

Abstract
The process of transcription initiation is the major target for regulation of gene expression in bacteria and is performed by a multi-subunit RNA polymerase enzyme (RNAp). A complex network of regulatory elements controls the activity of the RNAp to fine-tune transcriptional output. Thus, RNAp is a nexus for controlling bacterial gene expression at the transcription level. Many bacteriophages, viruses that infect bacteria, encode transcription factors that specifically target and modulate the activity of the host RNAp and, thereby, facilitate the acquisition of the host bacteria by the phage. Here, we describe the modus operandi of a T7 bacteriophage-encoded small protein called Gp2 and define Gp2 as a non-bacterial regulator of bacterial transcription.
Keywords: Gp2, RNA polymerase, T7 inhibition, bacterial transcription regulation, bacteriophage, σ factor
Introduction
Central to genetic regulation is transcription, the first step of gene expression at which DNA-directed synthesis of RNA occurs. Transcription regulation is key to developmental plasticity, homeostasis, adaptation and, ultimately, cell viability. The molecular machine responsible for all cellular RNA synthesis is the DNA-dependent RNA polymerase (RNAp). Controlling the activity of RNAp is central to the coordinated and temporal regulation of gene expression. Unlike eukaryotic genomes, all bacterial genomes encode a single multi-subunit RNAp, which is responsible for all cellular RNA synthesis. There appears to be a limited supply of RNAp in bacterial cells1 and a variety of mechanisms have evolved to modulate the activity of RNAp to fine-tune gene expression in response to environmental and intracellular changes.
The regulation of transcription predominantly occurs at the transcription initiation stage. For RNA synthesis to begin at a bacterial promoter, the catalytic five-subunit RNAp core (E, subunit composition α2ββ’ω) must reversibly associate with a promoter-specificity σ factor subunit to form an RNAp holoenzyme (Eσ). The σ factor confers promoter specificity upon the core RNAp. Most bacteria have several different σ factors, which direct the RNAp to the transcription of specific sets of genes and thereby help “program” the bacterial transcription machinery to adapt gene expression to suit a particular need. For example, in Escherichia coli, in which the mechanisms of transcription regulation are best-studied, the ratio of RNAp holoenzymes formed by the seven σ factors encoded in the genome controls gene expression pattern changes caused by environmental cues, such as elevated temperature or nutrient deprivation.1,2
All bacteria contain one primary σ factor that is essential for growth and is closely related to the primary σ70 of E. coli. E. coli RNAp containing σ70 (Eσ70) is responsible for transcription of “housekeeping” genes during exponential growth. A hallmark feature of primary σ factors is the unstructured, highly negatively charged and multifunctional N-terminal domain known as region 1.1 (R1.1). In addition to the primary σ factor, alternative σ factors are responsible for transcription of genes with functions associated with stress response, development and auxiliary metabolism.3,4 Modulation of σ factor activity and availability, accomplished, for example, through the binding of anti-σ factors,5 adds to the repertoire of mechanisms by which bacterial transcription is regulated and further illustrates the importance of σ factors in bacterial transcription regulation. Thus, the σ factor composition of the RNAp represents the primary mechanism by which bacterial transcription is regulated.
