

Abstract
Introduction
Th17 cells, while indispensable in host defense, may play pathogenic roles in many autoimmune diseases including rheumatoid arthritis (RA). However, the mechanisms by which human Th17 cells drive autoimmunity have not been fully defined. We assessed the potential of the human Th17 CD4 T cell subset to induce expression of cell-cell interaction molecules and inflammatory mediators by fibroblast-like synoviocytes (FLS), and the roles of.IFN-γ and IL-17 in these interactions.
Methods
Th1 or Th17 cells were induced from healthy adult donor CD4 T cells and were co-cultured with FLS for 48 hours with/without neutralization of IFN-γ, IL-17A, or both. Alternatively, FLS were treated only with IFN-γ or IL-17 for 48 hours. FLS expression of CD40, CD54, and MHC-II, as well as IL-6 and IL-8 secretion were assessed by surface staining followed by flow cytometry and ELISA respectively.
Results
Both Th1 and Th17 cells secreted IL-17 as well as IFN-γ, although IFN-γ production was much greater from Th1 cells. FLS expression of CD40, CD54, and MHC-II significantly increased upon co-culture with either Th1 or Th17 cells, and was largely due to the IFN-γ secreted by the T cells. Both T cell subsets induced IL-6 and IL-8 secretion by RA FLS. Neutralization of IL-17A did not reduce FLS expression of CD40, MHC-II or CD54, but did inhibit IL-6 and IL-8 secretion. Although IFN-γ was a weak inducer of IL-6 secretion and significantly inhibited IL-8 secretion from FLS when used as a single stimulus, neutralization of IFN-γ inhibited induction of FLS secretion of both cytokines in Th17/FLS co-cultures. The effects of Th17 cells on FLS were not entirely accounted for by IL-17 and IFN-γ, suggesting roles for additional cytokines secreted by these cells.
Conclusion
FLS cell-cell interaction molecules and soluble inflammatory mediators are differentially regulated by IFN-γ and IL-17, cytokines that are secreted by both human Th1 and Th17 cells. The effects of IFN-γ may depend in part on the particular milieu of other co-existing cytokines and cell-cell interaction signals. The potential benefit of therapeutic neutralization of either IL-17 or IFN-γ could depend on the relative proportion of these cytokines in the synovial compartment of an RA patient. Suppression of the differentiation of Th17 cells may hold more therapeutic potential than neutralization of a single cytokine produced by CD4 T cells.
Keywords: Rheumatoid arthritis, T lymphocytes, Fibroblast-like synoviocytes, Pathogenicity, IFN-γ, IL-17, cell-cell interaction molecules, IL-6, IL-8, IL-4
Introduction
Th17 cells play indispensable roles in host defense against certain extra-cellular bacteria and fungi, but also play central pathogenic roles in many human autoimmune diseases including rheumatoid arthritis (RA)[1-3]. Th17 cells induce pro-inflammatory cytokines which further commit naïve CD4 T cells to the Th17 lineage[4-6]. Induction of chemokine or chemokine receptors facilitates lymphocyte and neutrophil recruitment to the inflamed joint[7]. Th17 cells promote bone resorption by inducing RANKL in osteoblasts, which interact with osteoclasts expressing RANK[8]. Th17 cells facilitate angiogenesis and tissue destruction through induction of vascular endothelial growth factor[9] and matrix metalloproteinases[10] respectively. All of these mechanisms are highly relevant to RA pathogenesis.
Controversy persists, however, as to whether RA is driven by Th1 immunity, Th17 immunity, or both in light of murine models implicating roles of IFN-γ versus IL-17 in the pathogenesis of arthritis[11, 12], increasing recognition of IL-17(+)IFN-γ(+) cells in several murine and human autoimmune diseases, including arthritis[13-15], and a study showing lack of increased frequency of Th17 cells in RA patients as well as significantly fewer Th17 cells in the synovial fluid than in peripheral blood in RA[16]. The answer to this question, which might depend on the stage of disease, has not yet been clarified.
What further complicates our view is that Th17 cells represent heterogeneous populations of helper T lymphocytes. It is not clear which subsets of human Th17 cells drive autoimmune disease, such as IL-17(+) IL-10(-) cells[17], IL-17(+)IFN-γ(+) cells[13, 14], IL-17(+)IL-21(+) cells, or IL-17(+)IL-22(+) cells[17]. A study from patients with juvenile idiopathic arthritis showed that human Th17 cells converted to IL-17(+)IFN-γ(+) cells in an environment mimicking the synovial space, namely low TGF-β and high IL-12[15]. In a murine model of inflammatory bowel disease (IBD), IL-17(+)IFN-γ(+) cells contribute to IL-23 dependent intestinal inflammation[13]. In experimental autoimmune encephalomyelitis, Th17 cells induced in the absence of TGF-β co-express higher T-bet and IFN-γ and are more pathogenic than conventional Th17 cells induced in the presence of TGF-β[14].
In dissecting the pathogenicity of Th17 cells in RA, co-culture of T cells and FLS is a convenient and relevant system. A strong physical association between these two cell types has been documented, which is mediated by cell-cell adhesion molecules, such as LFA-1-ICAM-1(CD54) interaction[18, 19] and CD2-LFA3 interaction[20]. T cells induce adhesion molecules, such as CD54 or VCAM-1 on FLS, which requires direct cell-cell contact[21]. Analogous to T cell-professional antigen presenting cell (APC) interactions, T cells and FLS in co-culture interact in both antigen dependent and independent systems. FLS express a significant amount of MHC-II in vivo that may be functionally significant in antigen presentation to T cells[22]. The interaction of Staphylococcal enterotoxin A with MHC-II on FLS results in IL-6 and IL-8 expression[23]. FLS can present superantigens to T cells, inducing a proliferative response[24]. FLS are able to take up and present arthritogenic peptide autoantigens to HLA-DR4 restricted T cell hybridomas[25]. Furthermore, type II collagen-specific T cells, when stimulated by antigen, demonstrate augmented potential to induce production of pro-inflammatory cytokines by FLS; TNF-α, IL-15, and IL-18[26] as well as chemokines; IL-8, MCP-1, and MIP-1α[27]. Such T cells have higher potential to secrete IFN-γ and IL-17 upon co-culture with FLS than resting T cells. All of these features are in part dependent on cell-cell contact as well as CD40 ligation[26, 27]. Finally, the B7 family co-stimulatory molecule B7H3 expressed by FLS can send both stimulatory and inhibitory signals to T cells depending on the activation status of T cells[28].
The existence of multiple effector subsets of CD4+ T cells adds further complexity to the potential interactions of T cells with FLS. IFN-γ, a signature Th1 cytokine with significant pro-inflammatory potential, that can also be secreted by human Th17 cells up-regulates FLS expression of MHC-II[19, 29, 30], CD40[31], and CD54[19, 31-33]. IL-17 induces IL-6 and IL-8 production by FLS[34] and also augments the synthesis of these cytokines induced by co-culture of FLS with T cells[35]. Despite such extensive insights regarding T cell-FLS interactions, potentially different pathogenic functions of each Th subset have not been well defined.
Contrary to the initial speculation that each lineage of helper T lymphocytes (Th cells) represents a fixed phenotype tightly linked to expression of a lineage-specific transcription factor, it is now clear that Th cells can be redirected to other lineages depending on the cytokine milieu. Th17 cells can be reprogrammed to Th1 cells upon exposure to IL-12, both in mice[36] and humans[37]. One of the most important questions concerning human Th17 cell plasticity would be whether pathogenic human Th17 cells can be reprogrammed to a potentially less pathogenic phenotype through exposure to opposing cytokines, such as IL-4 or IL-2. In mice, IL-4 and IL-2 phosphorylate Stat6 and Stat5 respectively, both of which inhibit IL-17 expression. Our laboratory has shown that in vitro generated murine Th17 cells lose IL-17 expression upon exposure to IL-4, but become resistant to regulation by IL-4 after 3 weeks of Th17 polarization[38]. Whether similar phenomena will be observed with human T cells has not been clarified to date.
