47,49 although the extent to which these variants are present in immature mouse IHCs is unknown. Cav1.3 channels interact with a variety of regulatory proteins, which may also influence CDI.50 Rab 3-interacting molecule (RIM2α) inhibits CDI when coexpressed with Cav1.3 channels in HEK293 cells.51 RIM2α transcripts were detected in immature but not mature IHCs51 and so could be responsible for the more moderate CDI in mouse IHCs prior to hearing onset (Fig. 4). Other possibilities include Ca2+ binding proteins related to calmodulin (CaBPs),17,21,22 and various synaptic proteins,52 which also suppress inactivation of Cav1.3 in heterologous expression systems. Whether such Cav1.3 modulatory proteins undergo developmental changes in expression remains to be elucidated.
The BAPTA-sensitivity of CDI in immature IHCs (Figs. 7 and 8) contrasts with the notion that Cav1 channel CDI is largely mediated by local Ca2+ influx through single channels.53 In heterologous systems, Cav1.3 CDI is spared by high concentrations (10 mM) intracellular BAPTA due to a molecular determinant in the cytoplasmic N-terminal domain of α11.3 (N-terminal spatial Ca2+ transforming element, NSCaTE).29 Binding of the N-terminal lobe of calmodulin to NSCaTE is thought to alter the Ca2+ binding properties of calmodulin such that CDI is determined by rapid local increases in Ca2+ that cannot be buffered by BAPTA.29 Deletion of NSCaTE from α11.3 transforms the spatial selectivity of Cav1.3 CDI such that it can be blunted by BAPTA.29 While α11.3 splice variants lacking NSCaTE remain to be identified, their presence in immature but not mature IHCs could explain the BAPTA sensitivity of CDI in the former and not the latter. A second possibility relates to the increase in synaptic clustering of Cav1.3 channels in mouse IHCs after hearing onset.38,54 In frog auditory hair cells, synchronous multivesicular release occurs even in the presence of 10 mM BAPTA, which is attributed to a role of the ribbon as a barrier for Ca2+ diffusion.55 Cav1.3 channels anchored near the ribbon may therefore be influenced by rapidly accumulating Ca2+ nanodomains that promote single-channel CDI in mature IHCs. In contrast, a large fraction of Cav1.3 channels are extrasynaptically localized prior to hearing onset,38 where the absence of the ribbon may allow for faster Ca2+ diffusion away from open channels and slower elevations in Ca2+ that can be buffered by BAPTA.
Physiological significance of age-dependent changes in Cav1.3 properties and modulation
The distinct properties of Cav1.3 channels we describe in immature and mature mouse IHCs may support the varying roles of these channels during development. Prior to hearing onset, Cav1.3 channels mediate spontaneous Ca2+-dependent action potentials,11 which support exocytosis12,56 and synaptic activity in the auditory pathway.57 Raising the concentration of Ca2+ in the extracellular recording solution from 1.3 to 5 mM increases the amplitude and accelerates the upstroke and repolarization, and enhances the frequency of the presensory action potentials in mouse IHCs.56 Therefore, the increased ICa density and activation and stronger voltage-dependent activation in the immature compared with in mature IHCs (Fig. 2C and D; Table 1) may be required to maintain the shape and timing of the action potential waveforms. Cav1.3 Ca2+ influx enhances repolarization of the action potential through coupling to the activation of SK Ca2+ activation K+ channels.58 Based on the ability of BAPTA to suppress Cav1.3-dependent activation of SK channels, Cav1.3 channels may be localized ~40 nm from SK channels.58 This is in contrast to the tighter coupling of nicotinic acetylcholine receptors (~13 nm distance),58 which also conduct Ca2+ that activates SK channels.13,59 In this context, the reduced CDI prior to hearing onset (Fig. 4) would allow for more prolonged Ca2+ signals that support SK channel activation and efficient Ca2+ spiking. In addition, sustained ICa in immature IHCs may support activity-dependent gene transcription, a hallmark function of Cav1 channels in the central nervous system.60 Cav1.3 channels are required for normal levels of BK K+ channel transcription in mouse IHCs,16 and BK channel expression in muscle is regulated by the Ca2+-dependent gene transcription factor, nuclear factor of activated T-cells (NFAT3).61 Future studies analyzing gene expression differences in mature and immature IHCs would help identify Ca2+-regulated transcripts that may be important for IHC development.