The initial Eσ70-promoter complex, called the “closed” promoter complex (RPc) is transcriptionally inactive and must undergo large-scale conformational changes to form the transcriptionally active “open” promoter complex (RPo). In the RPo, the promoter DNA strands are locally melted, which results in a “transcription bubble” and the transcription start site on the template DNA strand is positioned at the RNAp active center (Fig. 1).6,7 An obligatory step for RPo formation to occur and to stably maintain the transcription bubble is the interaction between the double-stranded DNA downstream of the active center (dwDNA) with a structural feature of RNAp called the downstream DNA-binding channel (dwDBC), a part of the main DNA-binding channel (DBC) in the RNAp.7,8 However, in Eσ70, R1.1 occupies the dwDBC in the RPc and, therefore, for RPo to form R1.1 must be displaced from the dwDBC.9 In addition, during RPo formation, access to the main DNA-binding channel and dwDBC is controlled by large-scale movements of the β’ subunit: thus, with respect to accessibility of the DBC, RNAp can exist in a so-called “closed state” (in which the width of the DBC is insufficient to allow access of double-stranded DNA) or in an “open state” (in which the DBC is sufficiently wide to allow access of double-stranded DNA). The open state is required for RPo formation; however, once the DNA is loaded and unwound to form the transcription bubble, RNAp converts into the closed state, “locking” onto DNA.10
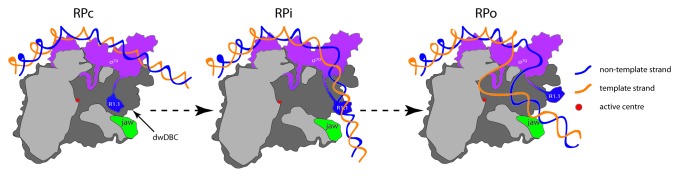
Figure 1. Cartoon depiction of the conversion of RPc to RPo (via intermediate complexes, RPi) at a typical σ70-dependent bacterial promoter (adapted from Haugen SP, Ross W, Gourse RL, 2008 and Murakami KS and Darst SA, 2003).6,42
In addition to σ factors, numerous transcription factors (TF) interact with the RNAp to ensure that the correct gene is expressed at an appropriate time and in the required amounts.11,12 The majority of bacterial TFs are DNA-binding proteins, which bind to specific regulatory sites, often located adjacent to promoters, and either interact with RNAp to facilitate the formation of the RPc and/or RPo or sterically prevent RNAp from binding to the promoter.11,12 A small subset of TFs affects transcription initiation in the absence of DNA binding and interacts with the RNAp directly.13,14 Low-molecular weight RNAp binding ligands6 and post-translational modification15 of the RNAp extend the repertoire of regulators that control bacterial gene expression at the transcriptional level. Not surprisingly, bacteriophages (phages), which are viruses that infect bacteria, encode specific TFs that modulate host transcription to favor phage development and/or for the transcription of the phage genome. We refer readers to comprehensive reviews by Nechaev and Severinov on the subject.16,17 Some phage-encoded TFs are potent inhibitors of the bacterial RNAp. Here, we discuss our current knowledge of one such inhibitor, called Gp2, which is encoded by the E. coli phage, T7. Our emerging understanding of the mechanism by which Gp2 inhibits the RNAp in a σ factor-dependent manner suggests that Gp2 is not a simple inhibitor of the bacterial RNAp but could also help reprogram bacterial transcription to favor T7 infection and progeny development.
The biological role of Gp2 during T7 phage infection of E. coli
Gp2, the 7 kDa product of T7 middle gene 2, was first isolated in 1974 by Hesselbach and Nakada from inactive RNAp purified from T7 infected E. coli cells and was later shown to be a potent inhibitor of E. coli Eσ70 in vitro using recombinant Gp2 and RNAp purified from non-infected host cells.18-21 Charge reversal amino acid substitutions at E1158 or E1188 in rpoC (gene encoding the catalytic β’ subunit of the RNAp) are non-permissive for T7 development in E. coli and define the binding site of Gp2, in the β’ jaw domain, a structurally flexible RNAp feature that contributes to the dwDBC.22
Gp2 plays an essential regulatory role during infection of E. coli by preventing interference between bacterial RNAp and single-subunit T7-encoded RNAp during transcription of the viral genome. Following adsorption of viral particles onto the E. coli cell, T7 phage injects only about ~0.9 kb of its ~40 kb genome into the cell.23 Entry of the remaining part of the genome occurs by a transcription-dependent process involving first the E. coli and then T7 RNAp.24 Translocation of the first 7 kb of the T7 genome into the E. coli cell is dependent upon bacterial RNAp that “pulls in” viral DNA as it transcribes from three strong Eσ70-dependent early promoters (A1–3).25 This region of the T7 genome contains all the early T7 genes, including gene 1, which encodes the T7 RNAp. The T7 RNAp then internalizes the remaining 33 kb of T7 DNA as it transcribes the middle and late T7 genes.26 Gp2, one of the first middle T7 proteins to be synthesized, functions to inhibit the E. coli RNAp so that it is unable to transcribe the middle and late regions of the T7 genome.27 The exact mechanism by which the E. coli RNAp transcription of middle and late T7 genes interferes with productive infection is not known; however, the most plausible model by which this could occur, proposed by Qimron et al., suggests that continuous transcription by the slow-moving bacterial RNAp into regions of the T7 genome normally transcribed by the fast-moving T7 RNAp causes the latter to pause inadvertently. A paused T7 RNAp elongation complex recruits phage DNA packaging machinery to aberrant sites, leading to production of less-than-unit length phage genomes.28 Consistent with this model, when Gp2 is rendered non-functional, either by mutation in gene 2 or by alteration of the Gp2-binding site in the β’ jaw domain, T7 infection of E. coli becomes arrested late in infection due to formation of defective viral particles.29-33
Does Gp2 regulate transcription of E. coli genes?