Here, we define roles of IFN-γ and IL-17 in human Th17 cell pathogenic functions relevant to RA, in inducing cell-cell interaction molecules and IL-6 and IL-8 secretion by FLS. We further report that IL-4 regulates both IFN-γ and IL-17 synthesis by in-vitro induced human Th17 cell even after multiple rounds of Th17 polarization.
Methods
Induction of mature dendritic cells (DCs.)
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy adult donors by Ficoll gradient and were incubated at 37 degrees in RPMI culture media with 10% FCS, 1% Penicillin/Streptomycin, and 2% L-glutamine (all from Lonza) in 10 cm Petri-dish for 2 hours. The adherent cells were cultured for 5 days in RPMI culture media with 10% FCS, 1% Penicillin/Streptomycin, and 2% L-glutamine in the presence of GM-CSF (20 ng/ml) and IL-4 (20 ng/ml) (both from Peprotech), transferred to 6 well plates, and further activated in the presence of TNF-α (5 ng/ml)(R and D Systems), IL-1 (5 ng/ml) (Peprotech), IL-6 (150 ng/ml) (Peprotech), and prostaglandin E2 (1 μg/ml) (Sigma-Aldrich) for 24 hours.
Induction of human Th1 or Th17 cells
CD4 T cells were isolated from healthy adult donors using a human CD4 T cells enrichment kit (Stemcell Technologies), and were cultured in RPMI culture media with 10%FCS, 1% penicillin/streptomycin, and 2% L-glutamine for 5 days, either in the presence of plate bound anti-CD3 (OKT3, produced from a hybridoma line obtained from ATCC), soluble anti-CD28 (1 μg/ml), anti-IL-4 (5 μg/ml) (both from Biolegend), and IL-12 (5 μg/ml) (R and D Systems) to induce Th1 cells; or autologous mature dendritic cells, soluble anti-CD3 (1 μg/ml), anti-IFN-γ (5 μg/ml) (Biolegend), anti-IL-4 (5 μg/ml), anti-TGF-β (10 μg/ml) (clone 1D11 from a hybridoma line obtained from ATCC), IL-1 (10 ng/ml), IL-6 (20 ng/ml), and IL-23 (10 ng/ml) (eBioscience) to induce Th17 cells.
Assessment of T cell phenotype
IFN-γ or IL-17 in culture supernatants was measured by ELISA (BD Biosciences). Concurrently, after FcR blocking and surface staining with APC conjugated anti-CD4 (Biolegend), IL-17, IFN-γ, and IL-4 in the CD4(+) population were stained with FITC-conjugated anti-IL-17 (eBioscience) and PE-conjugated anti-IFN-γ (eBioscience) and IL-4 (Biolegend). Flow cytometry analysis was done by FACS Caliber (Beckman Dickinson) to assess expression of IL-17 versus IFN-γ by the CD4 T cells.
Preparation of FLS
All procedures involving specimens obtained from human subjects were performed under a protocol approved by the University of Michigan Institutional Review Board. Patient specimens were obtained following written informed consent. FLS were obtained by collagenase (Worthington) digestion of human synovial tissue obtained at arthroplasty or synovectomy from rheumatoid arthritis (RA) or osteoarthritis (OA) joints. All RA patients fulfilled the 1987 ACR criteria for the classification of RA[39]. The diagnosis of OA was based on characteristic clinical and radiographic features, and confirmed by pathological findings at joint surgery. Cells were maintained in CMRL media with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine. Passage 4-8 FLS were used for experiments. A total of 5 RA lines and 6 OA lines were used in these experiments.
Co-culture of Th1 or Th17 cells with FLS and assessment of FLS phenotype
Th1 or Th17 cells were co-cultured with passage 4-8 FLS from patients with RA or OA in CMRL culture medium with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine with/without neutralization of IFN-γ(10 μg/ml), IL-17 (10 μg/ml) (R and D Systems), or both. After 48 hours co-culture, supernatants were saved to measure IL-6 and IL-8 by ELISA (BD Biosciences). T cells were washed off with cold RPMI medium. The FLS were detached in 3mM EDTA, stained with APC-conjugated anti-CD90 and PE-conjugated anti-MHC-II, CD40, and CD54 (all from Biolegend). Flow cytometry was done to assess the expression of MHC-II, CD40, and CD54 by CD90(+) cells.
Stimulation of FLS with pro-inflammatory cytokines
Passage 4-8 FLS were cultured in CMRL medium with 10% FCS, 1% Penicillin/Streptomycin, and 2% L-glutamine in the presence of either IFN-γ (1, 10, and 100 ng/ml, or 0.5, 5 and 50 ng/ml) (Thermo Scientific) or IL-17 (1 ng/ml, 10 ng/ml, or 100 ng/ml) (eBioscience) for 48 hours. The FLS were stained with PE-conjugated anti-MHC-II, CD40, and CD54 followed by flow cytometry analysis. IL-6 and IL-8 in the supernatants were measured by ELISA.
Statistical analysis
The Student t-test was performed for all of the statistical analyses of cytokine secretion, with correction for multiple comparisons. The paired t-test was used for analysis of flow cytometry data that was aggregated from replicate experiments, with correction for multiple comparisons. Error bars shown in the figures represent the standard error of the mean.
Results
Induction of Th1 cells and Th17 cells from human CD4 T cells
As shown in Fig 1 upper panels, polarization under Th1 conditions induced a differentiated T cell population that contained, in a representative experiment, 56% IFN-γ (+) cells. Th17 polarizing conditions induced 11% IL-17(+) cells and 30% IFN-γ (+) cells. Notably, almost 80% of the IL-17+ cells co-expressed IFN-γ. Measurement of IL-17 in culture supernatants revealed comparable amounts of IL-17 in Th1 and Th17 conditions whereas Th1 conditions led to a significantly higher amount of IFN-γ secretion than did Th17 conditions (Th1: 2979965.7 +/- 8056.344 pg/ml, Th17: 2366.487 +/- 18.709 pg/ml) (Fig 1 lower panels). Thus, in vitro polarized human T cell subsets displayed a mixed phenotype with respect to cytokine secretion, but the IFN-γ/IL-17 ratio was much greater in the cytokine output of Th1 cells compared to Th17 cells. Moreover, flow cytometry underestimated the amount of IL-17 secreted by Th1 cells.
Fig 1. Induction of Th1 cells and Th17 cells from human CD4 T cells.
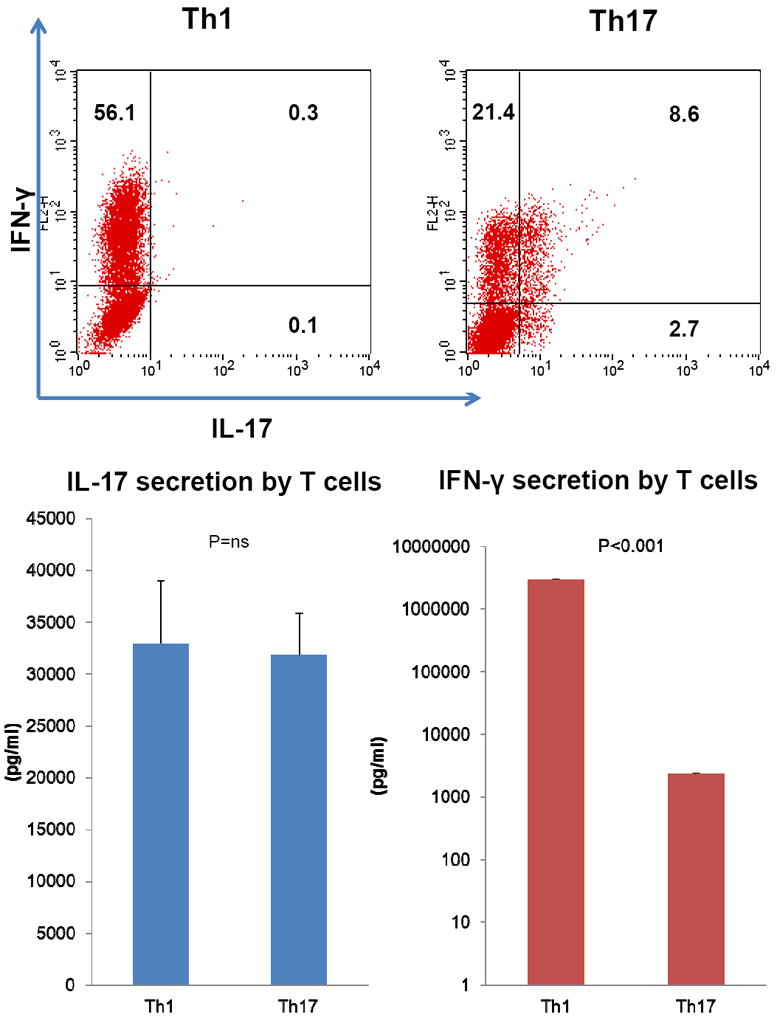
Human CD4 T cells were cultured for 5 days either under Th1 or Th17 polarizing conditions. Following FcR blocking, surface staining of CD4, and intracellular staining of IL-17 and IFN-γ, IL-17 (horizontal axis) versus IFN-γ expression (vertical axis) in the CD4(+) population was assessed by flow cytometry analysis. Representative data of three independent experiments is shown (Fig 1 upper panels). IFN-γ and IL-17 in both Th1 and Th17 polarizing cell culture supernatants were measured by ELISA (Fig 1 lower panels).