In mature IHCs, the developmental enhancement of Cav1.3 activation kinetics (Fig. 3) and CDI (Fig. 4) may improve the temporal aspects of sound coding. In paired, pre- and postsynaptic recording at the IHC synapse, faster Cav1.3 activation correlates with decreased latency of postsynaptic responses,62 which supports the ability of auditory nerve fibers to fire at particular times during low-frequency stimuli (phase-locking).63 While Cav1.3 CDI would be expected to limit exocytosis by restricting Ca2+ influx, CDI is expected to contribute modestly to depression at the IHC synapse.62,64,65 Rather, CDI may help shape Ca2+ signals required for appropriate rates of vesicle replenishment at the ribbon64,65 and/or help limit Ca2+ loads that could provoke metabolic stress.
In summary, we have described key functional distinctions in the properties of Cav1.3 channels in immature and mature mouse IHCs. An understanding of how these developmental differences in Cav1.3 Ca2+ signals contribute to the maturation and function of IHCs in the auditory pathway remains an important challenge for future studies.
Materials and Methods
Ethical approval
All procedures were approved by the Institutional Animal Care and Use Committee at the University of Iowa in accordance with National Institutes of Health guidelines.
Preparation of mouse cochlear tissue
C57bl/6 mice (Harlan Laboratories; P6–8 or P16–18 males or females) were euthanized with ketamine (100 mg/kg) and xylazine (9 mg/kg), the skull was opened, and the temporal bones were dissected out and immersed in dissection solution (MEM/Glutamax-1 (Invitrogen) supplemented with 10 mM HEPES) which was preincubated at 37°C in a humidified incubator with 5% CO2 . The apical turn was dissected in fresh dissection solution and the spiral ligament removed. The tissue was then secured under a glass pin on a 15 mm round coverslip in dissection solution and incubated for 2–5 h at 37°C in a humidified incubator with 5% CO2 prior to recording.
Electrophysiological recordings
IHCs in the apical cochlear turn were visualized on an upright microscope (BX51WI, Olympus) with a 40X water-immersion objective with DIC optics. The basolateral membrane of IHCs was subject to whole-cell patch clamp recording with electrodes pulled from thick-walled borosilicate glass capillaries (1B150F, Warner Instruments). Before recording, the viability of the IHCs was confirmed by the following morphological features: uniform cell shape with a narrow neck, basal location of the nucleus, membrane birefringence, and intact stereocilia. The internal solution contained (in mM): 100 Cs-gluconate, 30 TEA-Cl, 1 MgCl2, 4 MgATP, 0.3GTP, 5 HEPES and 2 EGTA; pH was adjusted to 7.35 with CsOH; osmolarity~305 mOsm. In some experiments, 10 mM BAPTA was substituted for 2 EGTA and Cs-gluconate was reduced to 92 mM. External solution contained (in mM): 105 NaCl, 2.8 KCl, 3 CsCl, 35 TEA-Cl, 5 CaCl2 or BaCl2, 1 MgCl2, 10 glucose, and 10 HEPES supplemented with MEM Vitamins and Amino Acids at 1X; pH was adjusted to 7.4 with TEA-OH; osmolarity~310 mOsm. Under these conditions, the free intracellular Ca2+ was estimated to be less than 0.5 nM (with 2 mM intracellular EGTA) or less than 0.1 nM (with 10 mM intracellular BAPTA). On the day of recording, 4-aminopyridine (10 mM), apamin (0.3 mM) and TTX (0.5 mM, for P6–8 IHCs) were added to the external solution. Electrode resistances were 3–5 MΩ in the external solution. Data were acquired at room temperature with a HEKA EPC-10 amplifier controlled by Patchmaster software (HEKA Elektronik). Leak subtraction was done online with a P/8 protocol. Series resistance was compensated with the patch clamp circuitry (50–70%); average uncompensated series resistance was 9.64 ± 0.38 (n = 121). Currents were low-pass filtered at 5 kHz and sampled at 20 kHz. Voltages were not corrected for the liquid junction potential of −7 mV in the external recording solution.