The effect of Gp2 on the transcription of E. coli genes is not known. It is conceivable that Gp2, instead of fully abolishing all host transcription, strategically modulates the transcription of E. coli genes in order to provide optimal conditions for the acquisition of E. coli by T7. The σ factor-specific inhibition of the E. coli RNAp by Gp2 (see below) is consistent with a view that the inhibition of E. coli RNAp by Gp2 could occur in a specific manner to benefit the phage. Plasmid-borne recombinant Gp2 produced in E. coli in the absence of T7 infection acts as a bacteriostatic agent.34 Microarray analysis (using Agilent microarrays consisting of 44,000 60-mer oligonucleotide probes that match E. coli MG1655 sequences at intervals of ~160 base pairs) of total bacterial RNA isolated at different times post-induction of Gp2 expression reveals that Gp2 does not fully abolish transcription in E. coli (even though the total number of Gp2 molecules per E. coli cell exceeds that of the total number of RNAp molecules by at least 2-fold under the assay conditions34). In fact, the transcript abundances of a total of 292, 535 and 1,067 genes are significantly differentially expressed (FDR corrected p value < 0.05, > 2-fold difference in expression level compared with control in which a functionally defective mutant version of Gp2 is expressed) at 30, 60 and 120 min, respectively, post-induction of Gp2 (Fig. 2A). Of the differentially expressed genes, approximately equal numbers are up- and downregulated at each time point. As can be seen from a Venn diagram presented in Figure 2B, 103 E. coli genes are commonly modulated by Gp2 over all three time points tested (Fig. 2B). It therefore seems that Gp2 is more than a general inhibitor of E. coli transcription and the specificity of its inhibitory action might allow T7 to successfully acquire the bacterial cell. The gene and regulon specificity of Gp2-mediated modulation of transcription in E. coli in the absence of T7 infection and its potential role during T7 infection deserves further attention and will be reported elsewhere. However, the degradation of E. coli genome by T7 middle genes 3 and 6 products (which encode an endo- and exonuclease, respectively) renders Gp2-mediated regulation of host genes doubtful or at best leaves a very small window of opportunity for such regulation.

Figure 2. (A) Bar chart displaying the number of E. coli genes which are up- or downregulated at 0, 30, 60 and 120 min post-induction of Gp2 expression. (B) Venn diagram illustrating the number of genes that are differentially expressed at 30, 60 and 120 post-induction of Gp2 expression.
Is the activity of T7 Gp2 (and that of its homologs) regulated during infection?
Gp2 homologs are found in several phages that belong to the Autographivirinae subfamily (formally known as the “T7 supergroup”), which are similar to T7 in their genomic organization and likely share a common strategy of infection. A recent communication by Klimuk et al. identified two Gp2 homologs (called Gp36 and Gp25.1) in Pseudomonas aeruginosa-infecting Autographivirinae subfamily ΦKMV-like phages that have an unusual N-terminal extension (Fig. 3A).35 Unlike T7 Gp2, recombinant forms of wild-type Gp36 and Gp25.1 are unable to inhibit the bacterial RNAp. However, removal of the N-terminal extension unmasks the ability of Gp36 and Gp25.1 to inhibit RNAp.35 Klimuk et al. propose that N-terminal extensions of Gp36 and Gp25.1 could represent receiver-like regulatory modules, which could allow these proteins to inhibit the bacterial RNAp at a precise time during infection. The nature of the signals that leads to this hypothetical regulation of Gp2 homologs Gp36 and Gp25.1 remains unidentified. It is also possible that host proteins can also influence the functionality of Gp2 during infection: Qimron et al. reported that overexpression of E. coli gene udk (which encodes an uridine/cytidine kinase) is non-permissive for T7 infection of E. coli.28 Shadrin et al. recently showed that overexpression of udk reduces the half-life of Gp2 in E. coli and in fact mimics the absence of Gp2 during T7 infection.36 In summary, given the essential nature of Gp2 and its homologs in T7 and other phages for successful acquisition of the bacterial host, it is conceivable that Gp2 and its homologs are subjected to some level of regulation during infection. However, the mechanisms that govern this regulation remain, as yet, elusive.