IFN-γ, but not IL-17, is contributory to human Th17 cell pathogenic potential in the induction of CD40, CD54, and MHC-II on FLS, and Th1 cells are more potent than Th17 cells in up-regulating these molecules
To gain insights into the roles of each pro-inflammatory cytokine in inducing these cell-cell interaction molecules on FLS, FLS were cultured with various concentrations of IFN-γ or IL-17 in the absence of T cells. As previously reported, IFN-γ significantly up-regulated the expression of CD40[31], CD54[19, 31-33], and MHC-II[29, 30] (Fig 2 and Supplemental Figure 1 upper panels). In contrast, IL-17 did not augment expression of any of these cell interaction molecules on FLS (Fig 2 and Supplemental Figure 1, lower panels).
Figure 2. IFN-γ, but not IL-17 induces CD40, CD54, and MHC-II on RA FLS.
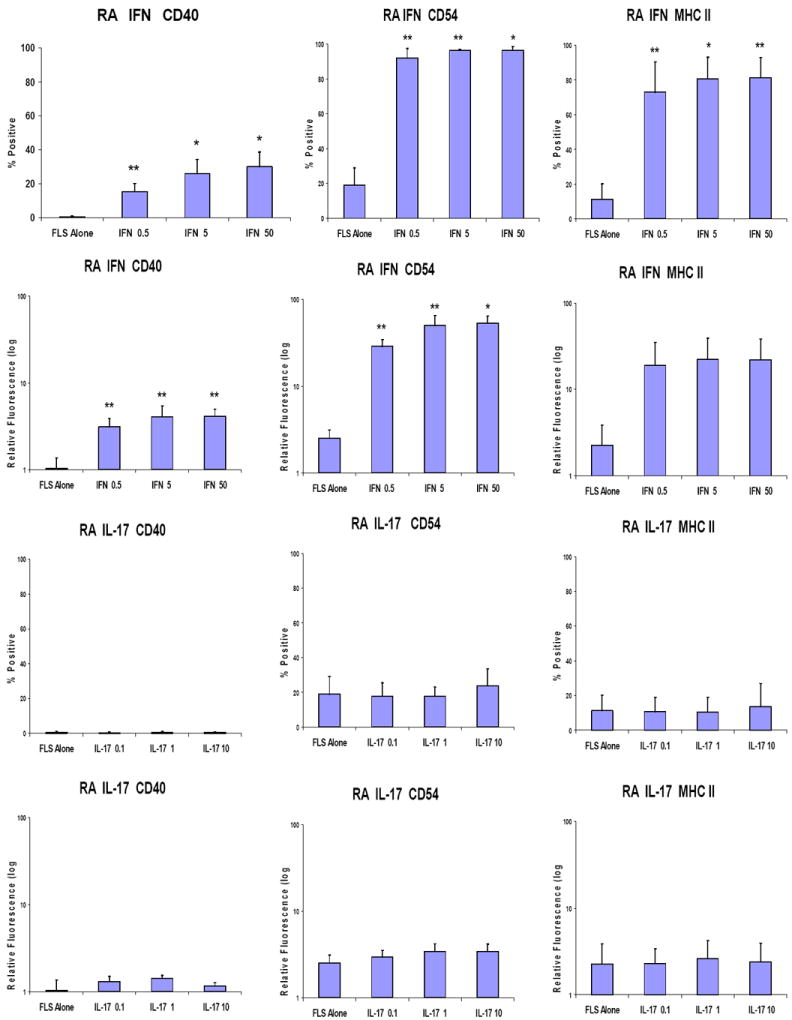
Passage 4-8 FLS from patients with RA were cultured in CMRL culture media with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine in the presence of either IFN-γ: 0.5 ng/ml, 5 ng/ml, or 50 ng/ml (upper panels) or IL-17: 0.1 ng/ml, 1 ng/ml, or 10 ng/ml (lower panels) for 48 hours. The FLS were stained with PE-conjugated anti-CD40, CD54, or MHC-II, followed by flow cytometry analysis. Combined results of 3 independent experiments are presented both by % expression above background or relative mean fluorescence. “FLS only” indicates FLS incubated for 48 hours without any cytokines. The data is expressed as the mean +/- standard error of the mean (** p<0.01, * p<0.05 by paired t testing compared to FLS only)
In co-culture experiments both Th17 and Th1 cells augmented expression of CD40, CD54 and MHC Class II on RA FLS (Figure 3) and OA FLS (data not shown). The magnitude of this effect was greater with Th1 cells, consistent with their higher production of IFN-γ. In co-cultures of RA FLS and Th17 cells, neutralization of IFN-γ reduced induction of CD40 and CD54 but no such effect was observed with neutralization of IL-17.
Figure 3. IFN-γ contributes to human Th17 cell pathogenic potential in inducing cell-cell interaction molecules on FLS.
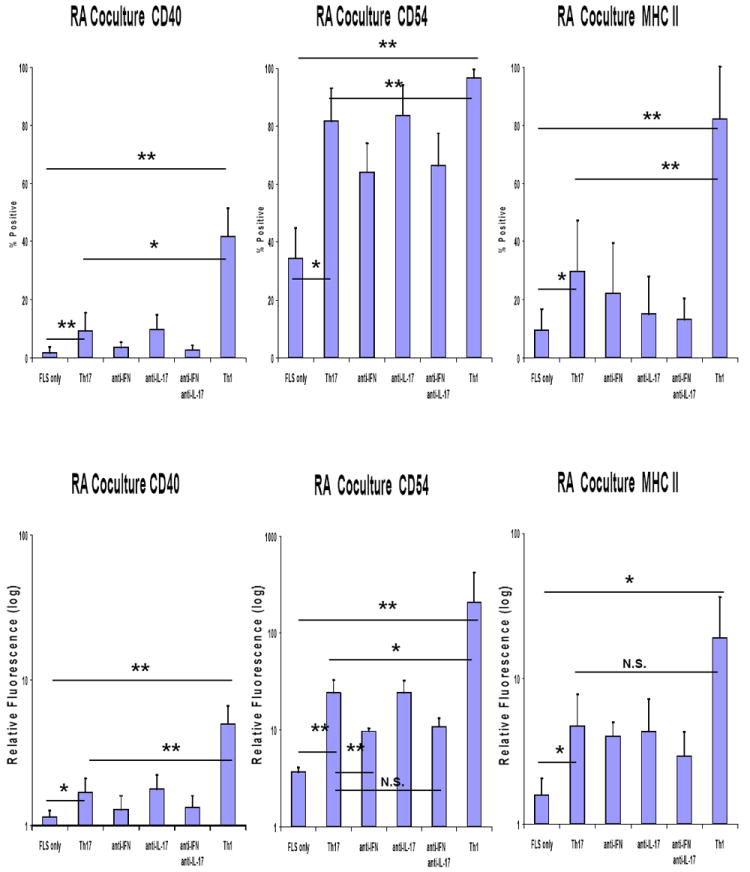
Human CD4 T cells cultured either under Th1 or Th17 polarizing condition were co-cultured with passage 4-8 FLS from patients with RA in CMRL culture medium with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine with/without neutralization of IFN-γ (10 μg/ml), IL-17 (10 μg/ml), or both. After 48 hours co-culture, T cells were washed off by cold RPMI medium. The FLS were detached by 3mM EDTA, stained with APC-conjugated anti-CD90 and PE-conjugated anti-CD40, CD54, and MHC-II. Flow cytometry was done to assess the expression of CD40, CD54, and MHC-II by CD90(+) cells. Combined results of 3 independent experiments are presented by % expression above background and by relative mean fluorescence. “FLS only” indicates FLS incubated for 48 hours without T cells. Error bars represent the standard error of the mean (** p<0.01, * p<0.05 by paired t testing).