Electrophysiological data were analyzed with custom routines in IgorPro software (Wavemetrics). Current-voltage (I-V) relationships were fit with the equation: I = [Gmax (V − Vrev)]/ [1 + exp [(Vh − V)/k], where I is the current, Gmax is maximum chord conductance, V is voltage, Vrev is reversal potential, Vh is half-maximal activation voltage, and k is the slope factor. Conductance-voltage relationships were fit with the equation; G/Gmax = 1/ [1 + exp [(Vh − V)/k]]. Time constants for activation and parameters for recovery from inactivation were obtained by fitting with the equation: I(t) = yo + A[exp (−t/τ)], where I(t) is current at time = t, y0 is the offset (asymptote), A is the amplitude, and τ is the time constant. The onset of ICa inactivation was fit with a double exponential function: I(t) = y0 + Afast [exp (−t/τ fast)] + Aslow [exp (−t/τ slow)]where I(t) is current at time = t, y0 is the offset (asymptote), Afast and Aslow are the amplitudes, and τ fast and τ slow are the time constants. Average data are expressed as mean ± SEM. Statistical comparisons were done as indicated using SigmaPlot software (Systat).
Supplementary Material
Acknowledgments
Support was provided by the NIH (DC009433, HL087120 to A.L. and DC010362 for support of the Iowa Center for Molecular Auditory Neuroscience) and a Carver Research Program of Excellence Award to A.L.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/24104
References
- 1.Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, et al. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–5. doi: 10.1016/S0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- 2.Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, et al. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α 1 subunits. J Cell Biol. 1993;123:949–62. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finkbeiner S, Greenberg ME. Ca2+ channel-regulated neuronal gene expression. J Neurobiol. 1998;37:171–89. doi: 10.1002/(SICI)1097-4695(199810)37:1<171::AID-NEU13>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 4.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. doi: 10.1016/0166-2236(95)93882-X. [DOI] [PubMed] [Google Scholar]
- 5.Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel α 1 subunit. Neuron. 1992;9:1099–115. doi: 10.1016/0896-6273(92)90069-P. [DOI] [PubMed] [Google Scholar]
- 6.Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP, et al. Immunochemical identification and subcellular distribution of the α 1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–18. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuchs PA, Evans MG, Murrow BW. Calcium currents in hair cells isolated from the cochlea of the chick. J Physiol. 1990;429:553–68. doi: 10.1113/jphysiol.1990.sp018272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts WM, Jacobs RA, Hudspeth AJ. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J Neurosci. 1990;10:3664–84. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, et al. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/S0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 10.Dou H, Vazquez AE, Namkung Y, Chu H, Cardell EL, Nie L, et al. Null mutation of α1D Ca2+ channel gene results in deafness but no vestibular defect in mice. J Assoc Res Otolaryngol. 2004;5:215–26. doi: 10.1007/s10162-003-4020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brandt A, Striessnig J, Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci. 2003;23:10832–40. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beutner D, Moser T. The presynaptic function of mouse cochlear inner hair cells during development of hearing. J Neurosci. 2001;21:4593–9. doi: 10.1523/JNEUROSCI.21-13-04593.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glowatzki E, Fuchs PA. Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science. 2000;288:2366–8. doi: 10.1126/science.288.5475.2366. [DOI] [PubMed] [Google Scholar]