Figure 3. (A) Sequence alignments of putative Gp2 homologs prepared using GENEDOC software, with T7 Gp2 and LKD16 Gp25b as queries. Sequences are displayed using the single amino acid code. The intensity of the background corresponds to the degree of conservation and the highly conserved R56 and R58 residues are highlighted in blue. The secondary structure features of T7 Gp2 are boxed and labeled above (see Fig. 3B). (B) Ribbon representation of the Gp2-β′ jaw fragment complex. Gp2 is shown in cyan and the β’ jaw fragment in green. The R56 and R58 residues of Gp2 and the E1188 residue of the β’ jaw, which are important for binding, are colored blue and red respectively and shown in stick representation. The amino acid residues of the NCS (E21, E34, D37, E38, E41, E44 and E53) in Gp2 and the extension of negatively charged residues (E1158, D1181, D1184, E1187 and E1188) in the β’ jaw are colored red. Gp2 residues E24 and F27 are shown in stick representation.
T7 Gp2: Insights from structure-function studies
A systematic alanine scanning mutagenesis analysis of T7 Gp2 revealed that two arginine residues, R56 and R58, are important for binding to and inhibition of E. coli Eσ70.37 Indeed, multiple protein sequence alignment of known Gp2 homologs (in the EBI database, January 2013) shows that R56 and R58 are identical in 33 and 26, respectively, out of 33 known Gp2 homologs (Fig. 3a). In the solution structure of Gp2, R56 and R58 are located on the β3 strand and are surface-exposed.37 In the structure of a complex of Gp2 with a fragment of the β’ jaw domain, the primary interface region is localized to the β3 strand of Gp2 and the two invariant arginine residues in Gp2 are located in the interface region in close proximity to amino acid E1188 in the β’ jaw domain38 (recall that alanine or charge reversal substitutions at E1188 prevents Gp2 from binding to the E. coli RNAp), thus providing a favorable ionic interaction across the interface (Fig. 3B. There is a conserved contiguous strip of seven negatively charged amino acids in Gp2, referred to as the negatively charged strip (NCS), on the side of the molecule opposing R56 and R58 (Fig. 3B). Analysis of the role of the NCS by mutagenesis reveals that the NCS is not important for the binding of Gp2 to E. coli RNAp, but the disruption of the NCS significantly attenuates the ability of Gp2 to inhibit RPo formation.39 An examination of the surface electrostatic properties of the Gp2-β’ jaw domain complex reveals that the NCS in Gp2 is extended by several negatively charged residues of the β’ jaw domain, thus underscoring the importance of the negatively charged patch in the mechanism by which Gp2 inhibits transcription initiation by the E. coli RNAp.38
As noted above, wild-type Gp2 is unable to inhibit the E. coli RNAp harboring the E1188K substitution. However, T7 phage carrying a triple mutation in gene 2 causing amino acid substitutions E24K, F27Y and R56C is able to productively infect cells carrying the E1188K substitution in the RNAp β’ subunit.28 In the context of the Gp2-β’ jaw domain structure, the E24K and F27Y substitutions are located in the middle and close to the end, respectively, of the loop connecting the β1 and β2 strands in Gp2, i.e., at the opposite side to the β3 strand, which contains the essential and invariant arginine residues R56 and R58 (Fig. 3B). Shadrin et al. demonstrated that the E24K and F27Y mutations facilitate the interaction between Gp2 and E. coli RNAp when the primary interaction interface between Gp2 and E. coli RNAp (i.e., β’ jaw domain) becomes compromised.34 Thus, it seems that the interface between Gp2 and the E. coli RNAp is at least bipartite and is made up of a primary interaction with the β’ jaw domain and an auxiliary interaction interface elsewhere on the RNAp. Together, these interfaces contribute to the very high affinity between Gp2 and the RNAp and the mechanism of RNAp inhibition by Gp2.