Contributions of IL-17 and IFN-γ to IL-6 and IL-8 secretion in a Th17 cell-FLS co-culture system
Both IL-6 and IL-8 are secreted by FLS and play important roles in RA[40]. To gain insight as to the roles of T cell-derived pro-inflammatory cytokine in inducing IL-6 and IL-8, FLS were treated with various concentrations of IFN-γ or IL-17. As expected, IL-17 augmented both IL-6 and IL-8 secretion in a dose dependent manner (Fig 4 and Supplemental Figure 2). In contrast, IFN-γ augmented IL-6 secretion inconsistently and to only a modest extent and suppressed IL-8 secretion in a dose dependent fashion (Fig 4 and Supplemental Figure 2).
Figure 4. IL-17 induces both IL-6 and IL-8 expression while IFN-γ weakly induces IL-6 but suppresses IL-8 expression by FLS.
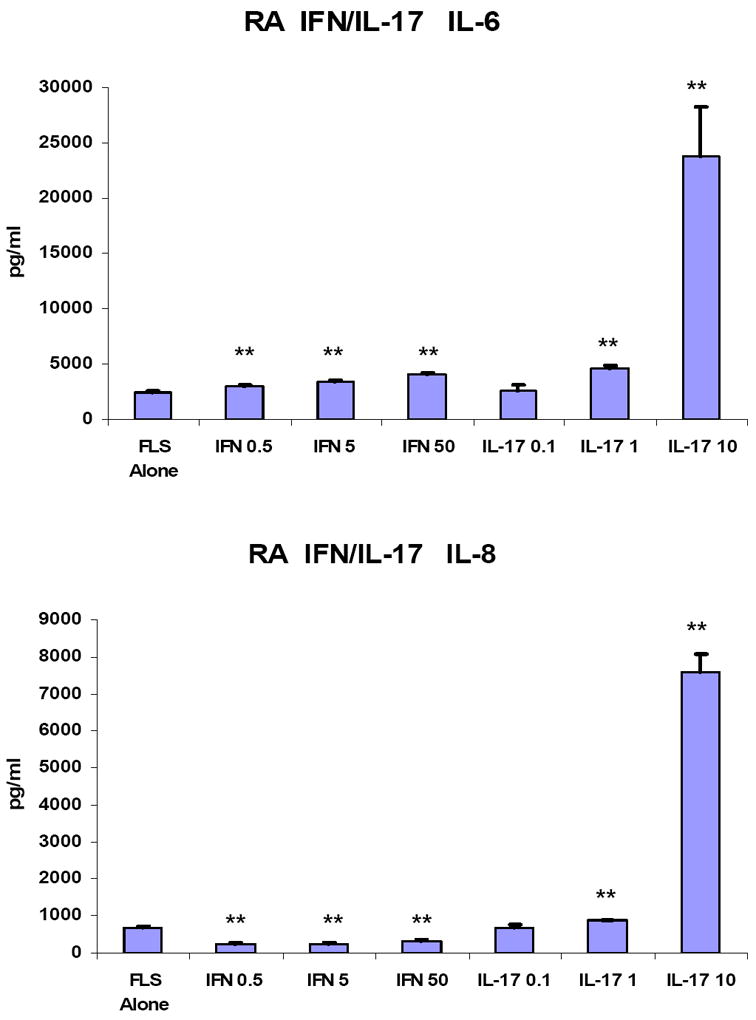
Passage 4-8 FLS from patients with RA were cultured in CMRL culture medium with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine in the presence of either IFN-γ (0.5 ng/ml, 5 ng/ml, or 50 ng/ml) or IL-17(0.1, 1 or 10 ng/ml) for 48 hours. IL-6 and IL-8 in the culture supernatants were measured by ELISA of triplicate wells. “FLS only” indicates FLS incubated for 48 hours without any cytokines. The results shown are from a single experiment representative of 3 identical experiments. Error bars are the standard error of the mean. Data was analyzed using the Student t test (**p<0.01 compared to FLS only).
In co-culture experiments, unexpected differences were observed between the responses of RA FLS versus OA FLS to Th17 cells. As expected, neutralization of IL-17 inhibited IL-6 and IL-8 secretion by the FLS (Figure 5). Unexpectedly, neutralization of IFN-γ also inhibited cytokine production by the RA FLS (Figure 5), but not the OA FLS (Supplemental Figure 3 and data not shown). These results suggest that the functional role of IFN-γ may be different in T cell interactions with RA compared to OA FLS, and may also be different in the context of other Th17 cytokines compared to when it is tested as a single stimulus.
Figure 5. IL-17 and IFN-γ contribute to IL-6 and IL-8 secretion in a Th17 cell-FLS co-culture system.
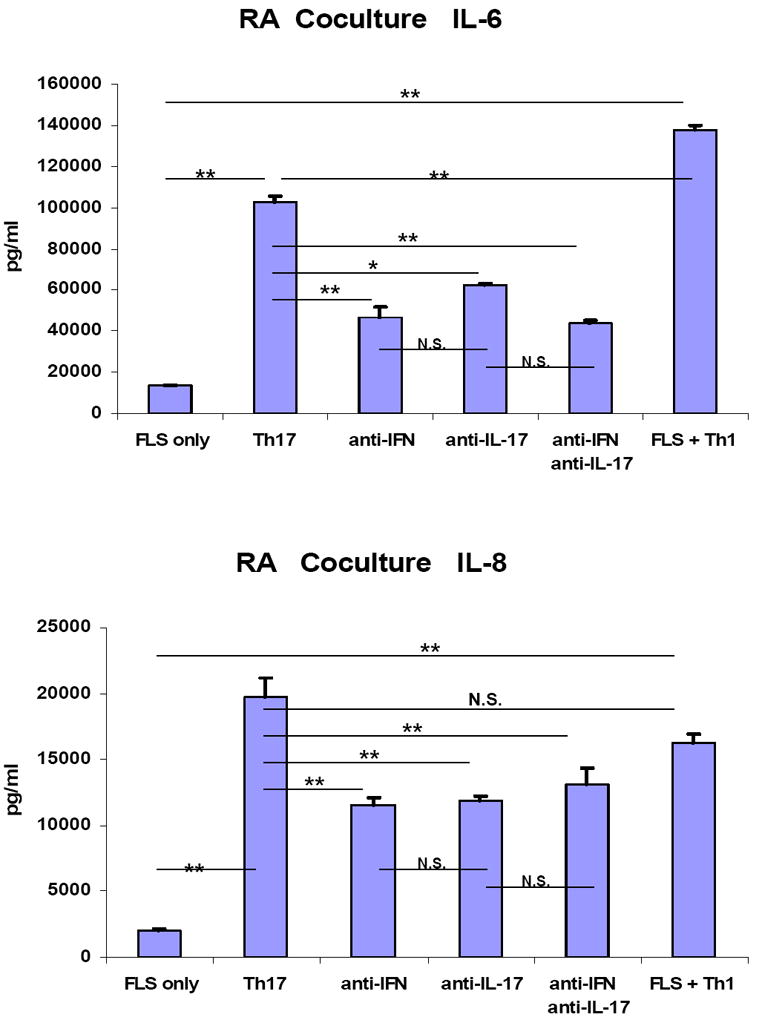
Human CD4 T cells cultured under Th17 polarizing condition were co-cultured with passage 4-8 FLS from patients with RA in CMRL culture medium with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine with/without neutralization of IFN-γ (10 μg/ml), IL-17(10 μg/ml), or both. After 48 hours co-culture, IL-6 and IL-8 in the co-culture supernatants were measured by ELISA from triplicate culture wells. Results of one experiment representative of 3 independent and identical experiments are presented. “FLS only” indicates FLS incubated for 48 hours without T cells. Error bars represent the standard error of the mean (* p<0.05, ** p<0.01, ns: not significant)
Moreover, in the co-culture system, IFN-γ neutralization did not augment IL-8 secretion as might be expected from the results in Fig 4 and Supplemental Figure 2, even from the OA FLS (Supplemental Figure 3). This may indicate that the inhibitory effect of IFN-γ on IL-8 secretion is negligible in the presence of an abundance of other IL-8 inducing pro-inflammatory mediators, such as IL-17. We confirmed that the amount of IL-6 and IL-8 secreted by T cells prior to the co-culture was negligible compared to the amount of cytokines detected in the co-culture supernatants, which further supports the inference that the major producers of IL-6 and IL-8 in this system were FLS (data not shown).