- 14.Johnson SL, Marcotti W, Kros CJ. Increase in efficiency and reduction in Ca2+ dependence of exocytosis during development of mouse inner hair cells. J Physiol. 2005;563:177–91. doi: 10.1113/jphysiol.2004.074740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kros CJ, Ruppersberg JP, Rüsch A. Expression of a potassium current in inner hair cells during development of hearing in mice. Nature. 1998;394:281–4. doi: 10.1038/28401. [DOI] [PubMed] [Google Scholar]
- 16.Nemzou N RM, Bulankina AV, Khimich D, Giese A, Moser T. Synaptic organization in cochlear inner hair cells deficient for the CaV1.3 (alpha1D) subunit of L-type Ca2+ channels. Neuroscience. 2006;141:1849–60. doi: 10.1016/j.neuroscience.2006.05.057. [DOI] [PubMed] [Google Scholar]
- 17.Grant L, Fuchs P. Calcium- and calmodulin-dependent inactivation of calcium channels in inner hair cells of the rat cochlea. J Neurophysiol. 2008;99:2183–93. doi: 10.1152/jn.01174.2007. [DOI] [PubMed] [Google Scholar]
- 18.Johnson SL, Marcotti W. Biophysical properties of CaV1.3 calcium channels in gerbil inner hair cells. J Physiol. 2008;586:1029–42. doi: 10.1113/jphysiol.2007.145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–60. doi: 10.1016/S0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- 20.Christel C, Lee A. Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim Biophys Acta. 2012;1820:1243–52. doi: 10.1016/j.bbagen.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang PS, Alseikhan BA, Hiel H, Grant L, Mori MX, Yang W, et al. Switching of Ca2+-dependent inactivation of Ca(v)1.3 channels by calcium binding proteins of auditory hair cells. J Neurosci. 2006;26:10677–89. doi: 10.1523/JNEUROSCI.3236-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui G, Meyer AC, Calin-Jageman I, Neef J, Haeseleer F, Moser T, et al. Ca2+-binding proteins tune Ca2+-feedback to Cav1.3 channels in mouse auditory hair cells. J Physiol. 2007;585:791–803. doi: 10.1113/jphysiol.2007.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiaho F, Nargeot J, Richard S. Repriming of L-type calcium currents revealed during early whole-cell patch-clamp recordings in rat ventricular cells. J Physiol. 1993;463:367–89. doi: 10.1113/jphysiol.1993.sp019599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cota G, Stefani E. Saturation of calcium channels and surface charge effects in skeletal muscle fibres of the frog. J Physiol. 1984;351:135–54. doi: 10.1113/jphysiol.1984.sp015238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Safa P, Boulter J, Hales TG. Functional properties of Cav1.3 (alpha1D) L-type Ca2+ channel splice variants expressed by rat brain and neuroendocrine GH3 cells. J Biol Chem. 2001;276:38727–37. doi: 10.1074/jbc.M103724200. [DOI] [PubMed] [Google Scholar]
- 26.Wang CL. A note on Ca2+ binding to calmodulin. Biochem Biophys Res Commun. 1985;130:426–30. doi: 10.1016/0006-291X(85)90434-6. [DOI] [PubMed] [Google Scholar]
- 27.Gregory FD, Bryan KE, Pangršič T, Calin-Jageman IE, Moser T, Lee A. Harmonin inhibits presynaptic Cav1.3 Ca²⁺ channels in mouse inner hair cells. Nat Neurosci. 2011;14:1109–11. doi: 10.1038/nn.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tadross MR, Dick IE, Yue DT. Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell. 2008;133:1228–40. doi: 10.1016/j.cell.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dick IE, Tadross MR, Liang H, Tay LH, Yang W, Yue DT. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–4. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee A, Scheuer T, Catterall WA. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci. 2000;20:6830–8. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, et al. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–9. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- 32.DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–9. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- 33.Kreiner L, Lee A. Endogenous and exogenous Ca2+ buffers differentially modulate Ca2+-dependent inactivation of Ca(v)2.1 Ca2+ channels. J Biol Chem. 2006;281:4691–8. doi: 10.1074/jbc.M511971200. [DOI] [PubMed] [Google Scholar]