The mechanism of E. coli RNAp inhibition by T7 Gp2—a multipronged strategy
It has been long known that Gp2 inhibits transcription initiation by the E. coli RNAp and that once the transcriptionally active RPo has formed, Gp2 is unable to bind to the E. coli RNAp.22 However, the precise step(s) inhibited by Gp2 during transcription initiation remained elusive. Several recent studies have significantly advanced our understanding of the precise mechanism of action of Gp2. Gp2 employs a multipronged strategy to inhibit Eσ70 by antagonizing several obligatory events en route to RPo formation (see above and Fig. 4A)10,34,37-40: (1) Gp2 sterically prevents the interaction between dwDNA and the β’ jaw domain, which is important for the formation and maintenance of the transcription bubble. The binding of Gp2 and dwDNA to the β’ jaw domain are mutually exclusive events, thus explaining why Gp2 cannot bind to the E. coli RNAp once the RPo has formed.40 In addition to steric hindrance, the extended negatively charged patch formed at the Gp2-β’ jaw domain interface may electrostatically repel the incoming DNA from binding in the dwDBC during RPo formation. (2) Gp2 appropriates R1.1 of σ70 to increase the efficiency of RPo formation inhibition. We envisage a model in which Gp2 repositions R1.1 and/or stabilizes R1.1 so that it can no longer be displaced from the dwDBC (recall that the displacement of R1.1 from the dwDBC is obligatory for RPo formation). The extended negatively charged patch of Gp2 may reposition negatively charged R1.1 and/or mimic the presence of R1.1 in the dwDBC. It is not known if Gp2 directly interacts with R1.1 and experiments are currently underway to address this. (3) The binding of Gp2 to the β’ jaw domain also results in a long-range, R1.1-dependent antagonistic effect on E. coli RNAp interactions with DNA around the RNAp active center. (4) Gp2 restricts the conformational flexibility in the E. coli RNAp that normally accompany RPo formation and induces RNAp to adopt a “closed state” conformation, thereby restricting even single-stranded DNA access to the RNAp active site. We envisage a model in which, when Gp2 is bound to the β’ jaw domain, the region surrounding and including the loop interconnecting the β1 and β2 of Gp2 interacts with the β subunit (which is located directly across from the β’ jaw domain on opposing side of the DBC) and, thereby, locks the E. coli RNAp in the “closed state” conformation (Fig. 4B).10

Figure 4. (A) Cartoon depiction (as in Fig. 1) of the multipronged strategy employed by Gp2 to inhibit transcription initiation by Eσ70. For interpretation of the numbering refer to the main text. (B) Cartoon illustrating how additional interactions of Gp2 with the β subunit lock RNAp in a ‘closed conformation.”
The multiple strategies used by Gp2 to inhibit transcription are best adapted to inhibit the RNAp associated with the housekeeping σ70 factor. This is not surprising since a key biological role of Gp2 is to inhibit Eσ70-dependent transcription initiation from the early A3 promoter.27 Consistent with the central role for σ70 R1.1 in the mechanism of inhibition of Eσ70, even though Gp2 binds to E. coli RNAp containing σ38 (Eσ38) and σ54 (Eσ54) with similar affinity and specificity as to Eσ70, Eσ38 and Eσ54 transcription is inhibited, respectively, poorly or not at all by Gp2.38,41 At σ54-dependent promoters, RPo formation depends upon the ATP-hydrolysis-dependent remodeling of the Eσ54 RPc by a specialized activator ATPase. Gp2 dissociates from the Eσ54 RPc during the ATP-hydrolysis-dependent conformational changes that accompany Eσ54 RPo formation.41 Thus, it seems that during RPo formation by Eσ54, the reaction equilibrium favors the formation of interactions between the β’ jaw domain and dwDNA when the activator ATPase drives forward RPo formation and, thereby, compromises Gp2 - β’ jaw interactions that results in the dissociation of Gp2 from Eσ54.