Discussion
Phenotypic features of human Th17 cells that drive autoimmune pathology have not been well defined. Specifically in RA, it has been a matter of debate whether IFN-γ or IL-17 drives synovial inflammation, given the presence of a substantial proportion of human Th17 cells co-expressing IFN-γ [15, 41] as well as animal studies showing discrepant findings concerning the roles of IFN-γ and IL-17 in the pathogenesis of arthritis[11, 12].
Understanding of the balance between IFN-γ and IL-17 is crucial to have a better insight into this paradigm. IFN-γ potently inhibits Th17 differentiation[42] through phosphorylation of Stat1. Whether the opposite is the case or not is controversial; however, IL-17 was shown to be protective in a mouse model of IBD, given that adoptive transfer of IL-17 deficient T cells was associated with a worse clinical outcome and higher IFN-γ expression in the inflamed colon, while IL-17 suppressed both T cell T-bet and IFN-γ expression in vitro[43].
In proteoglycan-induced arthritis (PGIA) in mice, although IFN-γ deficient mice develop less severe arthritis, they eventually succumb to arthritis[12]. Although IL-17 deficiency does not protect mice from PGIA, IFN-γ deficient or T bet deficient mice developed more severe PGIA than IFN-γ/IL-17 or T-bet/IL-17 double deficient mice[12]. In collagen induced arthritis (CIA), lack of IFN-γ signaling leads to severe disease. Neutralization of IFN-γ was beneficial at an earlier stage of CIA, but aggravated the disease at a later stage[11]. We have previously shown that neutralization of IFN-γ aggravated CIA, associated with increased IL-17 in the serum and joints. In this study, neutralization of IL-4 did not aggravate CIA unless IFN-γ was concomitantly neutralized[44]. One plausible interpretation of a series of such observations is that IL-17 mediated pathology is regulated by IFN-γ in PGIA and CIA.
Studies in other animal models of autoimmune diseases have provided important clues in dissecting the link between pathogenic Th1 and Th17 responses. Th17 cells are able to induce colitis upon transfer into immunodeficient mice, but many of them convert to Th1 cells[36]. Diabetogenic BDC2.5 CD4 T cells polarized in vitro to the Th17 phenotype lose IL-17 expression and express IFN-γ upon adoptive transfer into NOD-SCID mice, ultimately causing β-cell destruction and diabetes[45]. In humans, Th17 cells and IL-17(+)IFN-γ(+) cells from the inflamed joints in patients with juvenile idiopathic arthritis share distinct T cell receptor clonality[15], further supporting the conversion of Th17 cells into IL-17(+)IFN-γ(+) subset as a real phenomenon in view of the paucity of naïve CD4 T cell precursors in the inflamed joints. While Th17 to Th1 conversion has been reported in many murine and human autoimmune diseases as above, the opposite conversion has not been reported either in mice nor humans. This may be partly explained by epigenetic modifications of genes encoding the cytokines and transcription factors that are associated with each lineage. The T-bet locus in murine Th17 cells has both permissive and repressive histone modifications while both Rorc and Il17 loci in Th1 cells have exclusively repressive marks, which might support the concept that Th17 cells are poised to express IFN-γ whereas it is hard to induce IL-17 production in Th1 cells[46]. This notion is further supported by the observation that Th17 cells undergo dynamic epigenetic remodeling and acquire Th1 cell-like histone modifications across the ifng locus upon stimulation with IL-12[47]. Based on a series of such observations, we propose that an early stage of RA is primarily driven by Th17 cells, which gradually convert to Th17/Th1 cells and are finally replaced by Th1 cells during the progression of disease. Better understanding of such puzzling phenomena is directly relevant to defining a precise therapeutic window of either IL-17 or IFN-γ blockade in RA.
We herein report that the contribution of IFN-γ and IL-17 to activation of FLS by Th17 cells can vary depending on whether one is focusing on cell interaction molecule expression or soluble inflammatory mediator secretion; IFN-γ is essential for the former while IL-17 is more important for the latter. Expression of CD40, CD54 and Class II MHC are all likely crucial components of effective bidirectional T cell-FLS interaction. CD40 ligation augments proliferation, CD54 expression, and pro-inflammatory cytokine secretion by FLS[26, 27, 31]. In vivo, the importance of CD40 is further highlighted by the observation that a monoclonal antibody against CD40 ligand prevents the development of CIA, production of anti-collagen antibodies, synovial proliferation, and inflammatory cell infiltration to synovial tissues[48]. Except for cytokine activated T cells[49], CD54 plays a crucial role for robust T cell adherence to FLS in most systems studied to date[18, 19]. CD54 also plays a critical role in eliciting HLA-II restricted T cell responses[50]. As a result of such an efficient T cells-FLS interaction, T cells secrete TNF-α, IFN-γ, and IL-17, which in turn augment FLS secretion of IL-15, IL-6, and IL-8[51]. Membrane bound IL-15 on FLS stimulates T cells to secrete TNF-α, IFN-γ, and IL-17 in a cell contact dependent system, creating an auto-amplification loop of synovial inflammation[51]. Hence, the cell-cell interaction molecules and soluble inflammatory mediators both constitute a critical component of this inflammatory loop. Our observation that each component of FLS-T cell interactions is differentially regulated by potentially antagonistic T cell-derived cytokines, IFN-γ and IL-17, might indicate the presence of negative feedback systems to prevent unopposed inflammatory bursts mediated either by IFN-γ or IL-17.
The data in this report highlight the complexity of the interactions between FLS and T cell subsets. For example, IFN-γ directly inhibits IL-8 production by FLS, yet neutralization of IFN-γ in co-cultures of FLS with Th17 cells failed to augment IL-8 secretion. This apparently paradoxical result suggests that in the context of other pro-inflammatory cytokines, some of the regulatory potential of IFN-γ may be blocked or circumvented. Potentially, upregulation of FLS cell interaction molecules by IFN-γ augments the FLS response to IL-17. The data also indicate intrinsic differences in the responses of RA compared to OA FLS in co-culture with Th17 cells; neutralization studies suggested that RA compared to OA FLS appeared to be receiving a more positive signal from IFN-γ for IL-6 secretion. These FLS lines were derived from patients with established, generally late-stage disease, and could exhibit epigenetic changes resulting from long-standing disease activity. The FLS responses in the co-cultures could also be influenced by other cytokines that were not neutralized in these experiments, such as IL-17F and TNF.