- 34.Soong TW, DeMaria CD, Alvania RS, Zweifel LS, Liang MC, Mittman S, et al. Systematic identification of splice variants in human P/Q-type channel α1(2.1) subunits: implications for current density and Ca2+-dependent inactivation. J Neurosci. 2002;22:10142–52. doi: 10.1523/JNEUROSCI.22-23-10142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schnee ME, Ricci AJ. Biophysical and pharmacological characterization of voltage-gated calcium currents in turtle auditory hair cells. J Physiol. 2003;549:697–717. doi: 10.1113/jphysiol.2002.037481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hille B, ed. Ionic channels of excitable membranes, 2nd ed. Sunderland, MA: Sinauer Associates Inc., 1992. [Google Scholar]
- 37.Rodríguez-Contreras A, Yamoah EN. Effects of permeant ion concentrations on the gating of L-type Ca2+ channels in hair cells. Biophys J. 2003;84:3457–69. doi: 10.1016/S0006-3495(03)70066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zampini V, Johnson SL, Franz C, Lawrence ND, Münkner S, Engel J, et al. Elementary properties of CaV1.3 Ca(2+) channels expressed in mouse cochlear inner hair cells. J Physiol. 2010;588:187–99. doi: 10.1113/jphysiol.2009.181917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martini M, Rossi ML, Rubbini G, Rispoli G. Calcium currents in hair cells isolated from semicircular canals of the frog. Biophys J. 2000;78:1240–54. doi: 10.1016/S0006-3495(00)76681-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamaoka K, Yuki T, Kawase K, Munemori M, Seyama I. Temperature-sensitive intracellular Mg2+ block of L-type Ca2+ channels in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2002;282:H1092–101. doi: 10.1152/ajpheart.00585.2001. [DOI] [PubMed] [Google Scholar]
- 41.Tan BZ, Jiang F, Tan MY, Yu D, Huang H, Shen Y, et al. Functional characterization of alternative splicing in the C terminus of L-type CaV1.3 channels. J Biol Chem. 2011;286:42725–35. doi: 10.1074/jbc.M111.265207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu W, Lipscombe D. Neuronal Ca(V)1.3α(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–51. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klugbauer N, Welling A, Specht V, Seisenberger C, Hofmann F. L-type Ca2+ channels of the embryonic mouse heart. Eur J Pharmacol. 2002;447:279–84. doi: 10.1016/S0014-2999(02)01850-2. [DOI] [PubMed] [Google Scholar]
- 44.Lieb A, Scharinger A, Sartori S, Sinnegger-Brauns MJ, Striessnig J. Structural determinants of CaV1.3 L-type calcium channel gating. Channels (Austin) 2012;6:197–205. doi: 10.4161/chan.21002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bock G, Gebhart M, Scharinger A, Jangsangthong W, Busquet P, Poggiani C, et al. Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J Biol Chem. 2011;286:42736–48. doi: 10.1074/jbc.M111.269951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, et al. α 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001;276:22100–6. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- 47.Shen Y, Yu D, Hiel H, Liao P, Yue DT, Fuchs PA, et al. Alternative splicing of the Ca(v)1.3 channel IQ domain, a molecular switch for Ca2+-dependent inactivation within auditory hair cells. J Neurosci. 2006;26:10690–9. doi: 10.1523/JNEUROSCI.2093-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh A, Gebhart M, Fritsch R, Sinnegger-Brauns MJ, Poggiani C, Hoda JC, et al. Modulation of voltage- and Ca2+-dependent gating of CaV1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J Biol Chem. 2008;283:20733–44. doi: 10.1074/jbc.M802254200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang H, Tan BZ, Shen Y, Tao J, Jiang F, Sung YY, et al. RNA editing of the IQ domain in Ca(v)1.3 channels modulates their Ca²⁺-dependent inactivation. Neuron. 2012;73:304–16. doi: 10.1016/j.neuron.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calin-Jageman I, Lee A. Ca(v)1 L-type Ca2+ channel signaling complexes in neurons. J Neurochem. 2008;105:573–83. doi: 10.1111/j.1471-4159.2008.05286.x. [DOI] [PubMed] [Google Scholar]