Perspectives
Our studies on Gp2 have provided novel and unexpected insights into how transcription in E. coli is regulated by a non-bacterial TF and, thereby, uncovered new mechanisms by which the activity of the bacterial RNAp can be controlled. Owing to its small size, strong binding affinity to and multipronged mechanism of inhibition of the E. coli RNAp, it is conceivable that Gp2 could serve as an excellent platform for the development of new lead antibacterial compounds. Along similar lines, our global gene expression studies indicate that Gp2, in the absence of a T7 infection, functions like a bacteriostatic agent by strategically modulating transcription in E. coli. Therefore, genes that are selectively inhibited by Gp2 could potentially serve as novel antibacterial targets whose disruption could destabilize essential processes and lead to attenuation of bacterial growth. Research aligned to the latter two “uses” of Gp2 is currently underway in our laboratories. Since phages represent an absolute majority of all biological entities in the biosphere, emerging multidisciplinary studies on how phage encoded TFs, like Gp2, function will continue to empower and expand our knowledge of the mechanisms that govern bacterial transcription regulation and contribute to uncovering new paradigms of genetic control in bacteria.
Acknowledgments
This work is supported by grants from the BBSRC and Wellcome Trust to S.W. and NIH grant GM59295 and Molecular and Cellular Biology Program of the Russian Academy of Sciences to K.S. S.W is also a recipient of a BBSRC David Phillips Fellowship (BB/E023703). We are grateful to Andrey Shadrin and Daniel Brown for comments and useful suggestions on the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/24283
References
- 1.Ishihama A. Functional modulation of Escherichia coli RNA polymerase. Annu Rev Microbiol. 2000;54:499–518. doi: 10.1146/annurev.micro.54.1.499. [DOI] [PubMed] [Google Scholar]
- 2.Ishihama A. Prokaryotic genome regulation: multifactor promoters, multitarget regulators and hierarchic networks. FEMS Microbiol Rev. 2010;34:628–45. doi: 10.1111/j.1574-6976.2010.00227.x. [DOI] [PubMed] [Google Scholar]
- 3.Gruber TM, Gross CA. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu Rev Microbiol. 2003;57:441–66. doi: 10.1146/annurev.micro.57.030502.090913. [DOI] [PubMed] [Google Scholar]
- 4.Paget MS, Helmann JD. The sigma70 family of sigma factors. Genome Biol. 2003;4:203. doi: 10.1186/gb-2003-4-1-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helmann JD. Anti-sigma factors. Curr Opin Microbiol. 1999;2:135–41. doi: 10.1016/S1369-5274(99)80024-1. [DOI] [PubMed] [Google Scholar]
- 6.Haugen SP, Ross W, Gourse RL. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat Rev Microbiol. 2008;6:507–19. doi: 10.1038/nrmicro1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saecker RM, Record MT, Jr., Dehaseth PL. Mechanism of bacterial transcription initiation: RNA polymerase - promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J Mol Biol. 2011;412:754–71. doi: 10.1016/j.jmb.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mekler V, Minakhin L, Severinov K. A critical role of downstream RNA polymerase-promoter interactions in the formation of initiation complex. J Biol Chem. 2011;286:22600–8. doi: 10.1074/jbc.M111.247080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mekler V, Kortkhonjia E, Mukhopadhyay J, Knight J, Revyakin A, Kapanidis AN, et al. Structural organization of bacterial RNA polymerase holoenzyme and the RNA polymerase-promoter open complex. Cell. 2002;108:599–614. doi: 10.1016/S0092-8674(02)00667-0. [DOI] [PubMed] [Google Scholar]
- 10.Chakraborty A, Wang D, Ebright YW, Korlann Y, Kortkhonjia E, Kim T, et al. Opening and closing of the bacterial RNA polymerase clamp. Science. 2012;337:591–5. doi: 10.1126/science.1218716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee DJ, Minchin SD, Busby SJ. Activating transcription in bacteria. Annu Rev Microbiol. 2012;66:125–52. doi: 10.1146/annurev-micro-092611-150012. [DOI] [PubMed] [Google Scholar]