Our data may have clinical implications, since it suggests that blocking either cytokine alone might be detrimental depending on the context, particularly the relative proportion of each cytokine in the inflammatory milieu. In an abundance of IFN-γ, neutralization of IL-17 is potentially detrimental as this could relieve potential IL-17 mediated inhibition of T cell IFN-γ expression[43] and further augment the IFN-γ induced CD40, CD54, and MHC II expression by FLS. In view of the potential for FLS to serve as antigen-presenting cells for T cell responses to superantigens and even arthritogenic autoantigens,[24, 25] augmented expression of these molecules by FLS could indirectly enhance the pro-inflammatory cytokine milieu in the RA synovium. Alternatively, IL-17 neutralization may be more beneficial in the presence of a paucity of IFN-γ provided that there is not excessive expression of CD40, CD54, and MHC-II by FLS. In contrast, in an abundance of IL-17, neutralization of IFN-γ is likely to be detrimental as this could abrogate IFN-γ mediated suppression of T cell IL-17 expression and further augment IL-17 induced IL-6 and IL-8 secretion by FLS. Taking into account these insights as well as our hypothesis regarding the dynamically changing roles of Th17 versus Th1 cells during progression of RA, neutralization of IL-17 and IFN-γ may be more beneficial at its earlier and later stages respectively. In the “intermediate” stage, when both Th1 and Th17 immunity may play indispensable roles, one might need to neutralize both. However, in the actual patient care setting, assessment of such a complex issue may not be practical.
Alternatively, modulation of both Th1 and Th17 cell activation might serve as a more feasible approach in efficiently redirecting the uncontrolled immune system to tolerance. From this standpoint, it is quite promising that IL-4 can regulate both IL-17 and IFN-γ responses in the CIA mode.[44, 52] However, Th17 cells can become resistant to regulation by IL-4 after repeated in vivo or in vitro stimulation in mouse systems [38, 52] In preliminary experiments we have found that IL-4 was able to reverse both IFN-γ and IL-17 synthesis by human Th17 cells even after two rounds of Th17 polarization in vitro (data not shown).
While IL-23 is important in human Th17 differentiation[53, 54], it is plausible that IL-23 also plays an important role in stabilizing human Th17 lineage commitment given that the IL-23 receptor is induced in a Stat3[55] and ROR-γt[56] dependent manner, but not in naïve CD4 T cells, in mice. Of note, IL-23 renders murine Th17 cells resistant to regulation by one of the Th17-opposing cytokines, IL-27[57]. However, our initial results indicate that the regulatory effect of IL-4 was robust regardless of the concentrations of IL-23 during the Th17 polarization (data not shown). This suggests that IL-4 could serve as a potent immune-regulatory cytokine in Th17 (or Th17/Th1) driven autoimmune diseases such as RA. This concept is consistent with our previous observation that DCs genetically engineered to express IL-4 suppress both T cell IL-17 synthesis and collagen induced arthritis [52, 58]. However, much more work is required to examine whether IL-4 resistant Th17 cells exist in human immune-driven diseases such as RA.
In assessing the sensitivity of Th17 cells to regulatory cytokines, expression of transcription factors that control function of these cells is a critical issue. Stat3 and ROR-γt are both crucial transcription factors involved in murine Th17 differentiation. Stat3 induces ROR-γt[59]. Stat3 and ROR-γt cooperate with each other [55] and bind to the IL-17 promoter to induce IL-17 expression[60, 61]. Whether stability of these molecular interactions dynamically changes with increasing Th17 cell differentiation and ultimate senescence is an intriguing issue for further exploration.
Conclusions
Studies of T cell interactions with FLS have revealed that human Th17 cells secrete functionally significant amounts of IFN-γ as well as IL-17A. Two relevant components of synovial inflammation, cell-cell interaction molecules expressed by FLS and pro-inflammatory cytokines secreted by FLS, are differentially regulated by these potentially antagonistic T cell cytokines; IFN-γ is important for the former while IL-17 is more important for the latter. It is likely that other cytokines produced by Th17 and/or Th1 cells also contribute to the response of FLS, including IL-17F, TNF and GM-CSF. The complex balance and multiple effects of T cell cytokines on FLS and other cells in RA synovium create uncertainty regarding the potential benefit of blocking a single T cell-derived cytokine in the treatment of RA.
Supplementary Material
Passage 4-8 FLS from patients with OA were cultured in CMRL culture media with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine in the presence of either IFN-γ: 0.5 ng/ml, 5 ng/ml, or 50 ng/ml (upper panels) or IL-17: 0.1 ng/ml, 1 ng/ml, or 10 ng/ml (lower panels) for 48 hours. The FLS were stained with PE-conjugated anti-CD40, CD54, or MHC-II, followed by flow cytometry analysis. Combined results of 3 independent experiments are presented both by % expression above background or relative mean fluorescence. “FLS only” indicates FLS incubated for 48 hours without any cytokines. The data is expressed as the mean +/- standard error of the mean (** p<0.01, * p<0.05 by paired t testing compared to FLS only)
Passage 4-8 FLS from patients with OA were cultured in CMRL culture medium with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine in the presence of either IFN-γ (0.5 ng/ml, 5 ng/ml, or 50 ng/ml) or IL-17(0.1, 1 or 10 ng/ml in 2A and 1, 10, or 100 ng/ml in 2B) for 48 hours. IL-6 and IL-8 in the culture supernatants were measured by ELISA of triplicate wells. “FLS only” indicates FLS incubated for 48 hours without any cytokines. The results shown are from a single experiment representative of 3 identical experiments (2A), or from 2 independent and identical experiments (2B). Error bars are the standard error of the mean. Data was analyzed using the Student t test (* p<0.05 in 2A and <0.001 in 2B; **p<0.01 in 2A compared to FLS only).
Human CD4 T cells cultured under Th17 polarizing condition were co-cultured with passage 4-8 FLS from patients with OA in CMRL culture medium with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine with/without neutralizing antibodies against IFN-γ (10 μg/ml), IL-17(10 μg/ml), or both. After 48 hours co-culture, IL-6 and IL-8 in the co-culture supernatants were measured by ELISA from triplicate wells. Results of two independent experiments using CD4 T cells from two different human subjects are presented. “FLS only” indicates FLS incubated for 48 hours without T cells. (* p<0.001, ** p<0.01, *** p<0.05, ns: not significant).
Acknowledgments
We would like to thank Ms. Donna Cash for assisting with manuscript preparation. This work was supported by NIH grant AR38477 and by the University of Michigan Rheumatic Diseases Research Core Center.
List of Abbreviations
- APC
antigen presenting cell
- CIA
collagen-induced arthritis
- EDTA
Ethylenediaminetetraacetic acid
- ELISA
enzyme-linked immunosorbent assay
- FACS
fluorescence-activated cell sorting
- FcR
Fc receptor
- FCS
fetal calf serum
- FITC
fluorescein isothyocyanate
- FLS
fibroblast-like synoviocytes
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- HLA-DR4
human leukocyte antigen-DR4
- IBD
inflammatory bowel disease
- ICAM-1
intercellular adhesion molecule -1
- IFN-γ
Interferon gamma
- IL-6
Interleukin 6
- IL-8
Interleukin 8
- IL-17
Interleukin 17
- LFA-1
lymphocyte function-associated antigen-1
- LFA3
lymphocyte function-associated antigen-3
- MCP-1
monocyte chemotactic protein-1
- MHC-II
Major histocompatibility Complex II
- MIP-1α
macrophage inflammatory protein-1
- NOD-SCID mice
non obese diabetic-severe combined immunodeficiency disease
- OA
osteoarthritis
- PBMC
peripheral blood mononuclear cells
- PE
phycoerythrin
- PGIA
proteoglycan-induced arthritis
- RA
rheumatoid arthritis
- RANKL
receptor activator of nuclear factor kappa-B ligand
- RANK
receptor activator of nuclear factor kappa-B
- Rorc
transcription factor retinoic acid-related orphan receptor gt
- ROR-γt
transcription factor retinoic acid-related orphan receptor gt
- T-bet
transcription factor T box expressed in t cells
- Th cells
helper T lymphocytes
- Th1
T helper 1
- Th17
T helper 17
- VCAM-1
vascular cell adhesion molecule-1
Footnotes
Authors’ contributions
HK and JE performed the experiments and analyzed the data. HK prepared the initial draft of the manuscript. DAF reviewed the experimental design and data, and prepared the final version of the revised manuscript.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Hiroshi Kato, Email: katobonz@hotmail.com, Rheumatology Fellow, University of Michigan, Ann Arbor, MI.