- 51.Gebhart M, Juhasz-Vedres G, Zuccotti A, Brandt N, Engel J, Trockenbacher A, et al. Modulation of Cav1.3 Ca2+ channel gating by Rab3 interacting molecule. Mol Cell Neurosci. 2010;44:246–59. doi: 10.1016/j.mcn.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 52.Song H, Nie L, Rodriguez-Contreras A, Sheng ZH, Yamoah EN. Functional interaction of auxiliary subunits and synaptic proteins with Ca(v)1.3 may impart hair cell Ca2+ current properties. J Neurophysiol. 2003;89:1143–9. doi: 10.1152/jn.00482.2002. [DOI] [PubMed] [Google Scholar]
- 53.Yue DT, Backx PH, Imredy JP. Calcium-sensitive inactivation in the gating of single calcium channels. Science. 1990;250:1735–8. doi: 10.1126/science.2176745. [DOI] [PubMed] [Google Scholar]
- 54.Brandt A, Khimich D, Moser T. Few CaV1.3 channels regulate the exocytosis of a synaptic vesicle at the hair cell ribbon synapse. J Neurosci. 2005;25:11577–85. doi: 10.1523/JNEUROSCI.3411-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Graydon CW, Cho S, Li GL, Kachar B, von Gersdorff H. Sharp Ca²⁺ nanodomains beneath the ribbon promote highly synchronous multivesicular release at hair cell synapses. J Neurosci. 2011;31:16637–50. doi: 10.1523/JNEUROSCI.1866-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marcotti W, Johnson SL, Rusch A, Kros CJ. Sodium and calcium currents shape action potentials in immature mouse inner hair cells. J Physiol. 2003;552:743–61. doi: 10.1113/jphysiol.2003.043612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tritsch NX, Rodríguez-Contreras A, Crins TT, Wang HC, Borst JG, Bergles DE. Calcium action potentials in hair cells pattern auditory neuron activity before hearing onset. Nat Neurosci. 2010;13:1050–2. doi: 10.1038/nn.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marcotti W, Johnson SL, Kros CJ. A transiently expressed SK current sustains and modulates action potential activity in immature mouse inner hair cells. J Physiol. 2004;560:691–708. doi: 10.1113/jphysiol.2004.072868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oliver D, Klöcker N, Schuck J, Baukrowitz T, Ruppersberg JP, Fakler B. Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron. 2000;26:595–601. doi: 10.1016/S0896-6273(00)81197-6. [DOI] [PubMed] [Google Scholar]
- 60.Ma H, Cohen S, Li B, Tsien RW. Exploring the dominant role of Cav1 channels in signalling to the nucleus. Biosci Rep. 2012;33 doi: 10.1042/BSR20120099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Layne JJ, Werner ME, Hill-Eubanks DC, Nelson MT. NFATc3 regulates BK channel function in murine urinary bladder smooth muscle. Am J Physiol Cell Physiol. 2008;295:C611–23. doi: 10.1152/ajpcell.00435.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goutman JD. Transmitter release from cochlear hair cells is phase locked to cyclic stimuli of different intensities and frequencies. J Neurosci. 2012;32:17025–35a. doi: 10.1523/JNEUROSCI.0457-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rose JE, Brugge JF, Anderson DJ, Hind JE. Phase-locked response to low-frequency tones in single auditory nerve fibers of the squirrel monkey. J Neurophysiol. 1967;30:769–93. doi: 10.1152/jn.1967.30.4.769. [DOI] [PubMed] [Google Scholar]
- 64.Moser T, Beutner D. Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc Natl Acad Sci U S A. 2000;97:883–8. doi: 10.1073/pnas.97.2.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cho S, Li GL, von Gersdorff H. Recovery from short-term depression and facilitation is ultrafast and Ca2+ dependent at auditory hair cell synapses. J Neurosci. 2011;31:5682–92. doi: 10.1523/JNEUROSCI.5453-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.