- 12.Browning DF, Busby SJ. The regulation of bacterial transcription initiation. Nat Rev Microbiol. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- 13.Shah IM, Wolf RE., Jr. Novel protein--protein interaction between Escherichia coli SoxS and the DNA binding determinant of the RNA polymerase alpha subunit: SoxS functions as a co-sigma factor and redeploys RNA polymerase from UP-element-containing promoters to SoxS-dependent promoters during oxidative stress. J Mol Biol. 2004;343:513–32. doi: 10.1016/j.jmb.2004.08.057. [DOI] [PubMed] [Google Scholar]
- 14.Zafar MA, Sanchez-Alberola N, Wolf RE., Jr. Genetic evidence for a novel interaction between transcriptional activator SoxS and region 4 of the σ(70) subunit of RNA polymerase at class II SoxS-dependent promoters in Escherichia coli. J Mol Biol. 2011;407:333–53. doi: 10.1016/j.jmb.2010.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lima BP, Antelmann H, Gronau K, Chi BK, Becher D, Brinsmade SR, et al. Involvement of protein acetylation in glucose-induced transcription of a stress-responsive promoter. Mol Microbiol. 2011;81:1190–204. doi: 10.1111/j.1365-2958.2011.07742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nechaev S, Severinov K. Bacteriophage-induced modifications of host RNA polymerase. Annu Rev Microbiol. 2003;57:301–22. doi: 10.1146/annurev.micro.57.030502.090942. [DOI] [PubMed] [Google Scholar]
- 17.Nechaev S, Severinov K. The elusive object of desire--interactions of bacteriophages and their hosts. Curr Opin Microbiol. 2008;11:186–93. doi: 10.1016/j.mib.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hesselbach BA, Nakada D. “Host shutoff” function of bacteriophage T7: involvement of T7 gene 2 and gene 0.7 in the inactivation of Escherichia coli RNA polymerase. J Virol. 1977;24:736–45. doi: 10.1128/jvi.24.3.736-745.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hesselbach BA, Nakada D. Inactive complex formation between E. coli RNA polymerase and inhibitor protein purified from T7 phage infected cells. Nature. 1975;258:354–7. doi: 10.1038/258354a0. [DOI] [PubMed] [Google Scholar]
- 20.Hesselbach BA, Nakada D. I protein: bacteriophage T7-coded inhibitor of Escherichia coli RNA polymerase. J Virol. 1977;24:746–60. doi: 10.1128/jvi.24.3.746-760.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hesselbach BA, Yamada Y, Nakada D. Isolation of an inhibitor protein of E. coli RNA polymerase from T7 phage infected cell. Nature. 1974;252:71–4. doi: 10.1038/252071b0. [DOI] [PubMed] [Google Scholar]
- 22.Nechaev S, Severinov K. Inhibition of Escherichia coli RNA polymerase by bacteriophage T7 gene 2 protein. J Mol Biol. 1999;289:815–26. doi: 10.1006/jmbi.1999.2782. [DOI] [PubMed] [Google Scholar]
- 23.García LR, Molineux IJ. Rate of translocation of bacteriophage T7 DNA across the membranes of Escherichia coli. J Bacteriol. 1995;177:4066–76. doi: 10.1128/jb.177.14.4066-4076.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kemp P, Gupta M, Molineux IJ. Bacteriophage T7 DNA ejection into cells is initiated by an enzyme-like mechanism. Mol Microbiol. 2004;53:1251–65. doi: 10.1111/j.1365-2958.2004.04204.x. [DOI] [PubMed] [Google Scholar]
- 25.Zavriev SK, Shemyakin MF. RNA polymerase-dependent mechanism for the stepwise T7 phage DNA transport from the virion into E. coli. Nucleic Acids Res. 1982;10:1635–52. doi: 10.1093/nar/10.5.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chamberlin M, McGrath J, Waskell L. New RNA polymerase from Escherichia coli infected with bacteriophage T7. Nature. 1970;228:227–31. doi: 10.1038/228227a0. [DOI] [PubMed] [Google Scholar]