Judith Endres, Email: jendres@med.umich.edu, Research Associate, University of Michigan, Ann Arbor, MI.
David A. Fox, Professor of Internal Medicine, Division of Rheumatology, University of Michigan, Ann Arbor, MI
References
- 1.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 2.Bettelli E, Korn T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol. 2007;19:652–657. doi: 10.1016/j.coi.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Curr Opin Immunol. 2007;19:281–286. doi: 10.1016/j.coi.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 6.Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, Armitage RJ. Human IL-17: a novel cytokine derived from T cells. J Immunol. 1995;155:5483–5486. [PubMed] [Google Scholar]
- 7.Lundy SK, Sarkar S, Tesmer LA, Fox DA. Cells of the synovium in rheumatoid arthritis. T lymphocytes. Arthritis Res Ther. 2007;9:202. doi: 10.1186/ar2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Page G, Miossec P. RANK and RANKL expression as markers of dendritic cell-T cell interactions in paired samples of rheumatoid synovium and lymph nodes. Arthritis Rheum. 2005;52:2307–2312. doi: 10.1002/art.21211. [DOI] [PubMed] [Google Scholar]
- 9.Ryu S, Lee JH, Kim SI. IL-17 increased the production of vascular endothelial growth factor in rheumatoid arthritis synoviocytes. Clin Rheumatol. 2006;25:16–20. doi: 10.1007/s10067-005-1081-1. [DOI] [PubMed] [Google Scholar]
- 10.Sylvester J, Liacini A, Li WQ, Zafarullah M. Interleukin-17 signal transduction pathways implicated in inducing matrix metalloproteinase-3, -13 and aggrecanase-1 genes in articular chondrocytes. Cell Signal. 2004;16:469–476. doi: 10.1016/j.cellsig.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Boissier MC, Chiocchia G, Bessis N, Hajnal J, Garotta G, Nicoletti F, Fournier C. Biphasic effect of interferon-gamma in murine collagen-induced arthritis. Eur J Immunol. 1995;25:1184–1190. doi: 10.1002/eji.1830250508. [DOI] [PubMed] [Google Scholar]
- 12.Doodes PD, Cao Y, Hamel KM, Wang Y, Rodeghero RL, Mikecz K, Glant TT, Iwakura Y, Finnegan A. IFN-gamma regulates the requirement for IL-17 in proteoglycan-induced arthritis. J Immunol. 2010;184:1552–1559. doi: 10.4049/jimmunol.0902907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, Powrie F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279–288. doi: 10.1016/j.immuni.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, Evans JG, Cimaz R, Bajaj-Elliott M, Wedderburn LR. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci U S A. 2010;107:14751–14756. doi: 10.1073/pnas.1003852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, Hori A, Iwamoto Y, Yoshikai Y. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis. 2008;67:1299–1304. doi: 10.1136/ard.2007.080341. [DOI] [PubMed] [Google Scholar]
- 17.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 18.Nakatsuka K, Tanaka Y, Hubscher S, Abe M, Wake A, Saito K, Morimoto I, Eto S. Rheumatoid synovial fibroblasts are stimulated by the cellular adhesion to T cells through lymphocyte function associated antigen-1/intercellular adhesion molecule-1. J Rheumatol. 1997;24:458–464. [PubMed] [Google Scholar]
- 19.Krzesicki RF, Fleming WE, Winterrowd GE, Hatfield CA, Sanders ME, Chin JE. T lymphocyte adhesion to human synovial fibroblasts. Role of cytokines and the interaction between intercellular adhesion molecule 1 and CD11a/CD18. Arthritis Rheum. 1991;34:1245–1253. doi: 10.1002/art.1780341007. [DOI] [PubMed] [Google Scholar]
- 20.Haynes BF, Grover BJ, Whichard LP, Hale LP, Nunley JA, McCollum DE, Singer KH. Synovial microenvironment-T cell interactions. Human T cells bind to fibroblast-like synovial cells in vitro. Arthritis Rheum. 1988;31:947–955. doi: 10.1002/art.1780310802. [DOI] [PubMed] [Google Scholar]
- 21.Bombara MP, Webb DL, Conrad P, Marlor CW, Sarr T, Ranges GE, Aune TM, Greve JM, Blue ML. Cell contact between T cells and synovial fibroblasts causes induction of adhesion molecules and cytokines. J Leukoc Biol. 1993;54:399–406. doi: 10.1002/jlb.54.5.399. [DOI] [PubMed] [Google Scholar]
- 22.Zimmermann T, Kunisch E, Pfeiffer R, Hirth A, Stahl HD, Sack U, Laube A, Liesaus E, Roth A, Palombo-Kinne E, et al. Isolation and characterization of rheumatoid arthritis synovial fibroblasts from primary culture--primary culture cells markedly differ from fourth-passage cells. Arthritis Res. 2001;3:72–76. doi: 10.1186/ar142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mourad W, Mehindate K, Schall TJ, McColl SR. Engagement of major histocompatibility complex class II molecules by superantigen induces inflammatory cytokine gene expression in human rheumatoid fibroblast-like synoviocytes. J Exp Med. 1992;175:613–616. doi: 10.1084/jem.175.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai C, Diaz LA, Jr, Singer NG, Li LL, Kirsch AH, Mitra R, Nickoloff BJ, Crofford LJ, Fox DA. Responsiveness of human T lymphocytes to bacterial superantigens presented by cultured rheumatoid arthritis synoviocytes. Arthritis Rheum. 1996;39:125–136. doi: 10.1002/art.1780390117. [DOI] [PubMed] [Google Scholar]
- 25.Tran CN, Davis MJ, Tesmer LA, Endres JL, Motyl CD, Smuda C, Somers EC, Chung KC, Urquhart AG, Lundy SK, et al. Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum. 2007;56:1497–1506. doi: 10.1002/art.22573. [DOI] [PubMed] [Google Scholar]
- 26.Cho ML, Yoon CH, Hwang SY, Park MK, Min SY, Lee SH, Park SH, Kim HY. Effector function of type II collagen-stimulated T cells from rheumatoid arthritis patients: cross-talk between T cells and synovial fibroblasts. Arthritis Rheum. 2004;50:776–784. doi: 10.1002/art.20106. [DOI] [PubMed] [Google Scholar]
- 27.Min DJ, Cho ML, Lee SH, Min SY, Kim WU, Min JK, Park SH, Cho CS, Kim HY. Augmented production of chemokines by the interaction of type II collagen-reactive T cells with rheumatoid synovial fibroblasts. Arthritis Rheum. 2004;50:1146–1155. doi: 10.1002/art.20133. [DOI] [PubMed] [Google Scholar]
- 28.Tran CN, Thacker SG, Louie DM, Oliver J, White PT, Endres JL, Urquhart AG, Chung KC, Fox DA. Interactions of T cells with fibroblast-like synoviocytes: role of the B7 family costimulatory ligand B7-H3. J Immunol. 2008;180:2989–2998. doi: 10.4049/jimmunol.180.5.2989. [DOI] [PubMed] [Google Scholar]
- 29.Chin JE, Winterrowd GE, Krzesicki RF, Sanders ME. Role of cytokines in inflammatory synovitis. The coordinate regulation of intercellular adhesion molecule 1 and HLA class I and class II antigens in rheumatoid synovial fibroblasts. Arthritis Rheum. 1990;33:1776–1786. doi: 10.1002/art.1780331204. [DOI] [PubMed] [Google Scholar]
- 30.Amento EP, Bhan AK, McCullagh KG, Krane SM. Influences of gamma interferon on synovial fibroblast-like cells. Ia induction and inhibition of collagen synthesis. J Clin Invest. 1985;76:837–848. doi: 10.1172/JCI112041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yellin MJ, Winikoff S, Fortune SM, Baum D, Crow MK, Lederman S, Chess L. Ligation of CD40 on fibroblasts induces CD54 (ICAM-1) and CD106 (VCAM-1) up-regulation and IL-6 production and proliferation. J Leukoc Biol. 1995;58:209–216. doi: 10.1002/jlb.58.2.209. [DOI] [PubMed] [Google Scholar]