- 27.Savalia D, Robins W, Nechaev S, Molineux I, Severinov K. The role of the T7 Gp2 inhibitor of host RNA polymerase in phage development. J Mol Biol. 2010;402:118–26. doi: 10.1016/j.jmb.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qimron U, Kulczyk AW, Hamdan SM, Tabor S, Richardson CC. Inadequate inhibition of host RNA polymerase restricts T7 bacteriophage growth on hosts overexpressing udk. Mol Microbiol. 2008;67:448–57. doi: 10.1111/j.1365-2958.2007.06058.x. [DOI] [PubMed] [Google Scholar]
- 29.Chamberlin M. Isolation and characterization of prototrophic mutants of Escherichia coli unable to support the intracellular growth of T7. J Virol. 1974;14:509–16. doi: 10.1128/jvi.14.3.509-516.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LeClerc JE, Richardson CC. Gene 2 protein of bacteriophage T7: purification and requirement for packaging of T7 DNA in vitro. Proc Natl Acad Sci USA. 1979;76:4852–6. doi: 10.1073/pnas.76.10.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeWyngaert MA, Hinkle DC. Bacterial mutants affecting phage T7 DNA replication produce RNA polymerase resistant to inhibition by the T7 gene 2 protein. J Biol Chem. 1979;254:11247–53. [PubMed] [Google Scholar]
- 32.DeWyngaert MA, Hinkle DC. Characterization of the defects in bacteriophage T7 DNA synthesis during growth in the Escherichia coli mutant tsnB. J Virol. 1980;33:780–8. doi: 10.1128/jvi.33.2.780-788.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Center MS. Role of gene 2 in bacteriophage T7 DNA synthesis. J Virol. 1975;16:94–100. doi: 10.1128/jvi.16.1.94-100.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shadrin A, Sheppard C, Severinov K, Matthews S, Wigneshweraraj S. Substitutions in the Escherichia coli RNA polymerase inhibitor T7 Gp2 that allow inhibition of transcription when the primary interaction interface between Gp2 and RNA polymerase becomes compromised. Microbiology. 2012;158:2753–64. doi: 10.1099/mic.0.062547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klimuk E, Akulenko N, Makarova KS, Ceyssens PJ, Volchenkov I, Lavigne R, et al. Host RNA polymerase inhibitors encoded by ϕKMV-like phages of pseudomonas. Virology. 2013;436:67–74. doi: 10.1016/j.virol.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 36.Shadrin A, Sheppard C, Savalia D, Severinov K, Wigneshweraraj S. Overexpression of Escherichia coli udk mimics the absence of T7 Gp2 function and thereby abrogates successful infection by T7 phage. Microbiology. 2013;159:269–74. doi: 10.1099/mic.0.064527-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cámara B, Liu M, Reynolds J, Shadrin A, Liu B, Kwok K, et al. T7 phage protein Gp2 inhibits the Escherichia coli RNA polymerase by antagonizing stable DNA strand separation near the transcription start site. Proc Natl Acad Sci USA. 2010;107:2247–52. doi: 10.1073/pnas.0907908107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.James E, Liu M, Sheppard C, Mekler V, Cámara B, Liu B, et al. Structural and mechanistic basis for the inhibition of Escherichia coli RNA polymerase by T7 Gp2. Mol Cell. 2012;47:755–66. doi: 10.1016/j.molcel.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheppard C, Cámara B, Shadrin A, Akulenko N, Liu M, Baldwin G, et al. Inhibition of Escherichia coli RNAp by T7 Gp2 protein: role of negatively charged strip of amino acid residues in Gp2. J Mol Biol. 2011;407:623–32. doi: 10.1016/j.jmb.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Mekler V, Minakhin L, Sheppard C, Wigneshweraraj S, Severinov K. Molecular mechanism of transcription inhibition by phage T7 gp2 protein. J Mol Biol. 2011;413:1016–27. doi: 10.1016/j.jmb.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wigneshweraraj SR, Burrows PC, Nechaev S, Zenkin N, Severinov K, Buck M. Regulated communication between the upstream face of RNA polymerase and the beta’ subunit jaw domain. EMBO J. 2004;23:4264–74. doi: 10.1038/sj.emboj.7600407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murakami KS, Darst SA. Bacterial RNA polymerases: the wholo story. Curr Opin Struct Biol. 2003;13:31–9. doi: 10.1016/S0959-440X(02)00005-2. [DOI] [PubMed] [Google Scholar]