- 32.Morales-Ducret J, Wayner E, Elices MJ, Alvaro-Gracia JM, Zvaifler NJ, Firestein GS. Alpha 4/beta 1 integrin (VLA-4) ligands in arthritis. Vascular cell adhesion molecule-1 expression in synovium and on fibroblast-like synoviocytes. J Immunol. 1992;149:1424–1431. [PubMed] [Google Scholar]
- 33.Schlaak JF, Schwarting A, Knolle P, Meyer zum Buschenfelde KH, Mayet W. Effects of Th1 and Th2 cytokines on cytokine production and ICAM-1 expression on synovial fibroblasts. Ann Rheum Dis. 1995;54:560–565. doi: 10.1136/ard.54.7.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hwang SY, Kim JY, Kim KW, Park MK, Moon Y, Kim WU, Kim HY. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. 2004;6:R120–128. doi: 10.1186/ar1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamura Y, Gupta R, Morita Y, He X, Pai R, Endres J, Freiberg A, Chung K, Fox DA. Effector function of resting T cells: activation of synovial fibroblasts. J Immunol. 2001;166:2270–2275. doi: 10.4049/jimmunol.166.4.2270. [DOI] [PubMed] [Google Scholar]
- 36.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooney LA, Towery KL, Endres J, Fox DA. Sensitivity and resistance to regulation by IL-4 during Th17 maturation. The Journal of Immunology. 2011 doi: 10.4049/jimmunol.1002860. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 40.Harris ED., Jr Rheumatoid arthritis. Pathophysiology and implications for therapy. N Engl J Med. 1990;322:1277–1289. doi: 10.1056/NEJM199005033221805. [DOI] [PubMed] [Google Scholar]
- 41.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 42.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 43.O’Connor W, Jr, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, Kolls JK, Flavell RA. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarkar S, Cooney LA, White P, Dunlop DB, Endres J, Jorns JM, Wasco MJ, Fox DA. Regulation of pathogenic IL-17 responses in collagen-induced arthritis: roles of endogenous interferon-gamma and IL-4. Arthritis Res Ther. 2009;11:R158. doi: 10.1186/ar2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bending D, De La Pena H, Veldhoen M, Phillips JM, Uyttenhove C, Stockinger B, Cooke A. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Invest. 2009 doi: 10.1172/JCI37865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mukasa R, Balasubramani A, Lee YK, Whitley SK, Weaver BT, Shibata Y, Crawford GE, Hatton RD, Weaver CT. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity. 2010;32:616–627. doi: 10.1016/j.immuni.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Durie FH, Fava RA, Foy TM, Aruffo A, Ledbetter JA, Noelle RJ. Prevention of collagen-induced arthritis with an antibody to gp39, the ligand for CD40. Science. 1993;261:1328–1330. doi: 10.1126/science.7689748. [DOI] [PubMed] [Google Scholar]
- 49.Tran CN, Lundy SK, White PT, Endres JL, Motyl CD, Gupta R, Wilke CM, Shelden EA, Chung KC, Urquhart AG, Fox DA. Molecular interactions between T cells and fibroblast-like synoviocytes: role of membrane tumor necrosis factor-alpha on cytokine-activated T cells. Am J Pathol. 2007;171:1588–1598. doi: 10.2353/ajpath.2007.070004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altmann DM, Hogg N, Trowsdale J, Wilkinson D. Cotransfection of ICAM-1 and HLA-DR reconstitutes human antigen-presenting cell function in mouse L cells. Nature. 1989;338:512–514. doi: 10.1038/338512a0. [DOI] [PubMed] [Google Scholar]
- 51.Miranda-Carus ME, Balsa A, Benito-Miguel M, Perez de Ayala C, Martin-Mola E. IL-15 and the initiation of cell contact-dependent synovial fibroblast-T lymphocyte cross-talk in rheumatoid arthritis: effect of methotrexate. J Immunol. 2004;173:1463–1476. doi: 10.4049/jimmunol.173.2.1463. [DOI] [PubMed] [Google Scholar]
- 52.Sarkar S, Tesmer LA, Hindnavis V, Endres JL, Fox DA. Interleukin-17 as a molecular target in immune-mediated arthritis: immunoregulatory properties of genetically modified murine dendritic cells that secrete interleukin-4. Arthritis Rheum. 2007;56:89–100. doi: 10.1002/art.22311. [DOI] [PubMed] [Google Scholar]
- 53.Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 54.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 56.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 57.Diveu C, McGeachy MJ, Boniface K, Stumhofer JS, Sathe M, Joyce-Shaikh B, Chen Y, Tato CM, McClanahan TK, de Waal Malefyt R, et al. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J Immunol. 2009;182:5748–5756. doi: 10.4049/jimmunol.0801162. [DOI] [PubMed] [Google Scholar]
- 58.Morita Y, Yang J, Gupta R, Shimizu K, Shelden EA, Endres J, Mule JJ, McDonagh KT, Fox DA. Dendritic cells genetically engineered to express IL-4 inhibit murine collagen-induced arthritis. J Clin Invest. 2001;107:1275–1284. doi: 10.1172/JCI11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 60.Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O’Shea JJ. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. 2008;9:1297–1306. doi: 10.1038/ni.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Passage 4-8 FLS from patients with OA were cultured in CMRL culture media with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine in the presence of either IFN-γ: 0.5 ng/ml, 5 ng/ml, or 50 ng/ml (upper panels) or IL-17: 0.1 ng/ml, 1 ng/ml, or 10 ng/ml (lower panels) for 48 hours. The FLS were stained with PE-conjugated anti-CD40, CD54, or MHC-II, followed by flow cytometry analysis. Combined results of 3 independent experiments are presented both by % expression above background or relative mean fluorescence. “FLS only” indicates FLS incubated for 48 hours without any cytokines. The data is expressed as the mean +/- standard error of the mean (** p<0.01, * p<0.05 by paired t testing compared to FLS only)
Passage 4-8 FLS from patients with OA were cultured in CMRL culture medium with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine in the presence of either IFN-γ (0.5 ng/ml, 5 ng/ml, or 50 ng/ml) or IL-17(0.1, 1 or 10 ng/ml in 2A and 1, 10, or 100 ng/ml in 2B) for 48 hours. IL-6 and IL-8 in the culture supernatants were measured by ELISA of triplicate wells. “FLS only” indicates FLS incubated for 48 hours without any cytokines. The results shown are from a single experiment representative of 3 identical experiments (2A), or from 2 independent and identical experiments (2B). Error bars are the standard error of the mean. Data was analyzed using the Student t test (* p<0.05 in 2A and <0.001 in 2B; **p<0.01 in 2A compared to FLS only).
Human CD4 T cells cultured under Th17 polarizing condition were co-cultured with passage 4-8 FLS from patients with OA in CMRL culture medium with 10% FCS, 1% penicillin/streptomycin, and 2% L-glutamine with/without neutralizing antibodies against IFN-γ (10 μg/ml), IL-17(10 μg/ml), or both. After 48 hours co-culture, IL-6 and IL-8 in the co-culture supernatants were measured by ELISA from triplicate wells. Results of two independent experiments using CD4 T cells from two different human subjects are presented. “FLS only” indicates FLS incubated for 48 hours without T cells. (* p<0.001, ** p<0.01, *** p<0.05, ns: not significant).